New Perspectives on the Use of Cannabis in the Treatment of Psychiatric Disorders

 ,
, 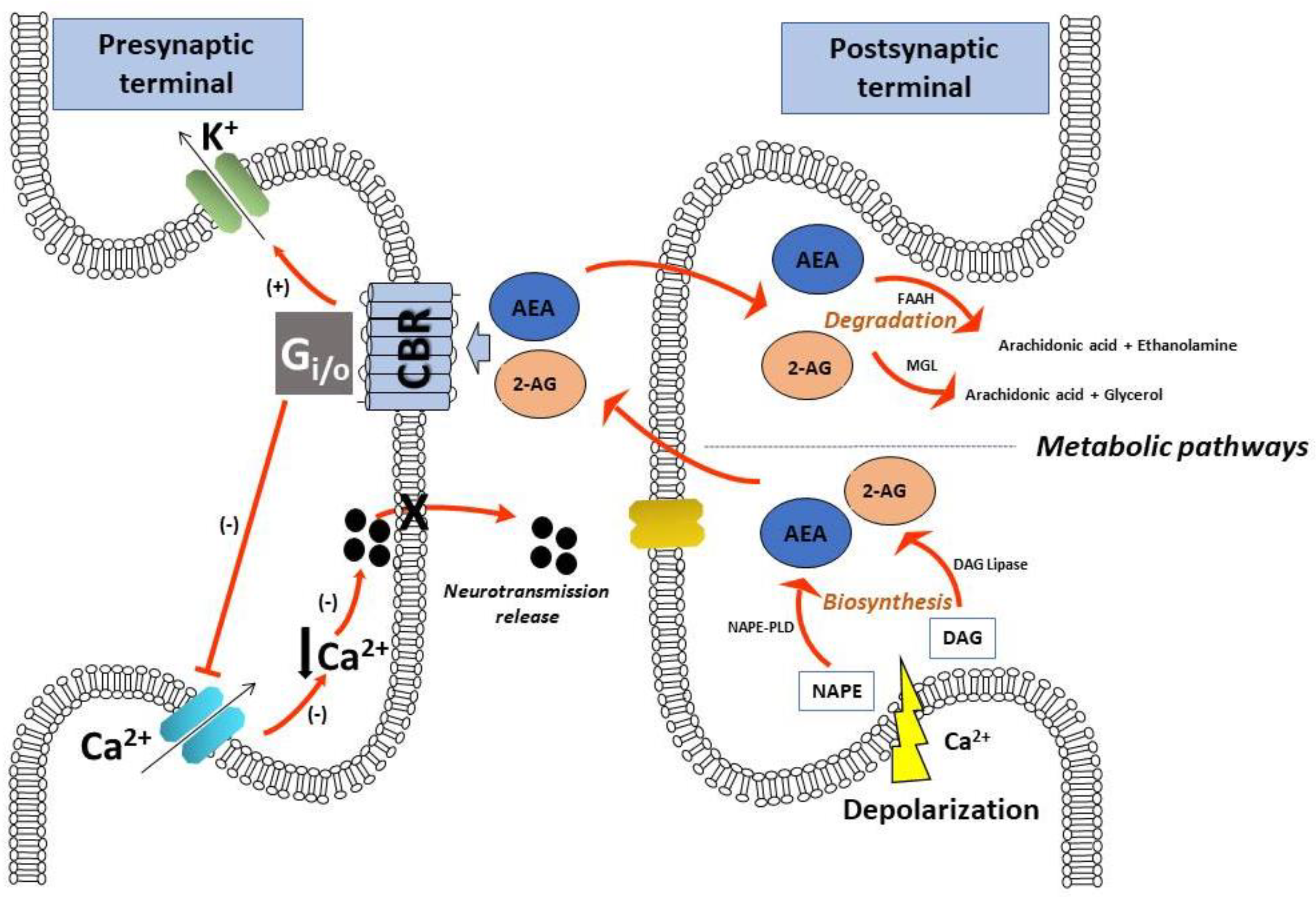{kind=link}
Abstract
1. Introduction
2. Search Methods
3. Therapeutic Potential of Cannabis Extracts on Psychiatric Disorders
3.1. Schizophrenia
3.1.1. Human Studies
3.1.2. Animal Studies
3.2. Anxiety Disorders
3.2.1. Human Studies
3.2.2. Animal Studies
3.3. Depression
3.3.1. Human Studies
3.3.2. Animal Studies
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Iversen, L.L. The Science of Marijuana, 2nd ed.; Oxford University Press: Oxford, UK, 2007; ISBN 978-0-19-532824-0. [Google Scholar]
- Zuardi, A.W. History of cannabis as a medicine: A review. Rev. Bras. Psiquiatr. 2006, 28, 153–157. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, W.B. New Remedy for Tetanus and Other Convulsive Disorders. Boston Med. Surg. J. 1840, 23, 153–155. [Google Scholar] [CrossRef]
- Moreau, J.-J. Du Hachisch et de L’Aliénationmentale, Étudespsychologiques, Par J. Moreau de Tours; Fortin, Masson et Cie: Paris, France, 1845. [Google Scholar]
- Grinspoon. Marihuana Reconsidered, 2nd ed.; Quick American Archives: Oakland, CA, USA, 1994; ISBN 978-0-932551-13-9. [Google Scholar]
- Brenneisen, R. Chemistry and Analysis of Phytocannabinoids and Other Cannabis Constituents. In Marijuana and the Cannabinoids; ElSohly, M.A., Ed.; Humana Press: Totowa, NJ, USA, 2007; pp. 17–49. ISBN 978-1-58829-456-2. [Google Scholar]
- Adams, R.; Hunt, M.; Clark, J.H. Structure of cannabidiol, a product isolated from the marihuana extract of Minnesota wild hemp. I. J. Am. Chem. Soc. 1940, 62, 196–200. [Google Scholar] [CrossRef]
- Mechoulam, R.; Shvo, Y. Hashish—I: The structure of cannabidiol. Tetrahedron 1963, 19, 2073–2078. [Google Scholar] [CrossRef]
- Gaoni, Y.; Mechoulam, R. Isolation, structure, and partial synthesis of an active constituent of hashish. J. Am. Chem. Soc. 1964, 86, 1646–1647. [Google Scholar] [CrossRef]
- Mechoulam, R.; Braun, P.; Gaoni, Y. A stereospecific synthesis of (-)-delta 1- and (-)-delta 1(6)-tetrahydrocannabinols. J. Am. Chem. Soc. 1967, 89, 4552–4554. [Google Scholar] [CrossRef] [PubMed]
- Leuschner, J.T.; Wing, D.R.; Harvey, D.J.; Brent, G.A.; Dempsey, C.E.; Watts, A.; Paton, W.D. The partitioning of delta 1-tetrahydrocannabinol into erythrocyte membranes in vivo and its effect on membrane fluidity. Experientia 1984, 40, 866–868. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C.; Scott, D.K.; Wilken, G.H. Regulation of adenylate cyclase by cannabinoid drugs. Insights based on thermodynamic studies. Biochem. Pharmacol. 1989, 38, 3297–3304. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Mackie, K. Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb. Exp. Pharmacol. 2005, 168, 299–325. [Google Scholar]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C. The cannabinoid receptors. Prostaglandins Other Lipid Mediat. 2002, 68–69, 619–631. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Receptors and channels targeted by synthetic cannabinoid receptor agonists and antagonists. Curr. Med. Chem. 2010, 17, 1360–1381. [Google Scholar] [CrossRef] [PubMed]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-Arachidonoylglycerol: A possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995, 215, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Marzo, V.D.; Petrocellis, L.D. Why do cannabinoid receptors have more than one endogenous ligand? Phil. Trans. R. Soc. B 2012, 367, 3216–3228. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Hanuš, L.O.; Pertwee, R.; Howlett, A.C. Early phytocannabinoid chemistry to endocannabinoids and beyond. Nat. Rev. Neurosci. 2014, 15, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Cravatt, B.F.; Demarest, K.; Patricelli, M.P.; Bracey, M.H.; Giang, D.K.; Martin, B.R.; Lichtman, A.H. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 2001, 98, 9371–9376. [Google Scholar] [CrossRef] [PubMed]
- Fegley, D.; Gaetani, S.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; Piomelli, D. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): Effects on anandamide and oleoylethanolamide deactivation. J. Pharmacol. Exp. Ther. 2005, 313, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Katona, I.; Freund, T.F. Multiple functions of endocannabinoid signaling in the brain. Annu. Rev. Neurosci. 2012, 35, 529–558. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ruiz, J.; Hernández, M.; Ramos, J.A. Cannabinoid-dopamine interaction in the pathophysiology and treatment of CNS disorders. CNS Neurosci. Ther. 2010, 16, e72–e91. [Google Scholar] [CrossRef] [PubMed]
