
Disrupted Sleep Homeostasis and Altered Expressions of Clock Genes in Rats with Chronic Lead Exposure

 ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Experimental Animals
2.3. Experimental Animal Model of Chronic Lead Toxication
2.4. Electroencephalography and Electromyography Recordings
2.5. Measurement of Blood Lead Level, Serum Creatinine Level and Daily Urine Amount
2.6. Analysis of Clock Gene Expression
2.7. Statistical Analysis
3. Results
3.1. The Blood Lead Level and Biochemical Assay
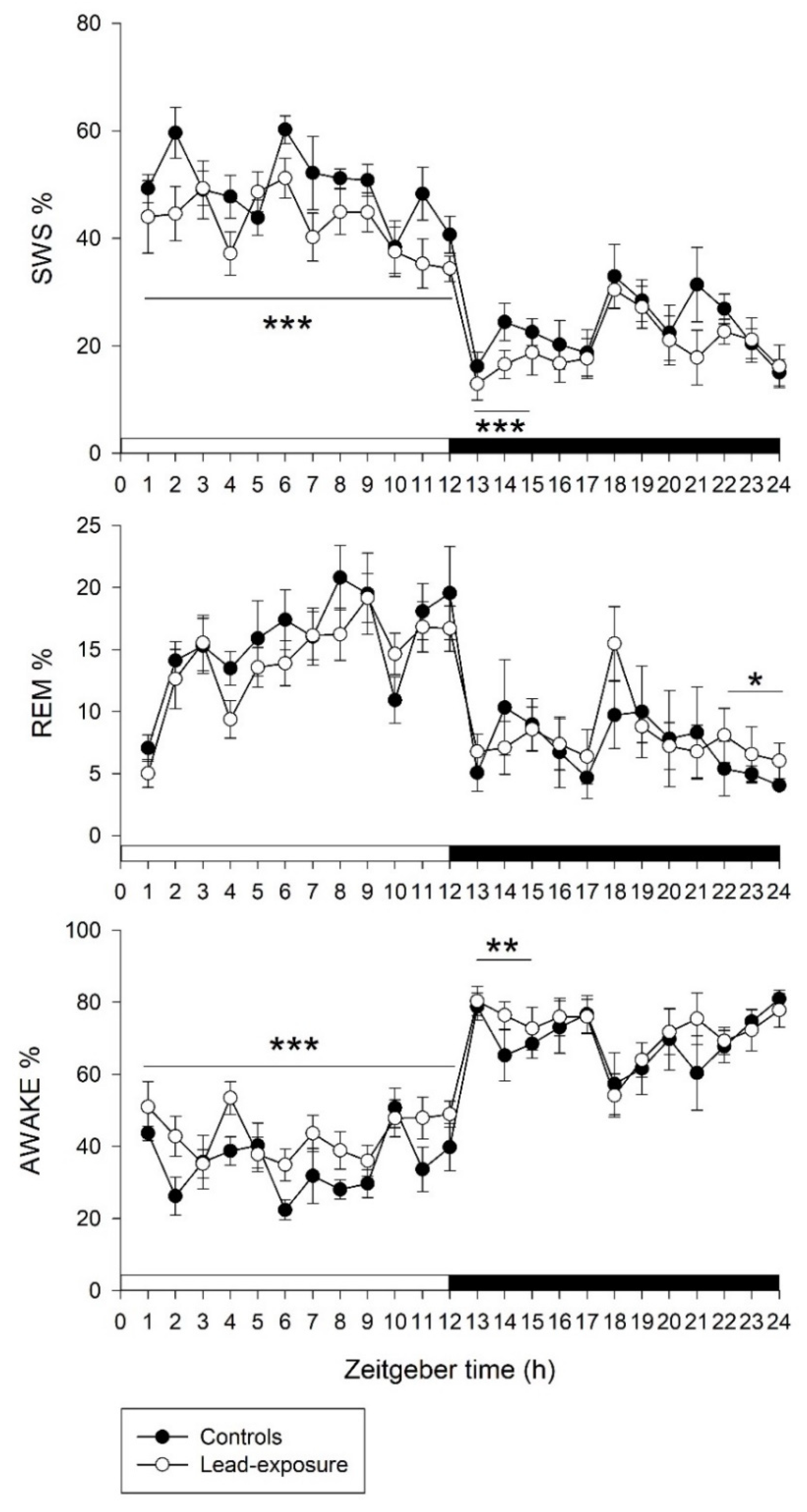
3.2. Alterations of Sleep–Wake Activities in Rats with Chronic Lead Poisoning
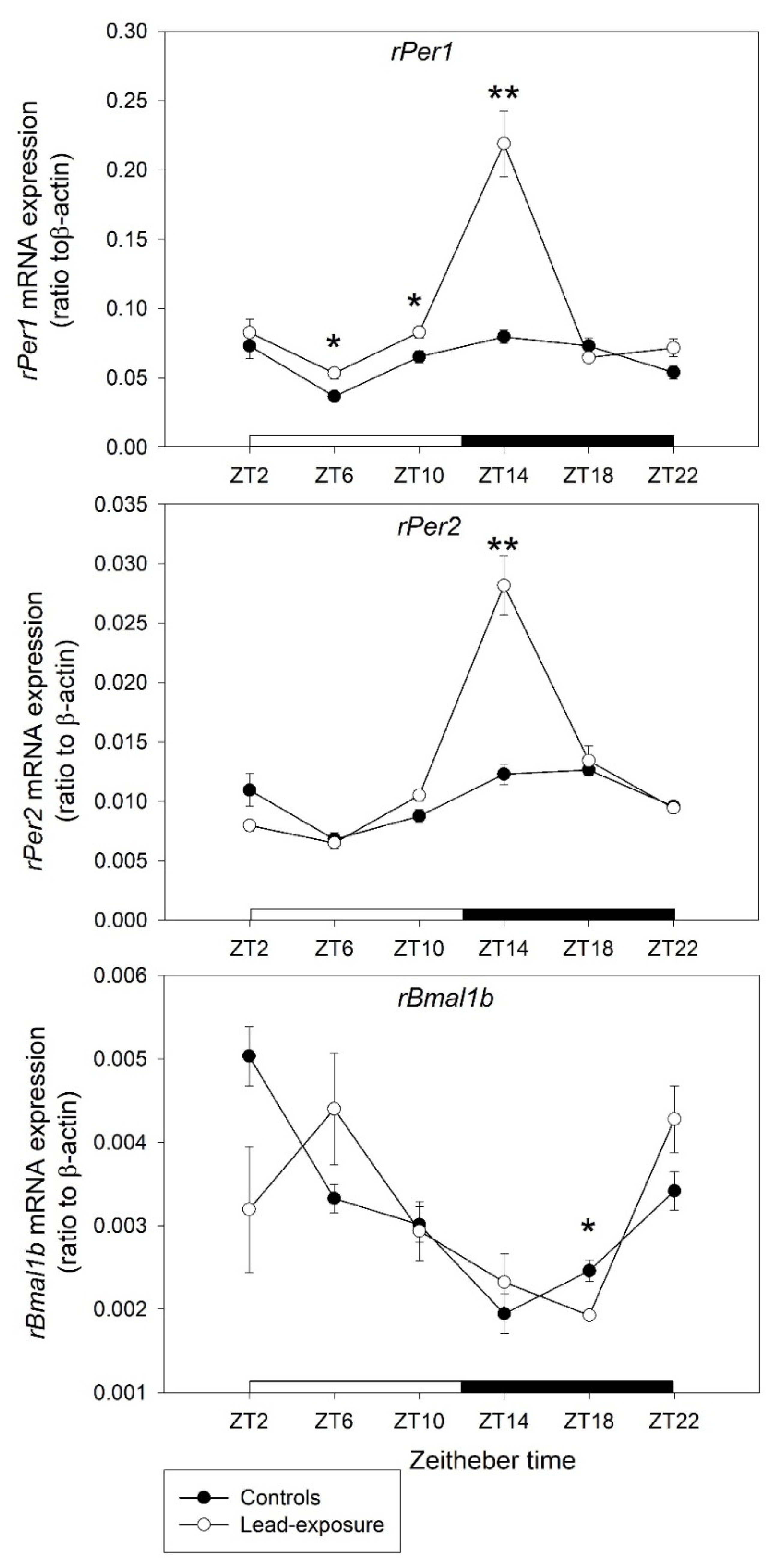
3.3. Alterations of the Clock Gene Expression in Rats with Chronic Lead Poisoning
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Senut, M.C.; Cingolani, P.; Sen, A.; Kruger, A.; Shaik, A.; Hirsch, H.; Suhr, S.T.; Ruden, D. Epigenetics of early-life lead exposure and effects on brain development. Epigenomics 2012, 4, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Patrick, L. Lead toxicity, a review of the literature. Part 1: Exposure, evaluation, and treatment. Altern. Med. Rev. 2006, 11, 2–22. [Google Scholar] [PubMed]
- Obeng-Gyasi, E. Lead Exposure and Oxidative Stress-A Life Course Approach in U.S. Adults. Toxics 2018, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Lanphear, B.P.; Rauch, S.; Auinger, P.; Allen, R.W.; Hornung, R.W. Low-level lead exposure and mortality in US adults: A population-based cohort study. Lancet Public Health 2018, 3, e177–e184. [Google Scholar] [CrossRef]
- Kuh, D.; Ben-Shlomo, Y.; Lynch, J.; Hallqvist, J.; Power, C. Life course epidemiology. J. Epidemiol. Community Health 2003, 57, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Landrigan, P.J.; Todd, A.C. Lead poisoning. West. J. Med. 1994, 161, 153–159. [Google Scholar] [PubMed]
- Mason, L.H.; Harp, J.P.; Han, D.Y. Pb neurotoxicity: Neuropsychological effects of lead toxicity. Biomed. Res. Int. 2014, 2014, 840547. [Google Scholar] [CrossRef]
- Chen, C.C.; Yen, H.W.; Lo, Y.H.; Chu, Y.H.; Chiu, Y.W.; Chuang, H.Y. The association of prolonged QT interval on electrocardiography and chronic lead exposure. J. Occup. Environ. Med. 2013, 55, 614–619. [Google Scholar] [CrossRef]
