Ovarian Cancers: Genetic Abnormalities, Tumor Heterogeneity and Progression, Clonal Evolution and Cancer Stem Cells



Abstract
1. Introduction
2. Genetic Abnormalities of Serous Ovarian Cancers
2.1. High-Grade Serous Ovarian Cancer
2.2. TP53 Mutations in HGSOCs
2.3. HRD in HGS-OvCa
2.4. Clonal Evolution of HGS-OvCa: Spatial and Temporal Heterogeneity
2.5. Familial HGS-OvCa
2.6. Gene Expression Profiling Studies of Serous Ovarian Cancer
2.7. Molecular Studies on Serous Ovarian Cancer Origin
3. Molecular Abnormalities of Ovarian Carcinosarcomas
4. Molecular Abnormalities of Low-Grade Serous Ovarian Carcinomas
5. Genetic Abnormalities of Mucinous, Endometrioid and Clear Cell Carcinoma
6. Molecular Abnormalities of Seromucinous Carcinomas of the Ovary
7. Molecular Abnormalities of Ovarian Granulosa Cell Tumors
8. Molecular Abnormalities in Ovarian Sertoli-Leydig Tumors
9. Molecular Abnormalities of the Small Cell Carcinoma of the Ovary of Hypercalcemic Type (SCCOHT)
10. Normal Ovarian Stem Cells
11. Ovarian Cancer Stem Cells
12. MiRNAs and Ovarian Cancer Stem Cells
13. Mouse Models of Human Epithelial Ovarian Cancer
14. Circulating Cell-Free DNA and Circulating Tumor Cells: The “Liquid Biopsies” in Ovarian Cancer Patients
15. Treatment Strategies for Ovarian Cancers
16. Conclusions
Author Contributions
Conflicts of Interest
References
- Jamal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar]
- Shih, I.M.; Kurman, R.J. Ovarian tumorigenesis: A proposed model based on morphological and molecular genetic analysis. Am. J. Pathol. 2004, 164, 1511–1518. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.M. The dualistic model of ovarian carcinogenesis revisited, revised, and expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: Review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [PubMed]
- Jones, S.; Wang, T.L.; Kurman, R.J.; Nakayama, K.; Velculescu, V.E.; Vogelstein, B.; Kinzler, K.W.; Papadopoulos, N.; Shih, I.M. Low-grade serous carcinomas of the ovary contain very few point mutations. J. Pathol. 2012, 226, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar]
- Huang, J.; Zhang, L.; Greshock, J.; Colligon, T.A.; Wang, Y.; Ward, R.; Katsaros, D.; Lassus, H.; Butzow, R.; Godwin, A.K.; et al. Frequent genetic abnormalities of the PI3K/AKT pathway in primary ovarian cancer predict patient outcome. Genes Chromosomes Cancer 2011, 50, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Carden, C.; Stewart, A.; Thavasu, P.; Kipps, E.; Pope, L.; Crespo, M.; Miranda, S.; Attard, G.; Garrett, M.D.; Clarke, P.A.; et al. The association of PI3 kinase signaling and chemo-resistance in advanced ovarian cancer. Mol. Cancer Ther. 2012, 11, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.C.; Santiago, I.; Trinh, A.; Xian, J.; Guo, A.; Sayal, K.; Jimenez-Linan, M.; Deen, S.; Driver, K.; Mack, M.; et al. Combined image and genomic analysis of high-grade serous ovarian cancer reveals PTEN loss a common driver event and prognostic classifier. Genome Biol. 2014, 15, 526. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Cheung, H.W.; Agarwalla, P.K.; Thomas, S.; Zektser, Y.; Karst, A.M.; Boehm, J.S.; Weir, B.A.; Berlin, A.M.; Zou, L.; et al. In vivo multiplexed interrogation of amplified genes identifies GAB2 as an ovarian cancer oncogene. Proc. Natl. Acad. Sci. USA 2014, 111, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Despierre, E.; Moisse, M.; Yesilyurt, B.; Sehouli, J.; Braicu, I.; Mahner, S.; Castillo-Tong, D.C.; Zeillinger, R.; Lambrechts, S.; Leunen, K.; et al. Somatic copy number alterations predict response to platinum therapy in epithelial ovarian cancer. Gynecol. Oncol. 2014, 135, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Sung, C.O.; Song, I.H.; Sohn, I. A distinctive ovarian cancer molecular subgroup characterized by poor prognosis and somatic focal copy number amplifications at chromosome 19. Gynecol. Oncol. 2014, 132, 343–350. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Alsop, K.; Etemadmoghadam, D.; Hondow, H.; Mikeska, T.; Dobrovic, A.; deFazio, A.; Australian Ovarian Cancer Study Group; Smyth, G.K.; Levine, D.A.; et al. Non equivalent gene expression and copy number alterations in high-grade serous ovarian cancers with BRCA1 and BRCA2mutations. Clin. Cancer Res. 2013, 19, 3474–3484. [Google Scholar] [CrossRef] [PubMed]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Chernick, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, T.; Zhang, Z.; Payne, S.H.; Zhang, B.; McDermott, E.; Zhou, J.Y.; Patyuk, V.A.; Chen, L.; Ray, D.; et al. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell 2016, 166, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.R.; Patel, C.B.; Willis, K.; Haghighiabyaneh, M.; Axelrod, J.; Tancioni, I.; Lu, D.; Bapat, J.; Young, S.; Cadassou, O.; et al. Haploinsufficiency networks identify targetable patterns of allelic deficiency in low mutation ovarian cancer. Nat. Commun. 2016, 8, 14423. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Marinelli, R.J.; Wang, P.L.; Green, A.E.; Nielsen, J.S.; Nelson, B.H.; Drescher, C.W.; Brown, P.O. ESRRA-C11orf20 is a recurrent gene fusion in serous ovarian carcinoma. PLoS Biol. 2011, 9, e1001156. [Google Scholar] [CrossRef] [PubMed]
- Micci, F.; Panagopoulos, J.; Thorsen, J.; Davidson, B.; Tropé, C.G.; Heim, S. Low frequency of ESRRA-C11orf20 fusion gene in ovarian carcinomas. PLoS Biol. 2014, 12, e1001284. [Google Scholar] [CrossRef] [PubMed]
- Kannan, K.; Coarfa, C.; Rajapakshe, K.; Hawkins, S.M.; Matzuk, M.M.; Milosavljevic, A.; Yen, L. CDKN2D-WDFY2 is cancer-specific fusion gene recurrent in high-grade serous ovarian carcinoma. PLoS Genet. 2014, 20, e1004216. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Hemann, M.T.; Bartel, D.P. Disrupting the pairing between let-7 and HMGA2 enhances oncogenic transformation. Science 2007, 315, 1576–1579. [Google Scholar] [CrossRef] [PubMed]
- Shell, S.; Park, S.M.; Radjabi, A.R.; Schickel, R.; Kistner, E.O.; Jewell, D.A.; Feig, C.; Lengyel, E.; Peter, M.E. Let-7 expression defines two differentiation stages of cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 11400–11405. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Liu, Z.; Gellert, L.; Zou, X.; Yang, G.; Lee, P.; Yang, X.; Wei, J.J. HMGA2: A biomarker significantly overexpressed in high-grade ovarian serous carcinoma. Mod. Pathol. 2010, 23, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Tan, Z.P.; Zhu, X.; Crosby, K.; Haack, H.; Ren, J.M.; Beausoleil, S.; Moritz, A.; Innocenti, G.; Rush, J.; et al. Identification of anaplastic lymphoma kinase as a potential therapeutic target in ovarian cancer. Cancer Res. 2012, 172, 3312–3323. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Lee, S.W.; Chun, S.M.; Kim, D.Y.; Kim, J.H.; Kim, K.R.; Kim, Y.T.; Nam, J.H.; van Hummelen, P.; MacConaill, L.E.; et al. Analysis and comparison of somatic mutations in paired primary and recurrent epithelial ovarian cancer samples. PLoS ONE 2014, 9, e99451. [Google Scholar] [CrossRef] [PubMed]
- Castellarin, M.; Milne, K.; Zeng, T.; Tse, K.; Mayo, M.; Zhao, Y.; Webb, J.R.; Watson, P.H.; Nelson, B.H.; Holt, R.A. Clonal evolution of high-grade serous ovarian carcinoma from primary to recurrent disease. J. Pathol. 2013, 229, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Patch, A.M.; Christie, E.; Etemadmoghadam, D.; Gursed, D.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.K.; Bashashati, A.; Anglesio, M.S.; Cochrane, D.; Grewal, D.; Ha, G.; McPherson, A.; Horlings, H.M.; Senz, J.; Prentice, L.; et al. Genomic consequences of aberrant DNA repair mechanisms stratify ovarian cancer histotypes. Nat. Genet. 2017, 49, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Vang, R.; Levine, D.A.; Soslow, R.A.; Zaloudek, C.; Shih, I.-M.; Kurman, R.J. Molecular alterations of TP53 are a defining feature of ovarian high-grade serous carcinoma: A rereview of cases lacking TP53 mutations in The Cancer Genome Atlas Ovarian Study. Int. J. Gynecol. Pathol. 2016, 35, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, L.; Nguyen, D.; Lu, H. TP53 mutations in epithelial ovarian cancer. Transl. Cancer Res. 2016, 5, 1–12. [Google Scholar] [CrossRef]
- Lee, Y.; Miron, A.; Drapkin, R.; Nucci, M.R.; Medeiros, F.; Saleemuddin, A.; Garber, J.; Birch, C.; Mou, H.; Gordon, R.W.; et al. A candidate precursor to serous carcinoma that originates in the distal fallopian tube. J. Pathol. 2007, 211, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Iwanicki, M.P.; Chen, H.Y.; Iavarone, C.; Zavantonakis, I.K.; Muranen, T.; Novak, M.; Ince, T.A.; Drapkin, R.; Brugge, J.J. Mutant p53 regulates ovarian cancer transformed phenotypes through autocrine matrix deposition. J. Clin. Investig. Insight 2016, 1, e86829. [Google Scholar] [CrossRef] [PubMed]
- Candeias, M.M.; Hagiwara, M.; Matsuda, M. Cancer-specific mutations in p53 induce the translation of Delta160p53 promoting tumorigenesis. EMBO Rep. 2016, 17, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.J.; Dwight, T.; Gill, A.J.; Dickson, K.A.; Zhu, Y.; Clarkson, A.; Gard, G.B.; Maideus, T.; Valmadre, S.; Clifton-Bligh, R.; et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunochemistry and massively parallel sequencing. Sci. Rep. 2016, 6, 26191. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Chon, S.M.; Kim, K.R.; Sohn, I.; Suug, C.O. Clinical prevalence of gain-of-function mutation of p53 in high-grade serous ovarian cancer. PLoS ONE 2013, 8, e72609. [Google Scholar]
- Brachovca, P.; Mereting, S.R.; Carlson, M.J.; Goodheart, M.J.; Button, A.M.; Mott, S.L.; Dai, D.; Thiel, K.W.; Devor, E.J.; Leslie, K.K. TP53 oncomorphic mutations predict resistance to platinum- and taxane-based standard chemotherapy in patients diagnosed with advanced ovarian carcinoma. Int. J. Oncol. 2015, 46, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Seagle, B.L.; Eng, K.H.; Daudapani, M.; Yeh, J.; Oduusi, K.; Shahabi, S. Survival of patients with structurally-grouped TP53 mutations in ovarian and breast cancers. Oncotarget 2015, 6, 18641–18652. [Google Scholar] [CrossRef] [PubMed]
- Rzpecka, I.; Szafron, L.; Stys, A.; Buijko, M.; Plisiecka-Halasa, M.; Madry, R.; Osuk, B.; Markowska, J.; Bidzinski, M.; Kupryjanczyk, J. High frequency of allelic loss at BRCA1 locus in ovarian cancers: Clinicopathologic and molecular associations. Cancer Genet. 2012, 205, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Kanchi, K.L.; Johnson, K.J.; Lu, C.; McLellan, M.D.; Leiserson, M.; Wendl, M.C.; Zhang, Q.; Koboldt, D.C.; Xie, M.; Kandoth, C.; et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat. Commun. 2014, 5, 3156. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.; Wubbenhorst, B.; Wenz, B.; De Sloover, D.; Pluta, J.; Emery, L.; Barrett, A.; Kraya, A.; Anastopoulos, I.; Yu, S.; et al. BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat. Commun. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Kostantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous recombination deficiency: Exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.; Shapira-Frommer, R.; Schmulzer, R.K. Olaparib monotherapy in patients with advanced cancer and a germline BRCA 1–2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancers: A preplanned prospective analysis outcomes by BRCA status in a randomized phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Leddermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA 1–2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomized, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: An updated analysis from a randomized, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol. 2016, 17, 1579–1589. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicenter, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL 3): A randomized, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Candido-dos-Reis, F.J.; Song, H.; Goode, A.L.; Cunningham, J.M.; Fridley, B.L.; Larson, M.C.; Alsop, K.; Dicks, E.; Harrington, P.; Ramus, S.J.; et al. Germline mutation in BRCA1 or BRCA2 and ten-year survival for women diagnosed with epithelial ovarian cancer. Clin. Cancer Res. 2015, 21, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Labidi-Galy, S.I.; Olivier, T.; Rodrigues, M.; Ferraioli, D.; Derbel, O.; Bodmer, A.; Petignat, P.; Rak, B.; Chopin, N.; Tredan, O.; et al. Location in BRCA2 gene and survival in patients with ovarian cancer. Clin. Cancer Res. 2017. [Google Scholar] [CrossRef]
- Bashashati, A.; Ha, G.; Ton, A.; Ding, J.; Prentice, L.M.; Roth, A.; Rosner, J.; Shumansky, K.; Kalloger, S.; Senz, J.; et al. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. J. Pathol. 2013, 231, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Hoogstraat, M.; de Patger, M.S.; Cirkel, G.A.; van Roosmalen, M.J.; Hasrkins, T.T.; Duran, K.; Kreeftmeijer, J.; Renkens, I.; Witteveen, P.O.; Lee, C.C.; et al. Genomic and transcriptomic plasticity in treatment-naïve ovarian cancer. Genome Res. 2014, 24, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Sukhbaatar, N.; Bachmayr-Heyda, A.; Auer, K.; Aust, S.; Deycmar, S.; Horvat, R.; Pils, D. Two different, mutually exclusively distributed, TP53 mutations in ovarian and peritoneal tumor tissues of a serous ovarian cancer patient: Indicative for tumor origin? Cold Sproing Harb. Mol. Case Stud. 2017, 3, a001461. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, R.F.; Ng, C.K.; Cooke, S.L.; Newman, S.; Temple, J.; Piskorz, A.M.; Gale, D.; Sayal, K.; Murtaza, M.; Baldwin, P.J.; et al. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: A phylogenetic analysis. PLoS Med. 2015, 12, e1001789. [Google Scholar] [CrossRef] [PubMed]