- Rubino, T.; Zamberletti, E.; Parolaro, D. Endocannabinoids and mental disorders. Handb. Exp. Pharmacol. 2015, 231, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Diagnostic and Statistical Manual of Mental Disorders (DSM–5). Available online: https://www.psychiatry.org/psychiatrists/practice/dsm (accessed on 30 July 2018).
- Harvey, P.D.; Koren, D.; Reichenberg, A.; Bowie, C.R. Negative symptoms and cognitive deficits: What is the nature of their relationship? Schizophr. Bull. 2006, 32, 250–258. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.; Saha, S.; Chant, D.; Welham, J. Schizophrenia: A concise overview of incidence, prevalence, and mortality. Epidemiol. Rev. 2008, 30, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Leweke, F.M.; Odorfer, T.M.; Bumb, J.M. Medical needs in the treatment of psychotic disorders. Handb. Exp. Pharmacol. 2012, 212, 165–185. [Google Scholar] [CrossRef]
- Patel, K.R.; Cherian, J.; Gohil, K.; Atkinson, D. Schizophrenia: Overview and treatment options. Pharm. Ther. 2014, 39, 638–645. [Google Scholar]
- Müller-Vahl, K.R.; Emrich, H.M. Cannabis and schizophrenia: Towards a cannabinoid hypothesis of schizophrenia. Expert Rev. Neurother. 2008, 8, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Ferretjans, R.; Moreira, F.A.; Teixeira, A.L.; Salgado, J.V. The endocannabinoid system and its role in schizophrenia: A systematic review of the literature. Rev. Bras. Psiquiatr. 2012, 34 (Suppl. 2), S163–S177. [Google Scholar] [CrossRef] [PubMed]
- Leweke, F.M.; Giuffrida, A.; Wurster, U.; Emrich, H.M.; Piomelli, D. Elevated endogenous cannabinoids in schizophrenia. Neuroreport 1999, 10, 1665–1669. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, N.; De Petrocellis, L.; Orlando, P.; Daniele, F.; Fezza, F.; Di Marzo, V. Endocannabinoid signalling in the blood of patients with schizophrenia. Lipids Health Dis. 2003, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, A.; Leweke, F.M.; Gerth, C.W.; Schreiber, D.; Koethe, D.; Faulhaber, J.; Klosterkötter, J.; Piomelli, D. Cerebrospinal anandamide levels are elevated in acute schizophrenia and are inversely correlated with psychotic symptoms. Neuropsychopharmacology 2004, 29, 2108–2114. [Google Scholar] [CrossRef] [PubMed]
- Leweke, F.M.; Giuffrida, A.; Koethe, D.; Schreiber, D.; Nolden, B.M.; Kranaster, L.; Neatby, M.A.; Schneider, M.; Gerth, C.W.; Hellmich, M.; et al. Anandamide levels in cerebrospinal fluid of first-episode schizophrenic patients: Impact of cannabis use. Schizophr. Res. 2007, 94, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Dean, B.; Sundram, S.; Bradbury, R.; Scarr, E.; Copolov, D. Studies on [3H]CP-55940 binding in the human central nervous system: Regional specific changes in density of cannabinoid-1 receptors associated with schizophrenia and cannabis use. Neuroscience 2001, 103, 9–15. [Google Scholar] [CrossRef]
- Newell, K.A.; Deng, C.; Huang, X.-F. Increased cannabinoid receptor density in the posterior cingulate cortex in schizophrenia. Exp. Brain Res. 2006, 172, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Zavitsanou, K.; Garrick, T.; Huang, X.F. Selective antagonist [3H]SR141716A binding to cannabinoid CB1 receptors is increased in the anterior cingulate cortex in schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2004, 28, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Eggan, S.M.; Hashimoto, T.; Lewis, D.A. Reduced cortical cannabinoid 1 receptor messenger RNA and protein expression in schizophrenia. Arch. Gen. Psychiatry 2008, 65, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Eggan, S.M.; Stoyak, S.R.; Verrico, C.D.; Lewis, D.A. Cannabinoid CB1 receptor immunoreactivity in the prefrontal cortex: Comparison of schizophrenia and major depressive disorder. Neuropsychopharmacology 2010, 35, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- Koethe, D.; Llenos, I.C.; Dulay, J.R.; Hoyer, C.; Torrey, E.F.; Leweke, F.M.; Weis, S. Expression of CB1 cannabinoid receptor in the anterior cingulate cortex in schizophrenia, bipolar disorder, and major depression. J. Neural Transm. 