- Liu, H.L.; Chuang, H.Y.; Hsu, C.N.; Lee, S.S.; Yang, C.C.; Liu, K.T. Effects of Vitamin D Receptor, Metallothionein 1A, and 2A Gene Polymorphisms on Toxicity of the Peripheral Nervous System in Chronically Lead-Exposed Workers. Int. J. Environ. Res. Public Health 2020, 17, 2909. [Google Scholar] [CrossRef]
- Altmann, L.; Weinsberg, F.; Sveinsson, K.; Lilienthal, H.; Wiegand, H.; Winneke, G. Impairment of long-term potentiation and learning following chronic lead exposure. Toxicol. Lett. 1993, 66, 105–112. [Google Scholar] [CrossRef]
- Wu, W.T.; Lin, Y.J.; Liou, S.H.; Yang, C.Y.; Cheng, K.F.; Tsai, P.J.; Wu, T.N. Brain cancer associated with environmental lead exposure: Evidence from implementation of a National Petrol-Lead Phase-Out Program (PLPOP) in Taiwan between 1979 and 2007. Environ. Int. 2012, 40, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Khan, K.M.; Al-Khaledi, G.; Khan, I.; Attur, S. Early postnatal lead exposure induces tau phosphorylation in the brain of young rats. Acta Biol. Hung. 2012, 63, 411–425. [Google Scholar] [CrossRef]
- Boskabady, M.; Marefati, N.; Farkhondeh, T.; Shakeri, F.; Farshbaf, A.; Boskabady, M.H. The effect of environmental lead exposure on human health and the contribution of inflammatory mechanisms, a review. Environ. Int. 2018, 120, 404–420. [Google Scholar] [CrossRef] [PubMed]
- Virgolini, M.B.; Aschner, M. Molecular Mechanisms of Lead Neurotoxicity. Adv. Neurotoxicol. 2021, 5, 159–213. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Choi, H.; Hwang, U.K.; Kang, J.C.; Kang, Y.J.; Kim, K.I.; Kim, J.H. Toxic effects of lead exposure on bioaccumulation, oxidative stress, neurotoxicity, and immune responses in fish: A review. Environ. Toxicol. Pharmacol. 2019, 68, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zeng, A.; Zheng, W.; Du, Y. Upregulation of zinc transporter 2 in the blood-CSF barrier following lead exposure. Exp. Biol. Med. 2013, 239, 202–212. [Google Scholar] [CrossRef]
- Simons, T.J. Lead-calcium interactions in cellular lead toxicity. Neurotoxicology 1993, 14, 77–85. [Google Scholar]
- Sanders, T.; Liu, Y.; Buchner, V.; Tchounwou, P.B. Neurotoxic effects and biomarkers of lead exposure: A review. Rev. Environ. Health 2009, 24, 15–45. [Google Scholar] [CrossRef]
- Liu, K.S.; Hao, J.H.; Zeng, Y.; Dai, F.C.; Gu, P.Q. Neurotoxicity and biomarkers of lead exposure: A review. Chin. Med. Sci. J. 2013, 28, 178–188. [Google Scholar] [CrossRef]