- Malek, J.A.; Mery, E.; Mashmoud, Y.A.; Al-Azwani, E.K.; Roger, L.; Huang, R.; Jouve, E.; Lis, R.; Thiery, J.P.; Querleu, D.; et al. Copy number variation analysis bofmatched ovarian primary tumors and peritoneal metastasis. PLoS ONE 2011, 6, e28561. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Rhee, J.K.; Hur, S.Y.; Lee, S.H.; Chung, Y.J.; Kim, T.M.; Lee, S.H. Intraindividual genomic heterogeneity of high-grade serous carcinoma of the ovary and clinical utility of ascetic cancer cells for mutation profiling. J. Pathol. 2017, 241, 57–66. [Google Scholar] [CrossRef] [PubMed]
- McPherson, M.; Roth, A.; Laks, E.; Masud, T.; Bashashati, A.; Zhang, A.; Ha, G.; Biele, J.; Yap, D.; Wan, A.; et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat. Genet. 2016, 48, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Pradeep, S.; Kim, S.W.; Wu, S.; Nishimura, M.; Chaluvally-Raghavan, P.; Miyake, T.; Pecot, C.V.; Kim, S.J.; Choi, H.J.; Bischoff, F.; et al. Hematogenous metastasis of ovarian cancer: Rethinking mode of spread. Cancer Cell 2014, 26, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Song, H.L.; Dicks, E.; Ramus, S.J.; Tyrer, J.P.; Intermaggio, M.P.; Hayward, J.; Edlund, C.K.; Conti, D.; Harrington, P.; Fraser, L.; et al. Contribution of germline mutations in the RAD51B, RD51C, and RAD51D genes to ovarian cancer in the population. J. Clin. Oncol. 2015, 33, 2901–2907. [Google Scholar] [CrossRef] [PubMed]
- Ramus, S.J.; Song, H.L.; Dicks, E.; Tyrer, J.P.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; Philpott, S.; et al. Germiline mutations in the BRIP1, PALB2, and NBN genes in women with ovarian cancer. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.M.; Cicek, M.S.; Larson, N.B.; Davila, J.; Wang, C.; Larson, M.C.; Song, H.; Dicks, E.M.; Harrington, P.; Wick, M.; et al. Clinical characteristics of ovarian cancer classified by BRCA1, BRCA2, and RAD51C status. Sci. Rep. 2014, 4, 4026. [Google Scholar] [CrossRef] [PubMed]
- Gabai-Kapara, E.; Lahad, A.; Kaufman, B.; Friedman, E.; Segev, S.; Renbaum, P.; Beeri, R.; Gal, M.; Grinshpun-Cohen, J.; Djemal, K.; et al. Population-based screening for breast and ovarian cancer risk due to BRCA1 and BRCA2. Proc. Natl. Acad. Sci. USA 2014, 39, 14205–14210. [Google Scholar] [CrossRef] [PubMed]
- Kuckenbaecher, K.; Ramus, S.; Tyrer, J.; Lee, A.; Shen, H.C.; Beesley, J.; Lawrenson, K.; McGuffog, L.; Healey, S.; Lee, J.M.; et al. Identification a six new susceptibility loci for invasive epithelial ovarian cancer. Nat. Genet. 2015, 47, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Kuckenbaecher, K.; McGuffog, L.; Barrowdale, D.; Lee, A.; Soucy, P.; Dennis, J.; Domchek, S.M.; Robson, M.; Spurdle, A.B.; Ramus, S.J.; et al. Evaluation of polygenic risk scores for breast and ovarian cancer risk prediction in BRCA1 and BRCA2 mutation carriers. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Phelan, C.M.; Kuckenbaecher, K.; Tyrer, J.P.; Kar, S.P.; Lawrenson, K.; Winham, S.J.; Dennis, J.; Pirie, A.; Riggan, M.J.; Chornokur, G.; et al. Identification of 12 new susceptibility loci for different histotypes of epithelial ovarian cancer. Nat. Genet. 2017, 49, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.X.; Ma, H.X.; Li, L.; Zang, R.Y.; Wang, C.; Song, F.J.; Shi, T.; Yu, D.; Yang, M.; Xue, W.; et al. Genome-wide association study identifies new susceptibility loci for epithelial ovarian cancer in Han Chinese women. Nat. Commun. 2014, 5, 4682. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.L.; Powell, C.B.; Chen, L.M.; Carter, J.; Bae Jump, V.L.; Parker, L.P.; Borowsky, M.E.; Gibb, R.K. Society of Gynecologic Oncology recommendations for the prevention of ovarian cancer. Cancer 2015, 121, 2108–2120. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.B.; Kenley, E.; Chen, L.M.; Crawford, B.; McLennan, J.; Zaloodak, C.; Kamoronij, M.; Beattie, M.; Ziegler, J. Risk reducing spang-oophorectomy in BRCA mutation carriers: Role of serial sectioning in the detection of occult malignancy. J. Clin. Oncol. 2005, 23, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Tothill, R.W.; Tinker, A.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [PubMed]
- Helland, A.; Anglesio, M.; George, J.; Cowin, P.A.; Johnstone, C.N.; House, C.M.; Sheppard, K.E.; Etemadmoghadam, D.; Melnyk, N.; Rustgi, A.K.; et al. Deregulation of MYCN, LIN28B and LET-7 in a molecular subtype of aggressive high-grade serous ovarian cancers. PLoS ONE 2011, 6, e18064. [Google Scholar] [CrossRef] [PubMed]
- Dressman, H.; Berchuck, A.; Chen, G.; Zhai, J.; Bild, A.; Sayer, R.; Cragun, J.; Clarke, J.; Whitaker, R.S.; Li, L.; Gray, J.; et al. An integrated genomic-based approach to individualized treatment of patients with advanced-stage ovarian cancer. J. Clin. Oncol. 2007, 25, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Crijns, A.; Fehrmann, R.; de Jong, S.; Gerbens, F.; Meersma, G.J.; Klip, H.G.; Hollema, H.; Hofstra, R.M.; te Meerman, G.J.; de Vries, E.G.; et al. Survival-related profile, pathways, and transcription factors in ovarian cancer. PlOS Med. 2009, 6, e1000024. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Tsumoda, T.; Shigemizu, D.; Fujiwara, H.; Hatal, M.; Tanaka K for the Japanese Serous Ovarian Cancer Study Group. High-risk ovarian cancer based on 126-gene expression signature is uniquely characterized by downregulation of antigen presentation pathway. Clin. Cancer Res. 2012, 18, 1374–1385. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; D’Andrea, A.; Kozono, D. A DNA repair pathway-focused score for projection of outcomes in ovarian cancer treated with platinum-based chemotherapy. J. Natl. Cancer Inst. 2012, 104, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Mok, S.; Bonome, T.; Vathipadiekal, V.; Bell, A.; Johnson, M.E.; Wong, K.K.; Park, D.C.; Hao, K.; Yip, D.K.; Donninger, H.; et al. A gene signature predictive for outcome in advanced ovarian cancer identifies a survival factor: Microfibril Associated Glycoprotein 2. Cancer Cell 2009, 16, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Stronach, E.A.; Alfraidi, A.; Rama, N.; Datler, C.; Studd, J.B.; Agarwal, R.; Guney, T.G.; Gourley, C.; Hennessy, B.T.; Mills, G.B.; et al. HDCA4 regulated STAT1 activation mediates platinum resistance in ovarian cancer. Cancer Res. 2011, 71, 4412–4422. [Google Scholar] [CrossRef] [PubMed]
- Stronach, E.A.; Chen, M.; Maginn, E.N.; Agarwal, R.; Mills, G.B.; Wasan, H.; Gabra, H. DNA-PK mediates AKT activation and apoptosis inhibition in clinically acquired platinum resistance. Neoplasia 2011, 13, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.; Tamayo, P.; Yang, J.Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Investig. 2013, 123, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Konecny, G.; Wang, C.; Hemidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.W.; Dowdy, S.; Cliby, W.; et al. Prognostic and therapeutic relevance of molecular subtypes ih high-grade serous ovarian cancer. J. Natl. Inst. Cancer 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, Y.; Sun, N.; Wang, D.; Boyd-Kirkup, J.; Dou, X.; Han, J.D. Integrating genomic, epigenomic, and transcriptomic features reveals modular signatures underlying poor prognosis in ovarian cancer. Cell Rep. 2013, 4, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.Z.; Miow, Q.H.; Huang, R.Y.; Wong, M.K.; Ye, J.; Lau, J.A.; Wu, M.C.; Bin Abdul Hadi, L.H.; Soong, R.; Choolani, M.; et al. Functional genomics identifies five distinct molecular subtypes with clinical relevance and pathways for growth control in epithelial ovarian cancer. EMBO Mol. Med. 2013, 5, 983–998. [Google Scholar] [CrossRef] [PubMed]
- Asad, M.; Wong, M.K.; Tan, T.Z.; Choolani, M.; Low, J.; Mori, S.; Virshup, D.; Thiery, J.P.; Huang, R.Y. FZD7 drives in vitro aggressiveness in Stem-A subtype of ovarian cancer via regulation on non-canonical Wnt/PCP pathway. Cell Death 2014, 5, e1346. [Google Scholar] [CrossRef] [PubMed]
- Riester, M.; Wei, W.; Waldron, L.; Culhane, A.C.; Trippa, L.; Oliva, E.; Kim, S.H.; Michor, F.; Huttenhower, C.; Parmigiani, G.; et al. Risk prediction for late-stage ovarian cancer by meta-analysis of 1525 patient samples. J. Natl. Cancer. Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Cheon, D.J.; Tong, Y.; Sim, M.S.; Dering, J.; Berel, D.; Cui, X.; Lester, J.; Beach, J.A.; Tighiouart, M.; Walts, A.E.; et al. A collagen-remodeling gene signature regulated by TGF-beta signaling is associated with metastasis and poor survival in serous ovarian cancer. Clin. Cancer Res. 2014, 20, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Leong, H.S.; Galletta, L.; Etamadmoghadam, D.; George, J.; The Australian Ovarian Cancer Study; Kobel, M.; Ramus, S.J.; Bowtell, D. Efficient molecular subtype classification of high-grade serous ovarian cancer. J. Pathol. 2015, 236, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Armasu, S.; Kalli, K.; Maurer, M.; Heinzen, E.; Keeny, G.; Ciliby, W.; Oberg, A.; Kaufmann, S.; Goode, E. Pooled clustering of high-grade serous ovarian cancer gene expression leads to novel consensus subtypes associated with survival and surgical outcomes. Clin. Cancer Res. 2017, 23, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Murakami, R.; Matsumura, N.; Mandai, M.; Yoshihara, K.; Hamanishi, J.; Yamaguchi, K.; Baba, T.; Koshiyama, M.; Enomoto, T.; Okamoto, A.; et al. Establishment of a novel histopathological classification of high-grade serous ovarian carcinoma correlated with prognostically distinct gene expression subtypes. Am. J. Pathol. 2016, 186, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Chen, L.L.; Zhou, X.H. Identifying prognostic signature in ovarian cancer using DirGenerank. Oncotarget 2017, 8, 46398–46413. [Google Scholar] [CrossRef] [PubMed]
- Matondo, A.; Jo, Y.H.; Shahid, M.; Choi, T.G.; Nguyen, M.N.; Nguyen, N.N.; Aketr, S.; Kang, I.; Ha, J.; Maeng, C.H.; et al. The prognostic 97 chemoresponse gene signature in ovarian cancer. Sci. Rep. 2017, 7, 96989. [Google Scholar] [CrossRef] [PubMed]
- Ovarian Tumor Tissue Analysis (OTTA) Consortium; Goode, E.L.; Block, M.S.; Kalli, K.R.; Vierkant, R.A.; Chen, W.; Fogarty, Z.C.; Gentry-Maharaj, A.; Toloczko, A.; Hein, A.; et al. Dose-response association of CD8+ tumor-infiltrating lymphocytes and survival time in high-grade serous ovarian cancer. JAMA Oncol. 2017, 3, e173290. [Google Scholar]
- Jimenez-Sanchez, A.; Memon, D.; Pourpe, S.; Veeraraghavan, H.; Li, Y.; Vargas, H.A.; Gill, M.B.; Park, K.J.; Zivanovic, O.; Konner, J.; et al. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell 2017, 170, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Lindermann, K.; Anderson, L.; Fereday, S.; Hung, J.; Alsop, K.; Tothill, R.W.; Gebski, V.; Kennedy, C.; Balleine, R.; et al. Serous ovarian and primary peritoneal cancers: A comparative analysis of clinic-pathological features, molecular subtypes and treatment outcome. Gynecol. Oncol. 2016, 142, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Kindelberger, D.W.; Lee, Y.; Miron, A.; Hirsch, M.S.; Feltmate, C.; Medeiros, F.; Callahan, M.J.; Garner, E.O.; Gordon, R.W.; Birch, C.; et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: Evidence for a causal relationship. Am. J. Surg. Pathol. 2007, 31, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Kurman, R.J.; Vang, R.; Sehdev, A.S.; Han, G.; Soslow, R.; Wang, T.L.; Shih, I.-M. TP53 mutations in serous intraepithelial carcinoma and concurrent pelvis high-grade serous carcinoma: Evidence supporting the clonal relationship of the two lesions. J. Pathol. 2012, 226, 421–426. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, A.S.; Stall, J.N.; Hovelson, D.H.; Cani, A.K.; Liu, C.J.; Tomlins, S.A.; Cho, K.R. Next-generation sequencing of tubal intra-epithelial carcinomas. JAMA Oncol. 2015, 1, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Ducie, J.; Dao, F.; Considine, M.; Olvera, N.; Show, P.; Kurman, R.J.; Shih, I.-M.; Soslow, R.A.; Cope, L.; Levine, D.A. Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nat. Commun. 2017, 8, 990. [Google Scholar] [CrossRef] [PubMed]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Wang, T.L.; Doberstein, K.; Bahadirli-Talbott, A.; Ayhan, A.; Sehdev, A.S.; Drapkin, R.; Kurman, R.J.; Shi, I.-M. CCNE1 amplification and centrosome number abnormality in serous tubal intraepithelial carcinoma: Further evidence supporting its role as a precursor of ovarian high-grade serous carcinoma. Mod. Pathol. 2016, 29, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- George, E.M.; Herzog, T.J.; Neugut, A.; Lu, J.S.; Burke, W.M.; Lewin, S.N.; Hershman, D.L.; Wright, J.D. Carcinosarcoma of the ovary: Natural history, patterns oif treatment, and outcome. Gynecol. Oncol. 2013, 131, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Ardighieri, L.; Mori, L.; Conzadori, S.; Bugatti, M.; Falchetti, M.; Donzell, C.M.; Ravaggi, A.; Odicino, P.E.; Facchetti, F. Identical TP53 mutations in pelvic carcinosarcomas and associated serous tubal intraepithelial carcinomas provide evidence of their clonal relatioship. Virchows Arch. 2016, 469, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Bellone, S.; Lopez, S.; Thakral, D.; Schwab, C.; English, D.P.; Black, J.; Cocco, E.; Choi, J.; Zammataro, L.; et al. Mutational landscape of uterine and ovarian carcinosarcomas implicates histone genes in epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2016, 113, 12238–12243. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Stransky, N.; McCord, C.L.; Cerami, E.; Lagowski, J.; Kelly, D.; Angiouli, S.D.; Sansen, M.; Kann, L.; Shukla, M.; et al. Genomic analyses of gynecologic carcinosarcomas reveal frequent mutations in chromatin remodeling genes. Nat. Commun. 2014, 5, 5006. [Google Scholar] [CrossRef] [PubMed]