2007, 114, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Urigüen, L.; García-Fuster, M.J.; Callado, L.F.; Morentin, B.; La Harpe, R.; Casadó, V.; Lluis, C.; Franco, R.; García-Sevilla, J.A.; Meana, J.J. Immunodensity and mRNA expression of A2A adenosine, D2 dopamine, and CB1 cannabinoid receptors in postmortem frontal cortex of subjects with schizophrenia: Effect of antipsychotic treatment. Psychopharmacology 2009, 206, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Ceccarini, J.; De Hert, M.; Van Winkel, R.; Peuskens, J.; Bormans, G.; Kranaster, L.; Enning, F.; Koethe, D.; Leweke, F.M.; Van Laere, K. Increased ventral striatal CB1 receptor binding is related to negative symptoms in drug-free patients with schizophrenia. NeuroImage 2013, 79, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.F.; Kuwabara, H.; Horti, A.G.; Raymont, V.; Brasic, J.; Guevara, M.; Ye, W.; Dannals, R.F.; Ravert, H.T.; Nandi, A.; et al. Quantification of cerebral cannabinoid receptors subtype 1 (CB1) in healthy subjects and schizophrenia by the novel PET radioligand [11C]OMAR. NeuroImage 2010, 52, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Horti, A.G.; Fan, H.; Kuwabara, H.; Hilton, J.; Ravert, H.T.; Holt, D.P.; Alexander, M.; Kumar, A.; Rahmim, A.; Scheffel, U.; et al. 11C-JHU75528: A radiotracer for PET imaging of CB1 cannabinoid receptors. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2006, 47, 1689–1696. [Google Scholar]
- Bioque, M.; García-Bueno, B.; MacDowell, K.S.; Meseguer, A.; Saiz, P.A.; Parellada, M.; Gonzalez-Pinto, A.; Rodriguez-Jimenez, R.; Lobo, A.; Leza, J.C.; et al. Peripheral endocannabinoid system dysregulation in first-episode psychosis. Neuropsychopharmacology 2013, 38, 2568–2577. [Google Scholar] [CrossRef] [PubMed]
- Ujike, H.; Morita, Y. New perspectives in the studies on endocannabinoid and cannabis: Cannabinoid receptors and schizophrenia. J. Pharmacol. Sci. 2004, 96, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Chavarría-Siles, I.; Contreras-Rojas, J.; Hare, E.; Walss-Bass, C.; Quezada, P.; Dassori, A.; Contreras, S.; Medina, R.; Ramírez, M.; Salazar, R.; et al. Cannabinoid receptor 1 gene (CNR1) and susceptibility to a quantitative phenotype for hebephrenic schizophrenia. Am. J. Med. Genet. Part B 2008, 147B, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.J.; Wang, Y.C.; Hong, C.J. Association study of a cannabinoid receptor gene (CNR1) polymorphism and schizophrenia. Psychiatr. Genet. 2000, 10, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Seifert, J.; Ossege, S.; Emrich, H.M.; Schneider, U.; Stuhrmann, M. No association of CNR1 gene variations with susceptibility to schizophrenia. Neurosci. Lett. 2007, 426, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.H.M.; Zammit, S.; Lingford-Hughes, A.; Barnes, T.R.E.; Jones, P.B.; Burke, M.; Lewis, G. Cannabis use and risk of psychotic or affective mental health outcomes: A systematic review. Lancet Lond. Engl. 2007, 370, 319–328. [Google Scholar] [CrossRef]
- Jiang, S.; Fu, Y.; Williams, J.; Wood, J.; Pandarinathan, L.; Avraham, S.; Makriyannis, A.; Avraham, S.; Avraham, H.K. Expression and function of cannabinoid receptors CB1 and CB2 and their cognate cannabinoid ligands in murine embryonic stem cells. PLoS ONE 2007, 2, e641. [Google Scholar] [CrossRef] [PubMed]
- Potvin, S.; Joyal, C.C.; Pelletier, J.; Stip, E. Contradictory cognitive capacities among substance-abusing patients with schizophrenia: A meta-analysis. Schizophr. Res. 2008, 100, 242–251. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, D.C.; Perry, E.; MacDougall, L.; Ammerman, Y.; Cooper, T.; Wu, Y.-T.; Braley, G.; Gueorguieva, R.; Krystal, J.H. The psychotomimetic effects of intravenous delta-9-tetrahydrocannabinol in healthy individuals: Implications for psychosis. Neuropsychopharmacology 2004, 29, 1558–1572. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, D.C.; Abi-Saab, W.M.; Madonick, S.; Forselius-Bielen, K.; Doersch, A.; Braley, G.; Gueorguieva, R.; Cooper, T.B.; Krystal, J.H. Delta-9-tetrahydrocannabinol effects in schizophrenia: Implications for cognition, psychosis, and addiction. Biol. Psychiatry 2005, 57, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Bossong, M.G.; Jager, G.; Bhattacharyya, S.; Allen, P. Acute and non-acute effects of cannabis on human memory function: A critical review of neuroimaging studies. Curr. Pharm. Des. 2014, 20, 2114–2125. [Google Scholar] [CrossRef] [PubMed]
- Murray, R.M.; Englund, A.; Abi-Dargham, A.; Lewis, D.A.; Di Forti, M.; Davies, C.; Sherif, M.; McGuire, P.; D’Souza, D.C. Cannabis-associated psychosis: Neural substrate and clinical impact. Neuropharmacology 2017, 124, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, G.; Karajgi, B.; McCarthy, R. Synthetic delta-9-tetrahydrocannabinol (dronabinol) can improve the symptoms of schizophrenia. J. Clin. Psychopharmacol. 2009, 29, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G.; Ross, R.A.; Craib, S.J.; Thomas, A. (−)-Cannabidiol antagonizes cannabinoid receptor agonists and noradrenaline in the mouse vas deferens. Eur. J. Pharmacol. 2002, 456, 99–106. [Google Scholar] [CrossRef]
- Bisogno, T.; Hanus, L.; De Petrocellis, L.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; Di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P. Cannabidiol is a partial agonist at dopamine D2High receptors, predicting its antipsychotic clinical dose. Transl. Psychiatry 2016, 6, e920. [Google Scholar] [CrossRef] [PubMed]