- White, L.D.; Cory-Slechta, D.A.; Gilbert, M.E.; Tiffany-Castiglioni, E.; Zawia, N.H.; Virgolini, M.; Rossi-George, A.; Lasley, S.M.; Qian, Y.C.; Basha, M.R. New and evolving concepts in the neurotoxicology of lead. Toxicol. Appl. Pharmacol. 2007, 225, 1–27. [Google Scholar] [CrossRef]
- Jansen, E.C.; Dunietz, G.L.; Dababneh, A.; Peterson, K.E.; Chervin, R.D.; Baek, J.; O’Brien, L.; Song, P.X.K.; Cantoral, A.; Hu, H.; et al. Cumulative Childhood Lead Levels in Relation to Sleep During Adolescence. J. Clin. Sleep Med. 2019, 15, 1443–1449. [Google Scholar] [CrossRef]
- Liu, J.; Liu, X.; Pak, V.; Wang, Y.; Yan, C.; Pinto-Martin, J.; Dinges, D. Early Blood Lead Levels and Sleep Disturbance in Preadolescence. Sleep 2015, 38, 1869–1874. [Google Scholar] [CrossRef]
- Shafiq-ur-Rehman. Circadian rhythm of stereotyped complex behaviours in rats in environmental lead exposure. Prog. Neuropsychopharmacol. Biol. Psychiatry 1999, 23, 149–159. [Google Scholar] [CrossRef]
- Lilis, R.; Valciukas, J.A.; Weber, J.P.; Malkin, J. Effects of low-level lead and arsenic exposure on copper smelter workers. Arch. Environ. Health 1985, 40, 38–47. [Google Scholar] [CrossRef]
- Rojas-Castaneda, J.C.; Vigueras-Villasenor, R.M.; Rojas, P.; Chavez-Saldana, M.; Gutierrez-Perez, O.; Montes, S.; Rios, C. Alterations induced by chronic lead exposure on the cells of circadian pacemaker of developing rats. Int. J. Exp. Pathol. 2011, 92, 243–250. [Google Scholar] [CrossRef]
- Allen, K.A. Is Prenatal Lead Exposure a Concern in Infancy? What Is the Evidence? Adv. Neonatal Care 2015, 15, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Giusi, G.; Alo, R.; Crudo, M.; Di Vito, A.; Facciolo, R.M.; Canonaco, M. Environmental stressors and neurobiological features of marine teleosts: Histamine receptors as targets. Crit. Rev. Toxicol. 2010, 40, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.Y.; Tain, Y.L.; Lee, Y.T.; Hsu, C.Y.; Chiou, T.T.; Huang, P.C.; Lee, C.T. Renin angiotensin system blockade ameliorates lead nephropathy. Biochem. Biophys. Res. Commun. 2013, 438, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Yi, P.L.; Jou, S.B.; Wu, Y.J.; Chang, F.C. Manipulation of Epileptiform Electrocorticograms (ECoGs) and Sleep in Rats and Mice by Acupuncture. J. Vis. Exp. 2016, 118, e54896. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.Y.; Chang, F.C.; Ng, H.Y.; Kuo, C.C.; Lee, Y.T.; Lu, C.Y.; Lee, C.T. Disrupted circadian rhythm in rats with nephrectomy-induced chronic kidney disease. Life Sci. 2012, 91, 127–131. [Google Scholar] [CrossRef]
