Supramolecular Solvent-Based Liquid Phase Microextraction Combined with Ion-Pairing Reversed-Phase HPLC for the Determination of Quats in Vegetable Samples



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Instrumentation
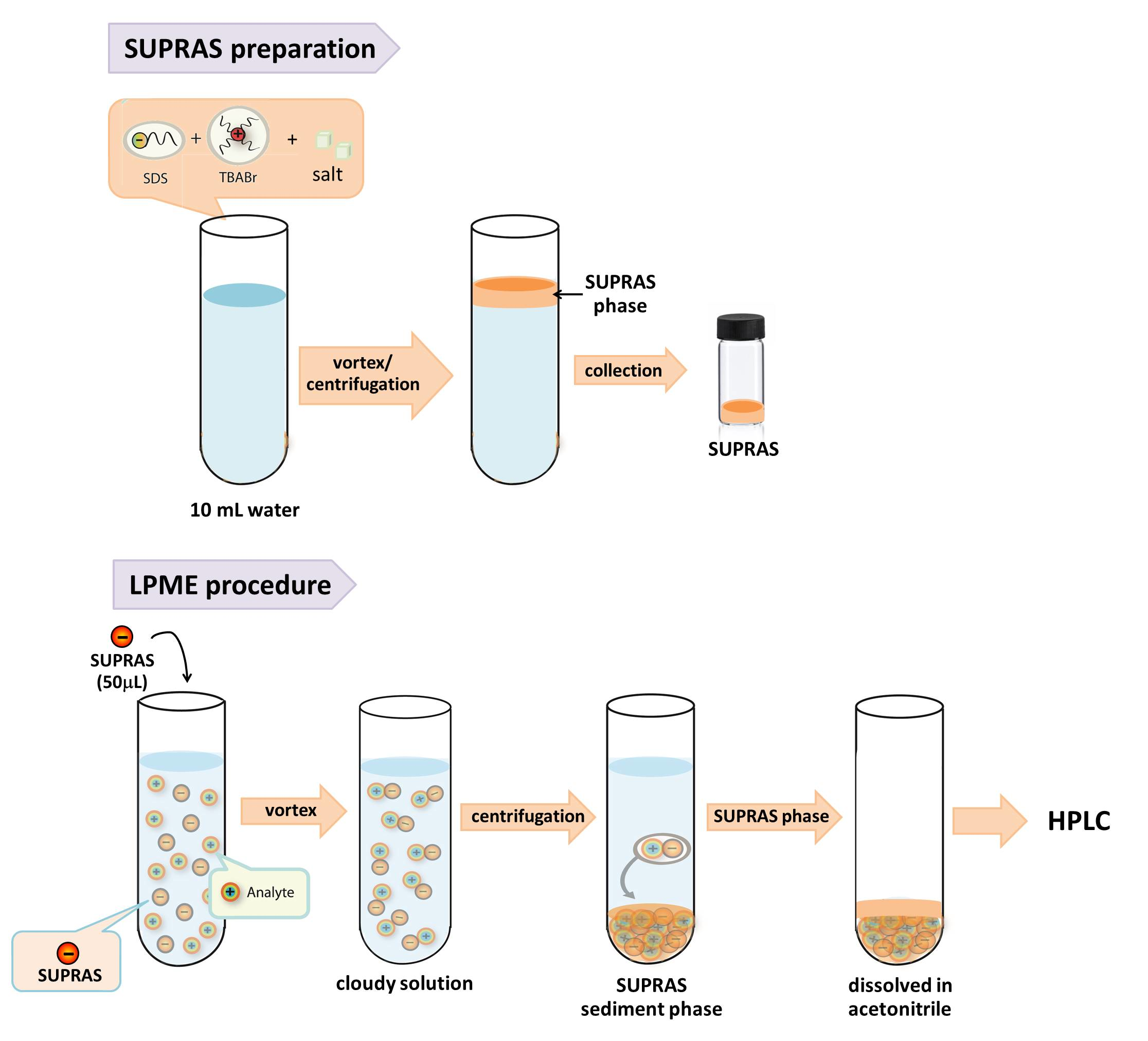
2.3. SUPRAS Preparation
2.4. LPME Procedure
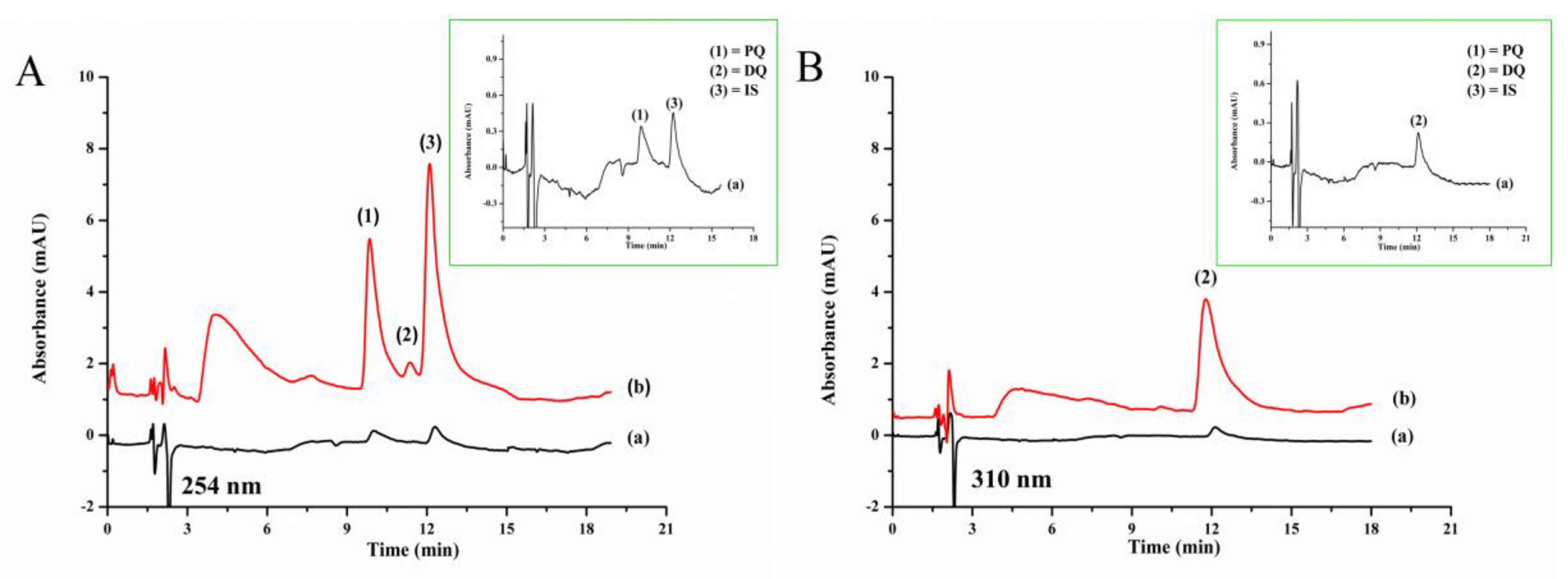
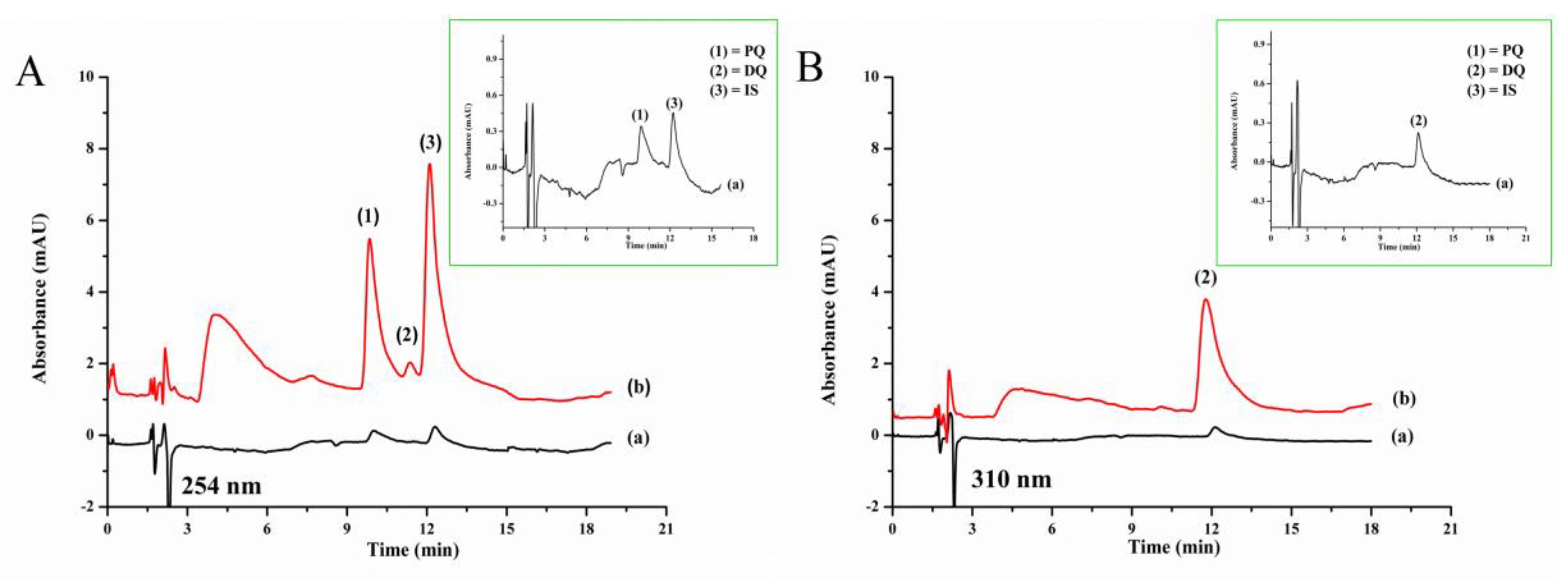
2.5. Chromatographic Separation of Quats
2.6. Sample Preparation
2.7. Validation Study
3. Results and Discussion
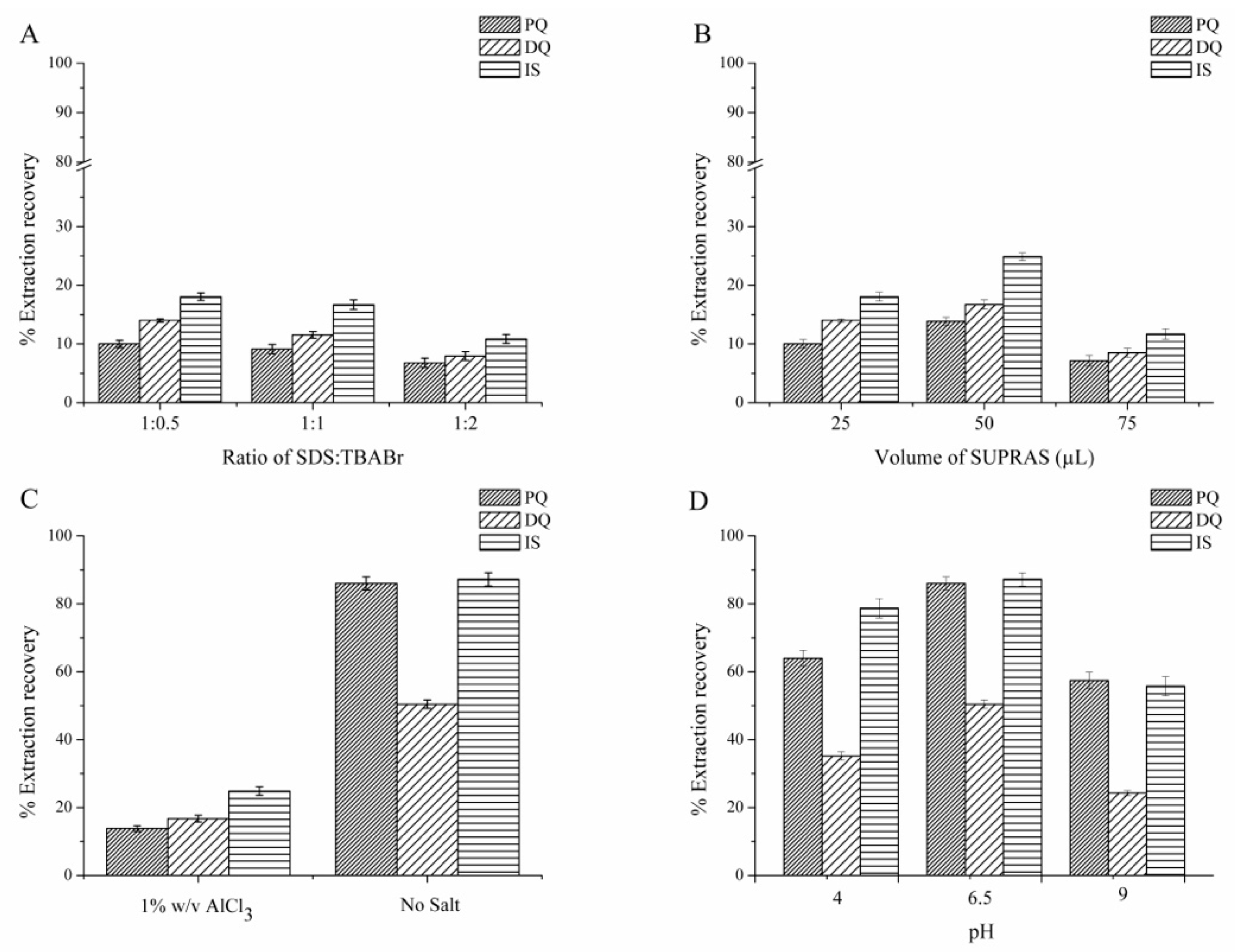
3.1. Optimization of SUPRAS-Based Liquid Phase Microextraction
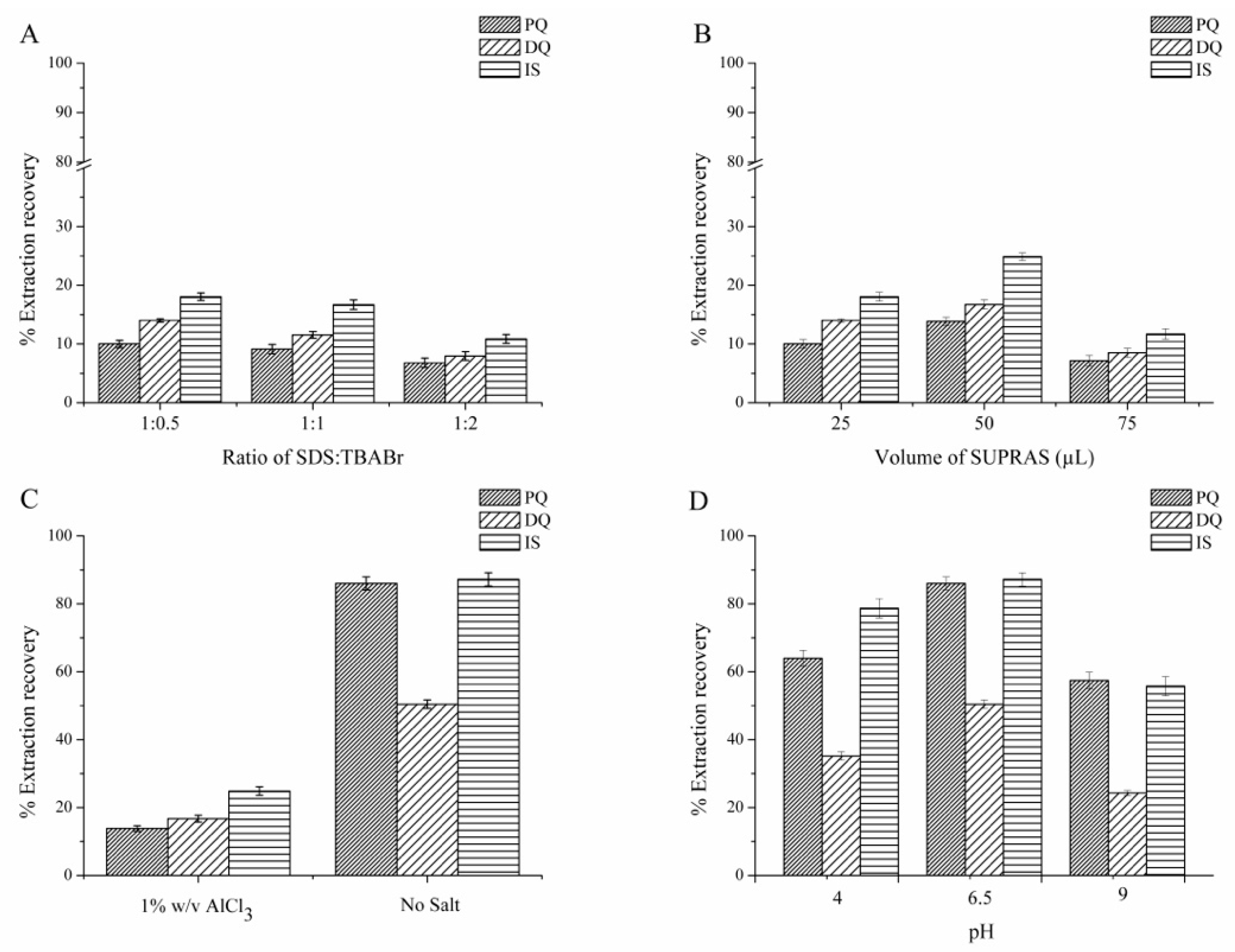
3.1.1. Effect of Surfactant Composition (SDS:TBABr)
3.1.2. Effect of SUPRAS Volume
3.1.3. Effect of Salt
3.1.4. Effect of pH
3.1.5. Effect of Vortex Time
3.1.6. Effect of Centrifugation Time
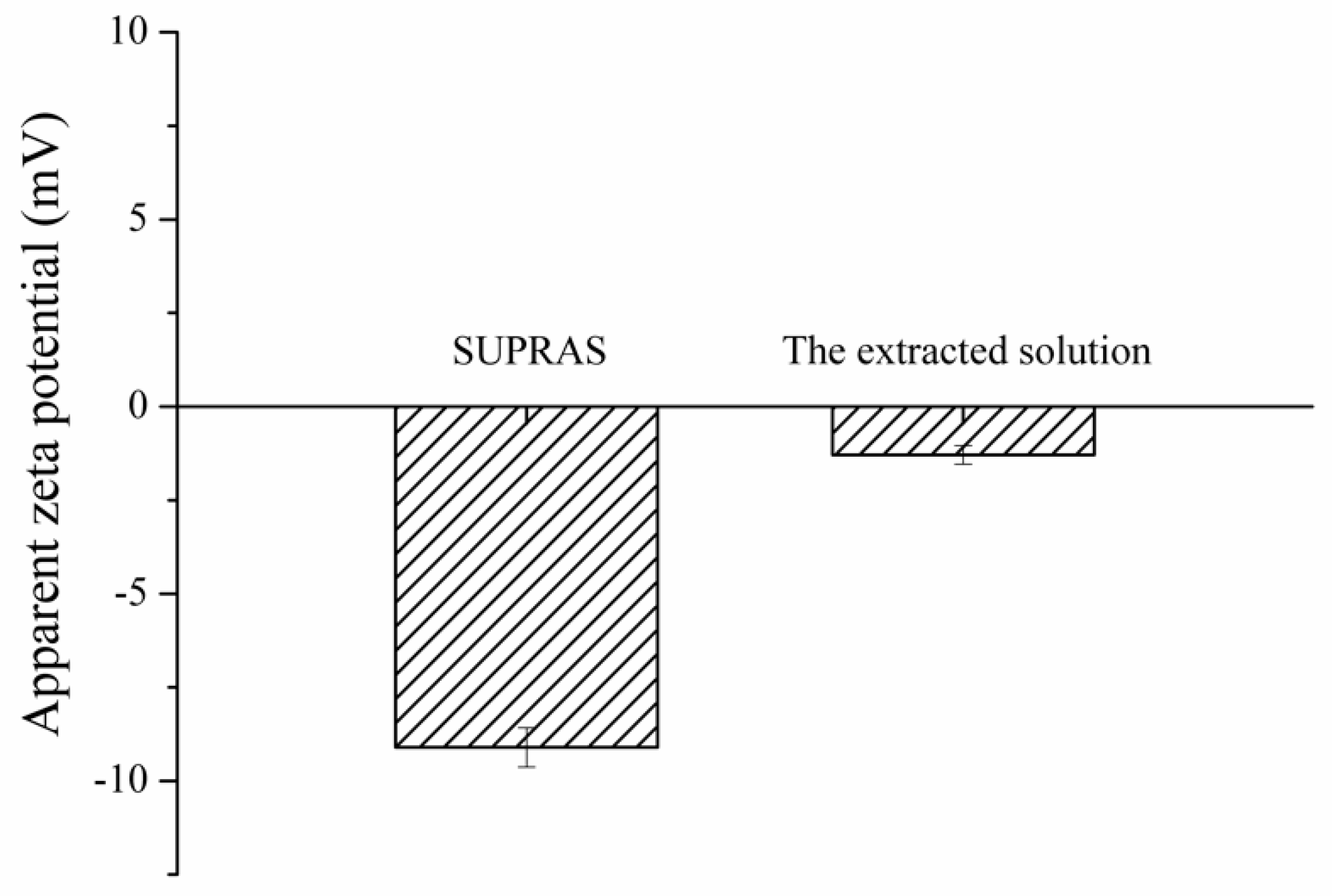
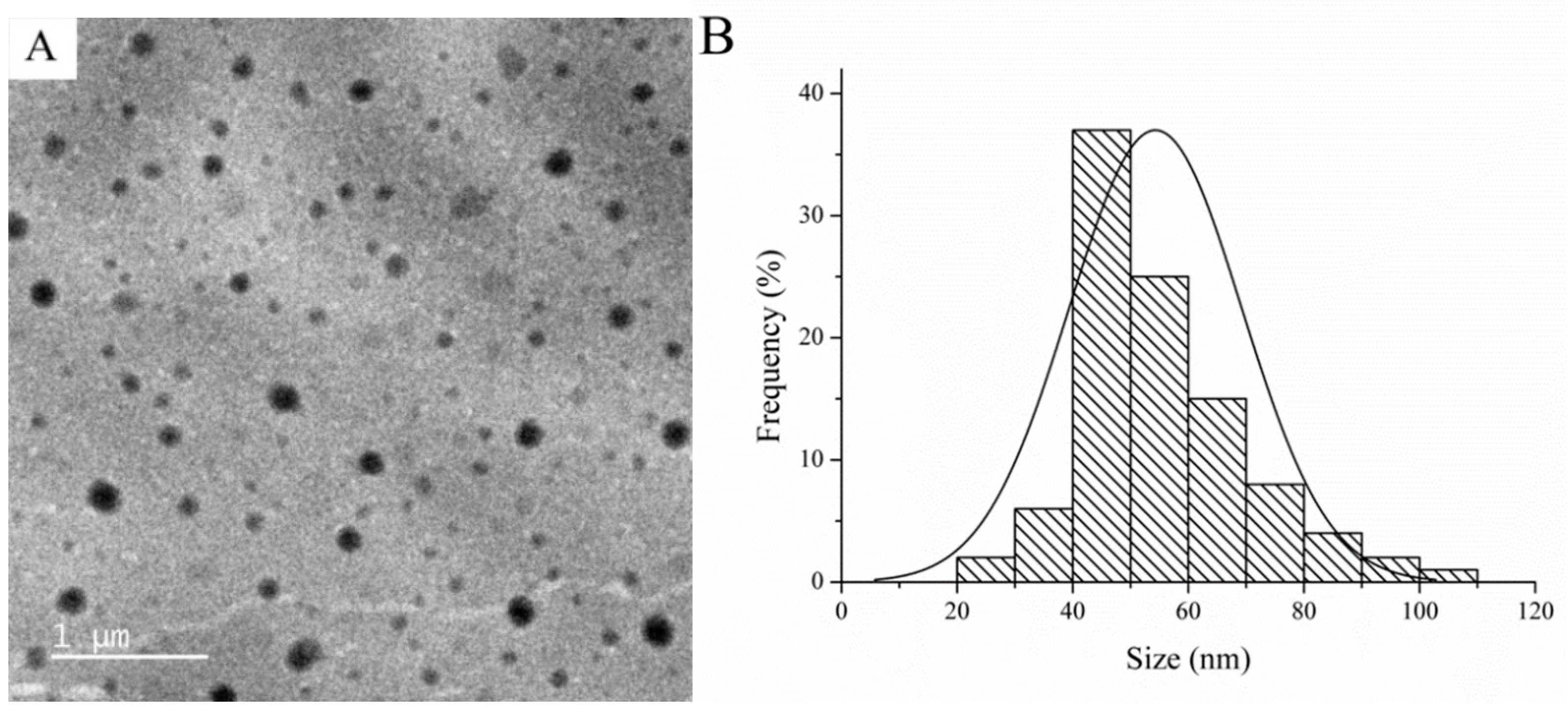
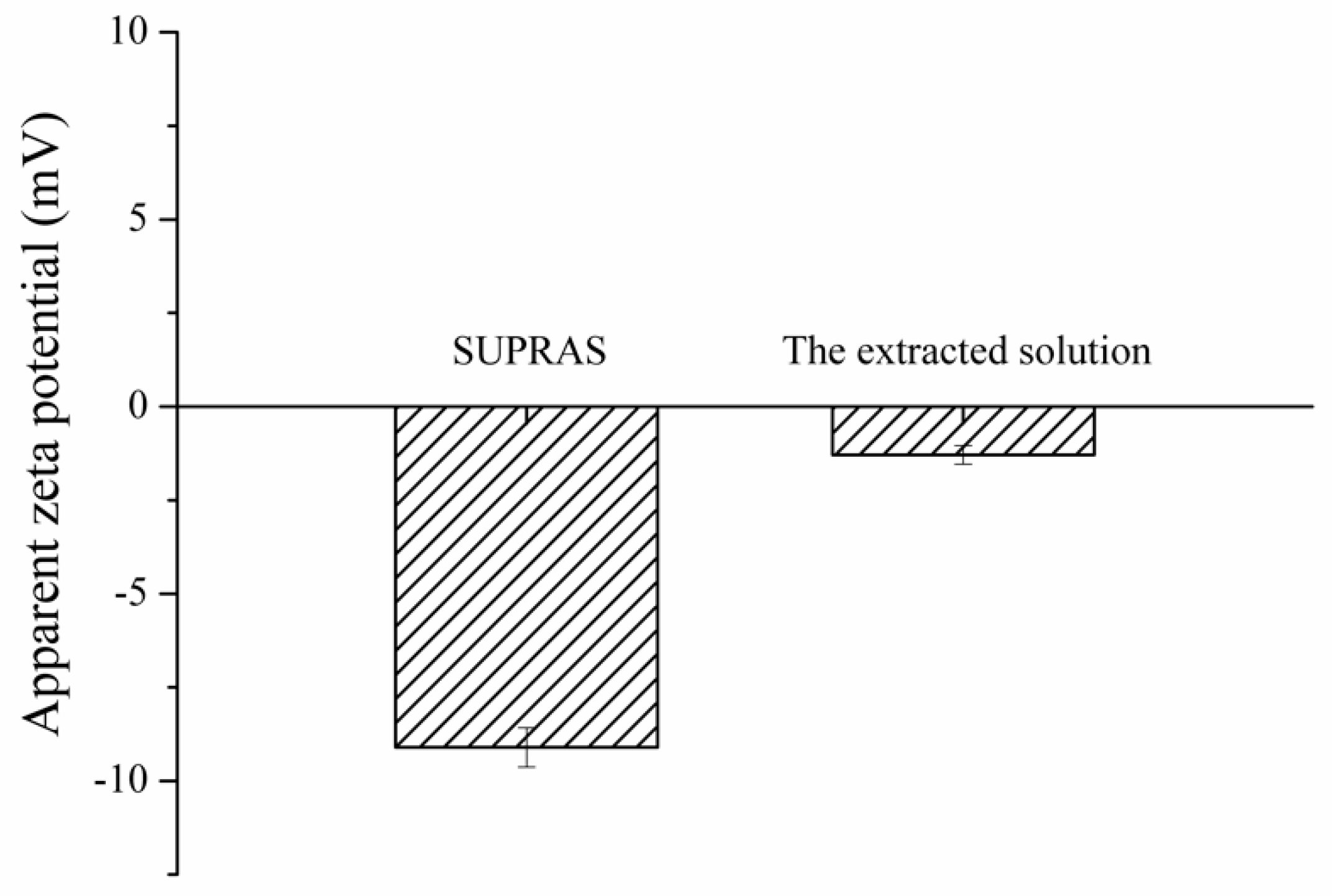
3.2. Proposed Extraction Mechanism
3.3. Analytical Performance and Method Validation
3.4. Comparison with Other Methods
3.5. Analysis of Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ariffin, M.M.; Anderson, R.A. LC/MS/MS analysis of quaternary ammonium drugs and herbicides in whole blood. J. Chromatogr. B 2006, 842, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Brunetto, M.R.; Morales, A.R.; Gallignani, M.; Burguera, J.L.; Burguera, M. Determination of paraquat in human blood plasma using reversed-phase ion-pair high-performance liquid chromatography with direct sample injection. Talanta 2003, 59, 913–921. [Google Scholar] [CrossRef]
- Grey, L.; Nguyen, B.; Yang, P. Liquid chromatography-electrospray ionization isotope dilution mass spectrometry analysis of paraquat and diquat using conventional and multilayer solid-phase extraction cartridges. J. Chromatogr. A 2002, 958, 25–33. [Google Scholar] [CrossRef]
- Castro, R.; Moyano, E.; Galceran, M.T. Ion-pair liquid chromatography–atmospheric pressure ionization mass spectrometry for the determination of quaternary ammonium herbicides. J. Chromatogr. A 1999, 830, 145–154. [Google Scholar] [CrossRef]
