
Toxicity of Glutathione-Binding Metals: A Review of Targets and Mechanisms


Abstract
:1. Introduction

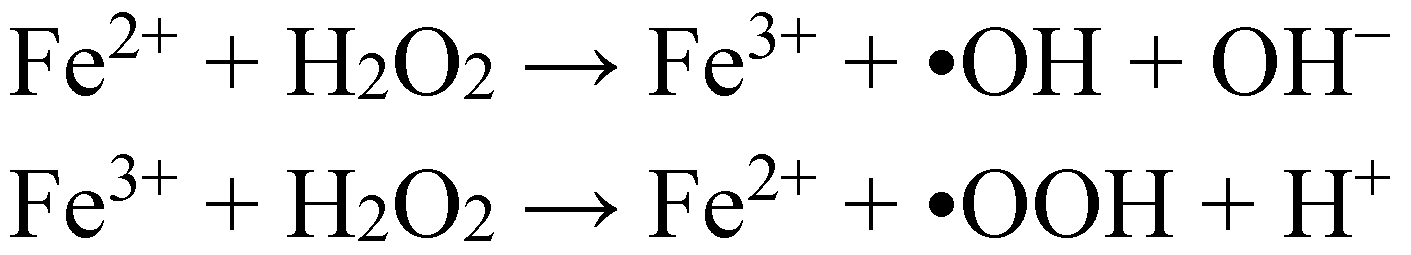
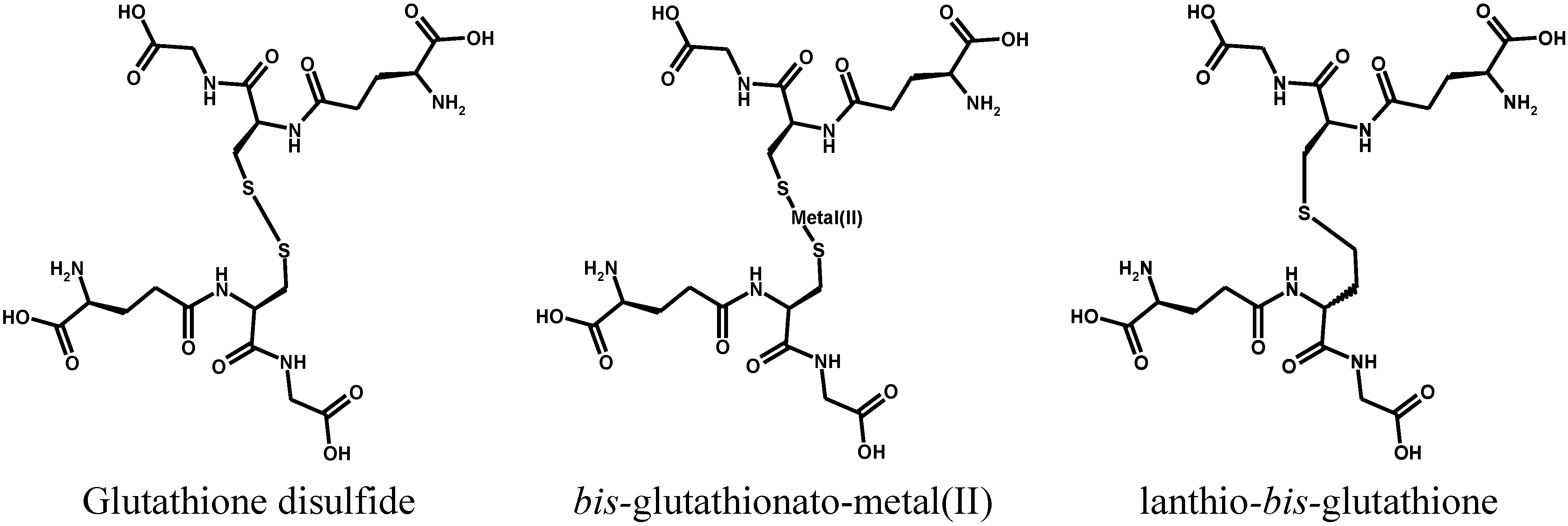
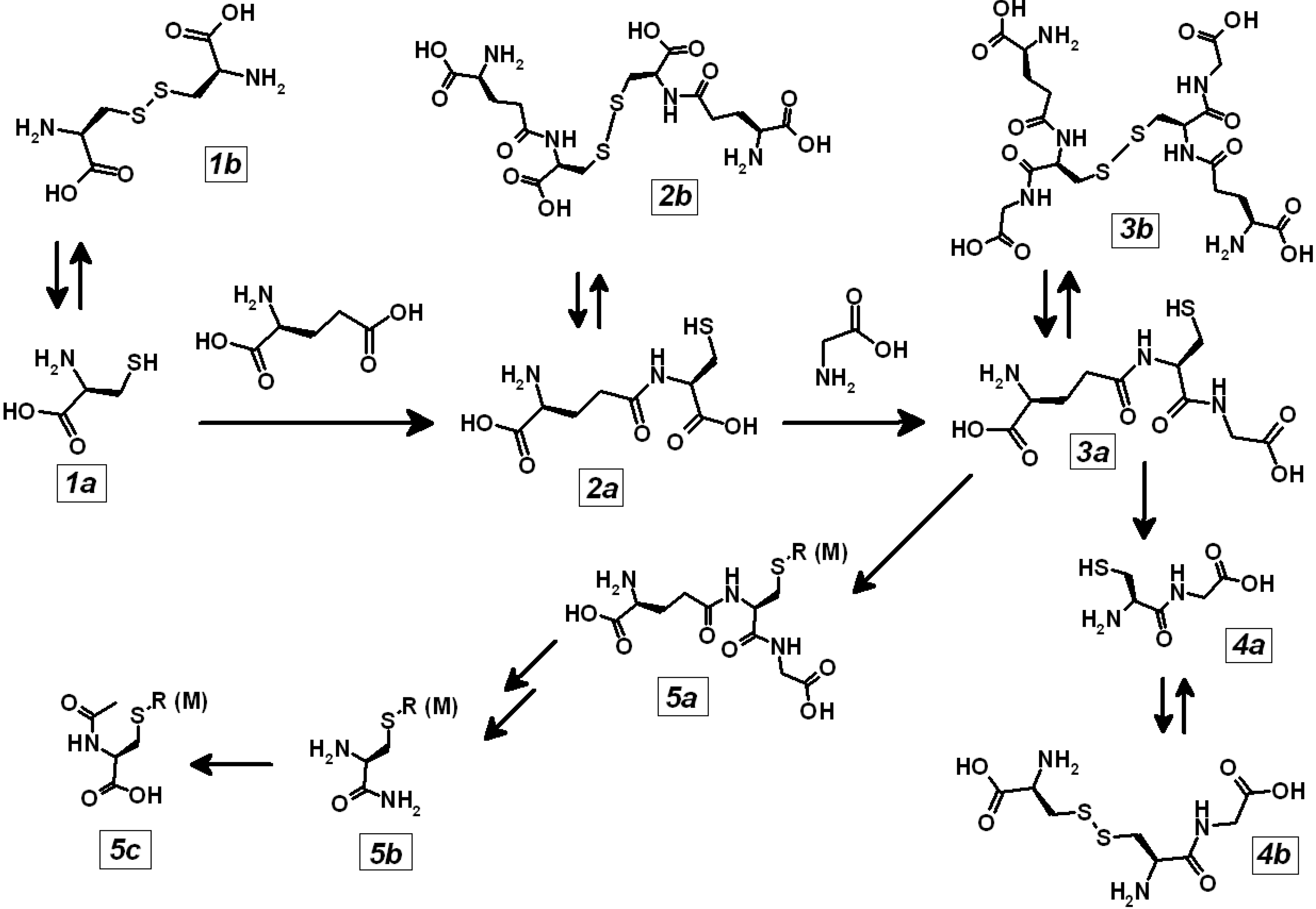
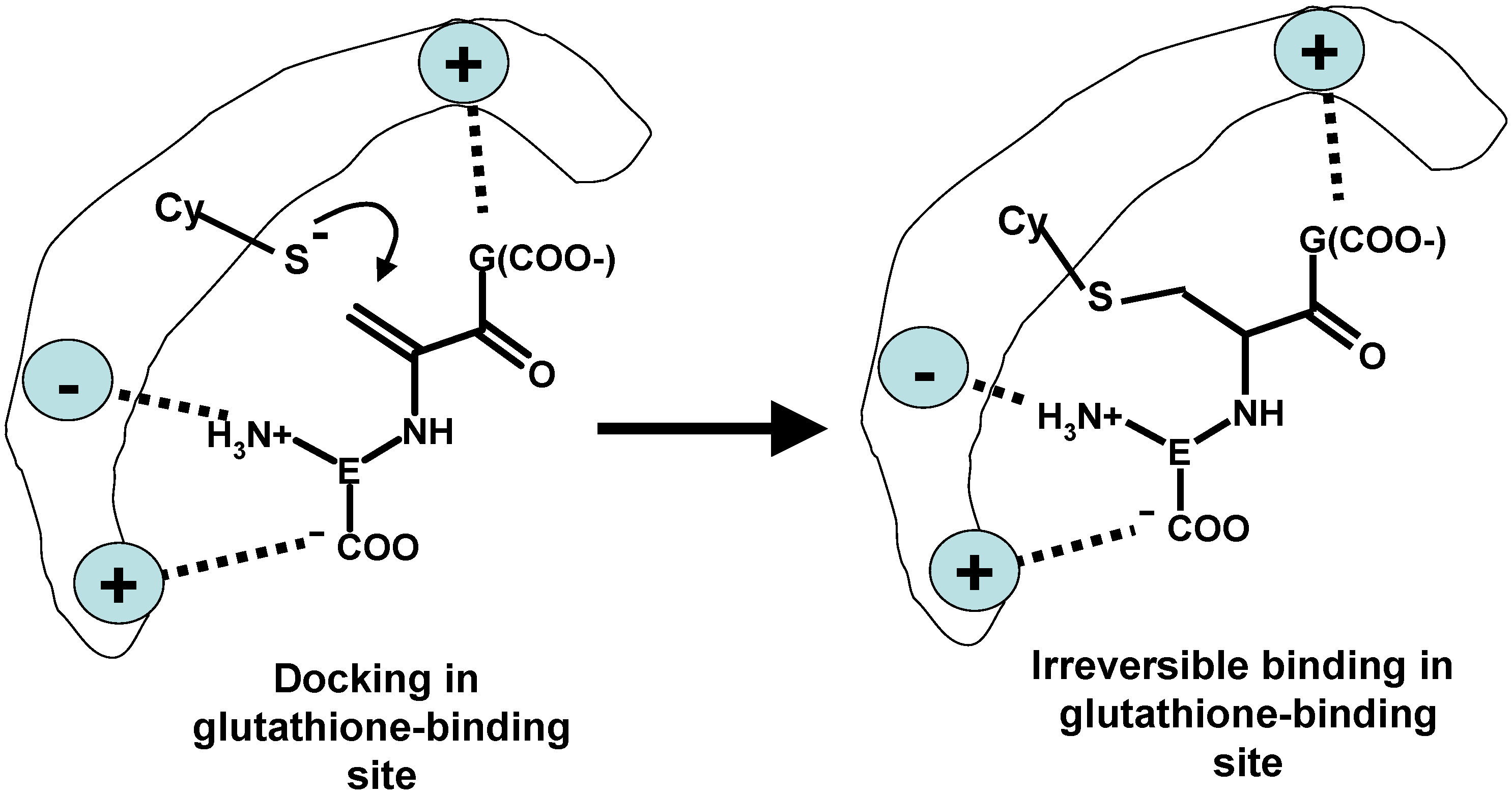
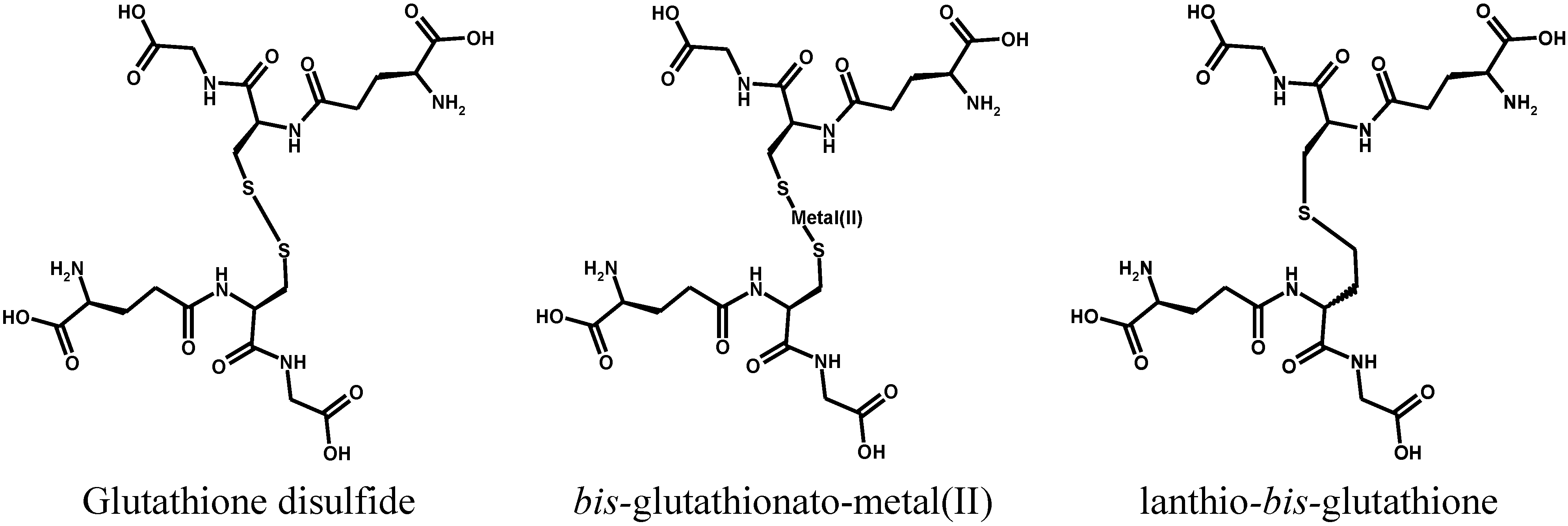
2. Metal Conjugates of Thiol Amino Acids and Peptides
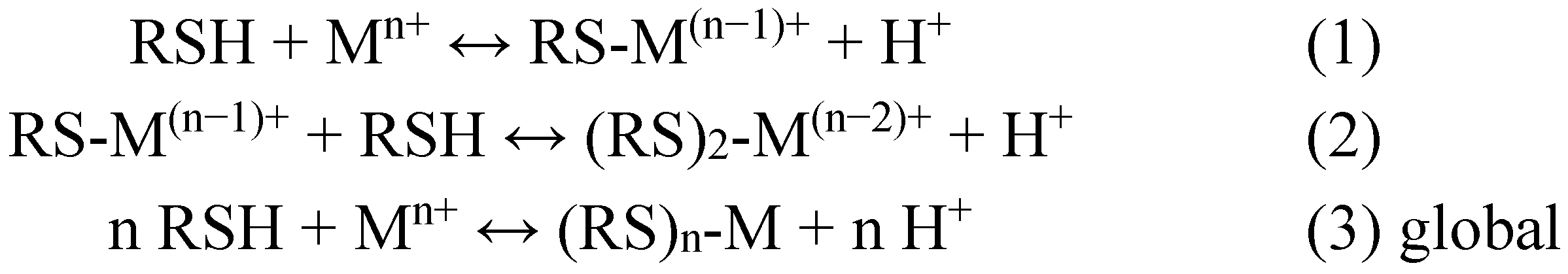