- Le Bon, O. Relationships between REM and NREM in the NREM-REM sleep cycle: A review on competing concepts. Sleep Med. 2020, 70, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Jou, S.B.; Tsai, C.J.; Fang, C.Y.; Yi, P.L.; Chang, F.C. Effects of N6-(4-hydroxybenzyl) adenine riboside in stress-induced insomnia in rodents. J. Sleep Res. 2021, 30, e13156. [Google Scholar] [CrossRef] [PubMed]
- Yi, P.L.; Tsai, C.H.; Lu, M.K.; Liu, H.J.; Chen, Y.C.; Chang, F.C. Interleukin-1beta mediates sleep alteration in rats with rotenone-induced parkinsonism. Sleep 2007, 30, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Yi, P.L.; Chen, Y.J.; Lin, C.T.; Chang, F.C. Occurrence of epilepsy at different zeitgeber times alters sleep homeostasis differently in rats. Sleep 2012, 35, 1651–1665. [Google Scholar] [CrossRef][Green Version]
- Baracchi, F.; Opp, M.R. Sleep–wake behavior and responses to sleep deprivation of mice lacking both interleukin-1 beta receptor 1 and tumor necrosis factor-alpha receptor 1. Brain Behav. Immun. 2008, 22, 982–993. [Google Scholar] [CrossRef]
- Vanderheyden, W.M.; George, S.A.; Urpa, L.; Kehoe, M.; Liberzon, I.; Poe, G.R. Sleep alterations following exposure to stress predict fear-associated memory impairments in a rodent model of PTSD. Exp. Brain Res. 2015, 233, 2335–2346. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.T.; Lien, Y.H.; Lai, L.W.; Chen, J.B.; Lin, C.R.; Chen, H.C. Increased renal calcium and magnesium transporter abundance in streptozotocin-induced diabetes mellitus. Kidney Int. 2006, 69, 1786–1791. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, S.K. Renal effects of environmental and occupational lead exposure. Indian J. Occup. Environ. Med. 2008, 12, 103–106. [Google Scholar] [CrossRef]
- Wedeen, R.P. The role of lead in renal failure. Clin. Exp. Dial. Apheresis 1982, 6, 113–146. [Google Scholar] [CrossRef]
- Dantzler, W.H.; Layton, A.T.; Layton, H.E.; Pannabecker, T.L. Urine-concentrating mechanism in the inner medulla: Function of the thin limbs of the loops of Henle. Clin. J. Am. Soc. Nephrol. 2014, 9, 1781–1789. [Google Scholar] [CrossRef]