- Carneiro, M.C.; Puignou, L.; Galceran, M.T. Comparison of silica and porous graphitic carbon as solid-phase extraction materials for the analysis of cationic herbicides in water by liquid chromatography and capillary electrophoresis. Anal. Chim. Acta 2000, 408, 263–269. [Google Scholar] [CrossRef]
- Lu, H.; Yu, J.; Wu, L.; Xing, J.; Wang, J.; Huang, P.; Zhang, J.; Xiao, H.; Gao, R. Optimized ultra-performance liquid chromatography tandem high resolution mass spectrometry method for the quantification of paraquat in plasma and urine. J. Chromatogr. B 2016, 1027, 96–102. [Google Scholar] [CrossRef]
- Pizzutti, I.R.; Vela, G.M.E.; Kok, A.D.; Scholten, J.M.; Dias, J.V.; Cardoso, C.D.; Concenço, G.; Vivian, R. Determination of paraquat and diquat: LC-MS method optimization and validation. Food Chem. 2016, 209, 248–255. [Google Scholar] [CrossRef]
- Garcia-Febrero, R.; Salvador, J.P.; Sanchez-Baeza, F.; Marco, M.P. Rapid method based on immunoassay for determination of paraquat residues in wheat, barley and potato. Food Control 2014, 41, 193–201. [Google Scholar] [CrossRef]
- Martínez Vidal, J.L.; Belmonte Vega, A.; Sánchez López, F.J.; Garrido Frenich, A. Application of internal quality control to the analysis of quaternary ammonium compounds in surface and groundwater from Andalusia (Spain) by liquid chromatography with mass spectrometry. J. Chromatogr. A 2004, 1050, 179–184. [Google Scholar] [CrossRef]
- Posecion, N.C.; Ostrea, E.M.; Bielawski, D.M. Quantitative determination of paraquat in meconium by sodium borohydride-nickel chloride chemical reduction and gas chromatography/mass spectrometry (GC/MS). J. Chromatogr. B 2008, 862, 93–99. [Google Scholar] [CrossRef]
- Zou, T.; He, P.; Cao, J.; Li, Z. Determination of paraquat in vegetables using HPLC-MS-MS. J. Chromatogr. Sci. 2015, 53, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Raina-Fulton, R. A review of methods for the analysis of orphan and difficult pesticides: Glyphosate, glufosinate, quaternary ammonium and phenoxy acid herbicides, and dithiocarbamate and