- Hahn, B. The potential of cannabidiol treatment for cannabis users with recent-onset psychosis. Schizophr. Bull. 2018, 44, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Mandolini, G.M.; Lazzaretti, M.; Pigoni, A.; Oldani, L.; Delvecchio, G.; Brambilla, P. Pharmacological properties of cannabidiol in the treatment of psychiatric disorders: A critical overview. Epidemiol. Psychiatr. Sci. 2018, 27, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Morrison, P.D.; Fusar-Poli, P.; Martin-Santos, R.; Borgwardt, S.; Winton-Brown, T.; Nosarti, C.; O’ Carroll, C.M.; Seal, M.; Allen, P.; et al. Opposite effects of delta-9-tetrahydrocannabinol and cannabidiol on human brain function and psychopathology. Neuropsychopharmacology 2010, 35, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Zuardi, A.W.; Morais, S.L.; Guimarães, F.S.; Mechoulam, R. Antipsychotic effect of cannabidiol. J. Clin. Psychiatry 1995, 56, 485–486. [Google Scholar] [PubMed]
- Zuardi, A.W.; Crippa, J.A.S.; Hallak, J.E.C.; Moreira, F.A.; Guimarães, F.S. Cannabidiol, a Cannabis sativa constituent, as an antipsychotic drug. Braz. J. Med. Biol. Res. Rev. 2006, 39, 421–429. [Google Scholar] [CrossRef]
- McGuire, P.; Robson, P.; Cubala, W.J.; Vasile, D.; Morrison, P.D.; Barron, R.; Taylor, A.; Wright, S. Cannabidiol (CBD) as an adjunctive therapy in schizophrenia: A multicenter randomized controlled trial. Am. J. Psychiatry 2018, 175, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Leweke, F.M.; Piomelli, D.; Pahlisch, F.; Muhl, D.; Gerth, C.W.; Hoyer, C.; Klosterkötter, J.; Hellmich, M.; Koethe, D. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl. Psychiatry 2012, 2, e94. [Google Scholar] [CrossRef] [PubMed]
- Beltramo, M.; de Fonseca, F.R.; Navarro, M.; Calignano, A.; Gorriti, M.A.; Grammatikopoulos, G.; Sadile, A.G.; Giuffrida, A.; Piomelli, D. Reversal of dopamine D2 receptor responses by an anandamide transport inhibitor. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 3401–3407. [Google Scholar] [CrossRef]
- Seillier, A.; Advani, T.; Cassano, T.; Hensler, J.G.; Giuffrida, A. Inhibition of fatty-acid amide hydrolase and CB1 receptor antagonism differentially affect behavioural responses in normal and PCP-treated rats. Int. J. Neuropsychopharmacol. 2010, 13, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Malone, D.T.; Kearn, C.S.; Chongue, L.; Mackie, K.; Taylor, D.A. Effect of social isolation on CB1 and D2 receptor and fatty acid amide hydrolase expression in rats. Neuroscience 2008, 152, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Vigano, D.; Guidali, C.; Petrosino, S.; Realini, N.; Rubino, T.; Di Marzo, V.; Parolaro, D. Involvement of the endocannabinoid system in phencyclidine-induced cognitive deficits modelling schizophrenia. Int. J. Neuropsychopharmacol. 2009, 12, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, H.; Horiuchi, Y.; Ishikawa, M.; Koga, M.; Imai, K.; Suzuki, Y.; Morikawa, M.; Inada, T.; Watanabe, Y.; Takahashi, M.; et al. Brain cannabinoid CB2 receptor in schizophrenia. Biol. Psychiatry 2010, 67, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Pedrazzi, J.F.C.; Issy, A.C.; Gomes, F.V.; Guimarães, F.S.; Del-Bel, E.A. Cannabidiol effects in the prepulse inhibition disruption induced by amphetamine. Psychopharmacology 2015, 232, 3057–3065. [Google Scholar] [CrossRef] [PubMed]
- Moreira, F.A.; Guimarães, F.S. Cannabidiol inhibits the hyperlocomotion induced by psychotomimetic drugs in mice. Eur. J. Pharmacol. 2005, 512, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Craske, M.G.; Stein, M.B. Anxiety. Lancet 2016, 388, 3048–3059. [Google Scholar] [CrossRef]
- Kessler, R.C.; Ruscio, A.M.; Shear, K.; Wittchen, H.-U. Epidemiology of anxiety disorders. Curr. Top. Behav. Neurosci. 2010, 2, 21–35. [Google Scholar] [PubMed]
- Bandelow, B.; Zohar, J.; Hollander, E.; Kasper, S.; Möller, H.-J.; WFSBP Task Force on Treatment Guidelines for Anxiety Obsessive-Compulsive Post-Traumatic Stress Disoders. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the pharmacological treatment of anxiety, obsessive-compulsive and post-traumatic stress disorders—First revision. World J. Biol. Psychiatry 2008, 9, 248–312. [Google Scholar] [CrossRef] [PubMed]