- Cherniack, A.D.; Shen, D.; Walter, V.; Stewart, C.; Murray, B.A.; Bowlby, R.; Ling, S.; Soslow, R.A.; Bruadduys, R.R.; Zuna, R.E.; et al. integrated molecular characterization of uterine carcinosarcoma. Cancer Cell 2017, 31, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.H.; Hsieh, T.H.; Hsu, C.Y.; Lin, H.Y.; Long, C.Y.; Cheng, K.H.; Tsai, E.M. KRAS mutation coupled with p53 loss is sufficient to induce ovarian carcinosarcoma in mice. Int. J. Cancer 2017, 140, 1860–1869. [Google Scholar] [CrossRef] [PubMed]
- Kaldawy, A.; Segev, Y.; Lavie, O.; Auslender, R.; Sopik, V.; Narod, S.A. Low-grade serous ovarian cancer: A review. Gynecol. Oncol. 2016, 143, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Grisham, R.N.; Sylvester, B.E.; Won, H.; McDermott, G.; DeLair, D.; Ramirez, R. Extreme outlier analysis identifies occult mitogen-actyivated protein kinase pathway mutations in patients with low-grade serous ovarian cancer. J. Clin. Oncol. 2015, 33, 4099–4105. [Google Scholar] [CrossRef] [PubMed]
- Grisham, R.N.; Iyer, G.; Garg, K.; Hyman, D.M.; Zhou, Q. BRAF mutation is associated with early stage disease and improved outcome in patients with low-grade serous ovarian cancer. Cancer 2015, 119, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Gershenson, D.M.; Sun, C.C.; Wong, K.K. Impact of mutational status on survival in low-grade serous carcinoma of the ovary or peritoneum. Br. J. Cancer 2015, 113, 1254–1258. [Google Scholar] [CrossRef] [PubMed]
- Emmanuel, C.; Chiew, Y.E.; George, J.; Etemadmoghadam, D.; Anglesio, M.S.; Sharma, R.; Russell, P.; Kennedy, C.; Fereday, S.; Hung, J.; et al. Genomic classification of serous ovarian cancer with adjacent borderline differentiates RAS pathway and TP53-mutant tumors and identifies NRAS as an ovarian driver. Clin. Cancer Res. 2014, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ardighieri, L.; Zeppernick, F.; Hannibal, C.G.; Vang, R.; Cope, L.; Junge, J.; Kjaer, S.K.; Kurman, R.J.; Shih, I.-M. Mutational analysis of BRAF and KRAS in ovarian serous borderline (atypical proliferative) tumours and associated peritoneal implants. J. Pathol. 2014, 232, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.M.; Anglesio, M.S.; Ryland, G.L.; Sharma, R.; Chiew, Y.; Rowley, S.M.; Doylee, M.; Li, J.; Gilks, B.; Moss, P.; et al. Molecular profiling of low grade serous ovarian tumours identifies novel candidate driver genes. Oncotarget 2015, 6, 37663–37672. [Google Scholar] [CrossRef] [PubMed]
- Etemadmoghadam, D.; Azar, W.; Lei, Y.; Moujaber, T.; Garsed, D.W.; Kennedy, C.J.; Fereday, S.; Mitchell, C.; Chiew, Y.E.; Hendley, J.; et al. EIF1AX and NRAS mutations co-occur and cooperate in low-grade serous ovarian carcinomas. Cancer Res. 2017, 77, 4268–4278. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.M.; Gorring, K.L.; Christie, M.; Rowley, S.M.; Bowtell, D.D.; Australian Ovarian Cancer Study Group; Campbell, I.G. Pre-invasive ovarian mucinous tumors are characterized by CDKN2A and RAS pathway aberrations. Clin. Cancer Res. 2012, 18, 5267–5277. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, M.Y.; Ruan, A.; Chen, C.K.; Liu, H.P.; Wang, C.J.; Chao, W.R.; Han, C.P. Multipoint Kras oncogene mutations potentially indicate mucinous carcinoma on the entire spectrum of mucinous ovarian neoplasms. Oncotarget 2016, 7, 82097–82103. [Google Scholar] [CrossRef] [PubMed]
- Ryland, G.L.; Hunter, S.M.; Doyle, M.A.; Rowley, S.M.; Christie, M.; Allan, P.E.; Bowtell, D.D.; Australian Ovarian Cancer Study Group; Gorring, K.L.; Campbell, I.G. RNF43 is a tumor suppressor gene mutated in mucinous tumours of the ovary. J. Pathol. 2013, 229, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Ryland, G.L.; Hunter, S.M.; Doyle, M.A.; Caramia, F.; Li, J.; Rowley, S.M.; Christie, M.; Allan, P.E.; Stephens, A.N.; Bowtell, D.D.; et al. Mutational landscape of mucinous ovarian carcinoma and its neoplastic precursors. Gen. Med. 2015, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, R.; Kommoss, S.; Winterhoff, B.J.; Kipp, B.R.; Garcia, J.J.; Voss, J.; Halling, K.; Karnezis, A.; Senz, J.; Yang, W.; et al. Targeted deep sequencing of mucinous ovarian tumors reveals multiple overlapping RAS-pathway activating mutations in borderline and cancerous neoplasms. BMC Cancer 2015, 15, 415. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Ayhan, A.; Shih, I.-M.; Seidman, J.D.; Kurman, R.J. Ovarian Brenner tumour: A morphologic and immunohistochemical analysis suggesting an origin from fallopian tube epithelium. Eur. J. Cancer 2013, 49, 3839–3849. [Google Scholar] [CrossRef] [PubMed]
- Cuatrecasas, M.; Catasus, L.; Palacios, J.; Prat, J. Transitional cell tumors of the ovary: A comparative clinopathologic, immunoihistochemical and molecular genetic analysis of Brenner tumors and transitional cell carccinomas. Am. J. Surg. Pathol. 2009, 33, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, E.; Ayhan, A.; Shih, I.; Seidman, J.D.; Kurman, R.J. The pathogenesis of atypical proliferative Brenner tumor: An immunohistochemical and molecular genetic analysis. Mod. Pathol. 2014, 27, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Tafe, L.J.; Muller, K.E.; Ananda, G.; Mitchell, T.; Spotlow, V.; Patterson, S.E.; Tsongalis, G.J.; Mockus, S.M. Molecular genetic analysis of ovarian Brenner tumors and associated mucinous epithelial neoplasms. Am. J. Pathol. 2016, 186, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Pfarr, N.; Darb-Esfahani, S.; Leichensering, J.; Taube, E.; Boxberg, M.; Braicu, I.; Jesinghaus, M.; Penzel, R.; Endris, V.; Noske, A.; et al. Mutational rpofiles of Brenner tumors show distinctive features uncoupling urothelial carcinomas and ovarian carcinoma with transitional cell histology. Genes Chrom. Cancer 2017, 56, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, R.; Shwartz, L.E.; Haley, L.; Lin, M.; Shih, I.-M.; Kurman, R.J. Clonally analysis of combined Brenner and mucinous tumors of the ovary reveals their monoclonal origin. J. Pathol. 2015, 23, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.R.; Kardia, S.; Shedden, K.; Kuick, R.; Michailidis, G.; Taylor, J.M.; Misek, D.E.; Wu, R.; Zhai, Y.; Darrah, D.M.; et al. Gene expression in ovarian cancer reflects both morphology and biological behavior, distinguishing clear cell from other poor-prognosis ovarian carcinomas. Cancer Res. 2002, 62, 4722–4729. [Google Scholar] [PubMed]
- Tan, D.S.; Lambros, M.D.; Rayter, S.; Natrajan, R.; Vatcheva, R.; Gao, Q.; Marchiò, C.; Geyer, F.C.; Savage, K.; Parry, S.; et al. PPM1D is a potential therapeutic target in ovarian clear cell carcinomas. Clin. Cancer Res. 2009, 15, 2269–2280. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.T.; Mao, T.L.; Chen, X.; Feng, Y.; Nakayama, K.; Wang, Y.; Glas, R.; Ma, M.J.; Kurman, R.J.; Shih, I.-M.; et al. DNA copy numbers profiles in affinity-purified ovarian clear cell carcinoma. Clin. Cancer Res. 2010, 16, 1997–2008. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, H.; Ito, Y.M.; Ohashi, Y.; Wong, K.K.; Hashiguchi, Y.; Welch, W.R.; Berkowitz, R.S.; Birrer, M.J.; Mok, S.C. Identification of overexpression and amplification of ABCF2 in clear cell ovarian adenocarcinomas by cDNA microarray analyses. Clin. Cancer Res. 2005, 11, 6880–6888. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Iravani, M.; McCluggage, G.; Lambros, M.B.; Milanezi, F.; Mackay, A.; Gourley, C.; Geyer, F.C.; Vatcheva, R.; Millar, J.; et al. Genomic analysis reveals the molecular heterogeneity of ovarian clear cell carcinomas. Clin. Cancer Res. 2011, 17, 1521–1534. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.T.; Mao, T.L.; Jones, S.; Veras, E.; Ayhan, A.; Wang, T.L.; Glas, R.; Slamon, D.; Velculescu, V.E.; Kuman, R.J.; et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am. J. Pathol. 2009, 174, 1597–1601. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, Y.; Tokunaga, H.; Saito, S.; Shomikawa, K.; Katsuoka, F.; Bin, L.; Kojima, K.; Nagasaki, M.; Yamamoto, M.; Yaegashi, N.; et al. Identification of somatic genetic alterations in ovarian clear cell carcinoma with next generation sequencing. Genes Chromosomes Cancer 2018, 57, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Murakami, R.; Matsumura, N.; Bown, J.B.; Higasa, K.; Tsutsumi, T.; Kamada, M.; Abou-Taleb, H.; Hosoe, Y.; Kitamura, S.; Yamaguchi, K.; et al. Exome sequencing landscape analysis in ovarian cklear cell carcinoma shed light on key chromosomal regions and mutation gene networks. Am. J. Pathol. 2017, 187, 2246–2258. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.C.; Ayhan, A.; Maeda, D.; Kim, K.R.; Clarke, B.A.; Shaw, P.; Chui, M.H.; Shih, I.-M.; Wang, T.L. Frequent somatic mutations of the telomerase reverse transcriptase promoter in ovarian clear cell carcinoma but not in other types of gynecologic malignancies. J. Pathol. 2014, 232, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Ayhan, A.; Kuhn, E.; Wu, R.C.; Ogawa, H.; Bahadirli-Talbott, A.; Mao, T.L.; Sugimura, H.; Shih, I.M.; Wang, T.L. CCNE1 copy-number gain and overexpression identify ovarian clear cell carcinoma with a poor prognosis. Mod. Pathol. 2017, 30, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, K.; Shah, S.; Al-Agha, O.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Wang, T.L.; Shih, I.M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Awedallah, A.; Xin, W. Loss of ARID1A/BAF250a expression in ovarian endometriosis and clear cell carcinoma. Int. J. Clin. Exp. Pathol. 2012, 5, 642–650. [Google Scholar] [PubMed]
- Anglesio, M.S.; Papadopoulos, N.; Ayhan, A.; Nazeran, T.M.; Noe, M.; Harlings, H.M.; Lum, A.; Joney, S.; Senza, J.; Seckin, T.; et al. Cancer-associated mutations in endometriosis without cancer. N. Engl. J. Med. 2017, 376, 1835–1848. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Tsuda, H.; Takano, M.; Tamai, S.; Matsubara, O. Loss of ARID1A protein expression occurs as an early event in ovarian clear-cell carcinoma development and frequently coexist with PIK3CA mutations. Mod. Pathol. 2012, 25, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, A.; Nakayama, K.; Rahman, M.T.; Rahman, M.; Katagiri, H.; Nakayama, N.; Ishikawa, M.; Ishibashi, T.; Iida, K.; Kobayashi, H.; et al. Loss of ARID1A expression is related to shorter progression-free survival and chemoresistance in ovarian clear cell carcinoma. Mod. Pathol. 2012, 25, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.; Wang, T.L.; Shih, L.M. ARID1A, a factor that promotes formation of SWI/SNF-mediated remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011, 71, 6718–6727. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.; Rahmanto, Y.S.; Wu, R.C.; Wang, Y.; Wang, T.L.; Shih, I.-M. Roles of deletion of Arid1A, a tumor suppressor, in mouse ovarian tumorigenesis. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Yee, D.; Damraruer, J.S.; Raab, J.R.; Schisler, J.C.; Wilkerson, M.D.; Didion, J.P.; Starmer, J.; Serber, D.; Yee, D.; Xiong, J.; et al. Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signaling. Nat. Commun. 2015, 6, 6118. [Google Scholar]
- Bitler, B.G.; Wu, S.; Park, P.H.; Hai, Y.; Aird, K.M.; Wang, Y.; Zhai, Y.; Kossenkov, A.V.; Vara-Ailor, A.; Rauscher, F.J.; et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat. Cell Biol. 2017, 19, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Tsuda, H.; Miyai, K.; Takano, M.; Tamai, S.; Matsubara, O. Accumulative copy number increase of MET drives tumor development and histological progression in a subset of ovarian clear-cell carcinomas. Mod. Pathol. 2012, 25, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Mandai, M.; Oura, T.; Matsumura, N.; Hamanishi, J.; Baba, T.; Matsui, S.; Murphy, S.K.; Konishi, I. Identification of an ovarian clear cell carcinoma gene signature that reflects inherent disease biology and carcinogenic processes. Oncogene 2010, 29, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Mandai, M.; Matsumura, N.; Yamaguchi, K.; Kondoh, H.; Amano, Y.; Baba, T.; Hamanishi, J.; Abiko, K.; Kosaka, K.; et al. Hepatocyte nuclear factor-1beta (HNF-1beta) promotes glucose uptake and glycolytic activity in ovarian clear cell carcinoma. Mol. Carcinog. 2015, 54, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Marquez, R.; Baggerly, K.; Patterson, A.; Liu, J.; Broaddus, R.; Frumovitz, M.; Atkinson, E.N.; Smith, D.I.; Hartmann, L.; Fishman, D.; et al. Patterns of gene expression in different histiotypes of epithelial ovarian cancer correlate with those in normal Fallopian tube, endometrium and colon. Clin. Cancer Res. 2005, 11, 6116–6126. [Google Scholar] [CrossRef] [PubMed]