- Campbell, I.G.; Darchia, N.; Higgins, L.M.; Dykan, I.V.; Davis, N.M.; de Bie, E.; Feinberg, I. Adolescent changes in homeostatic regulation of EEG activity in the delta and theta frequency bands during NREM sleep. Sleep 2011, 34, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.K.; Snyder, E.; Hall, J.; Randazzo, A.C.; Griffin, K.; Groeger, J.; Eisenstein, R.; Feren, S.D.; Dickey, P.; Schweitzer, P.K. Slow wave sleep enhancement with gaboxadol reduces daytime sleepiness during sleep restriction. Sleep 2008, 31, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Wurts, S.W.; Edgar, D.M. Circadian and homeostatic control of rapid eye movement (REM) sleep: Promotion of REM tendency by the suprachiasmatic nucleus. J. Neurosci. 2000, 20, 4300–4310. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Fukuda, K.; Takeuchi, T.; Inugami, M.; Miyasita, A. Sleep onset REM period appearance rate is affected by REM propensity in circadian rhythm in normal nocturnal sleep. Clin. Neurophysiol. 2000, 111, 428–433. [Google Scholar] [CrossRef]
- Pigeon, W.R.; Perlis, M.L. Sleep homeostasis in primary insomnia. Sleep Med. Rev. 2006, 10, 247–254. [Google Scholar] [CrossRef]
- Yasenkov, R.; Deboer, T. Circadian modulation of sleep in rodents. Prog. Brain Res. 2012, 199, 203–218. [Google Scholar] [CrossRef]
- Yasenkov, R.; Deboer, T. Circadian regulation of sleep and the sleep EEG under constant sleep pressure in the rat. Sleep 2010, 33, 631–641. [Google Scholar] [CrossRef]
- Kaskie, R.E.; Gill, K.M.; Ferrarelli, F. Reduced frontal slow wave density during sleep in first-episode psychosis. Schizophr. Res. 2019, 206, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Allada, R.; Bass, J. Circadian Mechanisms in Medicine. N. Engl. J. Med. 2021, 384, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Okamura, H. Gradients in the circadian expression of Per1 and Per2 genes in the rat suprachiasmatic nucleus. Eur. J. Neurosci. 2002, 15, 1153–1162. [Google Scholar] [CrossRef]
- Riddle, M.; Mezias, E.; Foley, D.; LeSauter, J.; Silver, R. Differential localization of PER1 and PER2 in the brain master circadian clock. Eur. J. Neurosci. 2017, 45, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Rijo-Ferreira, F.; Takahashi, J.S. Genomics of circadian rhythms in health and disease. Genome Med. 2019, 11, 82. [Google Scholar] [CrossRef]
- Sahar, S.; Sassone-Corsi, P. Metabolism and cancer: The circadian clock connection. Nat. Rev. Cancer 2009, 9, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Takekida, S.; Shigeyoshi, Y.; Okamura, H. Per1 and Per2 gene expression in the rat suprachiasmatic nucleus: Circadian profile and the compartment-specific response to light. Neuroscience 1999, 94, 141–150. [Google Scholar] [CrossRef]
