Hydrogen Peroxide Causes Cell Death via Increased Transcription of HOXB13 in Human Lung Epithelial A549 Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Measurement of Cytotoxicity
2.3. Measurement of mRNA Levels by Quantitative PCR
2.4. Immunoblotting
2.5. Construction of V5 Tag Fused HOXB13 (HOXB13-V5)-Expressing Plasmid
2.6. Measurement of Intercellular Reactive Oxygen Species
2.7. Statistical Analysis
3. Results
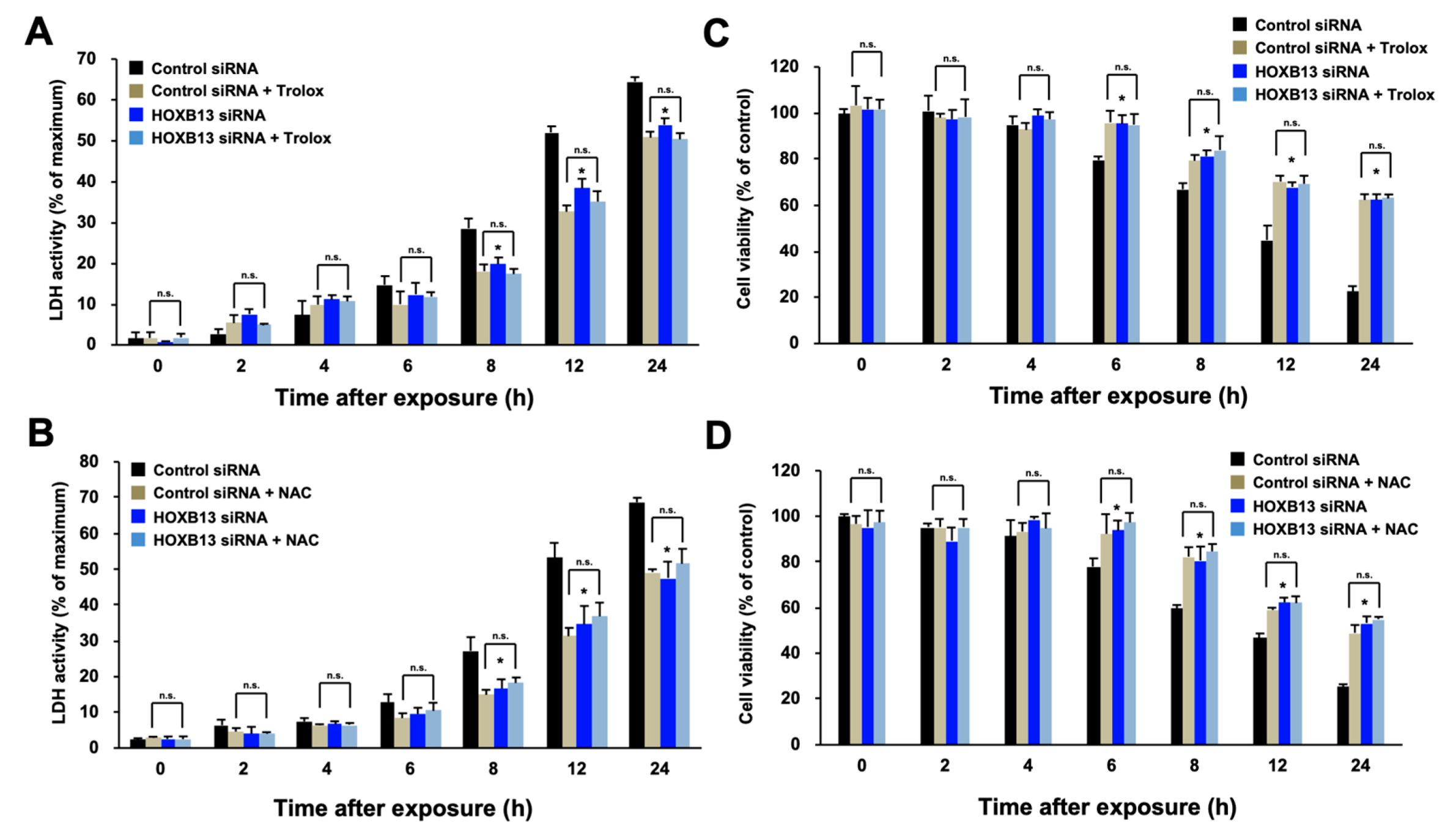
3.1. HOXB13 Is Involved in the Cell Death Caused by Hydrogen Peroxide through Enhancement of Oxidative Stress
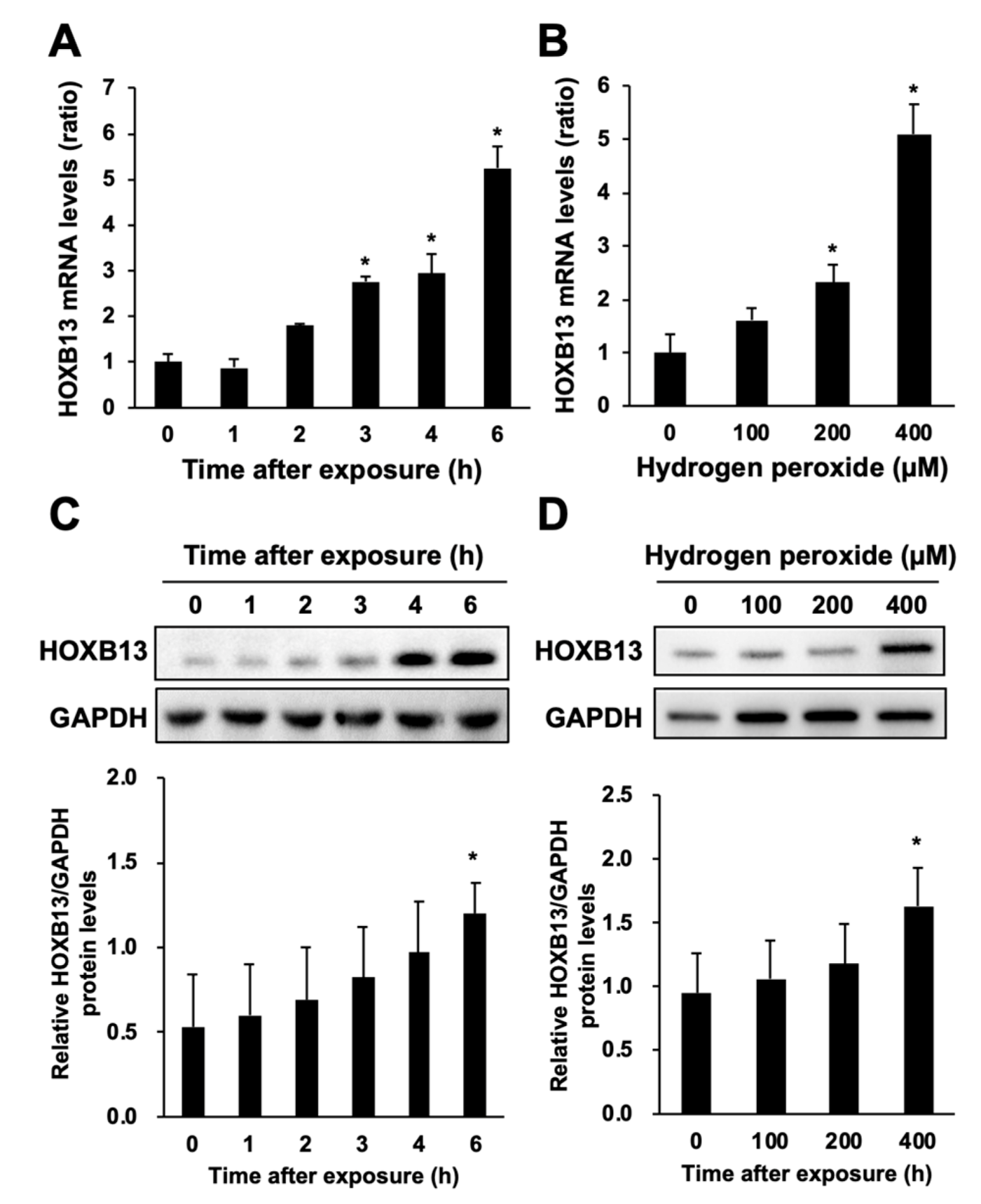
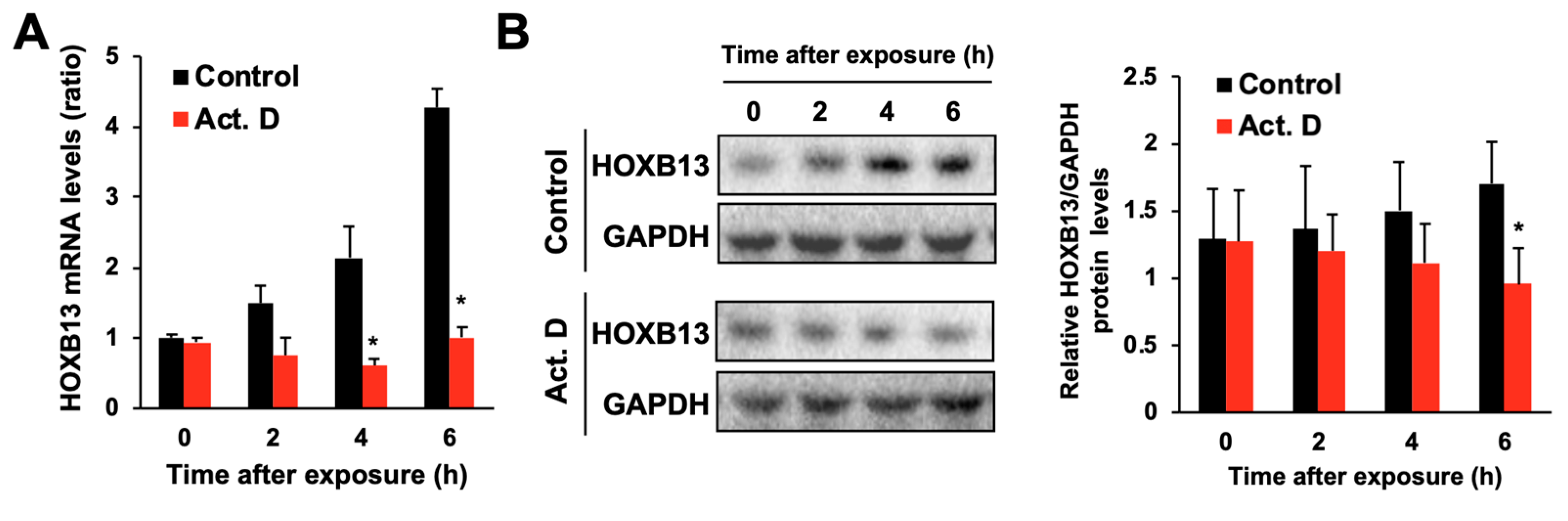
3.2. Hydrogen Peroxide Increases HOXB13 Protein Levels via Increasing Its Transcription
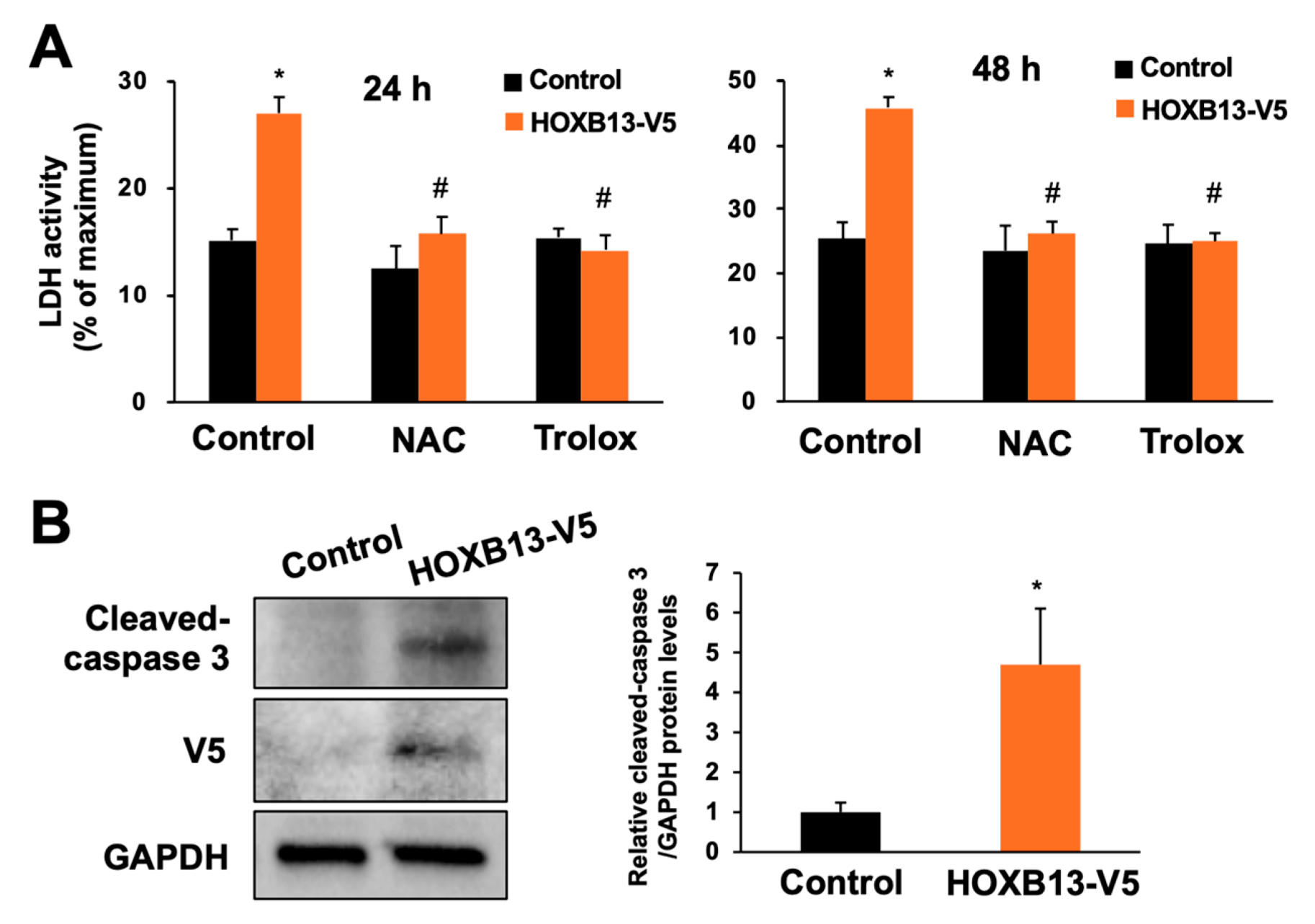
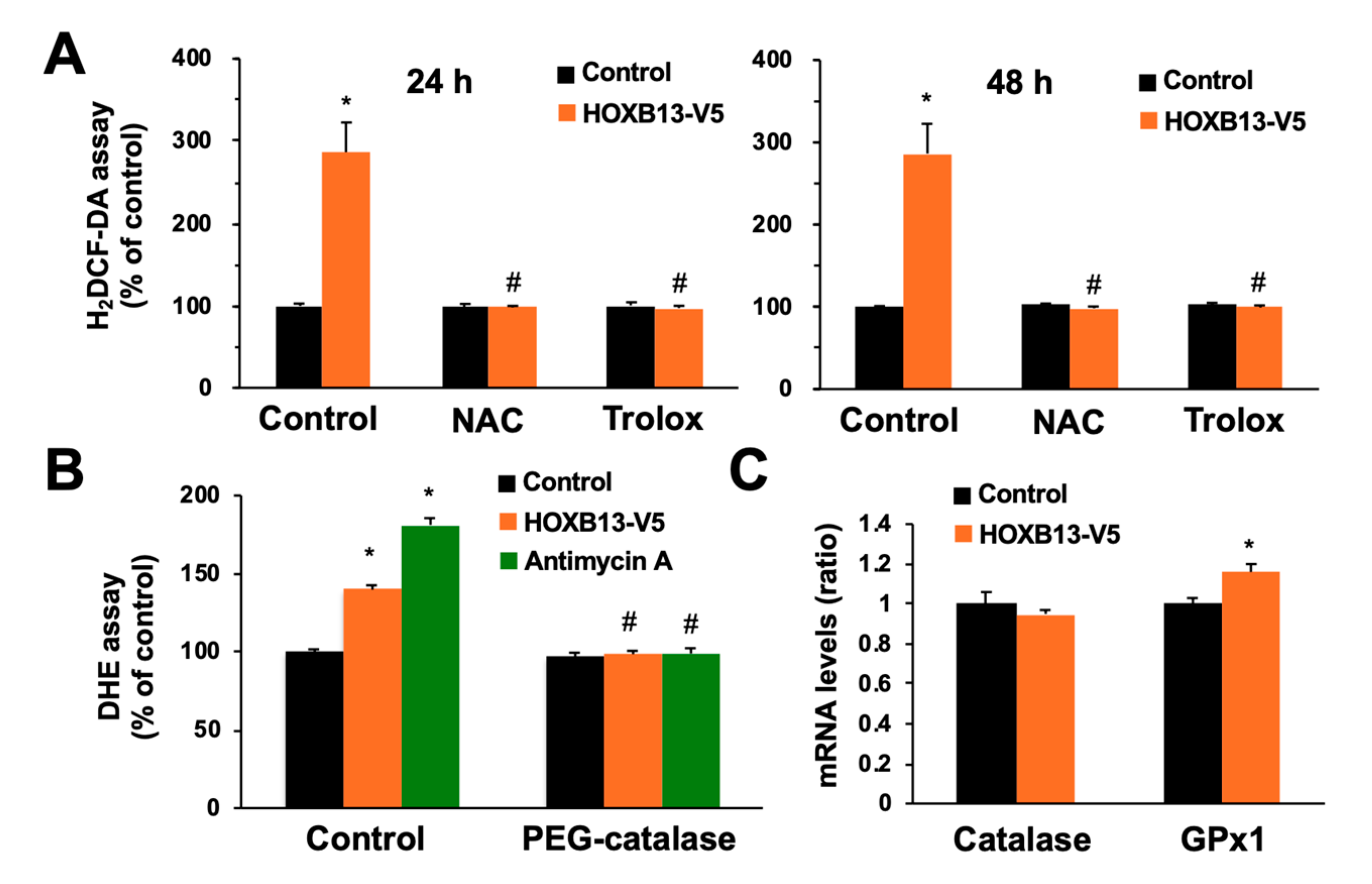
3.3. Overexpression of HOXB13 Caused Cell Death via Endogenous ROS Production
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- MacNee, W. Oxidative stress and lung inflammation in airways disease. Eur. J. Pharmacol. 2001, 429, 195–207. [Google Scholar] [CrossRef]