- Rubino, T.; Guidali, C.; Vigano, D.; Realini, N.; Valenti, M.; Massi, P.; Parolaro, D. CB1 receptor stimulation in specific brain areas differently modulate anxiety-related behaviour. Neuropharmacology 2008, 54, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Viveros, M.P.; Marco, E.M.; File, S.E. Endocannabinoid system and stress and anxiety responses. Pharmacol. Biochem. Behav. 2005, 81, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Turna, J.; Patterson, B.; Van Ameringen, M. Is cannabis treatment for anxiety, mood, and related disorders ready for prime time? Depress. Anxiety 2017, 34, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Herkenham, M.; Lynn, A.B.; Little, M.D.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. USA 1990, 87, 1932–1936. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Ledent, C.; Parmentier, M.; Maldonado, R.; Valverde, O. Involvement of CB1 cannabinoid receptors in emotional behaviour. Psychopharmacology 2002, 159, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Haller, J.; Varga, B.; Ledent, C.; Freund, T.F. CB1 cannabinoid receptors mediate anxiolytic effects: Convergent genetic and pharmacological evidence with CB1-specific agents. Behav. Pharmacol. 2004, 15, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Urigüen, L.; Pérez-Rial, S.; Ledent, C.; Palomo, T.; Manzanares, J. Impaired action of anxiolytic drugs in mice deficient in cannabinoid CB1 receptors. Neuropharmacology 2004, 46, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Mikics, E.; Vas, J.; Aliczki, M.; Halasz, J.; Haller, J. Interactions between the anxiogenic effects of CB1 gene disruption and 5-HT3 neurotransmission. Behav. Pharmacol. 2009, 20, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Marsicano, G.; Wotjak, C.T.; Azad, S.C.; Bisogno, T.; Rammes, G.; Cascio, M.G.; Hermann, H.; Tang, J.; Hofmann, C.; Zieglgänsberger, W.; et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature 2002, 418, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Rey, A.A.; Purrio, M.; Viveros, M.-P.; Lutz, B. Biphasic effects of cannabinoids in anxiety responses: CB1 and GABAB receptors in the balance of GABAergic and glutamatergic neurotransmission. Neuropsychopharmacology 2012, 37, 2624–2634. [Google Scholar] [CrossRef] [PubMed]
- Jacob, W.; Yassouridis, A.; Marsicano, G.; Monory, K.; Lutz, B.; Wotjak, C.T. Endocannabinoids render exploratory behaviour largely independent of the test aversiveness: Role of glutamatergic transmission. Genes Brain Behav. 2009, 8, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Morena, M.; Patel, S.; Bains, J.S.; Hill, M.N. Neurobiological interactions between stress and the endocannabinoid system. Neuropsychopharmacology 2016, 41, 80–102. [Google Scholar] [CrossRef] [PubMed]
- Dincheva, I.; Drysdale, A.T.; Hartley, C.A.; Johnson, D.C.; Jing, D.; King, E.C.; Ra, S.; Gray, J.M.; Yang, R.; DeGruccio, A.M.; et al. FAAH genetic variation enhances fronto-amygdala function in mouse and human. Nat. Commun. 2015, 6, 6395. [Google Scholar] [CrossRef] [PubMed]
- Kathuria, S.; Gaetani, S.; Fegley, D.; Valiño, F.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; La Rana, G.; Calignano, A.; et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 2003, 9, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Scherma, M.; Medalie, J.; Fratta, W.; Vadivel, S.K.; Makriyannis, A.; Piomelli, D.; Mikics, E.; Haller, J.; Yasar, S.; Tanda, G.; et al. The endogenous cannabinoid anandamide has effects on motivation and anxiety that are revealed by fatty acid amide hydrolase (FAAH) inhibition. Neuropharmacology 2008, 54, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Haller, J.; Barna, I.; Barsvari, B.; Gyimesi Pelczer, K.; Yasar, S.; Panlilio, L.V.; Goldberg, S. Interactions between environmental aversiveness and the anxiolytic effects of enhanced cannabinoid signaling by FAAH inhibition in rats. Psychopharmacology 2009, 204, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.J.; Williams, C.M.; Whalley, B.J.; Stephens, G.J. Phytocannabinoids as novel therapeutic agents in CNS disorders. Pharmacol. Ther. 2012, 133, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Buckner, J.D.; Schmidt, N.B.; Lang, A.R.; Small, J.W.; Schlauch, R.C.; Lewinsohn, P.M. Specificity of social anxiety disorder as a risk factor for alcohol and cannabis dependence. J. Psychiatr. Res. 2008, 42, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Metrik, J.; Bassett, S.S.; Aston, E.R.; Jackson, K.M.; Borsari, B. Medicinal versus recreational cannabis use among returning veterans. Transl. Issues Psychol. Sci. 2018, 4, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Passie, T.; Emrich, H.M.; Karst, M.; Brandt, S.D.; Halpern, J.H. Mitigation of post-traumatic stress symptoms by Cannabis resin: A review of the clinical and neurobiological evidence. Drug Test. Anal. 2012, 4, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Reznik, I. Post-traumatic stress disorder and medical cannabis use: A naturalistic observational study. Eur. Neuropsychopharmacol. 2012, 22, S363–S364. [Google Scholar] [CrossRef]