- Matsui, D.; Takekida, S.; Okamura, H. Molecular oscillation of Per1 and Per2 genes in the rodent brain: An in situ hybridization and molecular biological study. Kobe J. Med. Sci. 2005, 51, 85–93. [Google Scholar] [PubMed]
- Challet, E.; Poirel, V.J.; Malan, A.; Pevet, P. Light exposure during daytime modulates expression of Per1 and Per2 clock genes in the suprachiasmatic nuclei of mice. J. Neurosci. Res. 2003, 72, 629–637. [Google Scholar] [CrossRef]
- Marchetti, C. Molecular targets of lead in brain neurotoxicity. Neurotox. Res. 2003, 5, 221–236. [Google Scholar] [CrossRef]
- Cao, Y.; Liu, H.; Li, Q.; Wang, Q.; Zhang, W.; Chen, Y.; Wang, D.; Cai, Y. Effect of lead sulfide nanoparticles exposure on calcium homeostasis in rat hippocampus neurons. J. Inorg. Biochem. 2013, 126, 70–75. [Google Scholar] [CrossRef]
- Nahm, S.S.; Farnell, Y.Z.; Griffith, W.; Earnest, D.J. Circadian regulation and function of voltage-dependent calcium channels in the suprachiasmatic nucleus. J. Neurosci. 2005, 25, 9304–9308. [Google Scholar] [CrossRef]
- Kalinchuk, A.V.; McCarley, R.W.; Porkka-Heiskanen, T.; Basheer, R. The time course of adenosine, nitric oxide (NO) and inducible NO synthase changes in the brain with sleep loss and their role in the non-rapid eye movement sleep homeostatic cascade. J. Neurochem. 2011, 116, 260–272. [Google Scholar] [CrossRef]
- Obrietan, K.; Belousov, A.B.; Heller, H.C.; van den Pol, A.N. Adenosine pre- and postsynaptic modulation of glutamate-dependent calcium activity in hypothalamic neurons. J. Neurophysiol. 1995, 74, 2150–2162. [Google Scholar] [CrossRef]
- Ahnaou, A.; Ver Donck, L.; Drinkenburg, W.H. Blockade of the metabotropic glutamate (mGluR2) modulates arousal through vigilance states transitions: Evidence from sleep–wake EEG in rodents. Behav. Brain Res. 2014, 270, 56–67. [Google Scholar] [CrossRef]
- Cavas, M.; Scesa, G.; Navarro, J.F. Positive allosteric modulation of mGlu7 receptors by AMN082 affects sleep and wakefulness in the rat. Pharmacol. Biochem. Behav. 2013, 103, 756–763. [Google Scholar] [CrossRef]
- Greene, R.W. Role for neuronal nitric oxide synthase in sleep homeostasis and arousal. Proc. Natl. Acad. Sci. USA 2013, 110, 19982–19983. [Google Scholar] [CrossRef]