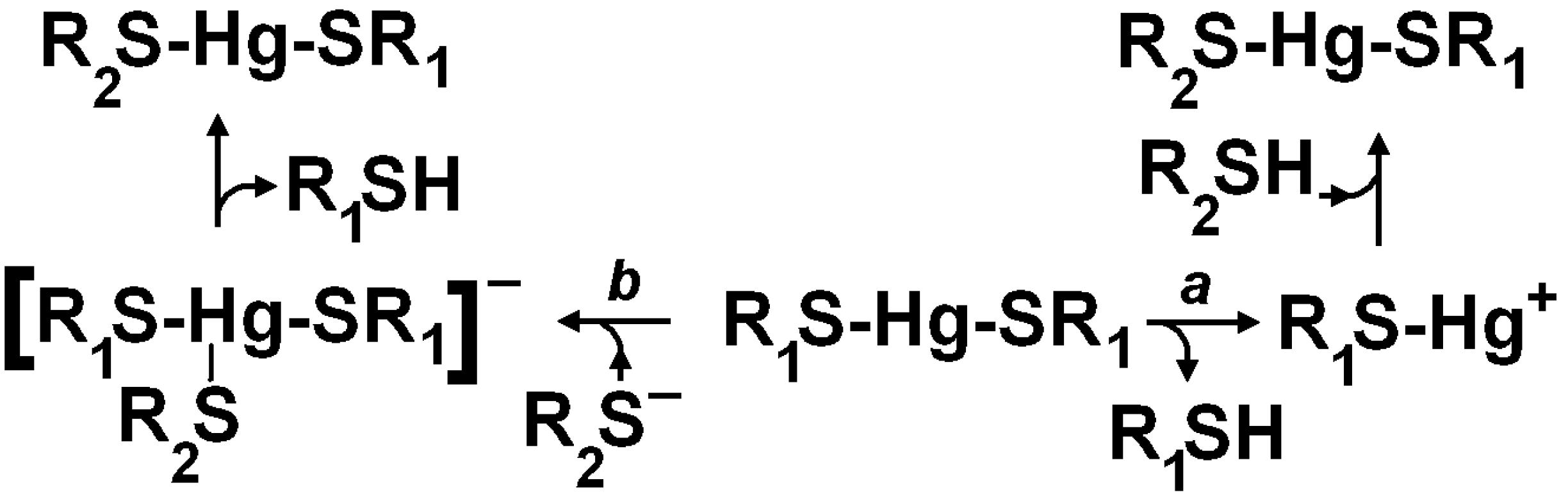
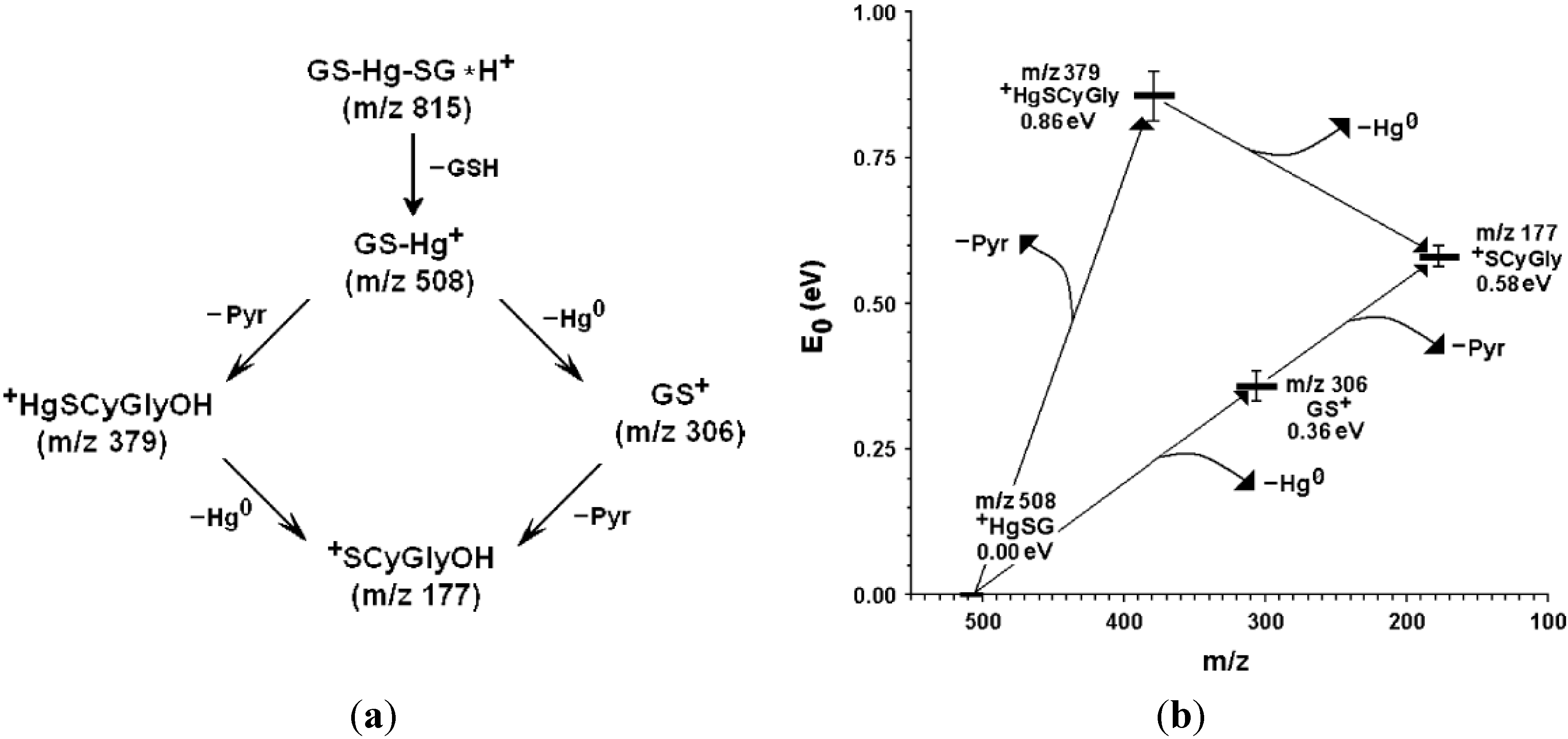
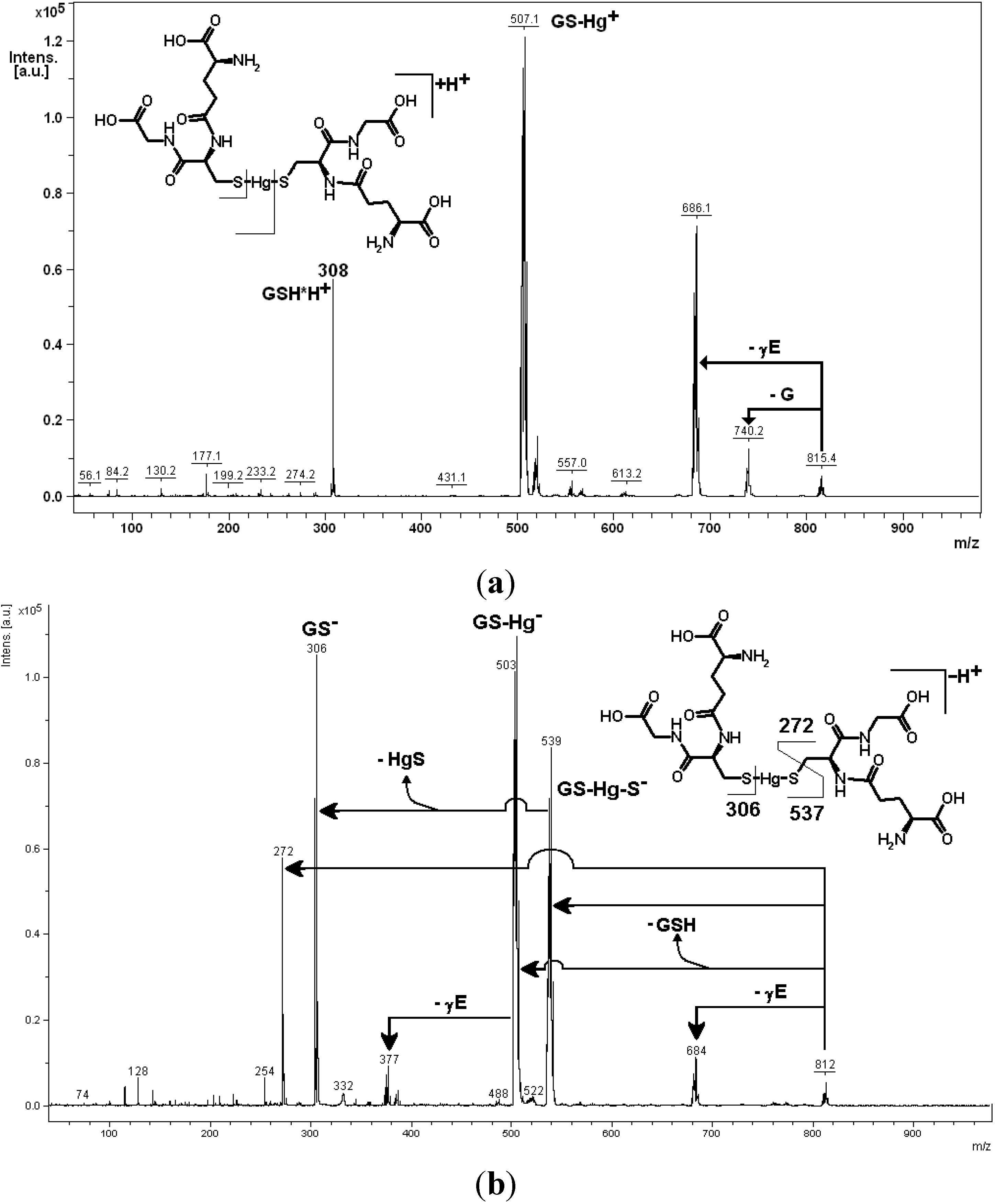
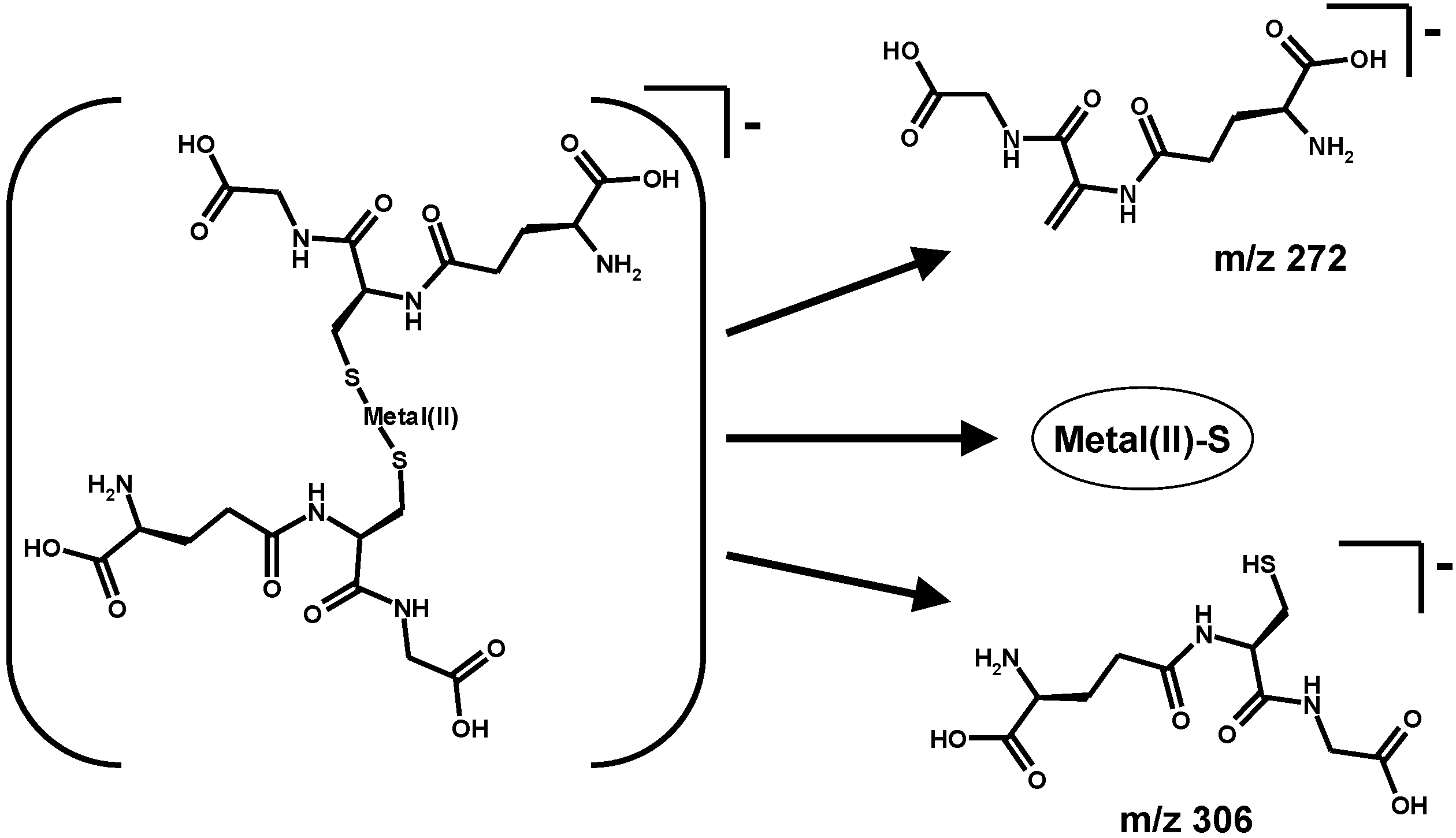
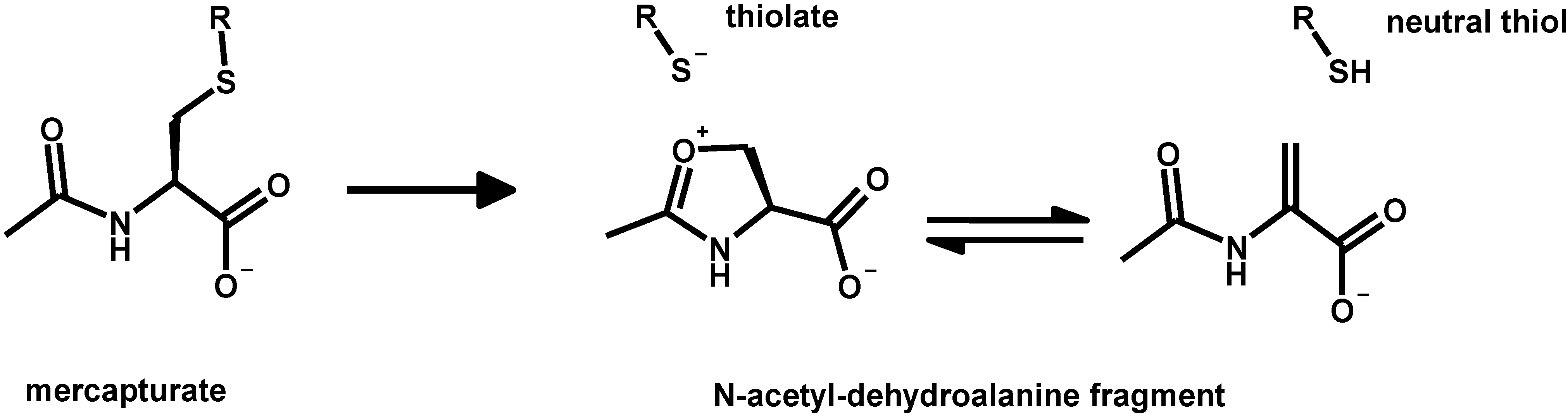
3. Oxidative Stress from Even-Electron Metal-Thiol Conjugates: A Case for Mercury
- (a)
- (b)
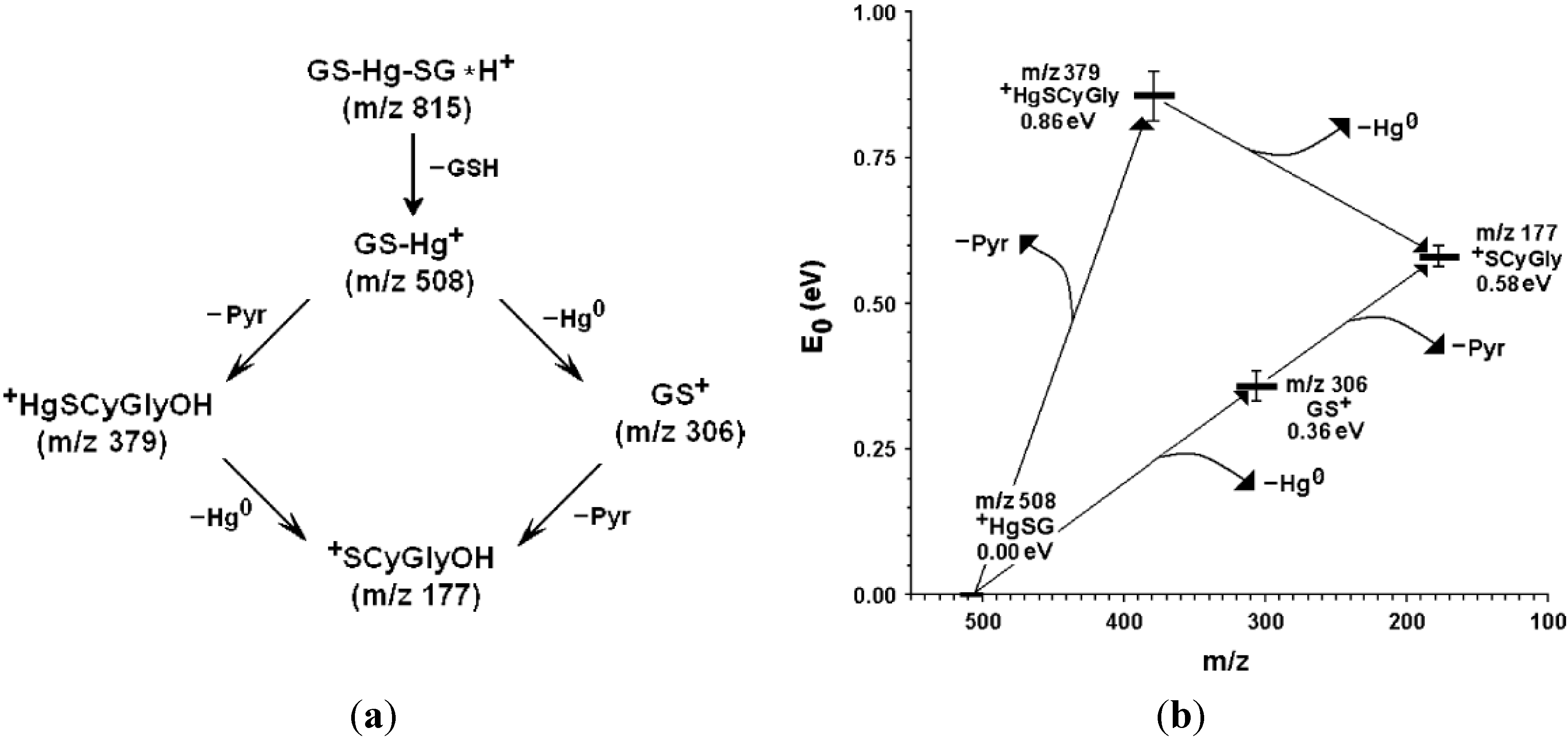
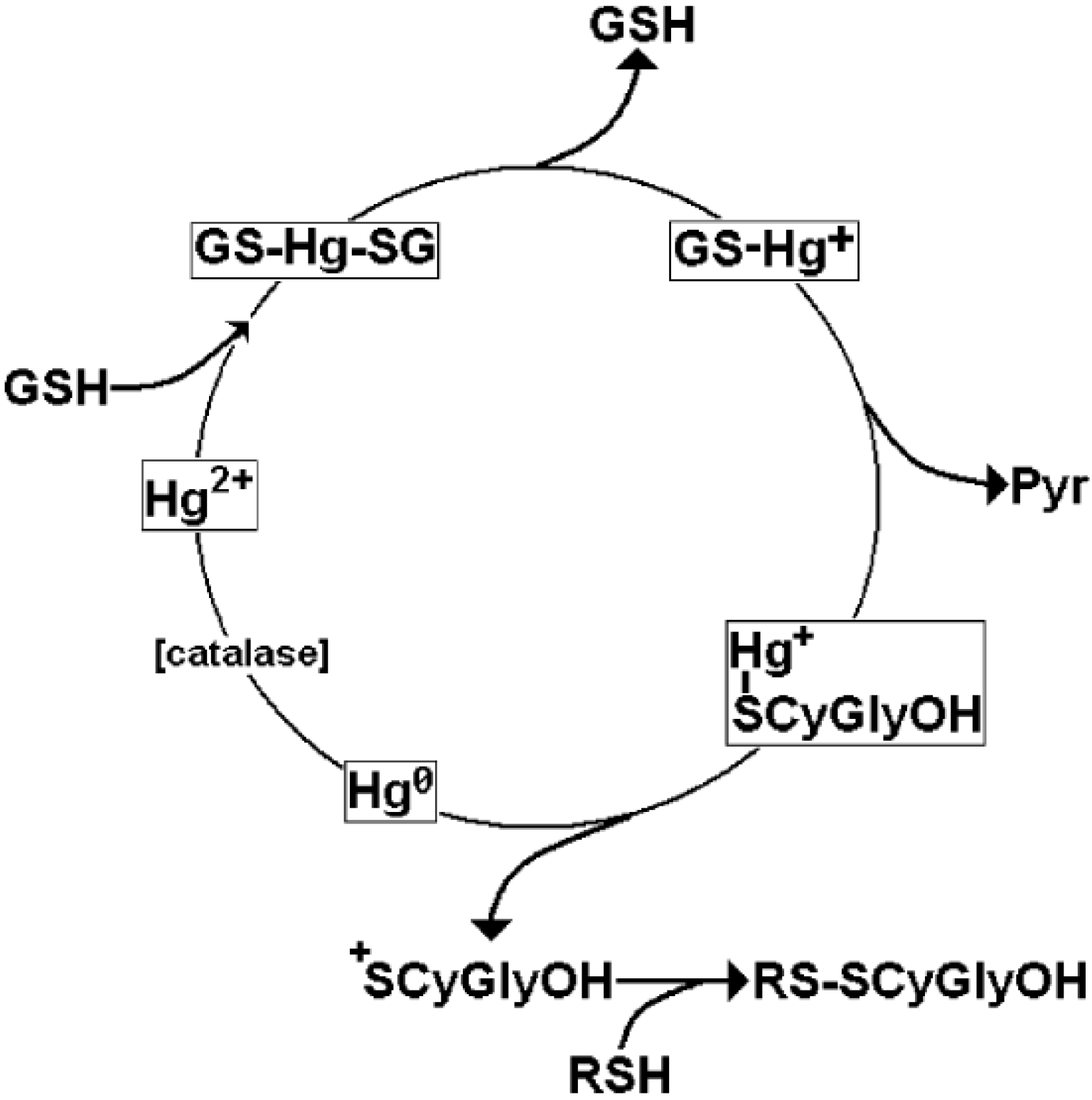