phthalimide fungicides. J. AOAC Int. 2014, 97, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Castro, R.; Moyano, E.; Galceran, M.T. On-line ion-pair solid-phase extraction-liquid chromatography-mass spectrometry for the analysis of quaternary ammonium herbicides. J. Chromatogr. A 2000, 11, 441–449. [Google Scholar] [CrossRef]
- Almeida, R.M.; Yonamine, M. Gas chromatographic-mass spectrometric method for the determination of the herbicides paraquat and diquat in plasma and urine samples. J. Chromatogr. B 2007, 853, 260–264. [Google Scholar] [CrossRef]
- Núñez, O.; Moyano, E.; Galceran, M.T. Capillary electrophoresis-mass spectrometry for the analysis of quaternary ammonium herbicides. J. Chromatogr. A 2002, 974, 243–255. [Google Scholar] [CrossRef]
- Mallat, E.; Barzen, C.; Abuknesha, R.; Gauglitz, G.; Barceló, D. Fast determination of paraquat residues in water by an optical immunosensor and validation using capillary electrophoresis-ultraviolet detection. Anal. Chim. Acta 2001, 427, 165–171. [Google Scholar] [CrossRef]
- Gill, R.; Qua, S.C.; Moffat, A.C. High-performance liquid chromatography of paraquat and diquat in urine with rapid sample preparation involving ion-pair extraction on disposable cartridges of octadecyl—silica. J. Chromatogr. A 1983, 255, 483–490. [Google Scholar] [CrossRef]
- Ito, S.; Nagata, T.; Kudo, K.; Kimura, K.; Imamura, T. Simultaneous determination of paraquat and diquat in human tissues by high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1993, 617, 119–123. [Google Scholar] [CrossRef]
- Whitehead, R.D., Jr.; Montesano, M.A.; Jayatilaka, N.K.; Buckley, B.; Winnik, B.; Needham, L.L.; Barr, D.B. Method for measurement of the quaternary amine compounds paraquat and diquat in human urine using high-performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B 2010, 878, 2548–2553. [Google Scholar] [CrossRef]
- Wang, K.C.; Chen, S.M.; Hsu, J.F.; Cheng, S.G.; Lee, C.K. Simultaneous detection and quantitation of highly water-soluble herbicides in serum using ion-pair liquid chromatography-tandem mass spectrometry. J. Chromatogr. B 2008, 876, 211–218. [Google Scholar] [CrossRef]