- Chow, C.W.; Herrera Abreu, M.T.; Suzuki, T.; Downey, G.P. Oxidative Stress and Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2003, 4, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Rogers, L.K.; Cismowski, M.J. Oxidative Stress in the Lung—The Essential Paradox. Curr. Opin. Toxicol. 2018, 7, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Vokurkova, M.; Xu, S.; Touyz, R.M. Reactive oxygen species, cell growth, cell cycle progression and vascular remodeling in hypertension. Future Cardiol. 2007, 3, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Kinnula, V.L.; Pääkkö, P.; Soini, Y. Antioxidant enzymes and redox regulating thiol proteins in malignancies of human lung. FEBS Lett. 2004, 569, 1–6. [Google Scholar] [CrossRef]
- Rahman, Q.; Abidi, P.; Afaq, F.; Schiffmann, D.; Mossman, B.T.; Kamp, D.W.; Athar, M. Glutathione redox system in oxidative lung injury. Crit. Rev. Toxicol. 1999, 29, 543–568. [Google Scholar] [CrossRef]
- Wispé, J.R.; Warner, B.B.; Clark, J.C.; Dey, C.R.; Neuman, J.; Glasser, S.W.; Crapo, J.D.; Chang, L.Y.; Whitsett, J.A. Human Mn-superoxide dismutase in pulmonary epithelial cells of transgenic mice confers protection from oxygen injury. J. Biol. Chem. 1992, 267, 23937–23941. [Google Scholar]