- Wamunyokoli, F.; Bonome, T.; Lee, J.Y.; Feltmate, C.M.; Welch, W.R.; Radonovich, M.; Pise-Masison, C.; Brady, J.; Hao, K.; Berkowitz, R.S.; et al. Expression profiling of mucinous tumors of the ovary identifies genes of clinopathological importance. Clin. Cancer Res. 2006, 12, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Stany, M.; Vathipadiekal, V.; Ozbun, L.; Stone, R.L.; Mok, S.C.; Xue, H.; Kagami, T.; Wang, Y.; McAlpine, J.N.; Bowtell, D.; et al. Identification of novel therapeutic targets in microdissected clear cell ovarian cancers. PLoS ONE 2011, 6, e21121. [Google Scholar] [CrossRef] [PubMed]
- Mc Conechy, M.; Farkkila, A.; Horling, H.; Tolhou, A.; Unkila-Kallio, L.; van Meurs, H.; Yang, W.; Rizenberg, N.; Andersson, N.; Zaby, K.; et al. Molecularly defined adult granulosa cell tumor of the ovary: The clinical phenotype. J. Natl. Cancer Inst. 2016, 108, djw134. [Google Scholar] [CrossRef] [PubMed]
- Rambau, P.F.; McIntyre, J.B.; Taylior, J.; Lee, S.; Ogilvie, T.; Sienko, A.; Morris, D.; Duggan, M.A.; McCluggage, W.G.; Kobel, M. Morphologic reproducibility, genotyping, and immunohistochemical profiling do not support a category of seromucinous carcinoma of the ovary. Am. J. Surg. Pathol. 2017, 41, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Kobel, M.; Senz, J.; Morin, R.D.; Clarke, B.A.; Wiegand, K.C.; Leung, G.; Zayed, A.; Mehl, E.; Kalloger, S.E.; et al. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N. Engl. J. Med. 2009, 360, 2719–2729. [Google Scholar] [CrossRef] [PubMed]
- Nolan, A.; Joseph, N.M.; Sangoi, A.R.; Rabban, J.; Zaludek, C.; Grag, K. FOXL2 status in granulosa theca cell tumors of the ovary. Int. J. Gynecol. Pathol. 2017, 36, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Caburet, S.; Anttonen, M.; Todecsini, A.L.; Unkila-Kallio, L.; Mestivier, D.; Butzow, R.; Veitia, R.A. Combined comparative genomic hybridization and transcriptomic analyses of ovarian granulosa cell tumors point to novel candidate driver genes. BMC Cancer 2015, 15, 251. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, S.; Anglesio, M.S.; Nazeran, T.M.; Lum, A.; Inone, M.; Ilda, Y.; Takano, H.; Nikaido, T.; Okamoto, A.; Huntsman, D.G. Clinical and genetic analysis of recurrent adult-type granulosa cell tumor of the ovary: Persistent preservation of heterozygous C.402>G FOXL2 mutation. PLoS ONE 2017, 12, e0178989. [Google Scholar] [CrossRef] [PubMed]
- Bessiere, L.; Todeschini, A.L.; Auguste, A.; Sarnacki, S.; Frotters, D.; Legoius, B.; Sultan, C.; Kalfa, N.; Galminche, L.; Veitia, R.A. A hot-spot of in-frame duplication activates the oncoprotein AKT1 in juvenile granulosa cell tumors. EBioMedicine 2015, 2, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Auguste, A.; Bessiere, L.; Todecshini, L.; Caburet, S.; Samacki, S.; Prat, J.; D’Angelo, E.; De La Grange, P.; Ariste, O.; Lemoine, F.; et al. Molecular analyses of juvenile granulosa cell tumors bearing AKT1 mutations provide insights into tumor biology and therapeutic leads. Hum. Mol. Genet. 2015, 24, 6687–6697. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Ebbert, K.; Cordeiro, M.H.; Romero, M.M.; Whelan, K.A.; Suarez, A.A.; Woodruff, T.K.; Kirita, T. Constitutive activation of PI3K in oocyte induces ovarian granulosa cell tumors. Cancer Res. 2016, 76, 3851–3861. [Google Scholar] [CrossRef] [PubMed]
- Kalfa, N.; Ecochard, A.; Patte, C. Activating mutations of the stimulatory g protein in juvenile ovarian granulosa cell tumors: A new prognostic factor? J. Clin. Endocrinol. Metab. 2006, 91, 1842–1847. [Google Scholar] [CrossRef] [PubMed]
- Emerson, R.E.; Wang, M.; Roth, L.M.; Zheng, W.; Abdul-Karim, F.W.; Liu, F.; Ulbright, T.M.; Eble, J.N.; Cheng, L. Molecular genetic evidence supporting the neoplastic nature of the Leydig component of ovarian Sertoli-Leydig cell tumors. Int. J. Gynecol. Pathol. 2007, 26, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Heravi-Moussavi, A.; Anglesio, M.S.; Cheng, G.S.-W.; Senz, J.; Winnie, Y.; Prentice, L.M.; Fejes, A.P.; Chow, C.; Tone, A.A.; Kalloger, S.; et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N. Engl. J. Med. 2012, 366, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, L.; Mattina, J.; Schonberger, S.; Murray, M.J.; Choong, J.S.; Hunstman, D.G.; Reis-Filho, J.S.; McCluggage, W.G.; Nicholson, J.C.; Coleman, N.; et al. DICER hotspot mutations in non-epithelial gonadal tumors. Br. J. Cancer 2013, 109, 2744–2750. [Google Scholar] [CrossRef] [PubMed]
- Anglesio, M.S.; Wang, Y.; Yang, W.; Senz, J.; Wan, A.; Heravi-Moussavi, A.; Salamanca, C.; Maines-Bandiera, S.; Hunstman, D.G.; Morin, G.B. Cancer-associated somatic DICER1 hotspot mutations cause defective miRNA processing and reverse-strand expression bias to predominantly mature 3p strands through loss of 5p strand cleavage. J. Pathol. 2013, 229, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, J.; Yang, W.; Mo, F.; Senz, J.; Yap, D.; Anglesio, M.; Gilks, B.; Morin, G.B.; Huntsman, D.G. The oncogenic roles of DICER1 RNase IIIb domain mutations in ovarian Sertoli-Leydig cell tumors. Neoplasia 2015, 17, 650–660. [Google Scholar] [CrossRef] [PubMed]
- De Kock, L.; Terzic, T.; McCluggage, W.G.; Stewart, C.J.R.; Show, P.; Foulkes, W.D.; Clarke, B.A. DICER1 mutations are consistently present in moderately and poorly differentiated Sertoli-Leydig tumors. Am. J. Surg. Pathol. 2017, 41, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Schulz, K.A.; Rednam, S.; Kamihara, J.; Doros, L.; Achatz, M.I.; Wasserman, J.; Diller, L.; Brugières, L.; Druker, H.; Schneider, K.A.; et al. PTEN, DICER1, FH, and their associated tumor susceptibility syndromes: Clinical features, genetics, and surveillance recommendations in childhood. Clin. Cancer Res. 2017, 23, e76–e82. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, L.; Carrott-Zhang, J.; Albrecht, S.; Fahiminiya, S.; Hamel, N.; Tomiak, E.; Grynspan, D.; Saloustros, E.; Nadaf, J.; Rivera, B.; et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat. Genet. 2014, 46, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Merritt, W.; Lin, Y.; Han, L.; Kamat, A.; Spannuth, W.; Schmandt, R.; Urbaner, D.; Pennacchio, L.; Cheng, J.F.; Nick, A.; et al. Dicer, Drosha and outcomes in patients with ovarian cancer. N. Engl. J. Med. 2008, 359, 2641–2650. [Google Scholar] [CrossRef] [PubMed]
- Wtkowski, L.; Goudie, C.; Ramos, P.; Boshari, T.; Brunet, J.S.; Karnezis, A.N.; Longy, M.; Knowst, J.A.; Saloustros, E.; MCCluggage, W.G.; et al. The influence of clinical and genetic factors on patients outcome in small cell carcinoma of the ovary, hypercalcemic type. Gynecol. Oncol. 2016, 141, 454–560. [Google Scholar] [CrossRef] [PubMed]
- Ramos, P.; Karneris, A.; Craig, D. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat. Genet. 2014, 46, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, P.; Mueller, J.J.; Olvera, N.; Dao, F.; Scott, S.N.; Shah, R.; Gao, J.; Schultz, N.; Gouen, M.; Soslow, R.A.; et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat. Genet. 2014, 46, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.I.; Chudnovsky, Y.; Duggan, B.; Zajchowski, D.; Greenbowe, J.; Ross, J.S.; Gay, L.M.; Ali, S.M.; Alvin, J.A. Comprehensive genomic profiling reveals inactivating sMARCA4 mutations and low tumor mutational burden in small cell carcinoma of the ovary, hypercalcemic-type. Gynecol. Oncol. 2017, 147, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Fahiminiya, S.; Witkowski, L.; Nafod, J.; Carrot-Zhang, J.; Goudie, C.; Hasselblatt, M.; Johann, P.; Kool, M.; Lee, R.S.; Gayden, T.; et al. Molecular analyses reveal close similarities between small cell carcinoma of the ovary, hypercalcemic type and atypical teratoid/rhabdoid tumor. Oncotarget 2015, 7, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Karnezis, A.; Wang, Y.; Ramos, P.; Hendricks, W.; Oliva, E.; D’Angelo, E.; Prat, J.; Nucci, M.; Nielsen, T.; Chow, C.; et al. Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcemic type. J. Pathol. 2016, 238, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Conlon, N.; Silva, A.; Guerra, E.; Jelinic, P.; Scglappe, B.A.; Olvera, N.; Mueller, J.; Tornos, C.; Jungbluth, A.; Young, R.H.; et al. Loss of SMARCA4 expression is both sensitive and specific for the diagnosis of small cell carcinoma of ovary, hypercalcemic type. Am. J. Surg. Pathol. 2016, 40, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Sterhl, J.D.; Wachter, D.L.; Fiedler, J. Pattern of SMARCB1 (INI1) and SMARCA4 (BRG1) in poorly differentiated endometrioid adenocarcinoma of the uterus: Analysis of a series with emphasis on a novel SMARCA4-deficient dedifferentiated rhabdoid variant. Ann. Diagn Pathol. 2015, 19, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, P.; Croce, S.; McClugagge, W.G. Loss of expression of SMARCA4 (BRG1), SMARCA2 (BRM) and SMARCAB1 (INI1) in undifferentiated carcinoma of the endometrium is not uncommon and is not always associated with rhabdoid morphology. Histopatology 2017, 70, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, P.; Schlappe, B.; Conlon, N.; Tseng, J.; Olvera, N.; Dao, F.; Mueller, J.; Hussein, Y.; Soslow, R.A.; Levine, D.A. Concomitant loss of SMARCA2 and SMARCA4 expression in small cell carcinoma of the ovary, hypercalcemic type. Mod. Pathol. 2016, 29, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Warholiuc, N.M.; Wigle, T.I.; Klaus, C.R.; Allain, C.J.; Raimondi, A. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef] [PubMed]
- Chan-Panebre, E.; Armstrong, K.; Drew, A.; Feldman, I.; Knutson, S.K.; Kuplast-Barr, K.; Roche, M.; Campbell, J.; Ho, P.; Copeland, R.A.; et al. selective killing of SMARCA2- and SMARCA-deficient small cell carcinoma of the ovary, hytpercalcemic tytpe cells by inhibition of E2ZH2: In vitro and in vivo preclinical models. Mol. Cancer Ther. 2017, 16, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, S.Y.; Karnezis, A.N.; Colborne, S.; Santos, N.D.; Lang, J.D.; Hendricks, W.P.; Orlando, K.A.; Yap, D.; Kommoss, F.; et al. The histone methyltransferase EZH2 is a therapeutic target in small cell carcinoma of the ovary, hypercalcemic type. J. Pathol. 2017, 242, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Kosho, T.; Miyake, N.; Carey, J.C. Coffin-Siris syndrome and related disordera involving components of the BAF (mSWI/SNF) complex: Historical review and recent advances using next generation sequencing. Am. J. Med. Genet. 2014, 166, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Errichiello, E.; Mustafa, N.; Vetro, A.; Notarangelo, L.D.; de Jonge, H.; Rinaldi, B.; Vergani, D.; Giglio, S.R.; Morbini, P.; Zuffardi, O. SMARCA4 inactivating mutations cause concomitant Coffin-Soris syndrome, microphtalmia and small-cell carcinoma of the ovary hypercalcemic type. J. Pathol. 2017, 243, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, L.; Donini, N.; Byler-Dann, R.; Knost, J.A.; Albrecht, S.; Berchuck, A.; McCluggage, W.G.; Hasselblatt, M. The hereditary nature of small cell carcinoma of the ovary, hypercalcemic type: Two new family cases. Fam. Cancer 2017, 16, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Canning, J.; Kaneko, T.; Pru, J.K.; Tilly, J.L. Germline stem cells and follicular renewal in the postnatal mammalian ovary. Nature 2004, 428, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Horan, C.J.; Williams, S.A. Oocyte stem cells: Fact or fantasy? Reproduction 2017, 154, R23–R35. [Google Scholar] [CrossRef] [PubMed]
- Zarate-Garcia, L.; Lane, S.; Jones, J. FACS-sorted putative oogonial stem cells from the ovary are neither DDX4-positive nor germ cells. Sci. Rep. 2016, 6, 27991. [Google Scholar] [CrossRef] [PubMed]
- Szotek, P.P.; Chang, H.L.; Brennand, K.; Fujino, A.; Pieretti-Vanmarcke, R.; Lo Celso, C.; Dombkowski, D.; Preffer, F.; Cohen, K.S.; Teixeira, J.; et al. Normal ovarian epithelial label-retaining cells exhibit stem/progenitor cell characteristics. Proc. Natl. Acad. Sci. USA 2008, 105, 12469–12473. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.L.; Pru, J.K. Long-term label retaining cells localize to distinct regions within the female reproductive epithelium. Cell Cycle 2013, 12, 2888–2898. [Google Scholar] [CrossRef] [PubMed]
- Paik, D.Y.; Janzen, D.M.; Schafenacker, A.M.; Velasco, V.S.; Shung, M.S.; Cheng, D.; Huang, J.; Witte, O.N.; Memarzadeh, S. Stem-like cells are concentrated in the distal end of the fallopian tube: A site for injury and serous cancer initiation. Stem Cells 2012, 30, 2487–2497. [Google Scholar] [CrossRef] [PubMed]
- Flesken-Nikitin, A.; Hwang, C.; Cheng, C.Y.; Michurina, T.V.; Enikolopov, G.; Nikitin, A.Y. Ovarian surface epithelium at the junction area contains a cancer-prone stem cell niche. Nature 2013, 495, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Seidman, J.D. Serous tubal intraepithelial carcinoma localizes to the tubal-peritoneal junction: A pivotal clue to the site of origin of extrauterine high-grade serous carcinoma (ovarian cancer). Int. J. Gynecol. Pathol. 2015, 34, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Schmoeckel, E.; Odai-Afotey, A.; Schleibheimer, M.; Rottmann, M.; Fesken-Nikitin, A.; Ellenson, L.H.; Kirchner, T.; Mayr, D.; Nikitin, A. LEF1 is preferentially expressed in the tubal-peritoneal junctions and is a reliable marker of tubal intraepithelial lesions. Mod. Pathol. 2017, 30, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Steg, A.; Bevis, K.; Katre, A.; Ziebarth, A.; Dobbin, Z.C.; Alvarez, R.D.; Zhang, K.; Conner, M.; Landen, C.N. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin. Cancer Res. 2011, 18, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed]