- Bassarab, P.; Williams, D.; Dean, J.R.; Ludkin, E.; Perry, J.J. Determination of quaternary ammonium compounds in seawater samples by solid-phase extraction and liquid chromatography-mass spectrometry. J. Chromatogr. B 2011, 1218, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Ruan, X.L.; Qiu, J.J.; Wu, C.; Huang, T.; Meng, R.B.; Lai, Y.Q. Magnetic single-walled carbon nanotubes-dispersive solid-phase extraction method combined with liquid chromatography-tandem mass spectrometry for the determination of paraquat in urine. J. Chromatogr. B 2014, 965, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Siangproh, W.; Somboonsuk, T.; Chailapakul, O.; Songsrirote, K. Novel colorimetric assay for paraquat detection on-silica bead using negatively charged silver nanoparticles. Talanta 2017, 174, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Liu, J.; Wang, C.; Liu, G.; Niu, X.; Shu, C.; Zhu, J. Fast determination of paraquat in plasma and urine samples by solid-phase microextraction and gas chromatography–mass spectrometry. J Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 944, 136–140. [Google Scholar] [CrossRef]
- Pico, Y.; Font, G.; Molto, J.C.; Manes, J. Solid-phase extraction of quaternary ammonium herbicides. J. Chromatogr. A 2000, 885, 251–271. [Google Scholar] [CrossRef]
- Asensio-Ramos, M.; Ravelo-Pérez, L.M.; González-Curbelo, M.A.; Hernández-Borges, J. Liquid phase microextraction applications in food analysis. J. Chromatogr. A 2011, 1218, 7415–7437. [Google Scholar] [CrossRef]
- Spietelun, A.; Marcinkowski, L.; Guardia, M.; Namieśnik, J. Green aspects, developments and perspectives of liquid phase microextraction techniques. Talanta 2014, 119, 34–45. [Google Scholar] [CrossRef]
- Hashemi, B.; Zohrabi, P.; Kim, K.H.; Shamsipur, M.; Deep, A.; Hong, J. Recent advances in liquid-phase microextraction techniques for the analysis of environmental pollutants. Trends Anal. Chem. 2017, 97, 83–95. [Google Scholar] [CrossRef]