- Satoh, M.; Fujimoto, S.; Haruna, Y.; Arakawa, S.; Horike, H.; Komai, N.; Sasaki, T.; Tsujioka, K.; Makino, H.; Kashihara, N. NAD(P)H oxidase and uncoupled nitric oxide synthase are major sources of glomerular superoxide in rats with experimental diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2005, 288, F1144–F1152. [Google Scholar] [CrossRef]
- Economides, K.D.; Zeltser, L.; Capecchi, M.R. Hoxb13 mutations cause overgrowth of caudal spinal cord and tail vertebrae. Dev. Biol. 2003, 256, 317–330. [Google Scholar] [CrossRef]
- Xiong, Y.; Kuang, W.; Lu, S.; Guo, H.; Wu, M.; Ye, M.; Wu, L. Long noncoding RNA HOXB13-AS1 regulates HOXB13 gene methylation by interacting with EZH2 in glioma. Cancer Med. 2018, 7, 4718–4728. [Google Scholar] [CrossRef]
- Bai-Qiang, S.; Chun-Dong, Z.; Ji-Chao, L.; Lei, W.; Dong-Qiu, D. HOXB13 expression and promoter methylation as a candidate biomarker in gastric cancer. Oncol Lett. 2018, 15, 8833–8840. [Google Scholar]