- Alvero, A.B.; Chen, R.; Fu, H.H.; Montagna, M.; Schwartz, P.E.; Rutherford, T.; Silasi, D.A.; Steffensen, K.D.; Waldstrom, M.; Visintin, I.; et al. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle 2009, 8, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Szotek, P.P.; Pieretti-Vanmarcke, R.; Masiakos, P.T.; Dinelescu, D.M.; Connolly, D.; Foster, R. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian inhibiting substance responsiveness. Proc. Natl. Acad. Sci. USA 2006, 103, 11154–11159. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Dombkowski, D.; Meirelles, K.; Pieretti-Vanmarcke, R.; Szotek, P.P.; Chang, H.L.; Preffer, F.I.; Mueller, P.R.; Teixeira, J.; MacLaughlin, D.T.; et al. Mullerian inhibiting substance preferentially inhibits stem/progenitors in human ovarian cancer cell lines compare with chemotherapeutics. Proc. Natl. Acad. Sci. USA 2009, 107, 18874–18879. [Google Scholar] [CrossRef] [PubMed]
- Slomiany, M.G.; Dai, L.; Tolliver, L.B.; Grass, G.D.; Zeng, Y.; Toole, B.P. Inhibition of functional hyaluronan-CD44 interactions in CD133-positive primary human ovarian carcinoma cells by small hyaluronan oligosaccharides. Clin. Cancer Res. 2009, 15, 7593–7601. [Google Scholar] [CrossRef] [PubMed]
- Curley, M.D.; Therrien, V.A.; Cummings, C.L.; Sergent, P.A.; Koulouris, C.R.; Friei, A.M. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells 2009, 27, 2875–2883. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Convery, P.A.; Matsumura, N.; Whitaker, R.S.; Kondoh, E.; Perry, T. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene 2009, 28, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Landen, C.N.; Goodman, B.; Katre, A.A.; Steg, A.D.; Nick, A.M.; Stone, R.L.; Miller, L.D.; Mejia, P.V.; Jennings, N.B.; Gershenson, D.M.; et al. Targeting aldehyde dehydrogenase cancer stem cells in ovarian cancer. Mol. Cancer Ther. 2010, 9, 3186–3199. [Google Scholar] [CrossRef] [PubMed]
- Bapat, S.A.; Mali, A.M.; Koppikar, C.B.; Kurrey, N.K. Stem and progenitor-like cells contribute to the aggressive behaviour of human epithelial ovarian cancer. Cancer Res. 2005, 65, 3025–3029. [Google Scholar] [CrossRef] [PubMed]
- Wani, A.A.; Sharma, N.; Shouche, Y.S.; Bapat, S.A. Nuclear-mitochondrial genomic profiling reveals a pattern of evolution in epithelial ovarian tumor stem cells. Oncogene 2006, 25, 6336–6344. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.M.; Shaw, P.A.; Gedye, C.; Bernardini, M.Q.; Neel, B.G.; Ailles, L.E. Phenotype heterogeneity and instability of human ovarian tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2011, 108, 6468–6473. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Liu, S.; Roh, M.; Vatan, L.; Szeliga, W.; Wei, S.; Banerjee, M.; Mao, Y.; Kotarski, J.; Wicha, M.S.; et al. Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int. J. Cancer 2011, 130, 29–39. [Google Scholar] [CrossRef] [PubMed]
- McLean, K.; Gong, Y.; Choi, Y.; Deng, N.; Yang, K.; Bai, S.; Cabrera, L.; Keller, E.; McCauley, L.; Cho, K.R.; et al. Human ovarian carcinoma-associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J. Clin. Investig. 2011, 121, 3206–3219. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.; Bai, S.; McLean, K.; Yang, K.; Griffith, K.; Thomas, D.; Ginestier, C.; Johnston, C.; Kueck, A.; Reynolds, R.K.; et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 2011, 71, 3991–4001. [Google Scholar] [CrossRef] [PubMed]
- Condello, S.; Morgan, C.A.; Nagdas, S.; Cao, L.; Turek, J.; Hurley, T.D.; Matei, D. Beta-catenin-regulated ALDH1A is a target in ovarian cancer spheroids. Oncogene 2014, 34, 2297–2308. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Ingram, P.N.; Coffman, L.; Iyengar, M.; Bai, S.; Thomas, D.G.; Buchanovic, R.J. Identifying and ovarian cancer hierarchy regulated by bone morphogenetic protein 2. Proc. Natl. Acad. Sci. USA 2105, 112, E6882–E6888. [Google Scholar] [CrossRef] [PubMed]
- Ruscito, I.; Cacsire Castillo-Tong, D.; Vergote, I.; Ignat, I.; Stanske, M.; Vanderstichele, A.; Ganapathi, R.N.; Glajzer, J.; Kunbe, H.; et al. Exploring the colnal evolution of CD133/aldehyde-dehydrogenase-1 (ALDH1)-positive cancer stem-like cells from primary to recurrent high-grade serous ovarian cancer (HGSOC). A study of the ovarian cancer therapy-innovative models prolong survival. Eur. J. Cancer 2017, 79, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Connor, J.; Al-Niaimi, A.; Rose, S.L.; Mohaian, A. Aldheyde dehydrogenase 1 (ALDH1A) expression by immunohistochemistry is associated with chemio-refractoriness in patients with high-grade ovarian serous carcinoma. Hum. Pathol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Sato, A.; Ohata, H.; Ikarashi, Y.; Takahashi, R.; Ochiya, T.; Yoshida, M.; Tsuda, H.; Onda, T.; Kato, T.; et al. Establishment and characterization of an in vitro model of ovarian cancer stem-like cells with an enhanced proliferatrive capacity. Cancer Res. 2015, 76, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Chui, M.H.; Wang, Y.; Wu, R.C.; Seidman, J.; Kurman, R.J.; Wang, T.L.; Shih, I.-M. Loss of ALDH1A expression in an early event in the pathogenesis of ovarian high-grade serous carcinoma. Mod. Pathol. 2015, 28, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Katz, E.; Shorecki, K.; Tzukerman, M. Niche–dependent tumorigenic capacity of malignant ovarian ascites-derived cancer cell subpopulations. Clin. Cancer Res. 2009, 15, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Tsukeman, M.; Rosenberg, T.; Reiter, I. The influence of a human embryonic stem cell-derived microenvironment on targeting of human solid tumor xenograft. Cancer Res. 2006, 66, 3792–3801. [Google Scholar]
- Abelson, S.; Shamai, Y.; Berger, L.; Shouval, R.; Shorecki, K.; Tzukerman, M. Intratumoral heterogeneity in the self-renewal and tumorigenic differentiation of ovarian cancer. Stem Cells 2012, 30, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Mor, G.; Alvero, A. The duplicitious origin of ovarian cancer. Rambam Naimonides Med. J. 2013, 4, e0006. [Google Scholar]
- Yokoyama, Y.; Zhu, H.; Lee, J.H.; Kossenkov, A.V.; Wu, S.Y.; Wickramasinghe, J.M.; Yin, X.; Palozola, K.C.; Gardini, A.; Showe, L.C.; et al. BET inhibitors suppress ALDH activity by targeting ALDH1A1 super-enhancer in ovarian cancer. Cancer Res. 2016, 76, 6320–6330. [Google Scholar] [CrossRef] [PubMed]
- House, C.D.; Jordan, E.; Hernandez, L.; Ozaki, M.; James, J.M.; Kim, M.; Kuhlak, M.J.; Batchelor, E.; Elloumi, F.; Cam, M.; et al. NF-kB promotes ovarian tumorigenesis via classical pathways that support proliferative cancer cells and alternative pathways that support ALDH+ cancer stem-like cells. Cancer Res. 2017, 77, 6927–6940. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Ingram, P.; Chen, Y.C.; Deng, N.; Pearson, A.; Niknafs, Y.S.; O’Hayer, P.; Wang, Y.; Zhang, Z.Y.; Boscolo, E.; et al. EGLF6 regulates the asymmetric division, maintenance, and metastasis of ALDH+ ovarian cancer cells. Cancer Res. 2016, 76, 6396–6409. [Google Scholar] [CrossRef] [PubMed]
- Chefetz-Menaker, I.; Yang, K.; Buchanovich, R. A novel ALDH1A selective inhibitor induces necroptosis in ovarian cancer stem-line cells. In Proceedings of the 107th Annual Meeting of the American Association for Cancer Research, New Orleans, LA, USA, 16–20 April 2016. [Google Scholar]
- Raghavan, S.; Mehta, P.; Ward, M.R.; Bregenzer, M.E.; Fleck, E.; Tan, L.; McLean, K.; Buchanovic, R.J.; Mehta, G. Personalized medicine-based approach to model patterns of chemoresistance and tumor recurrence using ovarian cancer stem cell spheroids. Clin. Cancer Res. 2017, 23, 6394–6945. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cui, B.; Lai, H.; Liu, G.; Ghia, E.M.; Widhopf, G.F.; Zhang, Z.; Wu, C.C.; Chen, L.; Wu, R.; et al. Ovarian cancer stem cells express ROR1, which can be targeted for anti-cancer stem cell therapy. Proc. Natl. Acad. Sci. USA 2014, 111, 17266–17271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qiu, J.; Yang, D.; Gao, L.; Su, Y. ROR1 expression correlated with poor clinical outcome in ovarian canecr. Sci. Rep. 2014, 4, 5811. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, A.; Goodpaster, T.; Randolpb-Habecker, J.; Hoffstrom, B.; Jalikis, F.; Koch, L.K.; Berger, C.; Kosasih, P.L.; Rajan, A.; Sommermeyer, D.; et al. Analysis of ROR1 protein expression in human canecr and normal tissues. Clin. Cancer Res. 2016, 23, 3061–3071. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, M.; D’Alterio, C.; Camerlingo, R.; Tirino, V.; Consales, C.; Riccio, A.; Ieranò, C.; Cecere, S.C.; Losito, N.S.; Greggi, S.; et al. Identification of a distinct population of CD1332+CXCR4+ cancer stem cells in ovarian cancer. Sci. Rep. 2015, 5, 10357. [Google Scholar] [CrossRef] [PubMed]
- Kasagi, Y.; Harada, Y.; Morodomi, Y.; Iwai, T.; Saito, S.; Yoshida, K.; Oki, E.; Saeki, H.; Ohgaki, K.; Sugiyama, M.; et al. Peritoneal dissemination requires an Sp1-dependent CXCR4/CXCL12 signaling axis and extracellular matrix-directed spheroid formation. Cancer Res. 2016, 76, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Figueras, A.; Alsina-Sanchis, E.; Lahiguera, A.; Abreu, M.; Muinelo-Romay, L.; Moreno-Bueno, G.; Casanovas, O.; Graupera, M.; Matias-Guiu, X.; Vidal, A.; et al. A role for CXCR4 in peritoneal and hematogenous ovarian cancer dissemination. Mol. Cancer Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Ning, G.; Howitt, B.E.; Mehra, K.; Wu, L.; Wang, X.; Hong, Y.; Kern, F.; Wei, T.S.; Zhang, T.; et al. In vitro and in vivo correlates of physiologiacal and neoplastic human Fallopian tube stem cells. J. Pathol. 2016, 238, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Ricci, F.; Bernasconi, S.; Perego, P.; Ganzinelli, M.; Russo, G.; Bono, F.; Mangioni, C.; Fruscio, R.; Signorelli, M.; Broggini, M.; et al. Ovarian carcinoma tumor-initiating cells have a mesenchymal phenotype. Cell Cycle 2012, 11, 1966–1976. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Alvero, A.B.; Craveiro, V.; Holmberg, J.C.; Fu, H.H.; Montagna, M.K.; Yang, Y.; Chefetz-Menaker, I.; Nuti, S.; Rossi, M.; et al. Constitutive proteosomal degradation of TWIST-1 in epithelial-ovarian cancer stem cells impacts differentiation and metastatic potential. Oncogene 2013, 32, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Lin, X.; Liu, Y.; Gong, W.; Ma, X.; Yu, Y.; Xie, Y.; Sun, X.; Feng, Y.; Janzen, V.; et al. Transformation of epithelial ovarian cancer stem like cells into mesenchymal lineage via EMT results in cellular heterogeneity and supports tumor engraftment. Mol. Med. 2012, 18, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Iwanichi, M.P.; Dadidowitz, R.A.; Ng, M.R.; Besser, A.; Muranen, T.; Merritt, M.; Danuser, G.; Ince, T.; Brugge, J.S. Ovarian cancer spheroids use myosin-generated force to clear the mesothelium. Cancer Discov. 2011, 1, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Dadidowitz, R.A.; Selfors, L.M.; Iwanichi, M.P.; Elias, K.M.; Karst, A.; Piao, H.; Ince, T.A.; Drage, M.G.; Dering, J.; Konecny, G.E.; et al. Mesenchymal gene program-expressing ovarian cancer spheroids exhibit enhanced mesothelial clearance. J. Clin. Investig. 2014, 124, 2611–2625. [Google Scholar] [CrossRef] [PubMed]
- Takai, M.; Terai, Y.; Kawaguchi, H.; Ashihara, K.; Fujiwara, S.; Tanaka, T.; Tsunetoh, S.; Tanaka, Y.; Sasaki, H.; Kanemura, M.; et al. The EMT (epithelial-mesenchymal transition)-related protein expression indicates the metastatic status and prognosis in patients with ovarian cancer. J. Ov. Res. 2014, 7, 76. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, E.; Mori, S.; Huang, R.Y.J.; Thiery, J.P. Epithelial-mesenchymaltransition spectrum quantification and its efficacy in deciphering survival and drug resonses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.; Holth, A.; Hellesylt, E.; Tan, T.Z.; Huang, R.Y.; Tropé, C.; Nesland, J.M.; Thiery, J.P. The clinical role of epithelial-mesenchymal transition and stem cell markers in advanced-stage ovarian serous carcinoma effusions. Hum. Pathol. 2015, 46, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Latifi, A.; Luwor, R.; Bilandric, M.; Nazaretian, S.; Stenvers, K.; Pyman, J.; Zhu, H.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Isolation and characterization of tumor cells from the ascites of ovarian cancer patients: Molecular phenotype and chemoresistant ovarian tumors. PLoS ONE 2012, 7, e46858. [Google Scholar] [CrossRef] [PubMed]
- Wea, N.; Wang, Y.; Wen, L.; Zhao, S.H.; Ai, Z.H.; Wang, Y.; Wu, B.; Lu, H.X.; Yang, H.; Liu, W.C.; et al. Overexpression of FOXM1 predicts poor prognosis and promotes cell proliferative migration and invasion in epithelial ovarian cancer. J. Transl. Med. 2014, 12, 134. [Google Scholar]
- Zhang, X.; Cheng, L.; Minn, K.; Madan, R.; Godwin, A.K.; Shridhar, V.; Chien, J. Targeting of mutant p53-induced FoxM1 with thiostrepton induces cytotoxicity and enhances carboplatin sensitivity in cancer cells. Oncotarget 2014, 5, 11365–11380. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.T.; Huang, Y.F.; Tsai, H.Y.; Chen, C.C.; Chang, C.H.; Huang, S.C.; Hsu, K.F.; Chou, C.Y. FOXM1 confers to epithelial-mesenchymal transition, stemness and chemoresistance in epithelial ovarian carcinoma cells. Oncotarget 2015, 6, 2349–2365. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.K.; Ip, C.K.; Mak, A.; Lai, H.C.; Wong, A.S. c-kit mediates chemoresistance and tumor-initiating capacity of ovarian cancers through activation of Wnt/beta-catenin-ATP binding cassette G2 signaling. Oncogene 2012, 32, 2767–2781. [Google Scholar]