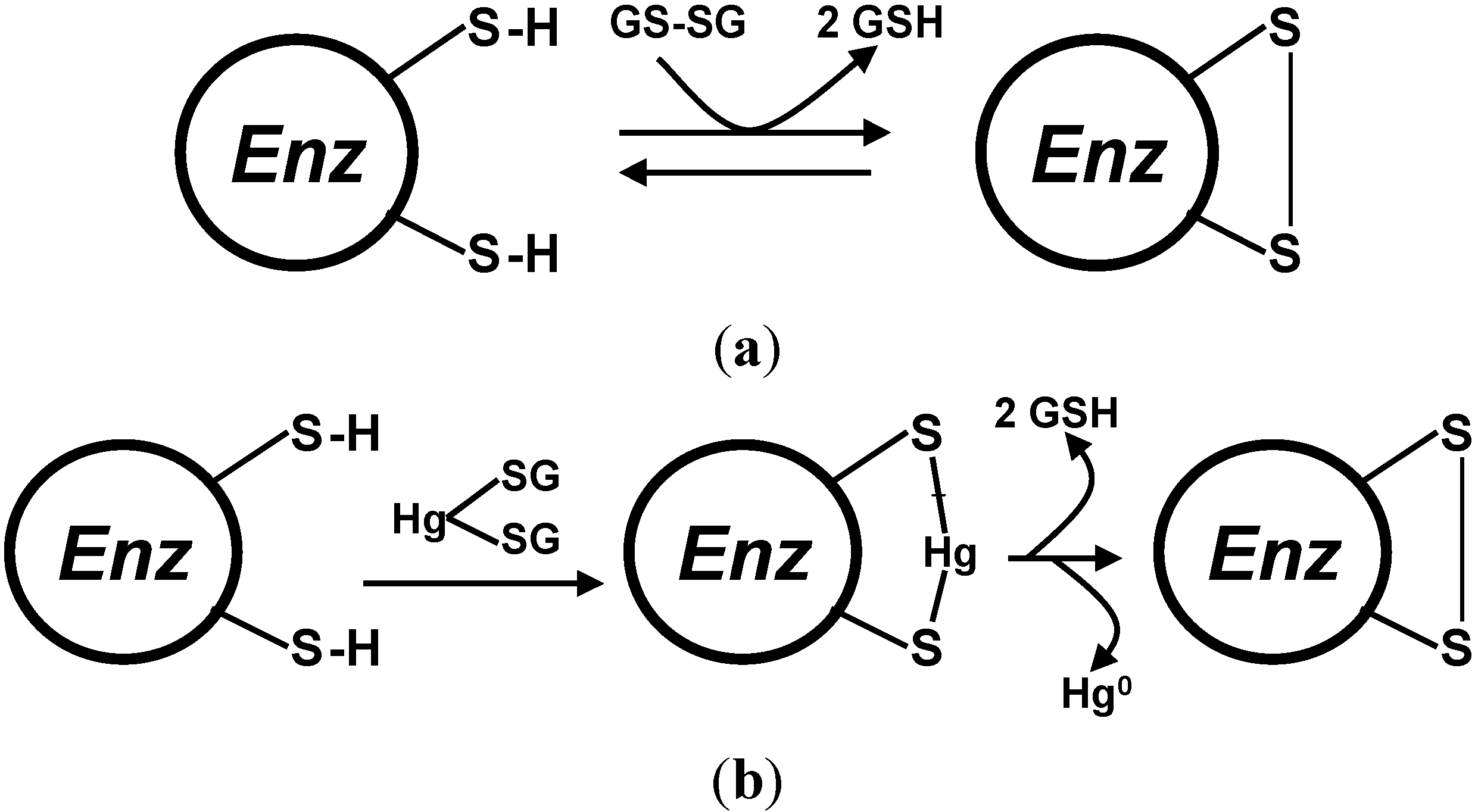
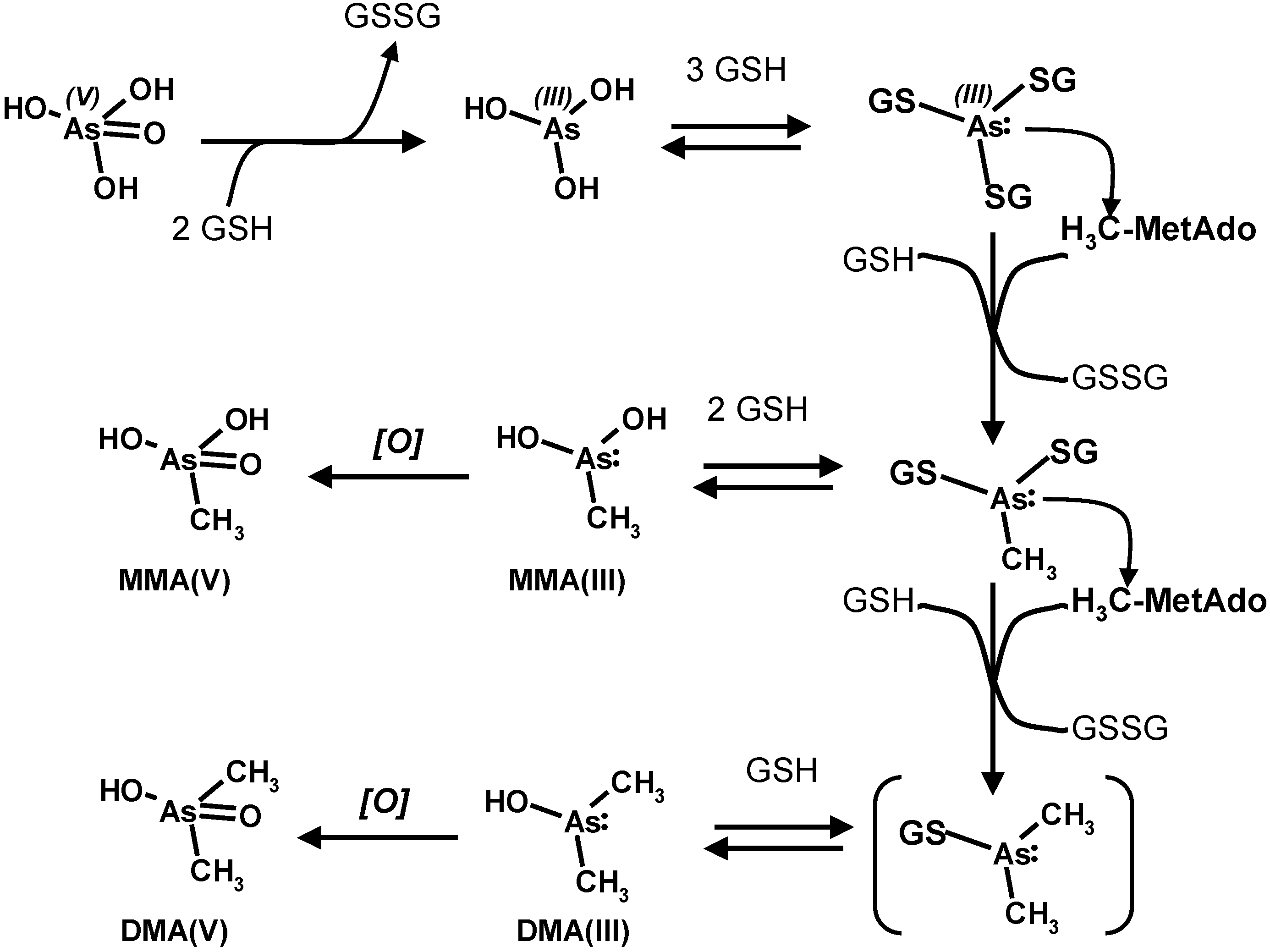
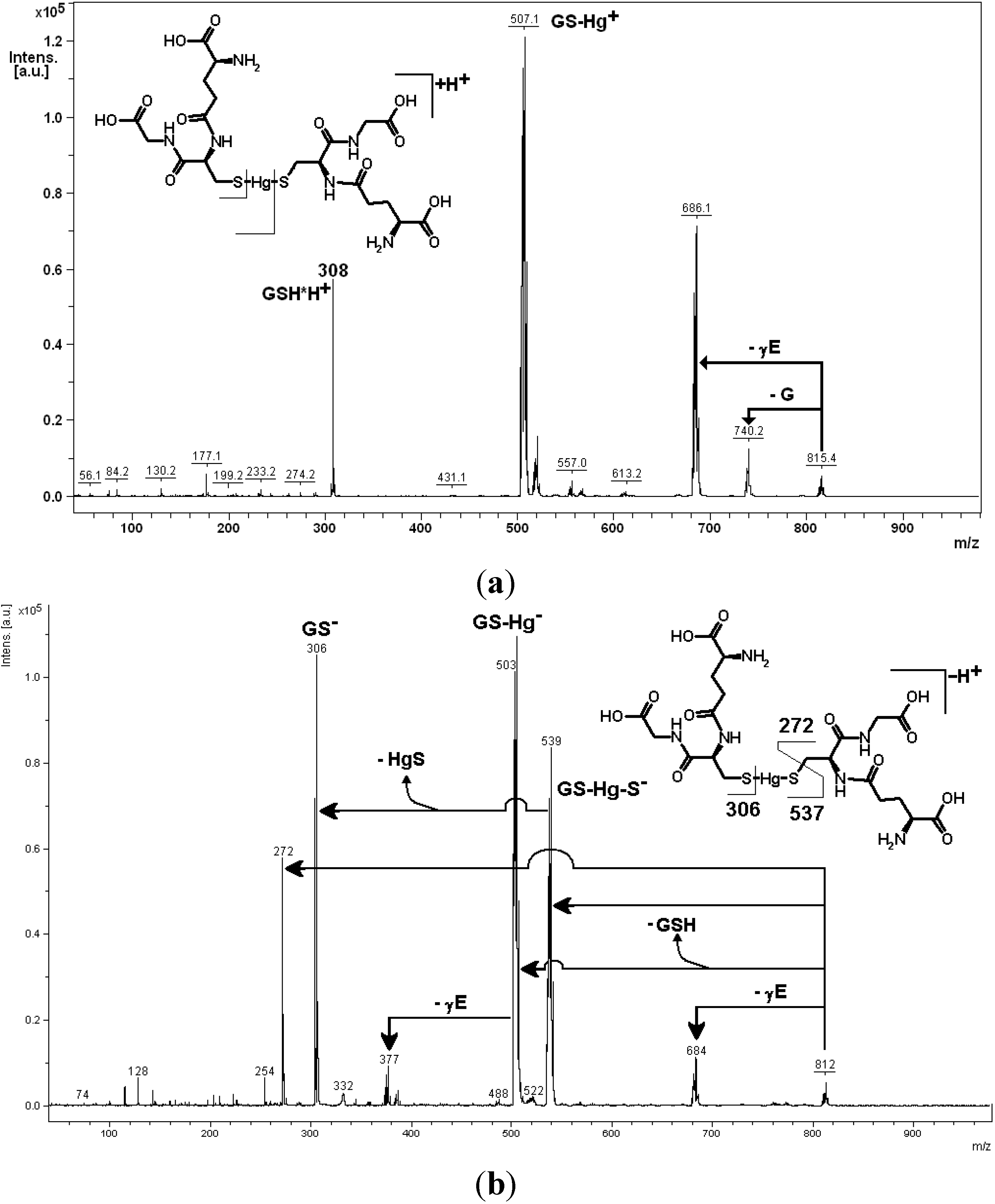
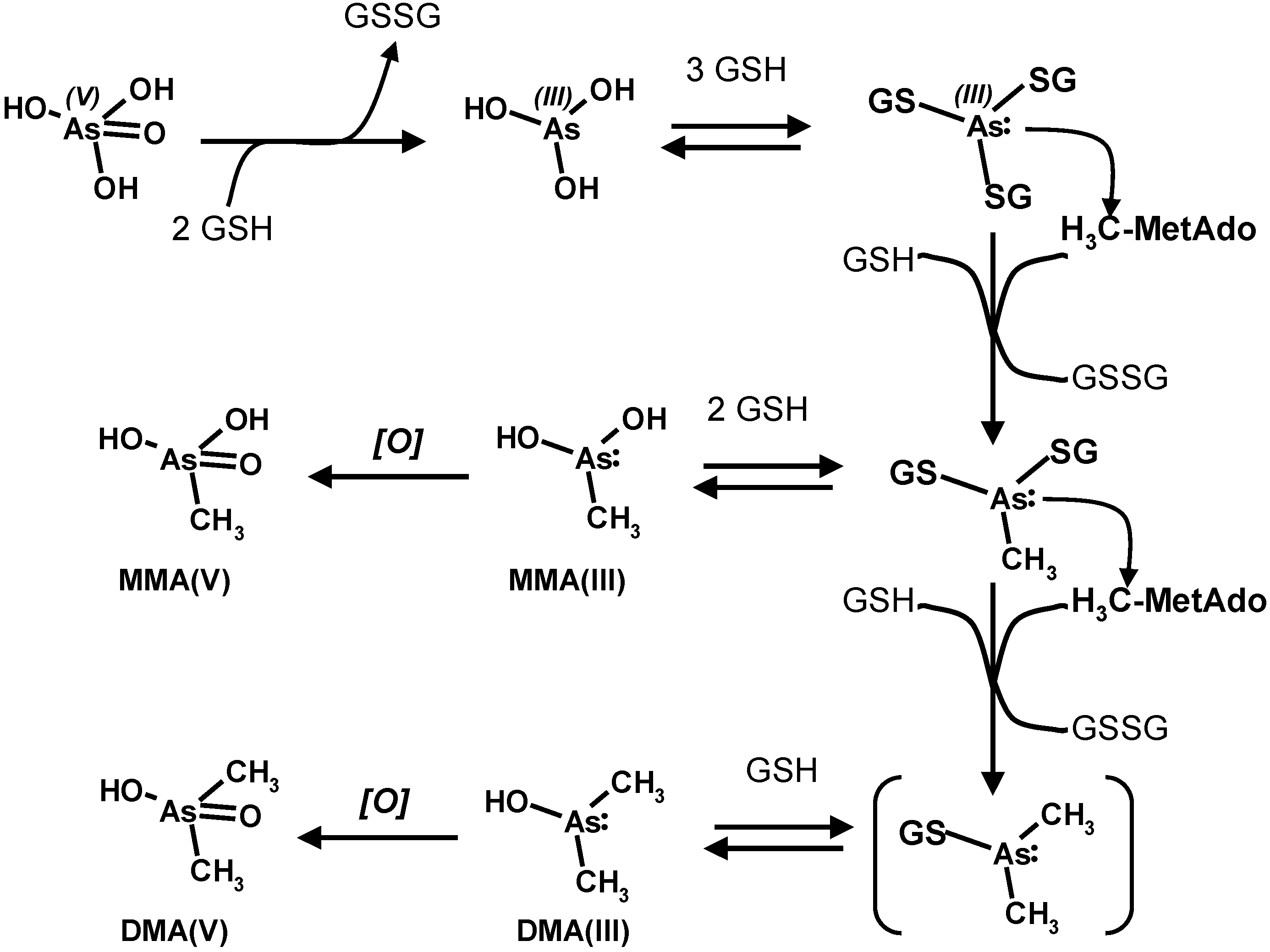
4. Oxidative Stress from Even-Electron Metal-Thiol Conjugates: A Case for Arsenic, Cadmium and Lead
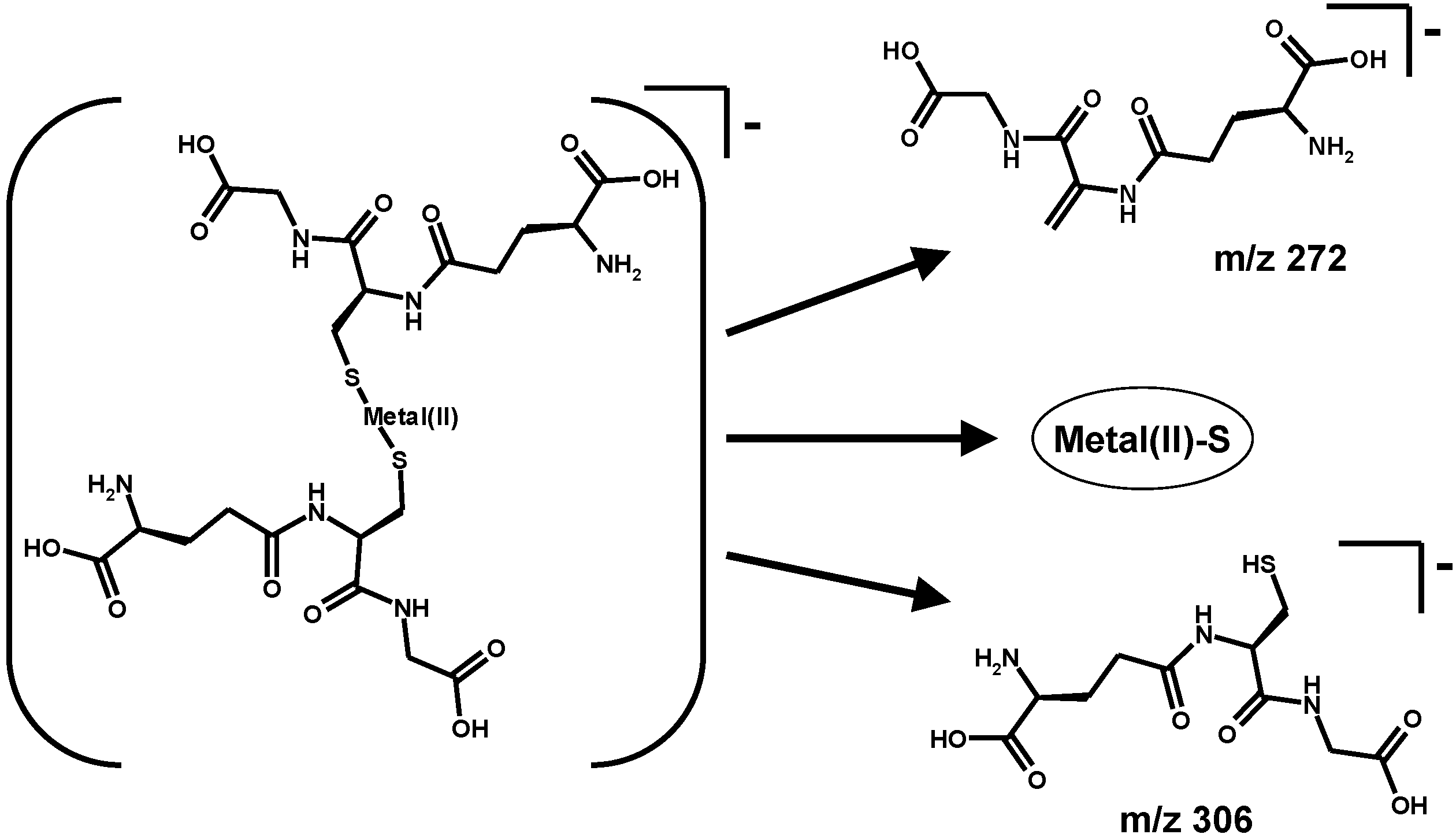
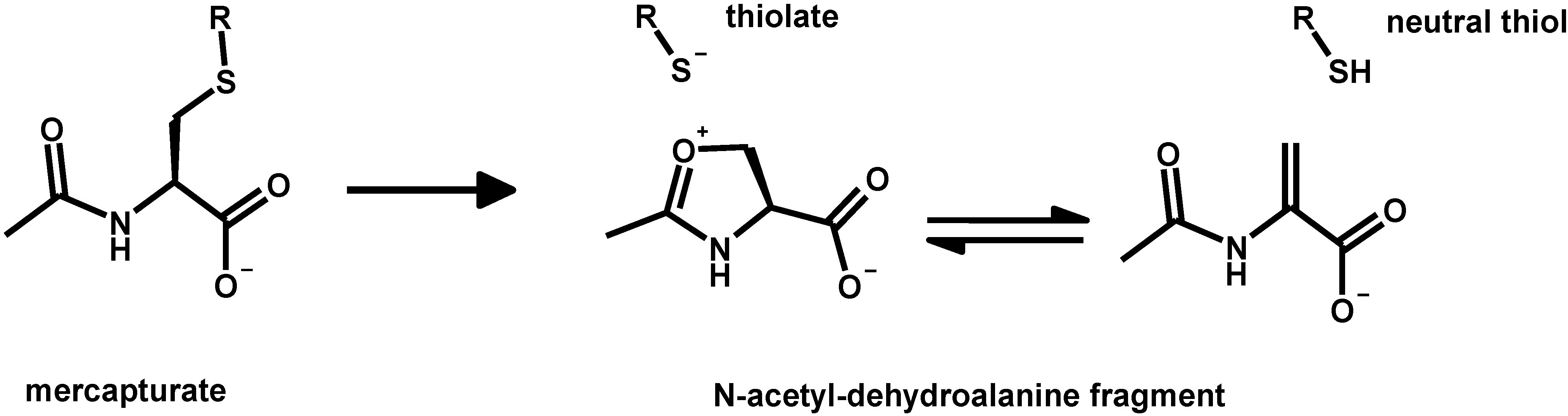
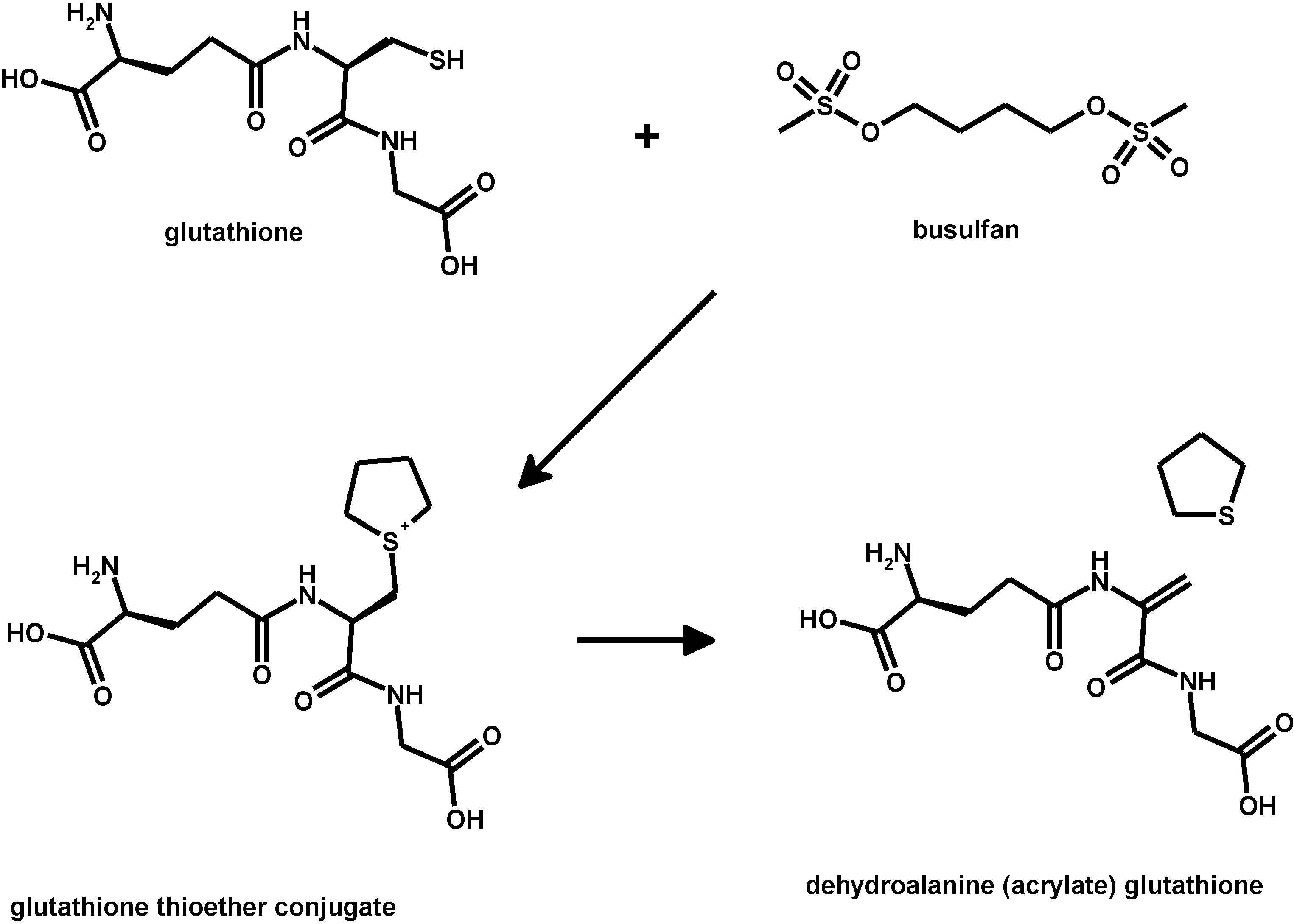