- Miao, J.; Wang, Z.; Provencher, H.; Muir, B.; Dahiya, S.; Carney, E.; Leong, C.O.; Sgroi, D.C.; Orsulic, S. HOXB13 promotes ovarian cancer progression. Proc. Natl. Acad. Sci. USA 2007, 104, 17093–17098. [Google Scholar] [CrossRef] [PubMed]
- Ewing, C.M.; Ray, A.M.; Lange, E.M.; Zuhlke, K.A.; Robbins, C.M.; Tembe, W.D.; Wiley, K.E.; Isaacs, S.D.; Johng, D.; Wang, Y.; et al. Germline Mutations in HOXB13 and Prostate-Cancer Risk. N. Engl. J. Med. 2012, 366, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Wang, P.; Li, S.; Song, J.; He, H.; Wang, Y.; Liu, Z.; Wang, F.; Bai, H.; Fang, W.; et al. HOXB13 networking with ABCG1/EZH2/Slug mediates metastasis and confers resistance to cisplatin in lung adenocarcinoma patients. Theranostics 2019, 9, 2084–2099. [Google Scholar] [CrossRef]
- Nakano, R.; Takahashi, T.; Naganuma, A.; Hwang, G.W. Knockdown of the gene for homeobox protein HOXB13 reduces toxicity of oxidative-stress inducers in HEK293 cells. J. Toxicol. Sci. 2013, 38, 821–822. [Google Scholar] [CrossRef]