- Rashidipour, M.; Heydari, R.; Maleki, A.; Mohammadi, E.; Davari, B. Salt-assisted liquid-liquid extraction coupled with reversed-phase dispersive liquid-liquid microextraction for sensitive HPLC determination of paraquat in environmental and food samples. J. Food Meas. Charact. 2019, 13, 269–276. [Google Scholar] [CrossRef]
- Hamamoto, T.; Katsuta, S. An ionic liquid-based microextraction method for ultra-high preconcentration of paraquat traces in water samples prior to HPLC determination. Anal. Sci. 2018, 34, 1439–1444. [Google Scholar] [CrossRef]
- Ballesteros-Gómez, A.; Sicilia, M.D.; Rubio, S. Supramolecular solvents in the extraction of organic compounds. A review. Anal. Chim. Acta 2010, 677, 108–130. [Google Scholar] [CrossRef] [PubMed]
- Accioni, F.; García-Gómez, D.; Girela, E.; Rubio, S. SUPRAS extraction approach for matrix-independent determination of amphetamine-type stimulants by LC-MS/MS. Talanta 2018, 182, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros-Gomez, A.; Lunar, L.; Sicilia, M.D.; Rubio, S. Hyphenating supramolecular solvents and liquid chromatography: Tips for efficient extraction and reliable determination of organics. Chromatographia 2019, 82, 111–124. [Google Scholar] [CrossRef]
- Salatti-Dorado, A.J.; García-Gómez, D.; Rodriguez-Ruiz, V.; Gueguen, V.; Pavon-Djavid, G.; Rubio, S. Multifunctional green supramolecular solvents for cost-effective production of highly stable astaxanthin-rich formulations from Haematococcus pluvialis. Food Chem. 2019, 279, 294–302. [Google Scholar] [CrossRef]
- Scheel, G.L.; Tarley, C.R.T. Feasibility of supramolecular solvent-based microextraction for simultaneous preconcentration of herbicides from natural waters with posterior determination by HPLC-DAD. Microchem. J. 2017, 133, 650–657. [Google Scholar] [CrossRef]
- Feizi, N.; Yamini, Y.; Moradi, M.; Karimi, M.; Salamat, Q.; Amanzadeh, H. A new generation of nano-structured supramolecular solvents based on propanol/gemini surfactant for liquid phase microextraction. Anal. Chim. Acta 2017, 953, 1–9. [Google Scholar] [CrossRef]