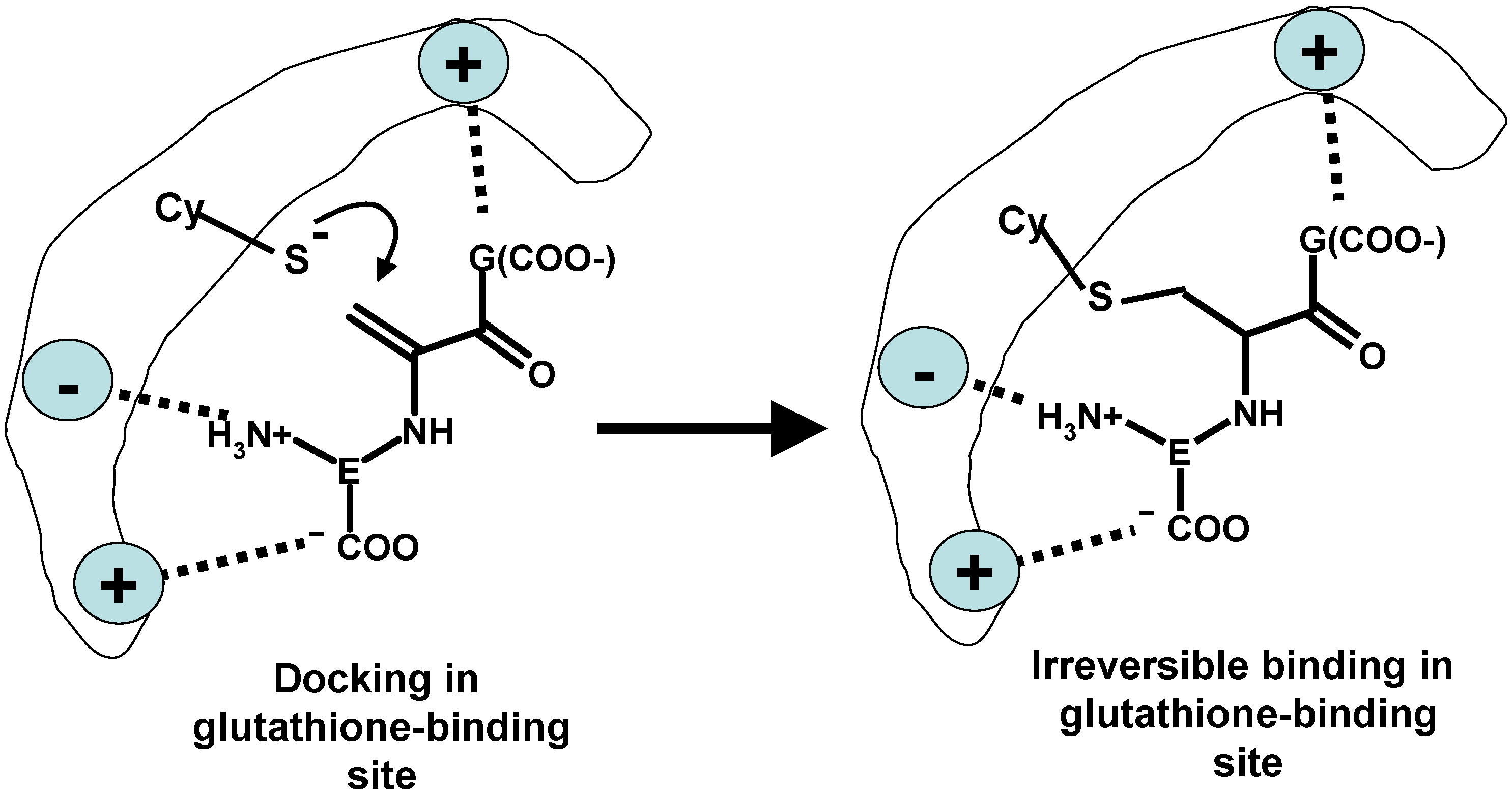
5. Oxidative Stress from Inactivation of Glutathione-Related Enzymes: A Non-Comprehensive Catalogue
5.1. Glutamate-Cysteine-Ligase
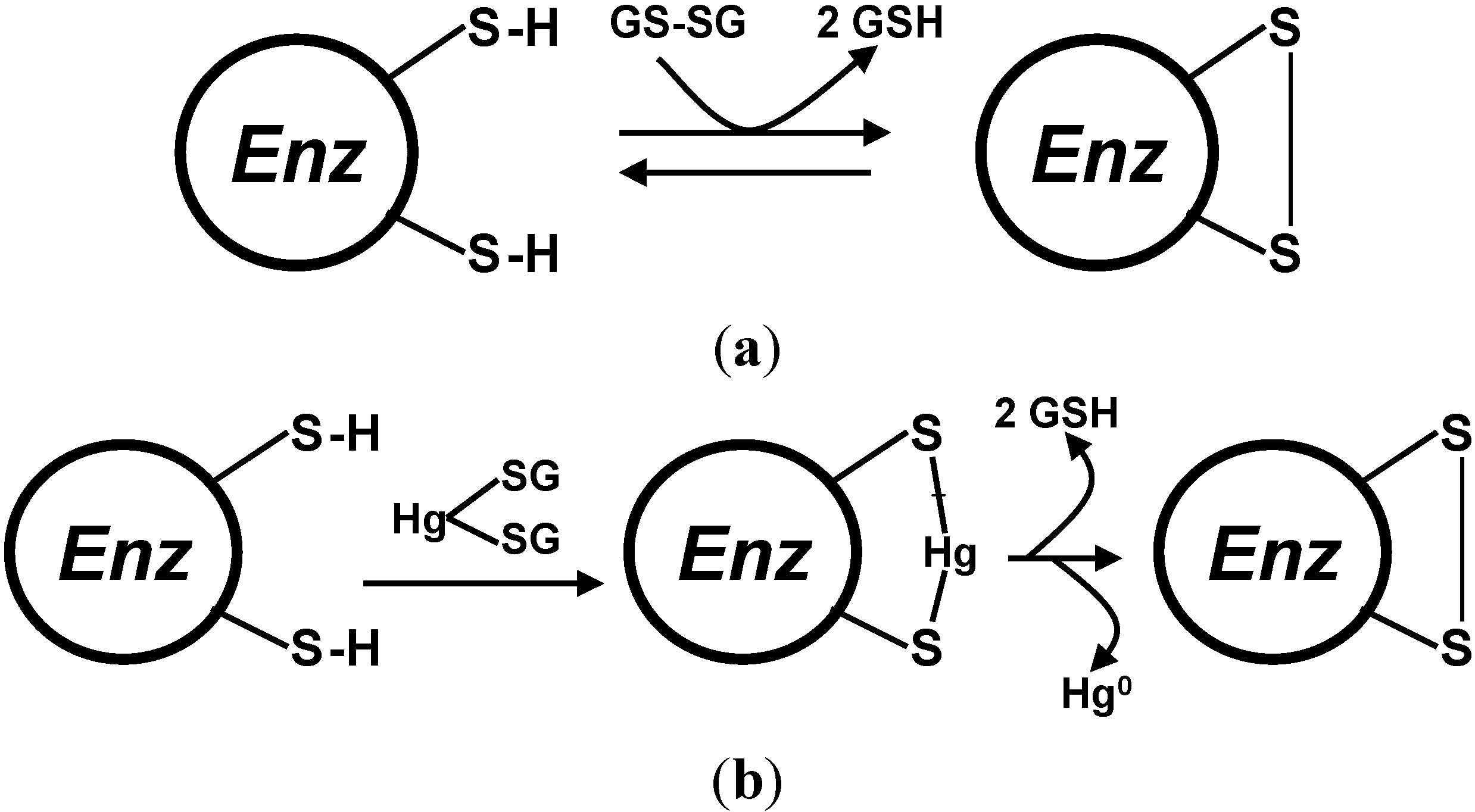
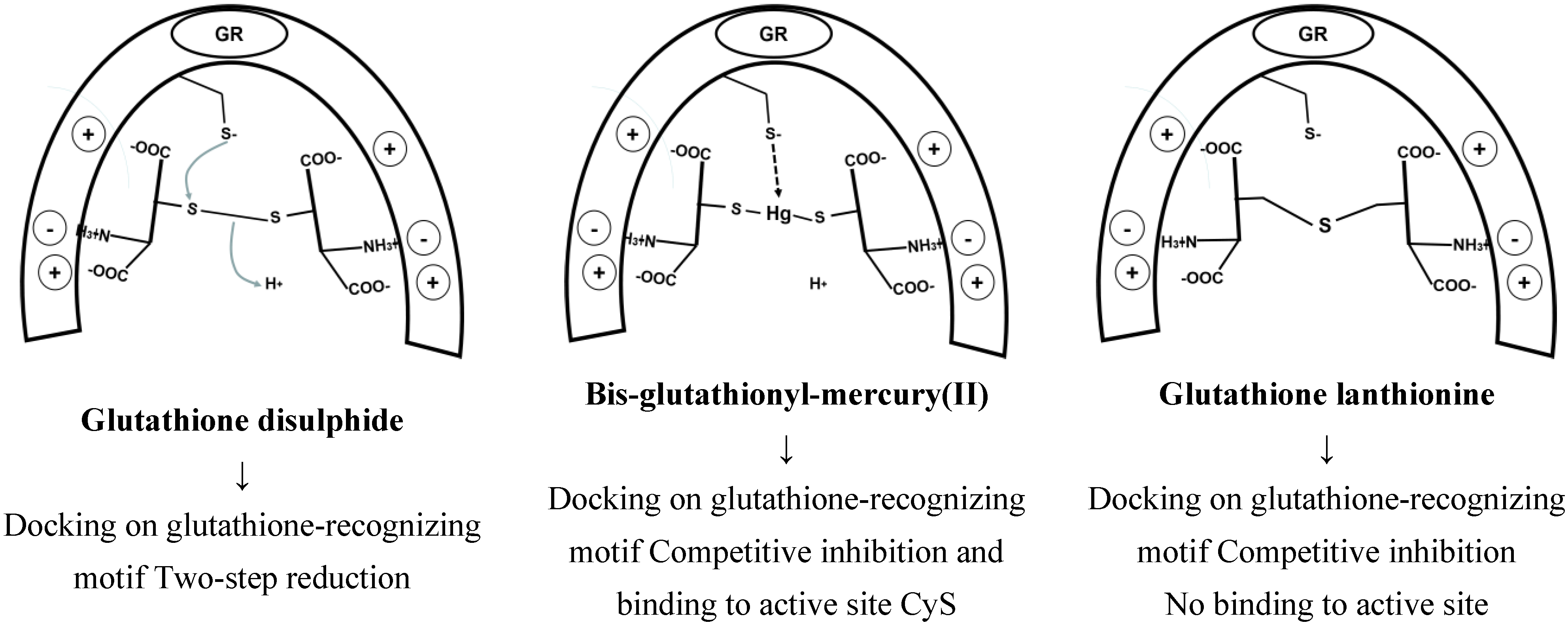
5.2. Glutathione Reductases

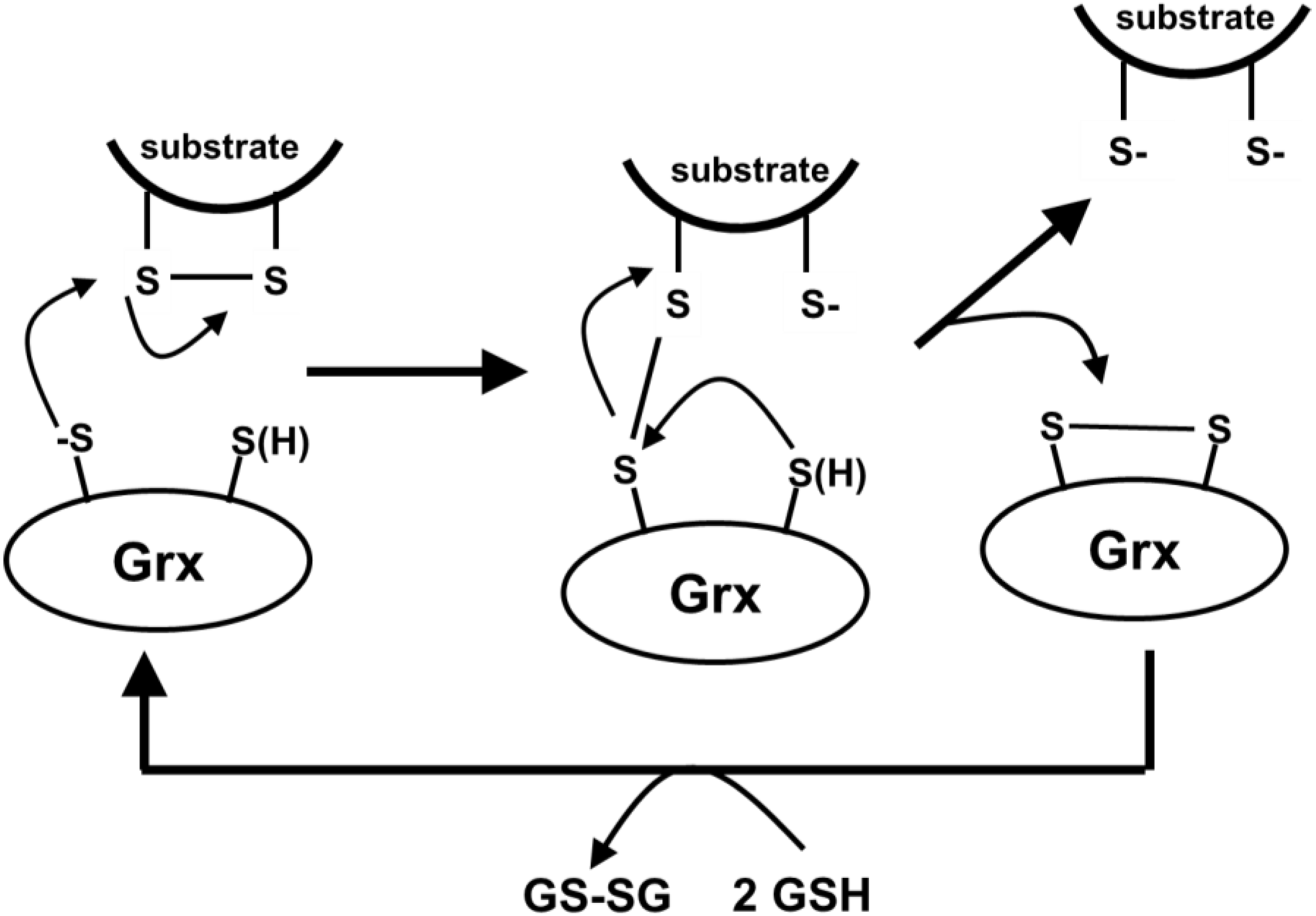
5.3. Glutaredoxins

5.4. Glutathione Peroxidases