- Tikoo, K.; Lau, S.S.; Monks, T.J. Histone H3 phosphorylation is coupled to poly(ADP-ribosylation) during reactive oxygen species-induced cell death in renal proximal tubular epithelial cells. Mol. Pharmacol. 2001, 60, 394–402. [Google Scholar] [CrossRef]
- Burtke, T.M.; Sandstrom, P.A. Oxidative stress as a mediator of apoptosis. Immunol. Today 1994, 15, 7–10. [Google Scholar]
- Kharroubi, I.; Ladrière, L.; Cardozo, A.K.; Dogusan, Z.; Cnop, M.; Eizirik, D.L. Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: Role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology 2004, 145, 5087–5096. [Google Scholar] [CrossRef]
- Kang, K.W.; Lee, S.J.; Kim, S.G. Molecular Mechanism of nrf2 activation by oxidative stress. Antioxid. Redox Signal. 2005, 7, 1664–1673. [Google Scholar] [CrossRef]
- Ahn, S.G.; Thiel, D.J. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 2003, 17, 516–528. [Google Scholar] [CrossRef]
- Wu, W.S. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Reactive Oxygen Species in Endothelial Function—From Disease to Adaptation–. Circ. J. 2015, 79, 1145–1155. [Google Scholar]
- Saito, S.; Ogawa, J.; Minamiya, Y. Pulmonary reexpansion causes xanthine oxidase-induced apoptosis in rat lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L400–L406. [Google Scholar] [CrossRef] [PubMed]
- Seno, T.; Inoue, N.; Gao, D.; Okuda, M.; Sumi, Y.; Matsui, K.; Yamada, S.; Hirata, K.I.; Kawashima, S.; Tawa, R.; et al. Involvement of NADH/NADPH Oxidase in Human Platelet ROS Production and xanthine oxidase. Thromb. Res. 2001, 103, 399–409. [Google Scholar] [CrossRef]
- Chang, S.; Jiang, X.; Zhao, C.; Lee, C.; Ferriero, D.M. Exogenous low dose hydrogen peroxide increases hypoxia-inducible factor-1alpha protein expression and induces preconditioning protection against ischemia in primary cortical neurons. Neurosci. Lett. 2008, 441, 134–138. [Google Scholar] [CrossRef]
- Chu, S.; Ferro, T.J. Identification of a hydrogen peroxide-induced PP1-JNK1-Sp1 signaling pathway for gene regulation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L983–L992. [Google Scholar] [CrossRef]
- Hamid, S.M.; Cicek, S.; Karamil, S.; Ozturk, M.B.; Debelec-Butuner, B.; Erbaykent-Tepedelen, B.; Varisli, L.; Gonen-Korkmaz, C.; Yorukoglu, K.; Korkmaz, K.S. HOXB13 contributes to G1/S and G2/M checkpoint controls in prostate. Mol. Cell. Endocrinol. 