- Roitman, P.; Mechoulam, R.; Cooper-Kazaz, R.; Shalev, A. Preliminary, open-label, pilot study of add-on oral Δ9-tetrahydrocannabinol in chronic post-traumatic stress disorder. Clin. Drug Investig. 2014, 34, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Neumeister, A.; Normandin, M.D.; Pietrzak, R.H.; Piomelli, D.; Zheng, M.Q.; Gujarro-Anton, A.; Potenza, M.N.; Bailey, C.R.; Lin, S.F.; Najafzadeh, S.; et al. Elevated brain cannabinoid CB1 receptor availability in post-traumatic stress disorder: A positron emission tomography study. Mol. Psychiatry 2013, 18, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H. Psychiatric symptoms in cannabis users. Br. J. Psychiatry 1993, 163, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Fusar-Poli, P.; Crippa, J.A.; Bhattacharyya, S.; Borgwardt, S.J.; Allen, P.; Martin-Santos, R.; Seal, M.; Surguladze, S.A.; O’Carrol, C.; Atakan, Z.; et al. Distinct effects of ∆9-tetrahydrocannabinol and cannabidiol on neural activation during emotional processing. Arch. Gen. Psychiatry 2009, 66, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Bossong, M.G.; van Hell, H.H.; Jager, G.; Kahn, R.S.; Ramsey, N.F.; Jansma, J.M. The endocannabinoid system and emotional processing: A pharmacological fMRI study with ∆9-tetrahydrocannabinol. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2013, 23, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Blessing, E.M.; Steenkamp, M.M.; Manzanares, J.; Marmar, C.R. Cannabidiol as a potential treatment for anxiety disorders. Neurother. J. Am. Soc. Exp. Neurother. 2015, 12, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Zuardi, A.W.; Shirakawa, I.; Finkelfarb, E.; Karniol, I.G. Action of cannabidiol on the anxiety and other effects produced by delta 9-THC in normal subjects. Psychopharmacology 1982, 76, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Zuardi, A.W.; Cosme, R.A.; Graeff, F.G.; Guimarães, F.S. Effects of ipsapirone and cannabidiol on human experimental anxiety. J. Psychopharmacol. 1993, 7, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, M.M.; Queiroz, R.H.C.; Chagas, M.H.N.; de Oliveira, D.C.G.; De Martinis, B.S.; Kapczinski, F.; Quevedo, J.; Roesler, R.; Schröder, N.; Nardi, A.E.; et al. Cannabidiol reduces the anxiety induced by simulated public speaking in treatment-naïve social phobia patients. Neuropsychopharmacology 2011, 36, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Crippa, J.A.; Zuardi, A.W.; Garrido, G.E.J.; Wichert-Ana, L.; Guarnieri, R.; Ferrari, L.; Azevedo-Marques, P.M.; Hallak, J.E.C.; McGuire, P.K.; Filho Busatto, G. Effects of cannabidiol (CBD) on regional cerebral blood flow. Neuropsychopharmacology 2004, 29, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Crippa, J.A.S.; Derenusson, G.N.; Ferrari, T.B.; Wichert-Ana, L.; Duran, F.L.S.; Martin-Santos, R.; Simões, M.V.; Bhattacharyya, S.; Fusar-Poli, P.; Atakan, Z.; et al. Neural basis of anxiolytic effects of cannabidiol (CBD) in generalized social anxiety disorder: A preliminary report. J. Psychopharmacol. 2011, 25, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, F.S.; Chiaretti, T.M.; Graeff, F.G.; Zuardi, A.W. Antianxiety effect of cannabidiol in the elevated plus-maze. Psychopharmacology 1990, 100, 558–559. [Google Scholar] [CrossRef] [PubMed]
- Onaivi, E.S.; Green, M.R.; Martin, B.R. Pharmacological characterization of cannabinoids in the elevated plus maze. J. Pharmacol. Exp. Ther. 1990, 253, 1002–1009. [Google Scholar] [PubMed]
- Moreira, F.A.; Aguiar, D.C.; Guimarães, F.S. Anxiolytic-like effect of cannabidiol in the rat Vogel conflict test. Prog. Neuropsychopharmacol. Biol. Psychiatry 2006, 30, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.C.; Guimarães, F.S. Involvement of 5HT1A receptors in the anxiolytic-like effects of cannabidiol injected into the dorsolateral periaqueductal gray of rats. Psychopharmacology 2008, 199, 223–230. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing, Inc.: Arlington, VA, USA, 2013; ISBN 978-0-89042-555-8. [Google Scholar]