5.5. Other Enzymes, Receptors and Regulatory Proteins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
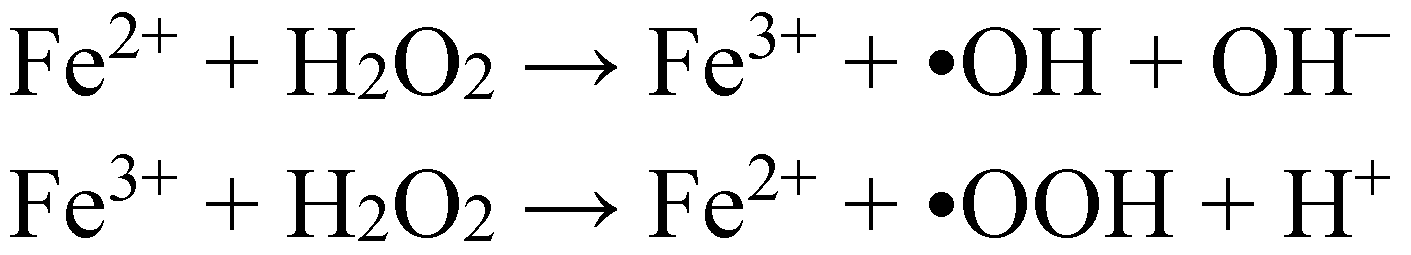{kind=link}
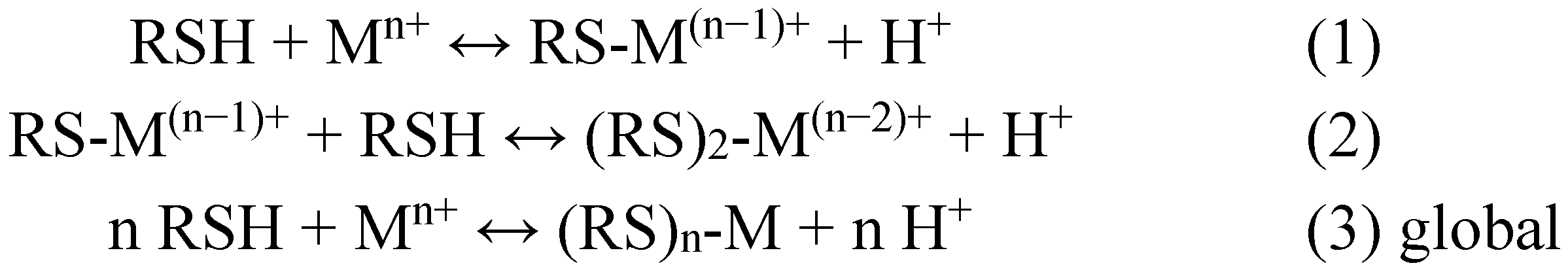{kind=link}
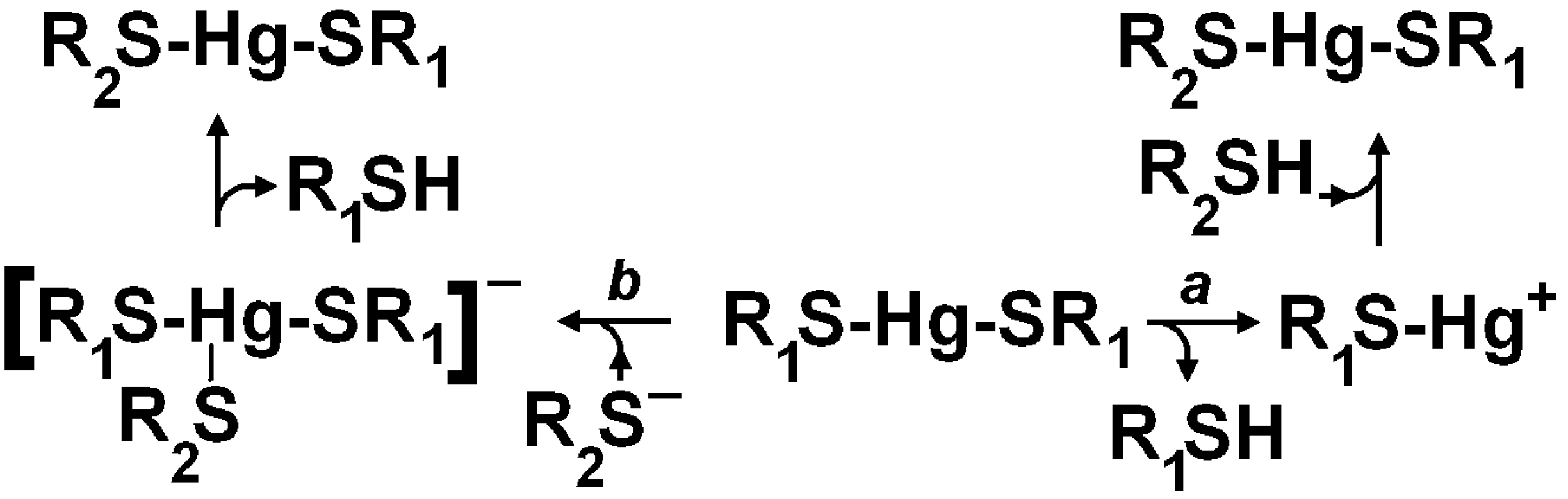{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Consequences of S-glutathionylation |
|---|---|
| glycolytic enzymes | alters enzymatic activity |