- Garcia-Arenas, G.; Claudio, L.; Perez-Severiano, F.; Rios, C. Lead acetate exposure inhibits nitric oxide synthase activity in capillary and synaptosomal fractions of mouse brain. Toxicol. Sci. 1999, 50, 244–248. [Google Scholar] [CrossRef]
- Crouzier, D.; Baubichon, D.; Bourbon, F.; Testylier, G. Acetylcholine release, EEG spectral analysis, sleep staging and body temperature studies: A multiparametric approach on freely moving rats. J. Neurosci. Methods 2006, 151, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Mike, A.; Pereira, E.F.; Albuquerque, E.X. Ca2+-sensitive inhibition by Pb2+ of alpha7-containing nicotinic acetylcholine receptors in hippocampal neurons. Brain Res. 2000, 873, 112–123. [Google Scholar] [CrossRef]
- Qiu, M.H.; Liu, W.; Qu, W.M.; Urade, Y.; Lu, J.; Huang, Z.L. The role of nucleus accumbens core/shell in sleep–wake regulation and their involvement in modafinil-induced arousal. PLoS ONE 2012, 7, e45471. [Google Scholar] [CrossRef] [PubMed]
- Nowak, P.; Szczerbak, G.; Nitka, D.; Kostrzewa, R.M.; Josko, J.; Brus, R. Cortical dopaminergic neurotransmission in rats intoxicated with lead during pregnancy. Nitric oxide and hydroxyl radicals formation involvement. Neurotoxicol. Teratol. 2008, 30, 428–432. [Google Scholar] [CrossRef]
- Hu, Q.; Fu, H.; Song, H.; Ren, T.; Li, L.; Ye, L.; Liu, T.; Dong, S. Low-level lead exposure attenuates the expression of three major isoforms of neural cell adhesion molecule. Neurotoxicology 2011, 32, 255–260. [Google Scholar] [CrossRef]
- Citri, A.; Malenka, R.C. Synaptic plasticity: Multiple forms, functions, and mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef] [PubMed]
- Black, M.A.; Deurveilher, S.; Seki, T.; Marsh, D.R.; Rutishauser, U.; Rafuse, V.F.; Semba, K. Role of polysialylated neural cell adhesion molecule in rapid eye movement sleep regulation in rats. Eur. J. Neurosci. 2009, 30, 2190–2204. [Google Scholar] [CrossRef]
- McCabe, M.J., Jr.; Singh, K.P.; Reiners, J.J., Jr. Low level lead exposure in vitro stimulates the proliferation and expansion of alloantigen-reactive CD4(high) T cells. Toxicol. Appl. Pharmacol. 2001, 177, 219–231. [Google Scholar] [CrossRef]
- Gast, H.; Muller, A.; Lopez, M.; Meier, D.; Huber, R.; Dechent, F.; Prinz, M.; Emmenegger, Y.; Franken, P.; Birchler, T.; et al. CD40 activation induces NREM sleep and modulates genes associated with sleep homeostasis. Brain Behav. Immun. 2013, 27, 133–144. [Google Scholar] [CrossRef]