- Moral, A.; Sicilia, M.D.; Rubio, S. Supramolecular solvent-based extraction of benzimidazolic fungicides from natural waters prior to their liquid chromatographic/fluorimetric determination. J. Chromatogr. A 2009, 1216, 3740–3745. [Google Scholar] [CrossRef]
- Caballo, C.; Sicilia, M.D.; Rubio, S. Fast, simple and efficient supramolecular solvent-based microextraction of mecoprop and dichlorprop in soils prior to their enantioselective determination by liquid chromatography–tandem mass spectrometry. Talanta 2014, 119, 46–52. [Google Scholar] [CrossRef]
- López-Jiménez, F.J.; Rosales-Marcano, M.; Rubio, S. Restricted access property supramolecular solvents for combined microextraction of endocrine disruptors in sediment and sample cleanup prior to their quantification by liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2013, 1303, 1–8. [Google Scholar] [CrossRef]
- López-Jiménez, F.J.; Rubio, S.; Pérez-Bendito, D. Supramolecular solvent-based microextraction of Sudan dyes in chilli-containing foodstuffs prior to their liquid chromatography-photodiode array determination. Food Chem. 2010, 121, 763–769. [Google Scholar] [CrossRef]
- Gissawong, N.; Boonchiangma, S.; Mukdasai, S.; Srijaranai, S. Vesicular supramolecular solvent-based microextraction followed by high performance liquid chromatographic analysis of tetracyclines. Talanta 2019, 200, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Kukusamude, C.; Quirino, J.P.; Srijaranai, S. A coacervative extraction based on single-chain and double-chain cationic surfactants. J. Chromatogr. A 2016, 1472, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Soisungnoen, S.; Burakham, R.; Srijaranai, S. Determination of organophosphorus pesticides using dispersive liquid–liquid microextraction combined with reversed electrode polarity stacking mode—micellar electrokinetic chromatography. Talanta 2012, 98, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Kukusamude, C.; Burakham, R.; Chailapakul, O.; Srijaranai, S. High performance liquid chromatography for the simultaneous analysis of penicillin residues in beef and milk using ion-paired extraction and binary water–acetonitrile mixture. Talanta 2012, 92, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Somsubsin, S.; Seebunrueng, K.; Boonchiangma, S.; Srijaranai, S. A simple solvent based microextraction for high performance liquid chromatographic analysis of aflatoxins in rice samples. Talanta 2018, 176, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Baeck, S.K.; Shin, Y.S.; Chung, H.S.; Pyo, M.Y. Comparison study of the extraction methods of paraquat in post-mortem human blood samples. Arch. Pharm. Res. 2007, 30, 235–239. [Google Scholar] [CrossRef]
- AOAC Official Methods of Analysis. Guidelines for Standard Method Performance Requirement; AOAC International: Rockville, MD, USA, 2016. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Linear Range (µg L−1) | Linear Equation | R2 | LOD (µg L−1) | LOQ (µg L−1) | EF |
|---|---|---|---|---|---|---|
| Diquat | 7–250 (100–5000) a | y = 0.0998x + 0.0633 (y = 0.0039x − 0.0986) a | 0.9996 (0.9990) a | 1.5 (25) a | 5.6 (80) a | 26 |
| Paraquat | 10–250 (100–5000) a | y = 0.1031x − 0.7353 (y = 0.0047x − 0.5519) a | 0.9986 (0.9976) a | 2.8 (40) a | 8.0 (100) a | 22 |
| Analyte | Sample | Sample Preparation | Extraction Solvent Volume (μL) | Extraction Time (min) | LOD (μg L−1) | Analytical Technique | Ref. |
|---|---|---|---|---|---|---|---|
| Paraquat | Blood | LLE (Chloroform- ethanol (7:3)) | 500 | 3 | 10 | HPLC–DAD (258 nm) | [47] |
| Paraquat, diquat | Water | ISFME (ILs) | 9.4 | 10 | 0.15–0.16 | HILIC–DAD (256, 310 nm) | [29] |
| Paraquat | Vegetable | SPE (weak cation exchanger) | 1000 | NR | 0.94 µg kg−1 | HILIC–MS/MS | [11] |
| Paraquat | Water, Vegetable | DSPE (silica gel) | - | 30 | 50 | Spectro photometry | [25] |
| Paraquat | Water, Soil, and Vegetable | SALLE-RP- DLLME | 2550 | 2.5 | 20 | HPLC–UV (257 nm) | [28] |
| Paraquat, diquat | Water, Vegetable | SPE (alkyl-silica and resin SPE cartridge) | 5000 | NR | 0.1–0.2 | LC–MS | [23] |
| Paraquat, diquat | Vegetable | LPME (SUPRAS) | 50 | 1 | 1.5–2.8 | HPLC–DAD (254, 310 nm) | This work |