- Pagotto, A.; Pilotto, G.; Mazzoldi, E.L.; Nicoletto, M.O.; Frezzini, S.; Pastò, A.; Amadori, A. Autophagy inhibition reduces chemoresistance and tumorigenic potential of human ovarian cancer stem cell. Cell Death Dis. 2017, 8, e2943. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Shin, S.J.; Chung, H.W.; Kwon, S.H.; Cha, S.D.; Lee, J.E.; Cho, C.H. Salinomycin reduces stemness and induces apoptosis on human ovarian cancer stem cell. J. Gynecol. Oncol. 2017, 28, e14. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Yan, X.; Liu, L.; Jiang, C.; Hou, S. Overexpression of the cancer stem cell marker CD117 predicts poor prognosis in epithelial ovarian cancer aptients: Evidence from meta-analysis. Oncol. Targets Ther. 2017, 10, 2951–2961. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2013, 33, 134. [Google Scholar] [CrossRef]
- Xiang, T.; Long, H.; He, L.; Han, X.; Lin, K.; Liang, Z.; Zhuo, W.; Xie, R.; Zhu, B. Interleukin-17 produced by tumor microenvironment promotes self-renewal of CD133+ cancer stem-like cells in ovarian cancer. Oncogene 2015, 34, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Latifi, A.; Abubaker, K.; Castrechini, N.; Ward, A.C.; Liongue, C.; Dobill, F.; Kumar, J.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Cisplatin treatment of primary and metastatic epithelial ovarian carcinomas generates residual cells with mesenchymal stem cell-like profile. J. Cell Biochem. 2011, 112, 2850–2864. [Google Scholar] [CrossRef] [PubMed]
- Meirelles, K.; Benedict, L.A.; Dormbkowski, D.; Pepin, D.; Preffer, F.I.; Teixeira, J.; Tanwar, P.S.; Young, R.H.; MacLaughlin, D.T.; Donahoe, P.K.; et al. Human ovarian cancer stem/progenitor cells are stimulated by doxorubicin but inhibited by Mullerian inhibiting substance. Proc. Natl. Acad. Sci. USA 2012, 109, 2358–2363. [Google Scholar] [CrossRef] [PubMed]
- Yo, Y.T.; Lin, Y.W.; Wang, Y.C.; Balch, C.; Huang, R.L.; Chan, M.W.; Sytwu, H.K.; Chen, C.K.; Chang, C.C.; Nephew, K.P.; et al. Growth inhibition of ovarian tumor-initiating cells by niclosamide. Mol. Cancer Ther. 2012, 11, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- McAuliffe, S.M.; Morgan, S.L.; Wyant, G.A.; Tran, L.T.; Muto, K.W.; Chen, Y.S.; Chin, K.T.; Partridge, J.C.; Poole, B.B.; Cheng, K.H.; et al. Targeting Notch, a key pathway of ovarian cancer stem cells sensitizes tumors to platinum therapy. Proc. Natl. Acad. Sci. USA 2012, 109, E2939–E2948. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.G.; Stoeck, A.; Guan, B.; Wu, R.C.; Zhu, H.; Blackshaw, S.; Shih, I.-M.; Wang, T.L. Notch3 interactoma analysis identified WWP2 as a negative regulator of Notch3 signaling in ovarian cancer. PLoS Genet. 2014, 10, e1004751. [Google Scholar] [CrossRef] [PubMed]
- Siu, M.K.Y.; Wong, E.S.Y.; Kong, D.S.H.; Chan, H.Y.; Jiang, L.; Wong, O.G.; Lam, E.W.; Chan, K.K.; Ngan, H.Y.; Le, X.F.; et al. Stem cell transcription factor NANOG controls cell migration and invasion via dysregulation of E-cadherin and FoxJ1 and contributes to adverse clinical outcome in ovarian cancers. Oncogene 2012, 32, 3500–3509. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Chung, Y.M.; Guan, M.; Ma, M.; Ma, J.; Berek, J.S.; Hu, M.C. Reprogramming ovarian and breast cancer cells into non-cancerous cells by low-dose metformin or SN-38 through FOXO3 activation. Sci. Rep. 2014, 4, 5810. [Google Scholar] [CrossRef] [PubMed]
- Bareiss, P.M.; Paczulla, A.; Wang, H.; Schairer, R.; Wiehr, S.; Kohlhofer, U.; Rothfuss, O.C.; Fischer, A.; Perner, S.; Staebler, A.; et al. SOX2 expression associates with stem cell state in human ovarian carcinoma. Cancer Res. 2013, 73, 5544–5555. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Hou, Y.; Huang, Z.; Cai, J.; Wang, Z. SOX2 is required to maintain cancer stem cells in ovarian cancer. Cancer Sci. 2017, 108, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Belotte, J.; Fletcher, N.M.; Alexis, M.; Morris, R.T.; Munkarah, A.R.; Diamond, M.P.; Saed, G.M. Sox2 gene amplification signifivcantly impacts overall survival in serous epithelial ovariuan cancer. Reprod. Sci. 2015, 22, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Hellnaer, K.; Miranda, F.; Chedom, D.F.; Herrero-Gonzalez, S.; Hayden, D.M.; Tearle, R.; Artibani, M.; Ranjbar, M.; Williams, R.; Gaitskell, K.; et al. Premalignant SOX2 overexpression in the fallopian tubes of ovarian cancer patients: Discovery and validation studies. EBioMedicine 2016, 10, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.J.; Kim, D.K.; Jang, I.H.; Choi, E.J.; Shih, S.H.; Lee, S.I.; Kwon, S.M.; Kim, K.H.; Suh, D.S.; Kim, J.H. Hypoxia-NOTCH1-SOX2 signalling is important for maintaining cancer stem cells in ovarian cancer. Oncotarget 2016, 7, 55624–55638. [Google Scholar] [CrossRef] [PubMed]
- Somasagara, R.R.; Tripathi, K.; Pencer, S.M.; Clark, D.W.; Barnett, R.; Bachaboina, L.; Scalici, J.; Rocconi, R.P.; Piazza, G.A.; Palle, K. Rad6 upregulation promotes stem cell-like characteristics and platinum resistance in ovarian cancer. Biochem. Biophys. Res. Commun. 2016, 469, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Somasagara, R.R.; Spencer, S.M.; Tripathi, K.; Clark, D.W.; Mani, C.; da Silva, L.M.; Scalici, J.; Kothayer, H.; Westwell, A.D.; Rocconi, R.P.; et al. RAD6 promotes DNA repair and stem cell signaling in ovarian cancer and is a promising therapeutic target to prevent and treat acquired chemoresistance. Oncogene 2017, 36, 6680–6690. [Google Scholar] [CrossRef] [PubMed]
- Sanders, M.A.; Haynes, B.; Nangia-Makker, P.; Polin, L.A.; Shekhar, M.P. Pharmacological targeting of RAD6 enzyme-mediated translesion synthesis overcomes resistance to platinum-based drugs. J. Biol. Chem. 2017, 292, 10347–10363. [Google Scholar] [CrossRef] [PubMed]
- Facciabene, A.; Peng, X.; Hagemann, J.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumor hypoxia promotes tolerance and angiogenesis via CCL28 and T reg cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Ma, Y.; Liu, J.; Trope, C.G.; Holm, R.; Nesland, J.; Suo, Z. The hypoxic microenvironment upgrades stem-like properties of ovarian cancer cells. BMC Cancer 2012, 12, 201. [Google Scholar] [CrossRef] [PubMed]
- Quin, Q.; Sun, Y.; Fei, M.; Zhang, J.; Jia, Y.; Gu, M.; Xia, R.; Chen, S.; Deng, A. Expression of putative stem marker nestin and CD133 in advanced serous ovarian cancer. Neoplasma 2012, 59, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guo, X.; Chang, D.Y.; Rosen, D.G.; Mercado-Uribe, I.; Liu, J. CD133 expression associated with poor prognosis in ovarian cancer. Mod. Pathol. 2012, 25, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.; Long, B.; Sullivan, P.; McClellan, S.; Finan, M.A.; Reed, E.; Shevde, L.; Rocconi, R.P. CD44+/CD24- ovarian cancer cells demonstrate cancer stem cell properties and correlate to survival. Clin. Exp. Metastasis 2012, 29, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, K.D.; Alvero, A.B.; Yang, Y.; Waldstrøm, M.; Hui, P.; Holmberg, J.C.; Silasi, D.A.; Jakobsen, A.; Rutherford, T.; Mor, G. Prevalence of epithelial ovarian cancer stem cells correlates with recurrence in early-stage ovarian cancer. J. Oncol. 2011, 2011, e62053. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Yo, Y.T.; Lee, H.Y.; Liao, Y.P.; Chao, T.K.; Su, P.H.; Lai, H.C. ALDH1-bright epithelial ovarian cancer cells are associated with CD44 expression, drug resistance, and poor clinical outcome. Am. J. Pathol. 2012, 180, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Liebscher, C.A.; Prinzler, J.; Sinn, B.V.; Budczies, J.; Denkert, C.; Noske, A.; Sehouli, J.; Braicu, E.I.; Dietel, M.; Darb-Esfahani, S. Aldheyde dehydrogenase 1/epidermal growth factor receptor coexpression is characteric of a highly aggressive, poor-prognosis subgroup of high-grade serous ovarian carcinoma. Hum. Pathol. 2013, 44, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Abubaker, K.; Findlay, J.; Quinn, M. Epithelial mesenchymal transition and cancer stem cell phenotypes facilitate chemoresistance in recurrent ovarian cancer. Curr. Cancer Drug Targets 2010, 10, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Craveiro, V.; Yong-Hartwich, Y.; Holmberg, J.C.; Joo, W.D.; Sumi, N.J.; Pizzonia, J.; Griffin, B.; Gill, S.K.; Silasi, D.A.; Azodi, M.; et al. Phenotypic modifications in ovarian cancer stem cells following Paclitaxel treatment. Cancer Med. 2013, 2, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, S.; Hersey, J.; Mellor, P.; Dai, W.; Santos-Silva, A.; Liber, D.; Luk, L.; Titley, I.; Carden, C.P.; Box, G. Ovarian cancer stem cells-like side populations are enriched following chemotherapy and overexpress EZH2. Mol. Cancer Ther. 2010, 10, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Wintzell, M.; Lofstedt, L.; Johannsson, J.; Pedersen, A.B.; Fuxe, J.; Shoshan, M. Repeated cisplatin treatment can lead to a multiresistant tumor cell population with stem cell features and sensitivity to 3-bromopyruvate. Cancer Biol. Ther. 2012, 13, 1454–1462. [Google Scholar] [CrossRef] [PubMed]
- Hosonuma, S.; Kobayashi, Y.; Kojo, S.; Wada, H.; Seino, K.; Kiguchi, K.; Ishizuka, B. Clinical significance of side population in ovarian cancer cells. Hum. Cell 2011, 24, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Abubaker, K.; Latifi, A.; Luwor, R.; Nazaretian, S.; Zhu, H.; Quinn, M.A.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Short-treatment single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol. Cancer 2013, 12, 24. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, T.; Hirohashi, Y.; Torigoe, T.; Yasuda, K.; Takahashi, A.; Asanuma, H.; Morita, R.; Mariya, T.; Asano, T.; Mizuuchi, M.; et al. ALDH1-high ovarian cancer stem-like cells can be isolated from serous and clear adenocarcinoma cells and ALDH1 high expression is associated with poor prognosis. PLoS ONE 2013, 8, e65158. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Suzuki, N.; Makino, H.; Furui, T.; Morii, E.; Aoki, H.; Kunisada, T.; Yano, M.; Kuji, S.; Hirashima, Y.; et al. Cancer stem-like cells of ovarian clear cell carcinoma are enriched in the ALDH-high population associated with an accelerated scavengening system in reactive oxygen species. Gynecol. Oncol. 2015, 137, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Kawana, K.; Adachi, K.; Fujimoto, A.; Yoshida, M.; Nakamura, H.; Nishida, H.; Inoue, T.; Taguchi, A.; Ogishima, J.; et al. Targeting glutamine metabolism and the focal adhesion kinase additively inhibits the mammalian target of the rapamycin pathway in spheroid cancer stem-like properties of ovarian clear cell carcinoma in vitro. Int. J. Oncol. 2017, 50, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Volinia, S.; Bonome, T.; Calin, G.A.; Greshock, J.; Yang, N.; Croce, C.M. Genomic and epigenetic alterations deregulate microRNA expression in human epithelial ovarian cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 7004–7009. [Google Scholar] [CrossRef] [PubMed]
- Van Jaarseld, M.; Helleman, J.; Berns, E.; Wiemer, E. MicroRNAs in ovarian cancer biology and therapy resistance. Int. J. Biochem. Cell Biol. 2010, 42, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Visone, R.R.; Di Leva, G.; Donati, V.; Petrocca, F.; Casalini, P.; Taccioli, C.; Violinia, S.; Liu, C.G.; Alder, H.; et al. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007, 67, 8699–8707. [Google Scholar] [CrossRef] [PubMed]
- Nam, E.J.; Yoon, H.; Kim, S.W.; Kim, Y.T.; Kim, J.H.; Kim, J.W.; Kim, S. MicroRNA expression profiles in serous ovarian carcinoma. Clin. Cancer Res. 2008, 14, 2690–2695. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-Cadherin repressors Zeb1 and Zeb2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, A.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Choi, P.W.; Ng, S.W. The functions of microRNA-200 family in ovarian cancer: Beyond epithelial-mesenchymal transition. Int. J. Mol. Sci. 2017, 18, 1207. [Google Scholar] [CrossRef] [PubMed]
- Mateescu, B.; Batista, L.; Cardon, M.; Gruosso, T.; de Feraudy, Y.; Mariani, O.; Nicolas, A.; Meyniel, J.P.; Cottu, P.; Sastre-Garau, X.; et al. miR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat. Med. 2011, 17, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Marchini, S.; Cavalieri, D.; Fruscio, R.; Calura, E.; Garavaglia, D.; Fuso Nerini, I.; Mangioni, C.; Cattoretti, G.; Clivio, L.; Beltrame, L.; et al. Association between miR-200c and the survival of patients with stage I epithelial ovarian cancer: A retrospective study of two independent tumor tissue collections. Lancet Oncol. 2011, 12, 273–285. [Google Scholar] [CrossRef]