- Han, M.-H.; Nestler, E.J. Neural substrates of depression and resilience. Neurother. J. Am. Soc. Exp. Neurother. 2017, 14, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Hardeveld, F.; Spijker, J.; De Graaf, R.; Hendriks, S.M.; Licht, C.M.M.; Nolen, W.A.; Penninx, B.W.J.H.; Beekman, A.T.F. Recurrence of major depressive disorder across different treatment settings: Results from the NESDA study. J. Affect. Disord. 2013, 147, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Smaga, I.; Bystrowska, B.; Gawliński, D.; Przegaliński, E.; Filip, M. The endocannabinoid/endovanilloid system and depression. Curr. Neuropharmacol. 2014, 12, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.N.; Miller, G.E.; Ho, W.-S.V.; Gorzalka, B.B.; Hillard, C.J. Serum endocannabinoid content is altered in females with depressive disorders: A preliminary report. Pharmacopsychiatry 2008, 41, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.N.; Miller, G.E.; Carrier, E.J.; Gorzalka, B.B.; Hillard, C.J. Circulating endocannabinoids and N-acyl ethanolamines are differentially regulated in major depression and following exposure to social stress. Psychoneuroendocrinology 2009, 34, 1257–1262. [Google Scholar] [CrossRef] [PubMed]
- Hungund, B.L.; Vinod, K.Y.; Kassir, S.A.; Basavarajappa, B.S.; Yalamanchili, R.; Cooper, T.B.; Mann, J.J.; Arango, V. Upregulation of CB1 receptors and agonist-stimulated [35S]GTPγS binding in the prefrontal cortex of depressed suicide victims. Mol. Psychiatry 2004, 9, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Le, T.; McGuire, J.; Xing, G.; Zhang, L.; Li, H.; Parker, C.C.; Johnson, L.R.; Ursano, R.J. Expression pattern of the cannabinoid receptor genes in the frontal cortex of mood disorder patients and mice selectively bred for high and low fear. J. Psychiatr. Res. 2012, 46, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, G.; Chase, D.; Pegg, E.; Downey, D.; Toth, Z.G.; Stones, K.; Platt, H.; Mekli, K.; Payton, A.; Elliott, R.; et al. CNR1 gene is associated with high neuroticism and low agreeableness and interacts with recent negative life events to predict current depressive symptoms. Neuropsychopharmacology 2009, 34, 2019–2027. [Google Scholar] [CrossRef] [PubMed]
- Domschke, K.; Dannlowski, U.; Ohrmann, P.; Lawford, B.; Bauer, J.; Kugel, H.; Heindel, W.; Young, R.; Morris, P.; Arolt, V.; et al. Cannabinoid receptor 1 (CNR1) gene: Impact on antidepressant treatment response and emotion processing in major depression. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2008, 18, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, P.; Bifulco, M.; Maina, G.; Tortorella, A.; Gazzerro, P.; Proto, M.C.; Di Filippo, C.; Monteleone, F.; Canestrelli, B.; Buonerba, G.; et al. Investigation of CNR1 and FAAH endocannabinoid gene polymorphisms in bipolar disorder and major depression. Pharmacol. Res. 2010, 61, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Mitjans, M.; Serretti, A.; Fabbri, C.; Gastó, C.; Catalán, R.; Fañanás, L.; Arias, B. Screening genetic variability at the CNR1 gene in both major depression etiology and clinical response to citalopram treatment. Psychopharmacology 2013, 227, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Grinspoon, L.; Bakalar, J.B. The use of cannabis as a mood stabilizer in bipolar disorder: Anecdotal evidence and the need for clinical research. J. Psychoactive Drugs 1998, 30, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Ashton, C.H.; Moore, P.B.; Gallagher, P.; Young, A.H. Cannabinoids in bipolar affective disorder: A review and discussion of their therapeutic potential. J. Psychopharmacol. 2005, 19, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.J.; Pope, H.G.; Brown, M.E. Do patients use marijuana as an antidepressant? Depression 1996, 4, 77–80. [Google Scholar] [CrossRef]
- Denson, T.F.; Earleywine, M. Decreased depression in marijuana users. Addict. Behav. 2006, 31, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Babson, K.A.; Boden, M.T.; Bonn-Miller, M.O. Sleep quality moderates the relation between depression symptoms and problematic cannabis use among medical cannabis users. Am. J. Drug Alcohol Abuse 2013, 39, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Walsh, Z.; Gonzalez, R.; Crosby, K.; Thiessen, M.S.; Carroll, C.; Bonn-Miller, M.O. Medical cannabis and mental health: A guided systematic review. Clin. Psychol. Rev. 2017, 51, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Gruber, S.A.; Sagar, K.A.; Dahlgren, M.K.; Olson, D.P.; Centorrino, F.; Lukas, S.E. Marijuana impacts mood in bipolar disorder: A pilot study. Ment. Health Subst. Use 2012, 5, 228–239. [Google Scholar] [CrossRef]