| Analyte | Spiked (mg kg−1) | Chinese Cabbage (n = 3) | Radish (n = 3) | Onion (n = 3) | Cabbage (n = 3) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Found (mg kg−1) | Recovery (%) | RSD (%) | Found (mg kg−1) | Recovery (%) | RSD (%) | Found (mg kg−1) | Recovery (%) | RSD (%) | Found (mg kg−1) | Recovery (%) | RSD (%) | ||
| Diquat | 0 | ND | - | - | ND | - | - | ND | - | - | ND | - | - |
| 0.12 | 0.11 | 91.7 | 2.0 | 0.11 | 91.7 | 4.8 | 0.12 | 99.3 | 4.8 | 0.11 | 91.7 | 2.5 | |
| 0.20 | 0.16 | 80.0 | 5.5 | 0.18 | 90.0 | 3.0 | 0.17 | 82.7 | 1.7 | 0.19 | 95.0 | 3.8 | |
| 0.40 | 0.36 | 90.0 | 2.2 | 0.39 | 97.5 | 3.8 | 0.41 | 103.9 | 5.2 | 0.38 | 95.0 | 1.6 | |
| Paraquat | 0 | ND | - | - | ND | - | - | ND | - | - | ND | - | - |
| 0.12 | 0.10 | 83.3 | 1.5 | 0.10 | 83.3 | 7.9 | 0.13 | 106.7 | 4.0 | 0.12 | 100.0 | 3.5 | |
| 0.20 | 0.17 | 85.0 | 2.5 | 0.17 | 85.0 | 5.8 | 0.17 | 85.9 | 2.5 | 0.21 | 105.0 | 2.9 | |
| 0.40 | 0.30 | 75.0 | 3.8 | 0.36 | 90.0 | 4.1 | 0.35 | 86.5 | 1.1 | 0.39 | 97.5 | 1.6 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hem, S.; Gissawong, N.; Srijaranai, S.; Boonchiangma, S. Supramolecular Solvent-Based Liquid Phase Microextraction Combined with Ion-Pairing Reversed-Phase HPLC for the Determination of Quats in Vegetable Samples. Toxics 2019, 7, 60. https://doi.org/10.3390/toxics7040060
Hem S, Gissawong N, Srijaranai S, Boonchiangma S. Supramolecular Solvent-Based Liquid Phase Microextraction Combined with Ion-Pairing Reversed-Phase HPLC for the Determination of Quats in Vegetable Samples. Toxics. 2019; 7(4):60. https://doi.org/10.3390/toxics7040060
Chicago/Turabian StyleHem, Sophon, Netsirin Gissawong, Supalax Srijaranai, and Suthasinee Boonchiangma. 2019. "Supramolecular Solvent-Based Liquid Phase Microextraction Combined with Ion-Pairing Reversed-Phase HPLC for the Determination of Quats in Vegetable Samples" Toxics 7, no. 4: 60. https://doi.org/10.3390/toxics7040060
APA StyleHem, S., Gissawong, N., Srijaranai, S., & Boonchiangma, S. (2019). Supramolecular Solvent-Based Liquid Phase Microextraction Combined with Ion-Pairing Reversed-Phase HPLC for the Determination of Quats in Vegetable Samples. Toxics, 7(4), 60. https://doi.org/10.3390/toxics7040060