- Pecot, C.V.; Rupaimoole, R.; Yang, D.; Akbani, R.; Ivan, C.; Lu, C.; Wu, S.; Han, H.D.; Shah, M.Y.; Rodriguez-Aguaayo, C.; et al. Tumor angiogenesis regulation by the miR-200 family. Nat. Commun. 2013, 4, 2427. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Guo, R.; Lin, M.; Zhou, B.; Wang, Y. Micro-RNAa inhibits CD133/1+ ovarian cancer stem cells migration and invasion by targeting E-cadherin repressor 2EB3. Gynecol. Oncol. 2011, 122, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Maihle, N.J.; Huang, Y. Pluripotency factors Lin 28 and Oct4 identify a sub-population of stem cell-like cells in ovarian cancer. Oncogene 2010, 29, 2153–2159. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lin, X.; Zhang, X.; Kaur, S.; Li, N.; Liang, S.; Lassus, H.; Wang, L.; Katsaros, D.; Montone, K.; et al. Double-negative feedback loop between reprogramming factor LIN28 and microRNA let-7 regulates aldehyde dehydrogenase 1-positive cancer stem cells. Cancer Res. 2010, 70, 9463–9472. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Kong, W.; He, L.; Zhao, J.J.; O’Donnell, J.D.; Wang, J.; Wenham, R.M.; Coppola, D.; Kruk, P.A.; Nicosia, S.V.; et al. MicroRNA expression profiling in human ovarian cancer: Mir-214 induces cell survival and cisplatin resistance by targetin PTEN. Cancer Res. 2008, 68, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Alvero, A.B.; Silasi, D.A.; Kelly, M.G.; Fest, S.; Visintin, I.; Leiser, A.; Schwartz, P.E.; Rutherford, T.; Mor, G.; et al. Regulation of IKKbeta by miR-199a affects NF-kappaB activity in ovarian cancer cells. Oncogene 2008, 27, 4712–4723. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Aguayo, C.; Monroig, P.; Rideis, R.; Bayraktar, E.; Almeida, M.I.; Ivan, C.; Fuentes-Mattei, E.; Rashed, M.; Chavez-Reyes, A.; Ozpolat, B.; et al. Regulation of hnRNPA1 by microRNAs controls the miR-18a-K-RAS axis in chemotherapy-resistant ovarian cancer. Cell Discov. 2017, 3, 17029. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liu, S.; Wang, G.; Wu, X.; Ding, Y.; Guo, G.; Jiang, J.; Cui, S. Expression of miR-136 is associated with the primary cisplatin resistant of human epithelial ovarian cancer. Oncol. Rep. 2015, 33, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.Y.; Kang, H.; Kim, T.H.; Kim, G.; Heo, J.H.; Kwon, A.Y.; Kim, S.; Jung, S.G.; An, H.J. MicroRNA-136 inhibits cancer stem cell activity and ehnaces the anti-tumor effect of paclitaxel against chemoresistant ovarian cancer cells by targeting Notch3. Cancer Lett. 2017, 386, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Nicoloso, M.; Arvizo, R.; Wang, E.; Cortez, A.; Rossi, S.; Calin, G.; Mukherjee, P. MIR-15° and MIR-16 control Bmi-1 expression in ovarian cancer. Cancer Res. 2009, 69, 9090–9095. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, S.K.; Mustafi, S.B.; Mangala, L.S.; Jiang, D.; Pradeep, S.; Rodriguez-Aguayo, C.; Ling, H.; Ivan, C.; Mukherjee, P.; Calin, G.; et al. Therapeutic evaluation of microRNA-15a and microRNA-16 in ovarian cancer. Oncotarget 2016, 7, 15093–15104. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Chen, R.; Alvero, A.B.; Fu, H.H.; Holmberg, J.; Glackin, C.; Rutherford, T.; Mor, G. TWISTing stemness, inflammation and proliferation of epithelial ovarian cancer cells through miR-199A2/214. Oncogene 2010, 29, 3545–3553. [Google Scholar] [CrossRef] [PubMed]
- Alvero, A.B.; Fu, H.H.; Holmberg, J.; Visintin, I.; Mor, G.; Morquina, C.C. Stem-like ovarian cancer cells can serve as tumor vascular progenitors. Stem Cells 2009, 27, 2405–2413. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Shao, M.; Schilder Guise, T.; Mohammed, K.S.; Matei, D. Tissue transglutaminase links TGF-beta, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene 2012, 31, 2521–2534. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Liu, T.; Wan, X.; Gao, Y.; Wang, H. MicroRNA-199a targets CD44 to suppress the tumoreginicity and multidrug resistance of ovarian cancer-initiating cells. FEBS J. 2012, 279, 2047–2059. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.X.; Xu, M.; Tan, L.; Yang, H.; Permuth-Wey, J.; Kruk, P.A.; Wenham, R.M.; Nicosia, S.V.; Lancaster, J.M.; Sellers, T.A.; et al. MicroRNA mir-214 regulates ovarian cancer cells stemness by targeting p53/Nanog. J. Biol. Chem. 2012, 287, 34970–34978. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Sun, Y.; Hu, L.; Zheng, H.; Ji, P.; Pecot, C.V.; Zhao, Y.; Reynolds, S.; Cheng, H.; Rupaimole, R.; et al. Integrated analyses identify a master microRNA regulatory network for the mesenchymal subtype in serous ovarian cancer. Cancer Cell 2013, 23, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Kanlikilicer, P.; Rashed, M.H.; Bayraktar, R.; Mitra, R.; Ivan, C.; Aslan, B.; Zhang, X.; Filant, J.; Silva, A.M.; Rodriguez-Aguayo, C.; et al. Ubiquitous release of exosomal tumor suppressor miR-6126 from ovarian cancer cells. Cancer Res. 2016, 76, 7194–7202. [Google Scholar] [CrossRef] [PubMed]
- Rashed, M.H.; Kanlikilicer, P.; Rodriguez-Aguayo, C.; Pichler, M.; Bayraktar, R.; Bayraktar, E.; Ivan, C.; Filant, J.; Silva, A.; Aslan, B.; et al. Exosomal miR-940 maintains SRC-mediated oncogenic activity in cancer cells: A possible role for exosomal disposal of tumor suppressor miRNAs. Oncotarget 2017, 8, 20145–20164. [Google Scholar] [CrossRef] [PubMed]
- Kinross, K.M.; Montgomery, K.G.; Kleinschmidt, M.; Waring, P.; Ivetac, I.; Tikoo, A.; Saad, M.; Hare, L.; Roh, V.; Mantamadiotis, T.; et al. An activating PIK3CA mutation coupled with PTEN loss is sufficient to initiate ovarian tumorigenesis in mice. J. Clin. Investig. 2012, 122, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Coffey, D.M.; Creighton, C.J.; Yu, Z.; Hawkins, S.M.; Matzuk, M.M. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, 3921–3926. [Google Scholar] [CrossRef] [PubMed]
- Perets, R.; Wyant, G.; Muto, K.; Bijron, J.G.; Poole, B.B.; Chin, K.T.; Chen, J.Y.; Ohman, A.W.; Stepule, C.D.; Kwak, S.; et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca; Tp53; Pten models. Cancer Cell 2013, 24, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Sherman-Baust, C.A.; Kuhn, E.; Valle, B.; Shih, I.-M.; Kurman, R.J.; Wang, T.L.; Amano, T.; Ko, M.S.; Miyoshi, I.; Araki, Y.; et al. A genetically engineered ovarian cancer mouse model based on fallopian transformation mimics human high-grade serous carcinoma development. J. Pathol. 2014, 233, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Wu, R.; Kuick, R.; Sessine, M.S.; Schulman, S.; Green, M.; Fearon, E.R.; Cho, K.R. High-grade serous carcinomas arise in the mouse oviduct via defects linked to the human disease. J. Pathol. 2017, 243, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E.; Burdette, J.E.; Kenny, H.A.; Matei, D.; Pilrose, J.; Haluska, P.; Nephew, K.P.; Hales, D.B.; Stack, M.S. Epithelial ovarian cancer experimental models. Oncogene 2014, 33, 3619–3633. [Google Scholar] [CrossRef] [PubMed]
- Weroha, J.; Becker, M.; Enderica-Gonzalez, S.; Harrington, S.C.; Oberg, A.L.; Maurer, M.J.; Perkins, S.E.; AlHilli, M.; Butler, K.A.; McKinstry, S.; et al. Tumorgrafts as in vivo surrogates for women with ovarian cancer. Clin. Cancer Res. 2014, 20, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.D.; Hartley, L.; Cook, M.; Heong, V.; Boehm, E.; McShane, L.; Pyman, J.; McNally, O.; Ananda, S.; Harrell, M.; et al. Molecular correlates of platinum response in human high-grade serous ovarian cancer patient-derived xenografts. Mol. Oncol. 2014, 8, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Dobbin, Z.; Katre, A.; Steg, A.D.; Erickson, B.K.; Shah, M.M.; Alvarez, R.D.; Conner, M.G.; Schneider, D.; Chen, D.; Landen, C.N. Using heterogeneity of the patient-derived xenograft model to identify the chemoresistant population in ovarian cancer. Oncotarget 2014, 5, 8750–8758. [Google Scholar] [CrossRef] [PubMed]
- Ricci, F.; Bizzaro, F.; Cesca, M.; Guffanti, F.; Ganzinelli, M.; Decio, A.; Ghilardi, C.; Perego, P.; Fruscio, R.; Buda, A.; et al. Patient-derived ovarian tumor xenografts recapitulate human clinicopathology and genetic alterations. Cancer Res. 2014, 74, 6980–6990. [Google Scholar] [CrossRef] [PubMed]
- Thu, K.L.; Papari-Zareei, M.; Stasny, V.; Song, K.; Peyton, M.; Martinez, V.D.; Zhang, Y.A.; Castro, I.B.; Varella-Garcia, M.; Liang, H.; et al. A comprehensively characterized cell line panel highly representative of clinical ovarian high-grade serous carcinomas. Oncotarget 2016, 8, 50489–50499. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Palakurthi, S.; Zeng, Q.; Zhou, S.; Ivanova, E.; Huang, W.; Zervantonakis, I.K.; Selfors, L.M.; Shen, Y.; Pritchard, C.C.; et al. Establishment of patient-derived tumor xenograft models of epithelial ovarian cancer for preclinical evaluation of novel therapeutics. Clin. Cancer Res. 2017, 23, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- George, E.; Kim, H.; Krepler, C.; Wenz, B.; Makvandi, M.; Tanyi, J.L.; Brown, E.; Zhang, R.; Brafford, P.; Jean, S.; et al. A patient-derived-xenograft platform to study BRCA-deficient ovarian cancers. JCI Insight 2017, 2, e89760. [Google Scholar] [CrossRef] [PubMed]
- Kenny, H.A.; Lal-Nag, M.; White, E.A.; Shen, M.; Chiang, C.Y.; Mitra, A.K.; Zhang, Y.; Curtis, M.; Schryver, E.M.; Bettis, S.; et al. Quantitative high throughput screening using a primary human three-dimensional organotypic culture predicts in vivo efficacy. Nat. Commun. 2015, 6, 6220. [Google Scholar] [CrossRef] [PubMed]
- Eoh, K.J.; Chung, Y.S.; Lee, S.H.; Park, S.A.; Kim, H.J.; Yang, W.; Lee, I.O.; Lee, J.Y.; Cho, H.; Chay, D.B.; et al. Comparison of clinical features and outcomes in epithelial ovarian cancer according to tumorigenicity in patients-derived xenograft models. Cancer Res. Treat. 2017. [Google Scholar] [CrossRef] [PubMed]
- Heo, E.J.; Cho, Y.J.; Cho, W.C.; Hong, J.E.; Leon, N.K.; Oh, D.Y.; Choi, Y.L.; Song, S.Y.; Choi, J.J.; Bae, D.S.; et al. Patient-derived xenograft models of epithelial ovarian cancer for preclinical studies. Cancer Res. Treat. 2017, 49, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Ohman, A.W.; Hasan, N.; Dinulescu, D.M. Advances in tumor screening, imaging and avatar technologies for high-grade serous ovarian cancer. Front. Oncol. 2014, 4, 322. [Google Scholar] [CrossRef] [PubMed]
- Zervantonakis, I.K.; Iavarone, C.; Chen, H.Y.; Selfors, L.M.; Palakurthi, S.; Liu, J.F.; Drapkin, R.; Matulonis, U.; Leverson, J.D.; Sampath, D.; et al. Systems analysis of apoptotic priming in ovarian cancer identifies vulnerabilities and predictors of drug response. Nat. Commun. 2017, 8, 365. [Google Scholar] [CrossRef] [PubMed]
- Kodama, M.; Kodama, T.; Newberg, J.Y.; Katayama, H.; Kobayashi, M.; Hanash, S.M.; Yoshihara, K.; Wei, Z.; Tien, J.C.; Rangel, R.; et al. In vivo loss-of-function screens identify KPNB1 as a new druggable oncogene in epithelial ovarian cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E7301–E7310. [Google Scholar] [CrossRef] [PubMed]
- Forshew, T.; Murataza, M.; Parkinson, G.; Gale, D.; Tsui, D.W.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification of cancer mutations by targeted DNA sequencing of plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef]
- Zhou, Q.; Li, W.; Leng, B.; Zheng, W.; He, Z.; Zuo, M.; Chen, A. Circulating cell free DNA as the diagnostic marker for ovarian cancer: A systematic review and meta-analysis. PLoS ONE 2016, 11, e0155495. [Google Scholar] [CrossRef] [PubMed]
- Vanderstichele, A.; Busschaert, P.; Smeets, D.; Landolfo, C.; Van Nisuwenhuysen, E.; Leunen, K.; Neven, P.; Amant, F.; Mahner, S.; Braica, E.I.; et al. Chromosomal instability in cell-free DNA, a highly specific biomarker for detection of ovarian cancer in women with adnexal mass. Clin. Cancer Res. 2017, 23, 2223–2231. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, K.D.; Madsen, C.V.; Andersen, R.F.; Waldstrom, M.; Adimi, P.; Jakobsen, A. Prognostic importance of cell-free DNA in chemotherapy resistant ovarian cancer treated with bevacizumab. Eur. J. Cancer 2014, 50, 2611–2618. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorr, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Zhang, L.; Chen, Y.; Qing, C. Circulating cell free-DNA and circulating tumor cells, the “liquid biopsies” in ovarian cancer. J. Ov. Res. 2017, 10, 75. [Google Scholar] [CrossRef] [PubMed]
- Chebouti, I.; Kasimiri-Bauer, S.; Buderath, P.; Winberger, P.; Hauch, S.; Kimming, R.; Kuhlmann, J.D. EMT-like circulating tumor cells in ovarian cancer patients are enriched by platinum-based chemotherapy. Oncotarget 2017, 8, 48820–48831. [Google Scholar] [CrossRef] [PubMed]
- Chebouti, I.; Kuhlmann, J.D.; Budeneth, P.; Weber, S.; Winberger, P.; Bokeloh, Y.; Hauch, S.; Kimming, R.; Kasimir-Bauer, S. ERCC1-expressing circulating tumor cells as a potential diagnostic tool for monitoring response to platinum-based chemotherapy and for predicting post-therapeutic outcome of ovarian cancer. Oncotarget 2017, 8, 24303–24313. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, I.S.; Menoin, U.; Ryan, A.; Gentry-Maharaj, A.; Burnell, M.; Kalsi, J.; Amso, N.; Apostodilous, S.; Benjamin, E.; Cruickshank, D.; et al. Ovarian cancer screening and mortality in the UK collaboratrive trial of ovarian cancer screening (UKCTOCS): A randomized controlled trial. Lancet 2016, 387, 945–956. [Google Scholar] [CrossRef]
- Renon, U.; McGuire, A.J.; Raikou, M.; Ryan, A.; Davies, S.K.; Burnell, M.; Genrty-Maharaj, A.; Kalsi, J.K.; Singh, N.; Anes, N.N.; et al. TRhe cost-effectiveness of screening for ovarian cancer: Results from the IK Collaboratrive Trial of Ovarian Cancer Screening (UKCTOCS). Br. J. Cancer 2017, 117, 619–627. [Google Scholar]