- Chen, J.; Li, M.; Lv, Q.; Chen, G.; Li, Y.; Li, S.; Mo, Y.; Ou, S.; Yuan, Z.; Huang, M.; et al. Blood lead level and its relationship to essential elements in preschool children from Nanning, China. J. Trace Elem. Med. Biol. 2015, 30, 137–141. [Google Scholar] [CrossRef]
- Mendelsohn, A.L.; Dreyer, B.P.; Fierman, A.H.; Rosen, C.M.; Legano, L.A.; Kruger, H.A.; Lim, S.W.; Courtlandt, C.D. Low-level lead exposure and behavior in early childhood. Pediatrics 1998, 101, E10. [Google Scholar] [CrossRef]
- Chambial, S.; Shukla, K.K.; Dwivedi, S.; Bhardwaj, P.; Sharma, P. Blood Lead Level (BLL) in the Adult Population of Jodhpur: A Pilot Study. Indian J. Clin. Biochem. 2015, 30, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.C.; Oh, S.Y.; Kwon, S.O.; Park, M.S.; Kim, H.; Leem, J.H.; Ha, E.H. Blood lead level modifies the association between dietary antioxidants and oxidative stress in an urban adult population. Br. J. Nutr. 2013, 109, 148–154. [Google Scholar] [CrossRef] [PubMed][Green Version]



{kind=link}
{kind=link}
| Term | Lead-Exposure Group (n = 6) | Control Group (n = 6) | p |
|---|---|---|---|
| Body weight (g) | 406.6 ± 25.8 | 412.1 ± 20.7 | 0.925 |
| Blood lead (μg/dL) | 15.30 ± 3.22 | 0.90 ± 0.10 | <0.001 |
| Serum creatinine (mg/dL) Daily urine amount (mL) | 0.39 ± 0.18 22.3 ± 5.1 | 0.23 ± 0.06 15.8 ± 4.9 | 0.039 0.017 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, C.-Y.; Chuang, Y.-C.; Chang, F.-C.; Chuang, H.-Y.; Chiou, T.T.-Y.; Lee, C.-T. Disrupted Sleep Homeostasis and Altered Expressions of Clock Genes in Rats with Chronic Lead Exposure. Toxics 2021, 9, 217. https://doi.org/10.3390/toxics9090217
Hsu C-Y, Chuang Y-C, Chang F-C, Chuang H-Y, Chiou TT-Y, Lee C-T. Disrupted Sleep Homeostasis and Altered Expressions of Clock Genes in Rats with Chronic Lead Exposure. Toxics. 2021; 9(9):217. https://doi.org/10.3390/toxics9090217
Chicago/Turabian StyleHsu, Chung-Yao, Yao-Chung Chuang, Fang-Chia Chang, Hung-Yi Chuang, Terry Ting-Yu Chiou, and Chien-Te Lee. 2021. "Disrupted Sleep Homeostasis and Altered Expressions of Clock Genes in Rats with Chronic Lead Exposure" Toxics 9, no. 9: 217. https://doi.org/10.3390/toxics9090217
APA StyleHsu, C.-Y., Chuang, Y.-C., Chang, F.-C., Chuang, H.-Y., Chiou, T. T.-Y., & Lee, C.-T. (2021). Disrupted Sleep Homeostasis and Altered Expressions of Clock Genes in Rats with Chronic Lead Exposure. Toxics, 9(9), 217. https://doi.org/10.3390/toxics9090217