- Ringen, P.A.; Vaskinn, A.; Sundet, K.; Engh, J.A.; Jónsdóttir, H.; Simonsen, C.; Friis, S.; Opjordsmoen, S.; Melle, I.; Andreassen, O.A. Opposite relationships between cannabis use and neurocognitive functioning in bipolar disorder and schizophrenia. Psychol. Med. 2010, 40, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Braga, R.J.; Burdick, K.E.; DeRosse, P.; Malhotra, A.K. Cognitive and clinical outcomes associated with cannabis use in patients with bipolar I disorder. Psychiatry Res. 2012, 200, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Lev-Ran, S.; Roerecke, M.; Le Foll, B.; George, T.P.; McKenzie, K.; Rehm, J. The association between cannabis use and depression: A systematic review and meta-analysis of longitudinal studies. Psychol. Med. 2014, 44, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Aspis, I.; Feingold, D.; Weiser, M.; Rehm, J.; Shoval, G.; Lev-Ran, S. Cannabis use and mental health-related quality of life among individuals with depressive disorders. Psychiatry Res. 2015, 230, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.; Winsper, C.; Marwaha, S.; Gilbert, E.; Broome, M.; Singh, S.P. Cannabis use and mania symptoms: A systematic review and meta-analysis. J. Affect. Disord. 2015, 171, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Parolaro, D.; Realini, N.; Vigano, D.; Guidali, C.; Rubino, T. The endocannabinoid system and psychiatric disorders. Exp. Neurol. 2010, 224, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.N.; Patel, S.; Carrier, E.J.; Rademacher, D.J.; Ormerod, B.K.; Hillard, C.J.; Gorzalka, B.B. Downregulation of endocannabinoid signaling in the hippocampus following chronic unpredictable stress. Neuropsychopharmacology 2005, 30, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.N.; Carrier, E.J.; McLaughlin, R.J.; Morrish, A.C.; Meier, S.E.; Hillard, C.J.; Gorzalka, B.B. Regional alterations in the endocannabinoid system in an animal model of depression: Effects of concurrent antidepressant treatment. J. Neurochem. 2008, 106, 2322–2336. [Google Scholar] [CrossRef] [PubMed]
- Bortolato, M.; Mangieri, R.A.; Fu, J.; Kim, J.H.; Arguello, O.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; Piomelli, D. Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol. Psychiatry 2007, 62, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.N.; Ho, W.-S.V.; Hillard, C.J.; Gorzalka, B.B. Differential effects of the antidepressants tranylcypromine and fluoxetine on limbic cannabinoid receptor binding and endocannabinoid contents. J. Neural Transm. 2008, 115, 1673–1679. [Google Scholar] [CrossRef] [PubMed]
- Egashira, N.; Matsuda, T.; Koushi, E.; Higashihara, F.; Mishima, K.; Chidori, S.; Hasebe, N.; Iwasaki, K.; Nishimura, R.; Oishi, R.; et al. ∆9-tetrahydrocannabinol prolongs the immobility time in the mouse forced swim test: Involvement of cannabinoid CB1 receptor and serotonergic system. Eur. J. Pharmacol. 2008, 589, 117–121. [Google Scholar] [CrossRef] [PubMed]
- El-Alfy, A.T.; Ivey, K.; Robinson, K.; Ahmed, S.; Radwan, M.; Slade, D.; Khan, I.; ElSohly, M.; Ross, S. Antidepressant-like effect of ∆9-tetrahydrocannabinol and other cannabinoids isolated from Cannabis sativa L. Pharmacol. Biochem. Behav. 2010, 95, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Sartim, A.G.; Guimarães, F.S.; Joca, S.R.L. Antidepressant-like effect of cannabidiol injection into the ventral medial prefrontal cortex—Possible involvement of 5-HT1A and CB1 receptors. Behav. Brain Res. 2016, 303, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Di Forti, M.; Morgan, C.; Dazzan, P.; Pariante, C.; Mondelli, V.; Marques, T.R.; Handley, R.; Luzi, S.; Russo, M.; Paparelli, A.; et al. High-potency cannabis and the risk of psychosis. Br. J. Psychiatry 2009, 195, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Schubart, C.D.; Sommer, I.E.; van Gastel, W.A.; Goetgebuer, R.L.; Kahn, R.S.; Boks, M.P. Cannabis with high cannabidiol content is associated with fewer psychotic experiences. Schizophr. Res. 2011, 130, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.J.; Curran, H.V. Eff ects of cannabidiol on schizophrenia like symptoms in people who use cannabis. Br. J. Psychiatry 2008, 192, 306–307. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scherma, M.; Masia, P.; Deidda, M.; Fratta, W.; Tanda, G.; Fadda, P. New Perspectives on the Use of Cannabis in the Treatment of Psychiatric Disorders. Medicines 2018, 5, 107. https://doi.org/10.3390/medicines5040107
Scherma M, Masia P, Deidda M, Fratta W, Tanda G, Fadda P. New Perspectives on the Use of Cannabis in the Treatment of Psychiatric Disorders. Medicines. 2018; 5(4):107. https://doi.org/10.3390/medicines5040107
Chicago/Turabian StyleScherma, Maria, Paolo Masia, Matteo Deidda, Walter Fratta, Gianluigi Tanda, and Paola Fadda. 2018. "New Perspectives on the Use of Cannabis in the Treatment of Psychiatric Disorders" Medicines 5, no. 4: 107. https://doi.org/10.3390/medicines5040107
APA StyleScherma, M., Masia, P., Deidda, M., Fratta, W., Tanda, G., & Fadda, P. (2018). New Perspectives on the Use of Cannabis in the Treatment of Psychiatric Disorders. Medicines, 5(4), 107. https://doi.org/10.3390/medicines5040107