- Mc Laughlin, J.R.; Rosen, B.; Moody, J.; Pal, T.; Fan, I.; Shaw, P.A.; Risch, H.A.; Sellers, T.A.; Sun, P.; Naod, S.A. Long-term ovarian cancer survival associated with mutation in BRCA1 or BRCA2. J. Natl. Cancer Inst. 2013, 105, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Kotsopoulos, J.; Rosen, B.; Fan, I.; Moody, J.; McLaughlin, J.R.; Risch, H.; May, T.; Sun, P.; Narod, S.A. Ten-year survival after epithelial ovarian cancer is not associated with BRCA mutation status. Gynecol. Oncol. 2016, 140, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Rosen, B.; Fan, I.; Ivanova, A.; McLaughlin, J.R.; Risch, H.; Narod, S.A.; Kotsopoulos, J. Epidemiologic factors that predict long-term survival following a diagnosis of epithelial ovarian cancer. Br. J. Cancer 2017, 116, 964–971. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Barry, W.T.; Birrer, M. Combination Cediranib and Olaparib versus Olaparib alone for women with recurrent platinum-sensitive ovarian cancer: A randomized phase 2 study. Lancet Oncol. 2014, 15, 1207–1214. [Google Scholar] [CrossRef]
- Liu, J.F.; Barry, W.; Barrer, M. Overall survival and updated progression-free survival results from a randomized phase 2 trial comparing the combination of olaparib and cediranib against olaparib alone in recurrent platinum-sensitive ovarian cancer. J. Clin. Oncol. 2017, 35, 1686–1694. [Google Scholar]
- Ivy, S.P.; Liu, J.F.; Lee, J.M. Cediranib, a pan-VEGFR inhibitor, and olaparib, a PARP inhibitor, in combination therapy for high grade serous ovarian cancer. Expert Opin. Investig. Drugs 2016, 25, 597–611. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.; Kim, M.; Gilbert, C.A.; Simpkins, F.; Ince, T.A.; Slinyerland, J.N. VEGFA activates an epigenetic pathway upregulating ovarian cancer-initiating cells. EMBO Mol. Med. 2017, 9, 304–318. [Google Scholar] [CrossRef] [PubMed]
- Momeny, M.; Sabourinejad, Z.; Zuzzinrad, G.; Maghaddaskho, F.; Eyvani, H.; Yousefi, H.; Mirshahvaladi, S.; Poursani, E.M.; Barghi, F.; Poursheikhani, A.; et al. Anti-tumor activity of tivozanib, a pan-inhibitor of VEGF receptors, in therapy-resistant ovarian ovarian carcinoma cells. Sci. Rep. 2017, 7, 45954. [Google Scholar] [CrossRef] [PubMed]
- O’Kane, G.; Connor, A.; Gallinger, S. Characterization, detection, and treatment approaches for homologous recombination deficiency. Trends Mol. Med. 2017, 23, 1121–1137. [Google Scholar] [CrossRef] [PubMed]
- Jim, J.; Tan, D. Understanding resistance mechanisms and expanding the therapeutic utility of PARP inhibitors. Cancers 2017, 9, 109. [Google Scholar]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Kazakashev, S.; Zhu, H.; Yokoyama, Y.; Zhao, B.; Fatkudinov, N.; Kronenkov, A.V.; Wilson, A.J.; Simpkins, F.; Speicher, D.; Khabele, D.; et al. BET bromodomain inhibition synergizes with PARP inhibitor in epithelial ovarian cancer. Cell Rep. 2017, 21, 3398–3405. [Google Scholar] [CrossRef] [PubMed]
- Yong, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Kanska, J.; Zakhour, M.; Taylor-Harding, B.; Karlan, B.Y.; Wiedemeyer, W.R. Cyclin E as a potential therapeutic target in high grade serous ovarian cancer. Gynecol. Oncol. 2016, 143, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synott, N.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Heong, V.; Ngoi, N.; Tan, D.S. Update on immune checkpoint inhibitors in gynecological cancers. J. Gynecol. Oncol. 2017, 28, e20. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, S.L.; Secord, A.A.; Monk, B. The role of immune checkpoint inhibition in the treatment of ovarian cancer. Gynecol. Oncol. Res. Pract. 2016, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Tsafou, K.; Villa, A.; Kowalczyk, K.; Rakowniknow, R.; Bartalot, G.; Confalonieri, S.; Brumak, S.; Jensen, L.J.; Brunak, S.; Jensen, L.J.; et al. Phosphoproteomics of primary cells reveals druggable kinase signatures in ovarian cancer. Cell Rep. 2017, 18, 3242–3256. [Google Scholar]
- Battle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Mati, D.; Cheng, J.X. Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell 2017, 20, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Bohm, S.; Faruqi, A.; Said, I.; Lockley, M.; Bocknak, E.; Jeyarajah, A.; Fritzpatrick, A.; Ennis, D.; Dowe, T.; Santos, J.L.; et al. Chemotherapy response score: Development and validation of a system to quantify histopathologic response to neoadjuvant chemotherapy in tubo-ovarian high grade serous carcinoma. J. Clin. Oncol. 2015, 33, 2457–2463. [Google Scholar] [CrossRef] [PubMed]



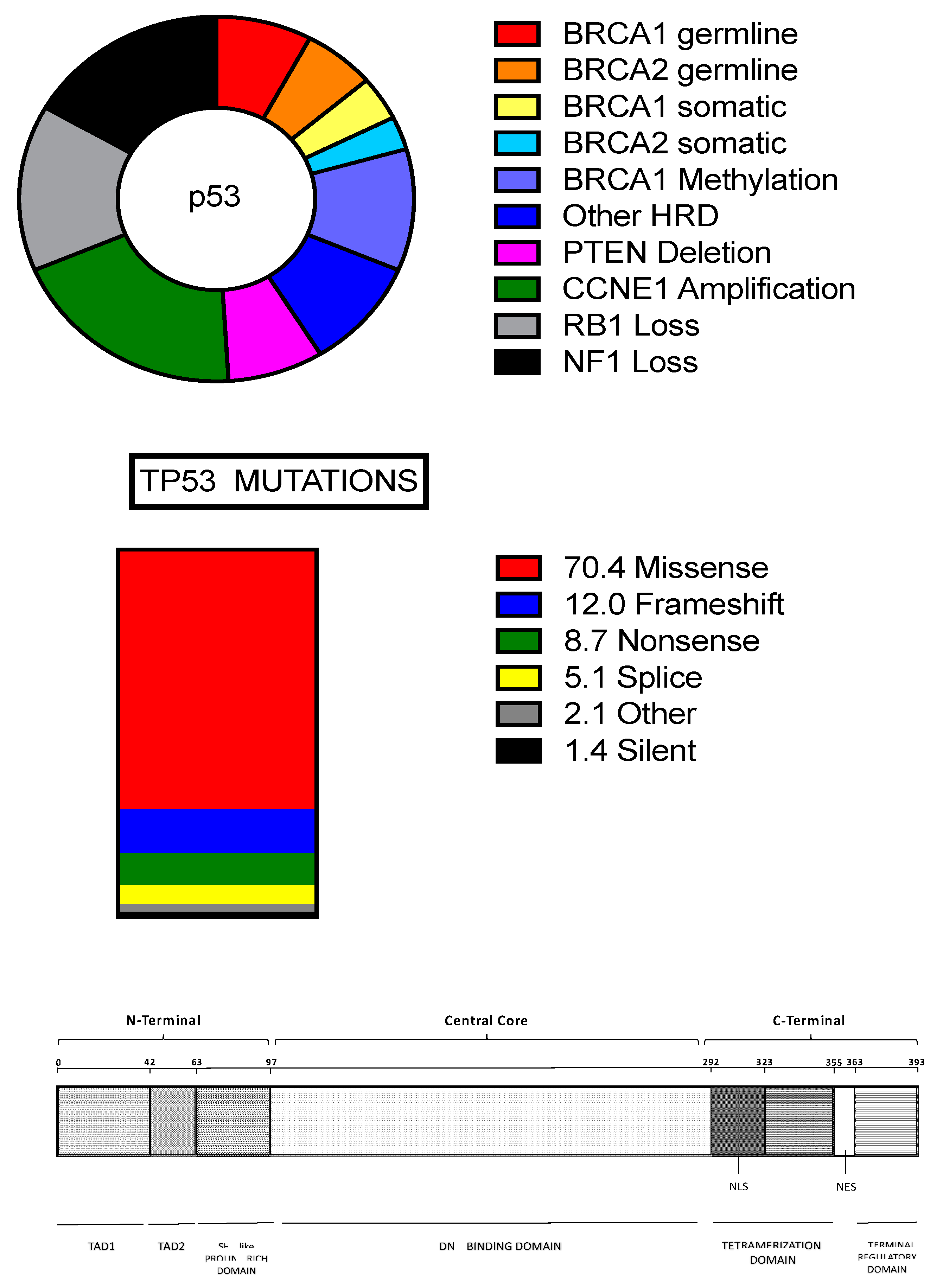{kind=link}
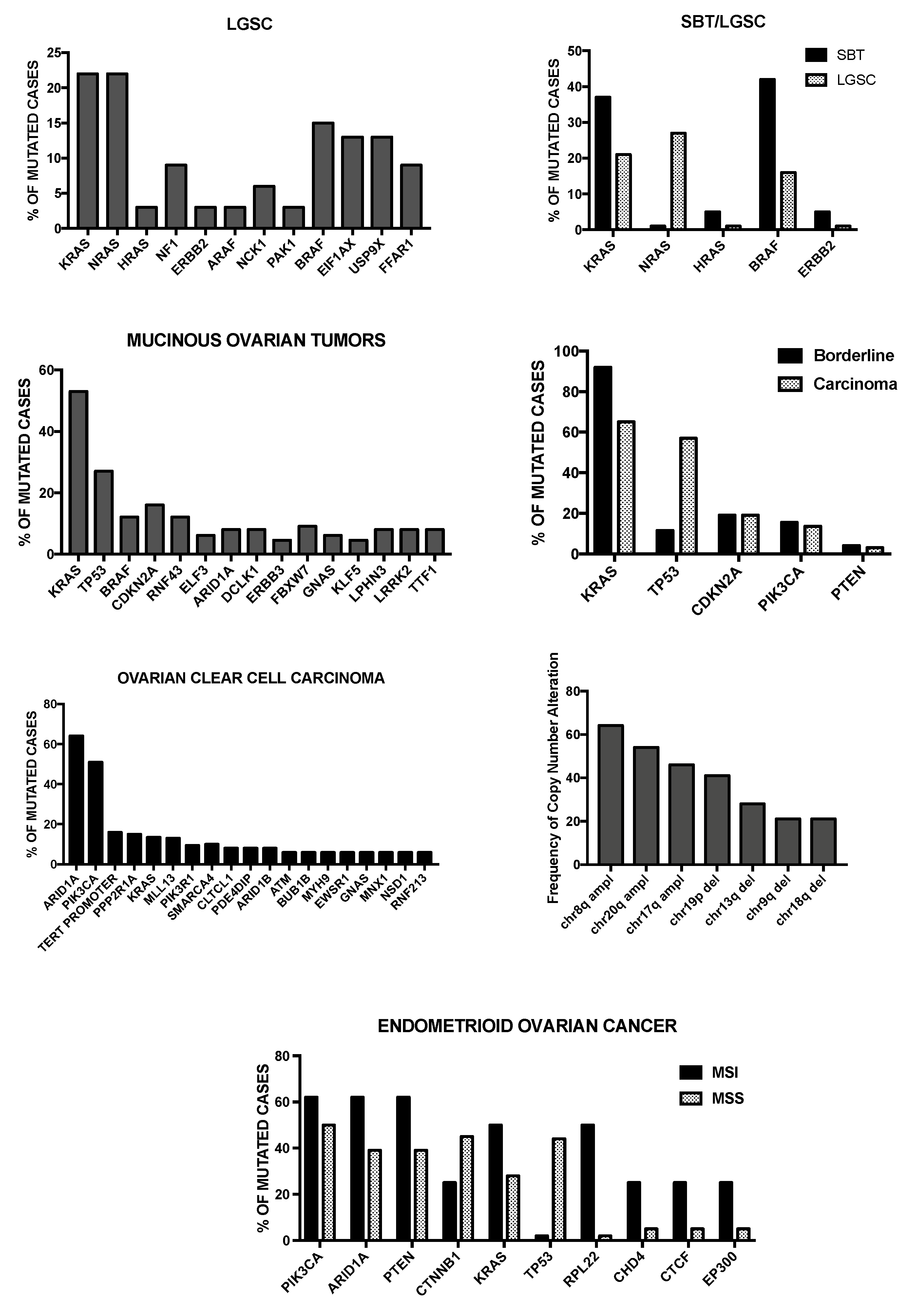{kind=link}
| Tumor | Type | Cells of Origin | Precursor Lesion | More Frequent Mutations | Familial Risk |
|---|---|---|---|---|---|
| Endometrioid Cancer | I | Endometrial epithelial cells | Endometrioid borderline tumor | ARID1A, PIK3CA, TERT | Lynch syndrome |
| Ovarian Clear Cell Carcinoma | I | Endometrial epithelial cells | Endometrioid borderline tumor | ARID1A, PIK3CA, PTEN, CTNNB1, KRAS, TP53, RPL22 | Lynch syndrome |
| Mucinous Carcinoma | I | Unknown | Cystadenoma, Mucine borderline tumors, Brenner Tumors | KRAS, TP53, CDKN2A, BRAF, RNF43 | Unknown |
| Brenner Tumor | I | Transitional-like cells of fallopian tube | Benign Brenner tumor | Sporadic point mutations MYC amplification | Unknown |
| Low-Grade Serous Carcinoma | I | Fallopian tube progenitor cell or secretory epithelial cell | Serous borderline tumors | KRAS, NRAS, BRAF, EIF1AX, USP9X, FFAR1, NF1, HRAS | Unknown |
| Seromucinous Carcinoma | I | The same as for endometrioid, Low-grade serous and mucinous carcinomas | The same as for endometrioid, Low-grade serous and mucinous carcinomas | KRAS, PIK3CA, PTEN, ARID1A | Unknown |
| High-Grade Serous Ovarian Cancer | II | Fallopian tube progenitor cell or secretory epithelial cell | STIC, SCOUT, TP53 signature | TP53, BRCA1, BRCA2, CNAs of CCNE1 amplification, PTEN deletion, RB1 and NF1 loss | BRCA1, BRCA2, BRIP1, PALB2, RAD51C and RAD541D |
| Ovarian Carcinosarcoma or Malignant MullerianMixed Tumors | II | Unknown | Unknown | TP53, PI3KCA, PPP2R1A, KRAS, PTEN, CHD4, BCOR, histone H2A and H2B | Unknown |
| Ovarian Granulosa Cell Tumors | Adult Juvenile | Sex-cord stromal Cells (granulosa cells) | Unknown | FOX2L in 87% of adult-type patients | Unknown |
| Sertoli-Leydig Cell Tumor | NA | Granulosa cells or other Stromal cells | None | DICER1 | DICER1 syndrome |
| Small Cell Carcinoma of The Ovary, hypercalcemic Type | NA | Rare resident ovarian Cells | None | SMARCA4 | RTPS2 (SMARCA4) |
| Fibroma | NA | Ovarian stromal cells | None | None |
| Primary Tumor | FIGO | Tumor Extension | 5-Year Survival Rate |
|---|---|---|---|
| T0 | No evidence of primary tumor | N.A. | |
| T1 | I | Tumor limited to the ovaries (one or both) | 85–94% |
| T1a | IA | Tumor limited to one ovary; capsule intact, no tumor on ovarian surface; no malignant cells in ascites or peritoneal washings. | 94% |
| T1b | IB | Tumor limited to both ovaries; capsule intact, no tumor on ovarian surface; no malignant cells in ascites or peritoneal washings. | 92% |
| T1c | IC | Tumor limited to one or both ovaries with any of the following: capsule ruptures, tumor on ovarian surface, malignant cells in ascites or peritoneal washings. | 85% |
| T2 | II | Tumor involves one or both ovaries with pelvic extension. | 69–78% |
| T2a | IIA | Extension and/or implants in the uterus and/other tube(s); no malignant cells in ascites or peritoneal washings. | 78% |
| T2b | IIB | Extension and/or implants in other pelvic tissues; no malignant cells in ascites or peritoneal washings. | 73% |
| T2c | IIC | Pelvic extension and/or implants (T2a or T2b) with malignant cells in ascites or peritoneal washings. | 69% |
| T3 | III | Tumor involves one or both ovaries with microscopically confirmed peritoneal metastasis outside the pelvis. | 17–59% |
| T3a | IIIA | Microscopic peritoneal metastasis beyond the pelvis (no macroscopic tumor). | 59% |
| T3b | IIIB | Macroscopic peritoneal metastasis beyond the pelvis 2 cm or less in greatest dimension. | 39% |
| T3c | IIIC | Macroscopic peritoneal metastasis beyond the pelvis >2 cm in greatest dimension and/or regional lymph node metastasis. | 17% |
| T4 | IV | Malignant cells in pleural effusion, parenchymal metastases and or metastases to extra-abdominal organs (including inguinal lymph nodes and lymph nodes outside of the abdominal cavity). | 12% |
| Subtype | Main Features | Genes Expressed | Main Pathways | Clinical Outcome |
|---|---|---|---|---|
| C1/Mesenchymal (28%) | Extensive myofibroblast infiltration (desmoplasia). Mesenchymal gene expression signature | ↑ COLL11A1, CXCL14, POSTN, SNAIL2, VCAN, ZEB1 | Focal adhesion ECM receptor interaction JAK-STAT signaling TGF-β signaling VEGF signaling Fibroblast signature EMT/Stem cell | Negative prognosis |
| C2/Immunoreactive (21.5%) | Extensive intratumoral T lymphocyte infiltration | ↑ CXCL10, CXCL11, PSMB8, PSMB9, TAP1 | T-cell receptor signaling Toll-like receptor Antigen presentation machinery | Better prognosis |
| C5/Proliferative (20.5%) | Low expression of differentiation markers, limited inflammatory infiltration, activation of signaling pathways involving oncogenic and stem cell factors Lower BRCA1-2 mutation rate | ↑ HMGA2, SALL2, SOX11, TCF7L1 | Cell cycle NOTCH signaling | Negative prognosis |
| C4/Differentiated (17.5%) | Gene signature resembling serous borderline tumors | ↑ COLEC11, DEFB1, ITGB4, MGLL, MLPH, STAR | Ribosome Metabolism Cytochrome p450 | Intermediate prognosis |
| C4/Anti-mesenchymal (12.5%) | Downregulation of the genes typically upregulated in the mesenchymal subtype Lower BRCA1-2 mutation rate | ↓ COLL11A1, DCN, FAP, POSTN, VCAN, ZEB1 | Oxidative phosphorylation Peroxisome Butanoate metabolism | Better Prognosis |
| Marker | Chromosome Location | Biochemical Properties | Function in Ovarian CSC Biology | Number of Tumorigenic Cells |
|---|---|---|---|---|
| CD133 | 4p15.32 | Transmembrane glycoprotein It induces PI3K/AKT pathway | Tumor sphere formation, Tumor-initiating capacity, Self-renewal. Chemoresistance. High CD133 expression is a negative prognostic factor. | 102 CD133+ |
| CD44 | 11p13 | Transmembrane glycoprotein Hyaluronic acid Receptor It stimulates EGFR-Ras-ERK | Tumor sphere formation, Tumor-initiating capacity, Self-renewal. High CD44 expression is associated with TNM stage and survival. CD44 promotes epithelial-mesenchymal transition. | 102 CD44+ |
| CD24 | 6q21 | Transmembrane glycoprotein It activates STAT3 | Tumor sphere formation, Tumor-initiating capacity, Chemoresistance. It is involved in ovarian cancer metastasis. High CD24 is a negative prognostic factor. | 5 × 103 CD24+ |
| CD117 | 4q12 | Transmembrane glycoprotein Kit-Ligand Receptor | Tumor sphere formation, Tumor-initiating capacity (particularly CD117+/CD44+), Self-renewal. Chemoresistance through Wnt/β-catenin activation. High CD177 expression is a negative prognostic factor. | 103 CD117+ 102 CD44+/CD117+ |
| CXCR4 | 2q22.1 | Transmembrane glycoprotein SDF-1α Receptor | Tumor sphere formation, Tumor-initiating capacity. CXCR4+/CD133+ cells are chemoresistant. The number of CXCR4+/CD133+ cells is highly variable in primary tumors. | 103 CXCR4+/CD133+ |
| CD151 | 11p15.5 | Membrane Protein Tetraspanin | Tumor sphere formation, Tumor-initiating capacity. It is involved in epithelial-mesenchymal transition and induces chemoresistance. High CD151 expression is associated with poor prognosis. | N.D. |
| EpCAM | 2p21 | Transmembrane Glycoprotein Epithelial Cell Adhesion | CD44+/CD24+/EpCAM+ cells promote tumor sphere formation, have tumor-initiating capacity, are chemoresistant. High EpCAM expression is associated with poor prognosis. | 102 cells |
| ALDH1 | 9q21.13 | Enzymatic activity Aldehyde Dehydrogenase | Tumor sphere formation, Tumor-initiating capacity. Total ALDH1 expression correlates with a favorable prognosis. CD133+/ALDH1+ cells have a negative prognostic impact. | N.D. |
| SOX2 | 3q26.33 | Transcription Factor It maintains pluripotency and Self-renewal in embryonic Stem cells. | It promotes migration and invasion of ovarian cancer cells. It increases the expression of CSC markers, tumor sphere formation, tumor-initiating capacity. Its expression in HGS-OvCa is associated with negative prognosis. | N.A. |
| OCT4 | 6p21.33 | Transcription Factor Octamer-Binding Transcription Factor 4 | It is required for self-renewal capacity of CSCs. It induces epithelial-mesenchymal transition. It is involved in chemoresistance. | N.A. |
| NANOG | 12p13.31 | Transcription Factor Nanog Homeobox | It is required for CSC tumorigenic activity. It is involved in both epithelial-mesenchymal transition and in chemoresistance through STAT3 activation. High NANOG is a negative prognostic factor. | N.A. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Testa, U.; Petrucci, E.; Pasquini, L.; Castelli, G.; Pelosi, E. Ovarian Cancers: Genetic Abnormalities, Tumor Heterogeneity and Progression, Clonal Evolution and Cancer Stem Cells. Medicines 2018, 5, 16. https://doi.org/10.3390/medicines5010016
Testa U, Petrucci E, Pasquini L, Castelli G, Pelosi E. Ovarian Cancers: Genetic Abnormalities, Tumor Heterogeneity and Progression, Clonal Evolution and Cancer Stem Cells. Medicines. 2018; 5(1):16. https://doi.org/10.3390/medicines5010016
Chicago/Turabian StyleTesta, Ugo, Eleonora Petrucci, Luca Pasquini, Germana Castelli, and Elvira Pelosi. 2018. "Ovarian Cancers: Genetic Abnormalities, Tumor Heterogeneity and Progression, Clonal Evolution and Cancer Stem Cells" Medicines 5, no. 1: 16. https://doi.org/10.3390/medicines5010016
APA StyleTesta, U., Petrucci, E., Pasquini, L., Castelli, G., & Pelosi, E. (2018). Ovarian Cancers: Genetic Abnormalities, Tumor Heterogeneity and Progression, Clonal Evolution and Cancer Stem Cells. Medicines, 5(1), 16. https://doi.org/10.3390/medicines5010016