2014, 383, 38–47. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Endo, N.; Toyama, T.; Naganuma, A.; Saito, Y.; Hwang, G.-W. Hydrogen Peroxide Causes Cell Death via Increased Transcription of HOXB13 in Human Lung Epithelial A549 Cells. Toxics 2020, 8, 78. https://doi.org/10.3390/toxics8040078
Endo N, Toyama T, Naganuma A, Saito Y, Hwang G-W. Hydrogen Peroxide Causes Cell Death via Increased Transcription of HOXB13 in Human Lung Epithelial A549 Cells. Toxics. 2020; 8(4):78. https://doi.org/10.3390/toxics8040078
Chicago/Turabian StyleEndo, Naoki, Takashi Toyama, Akira Naganuma, Yoshiro Saito, and Gi-Wook Hwang. 2020. "Hydrogen Peroxide Causes Cell Death via Increased Transcription of HOXB13 in Human Lung Epithelial A549 Cells" Toxics 8, no. 4: 78. https://doi.org/10.3390/toxics8040078
APA StyleEndo, N., Toyama, T., Naganuma, A., Saito, Y., & Hwang, G.-W. (2020). Hydrogen Peroxide Causes Cell Death via Increased Transcription of HOXB13 in Human Lung Epithelial A549 Cells. Toxics, 8(4), 78. https://doi.org/10.3390/toxics8040078