| glyceraldehyde-3-phosphate dehydrogenase | inactivated by S-glutathionylation |
| isocitrate dehydrogenases (ICDHs1; EC 1.1.1.41 and EC 1.1.1.42) | regulated by S-glutathionylation |
| creatine kinase | inactivation |
| carbonic anhydrase III | reversible regulation of phosphatase activity |
| components of the respiratory chain | formation of ROS |
| reversible glutathionylation of complex I increases mitochondrial superoxide formation | |
| Actin | polymerization and effect on cytoskeleton |
| membrane receptors, transporters and ion channels | alters ion and metabolite transport |
| causes cell death | |
| several protein kinases and phosphatases | alters signal transduction |
| protein kinase C (12), guanylate cyclase | alters enzymatic activity |
| nuclear factor I | alters signal transduction |
| Ras | alters signal transduction |
| c-Jun | redox-regulated by mechanisms that include protein S-thiolation |
| ubiquitin-activating enzymes | when cells are exposed to oxidants are glutathionylated, with a concomitant decrease in the ubiquitinination pathway |
| NFκB | redox-induced inhibition of DNA binding; cell death |
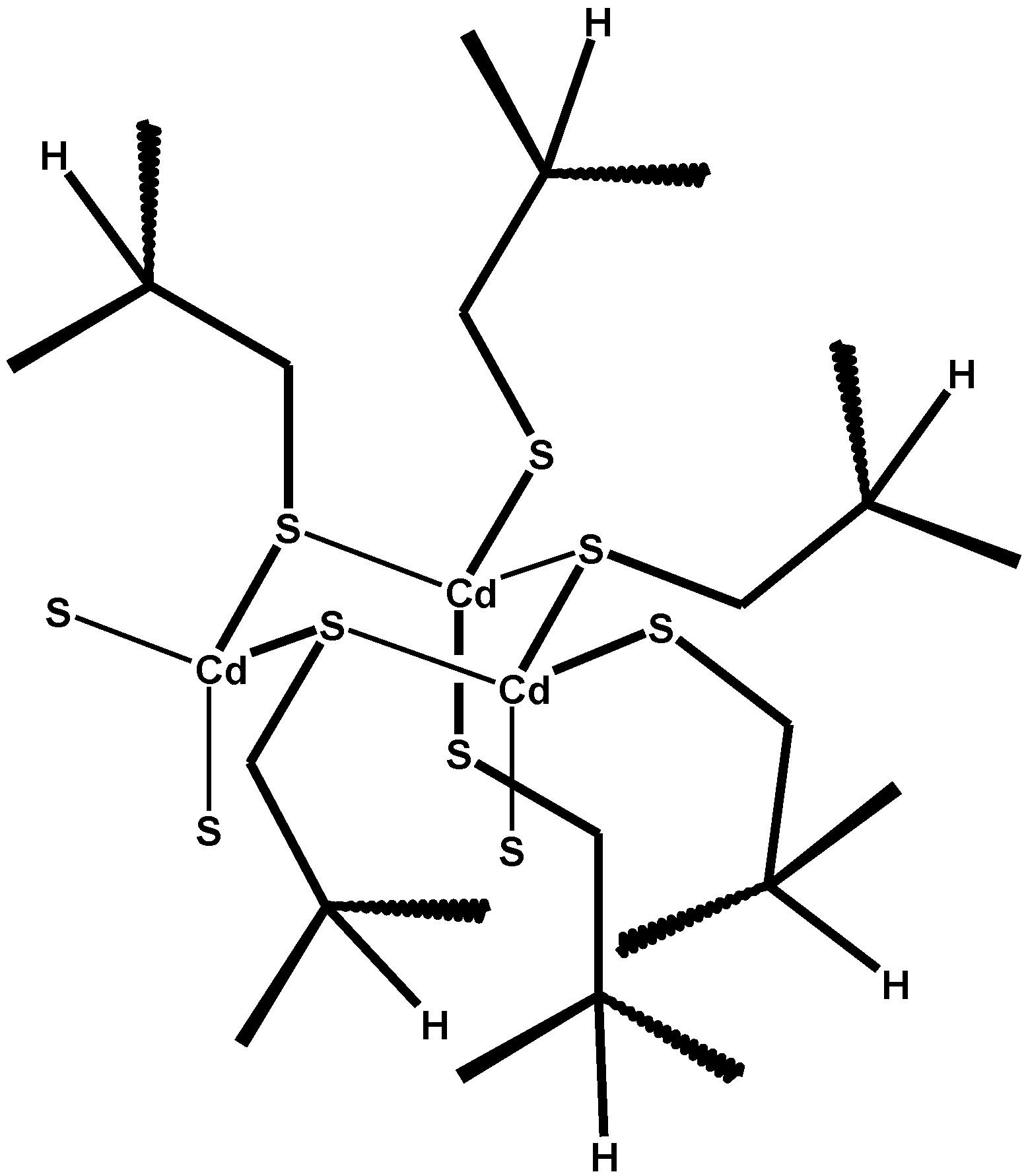
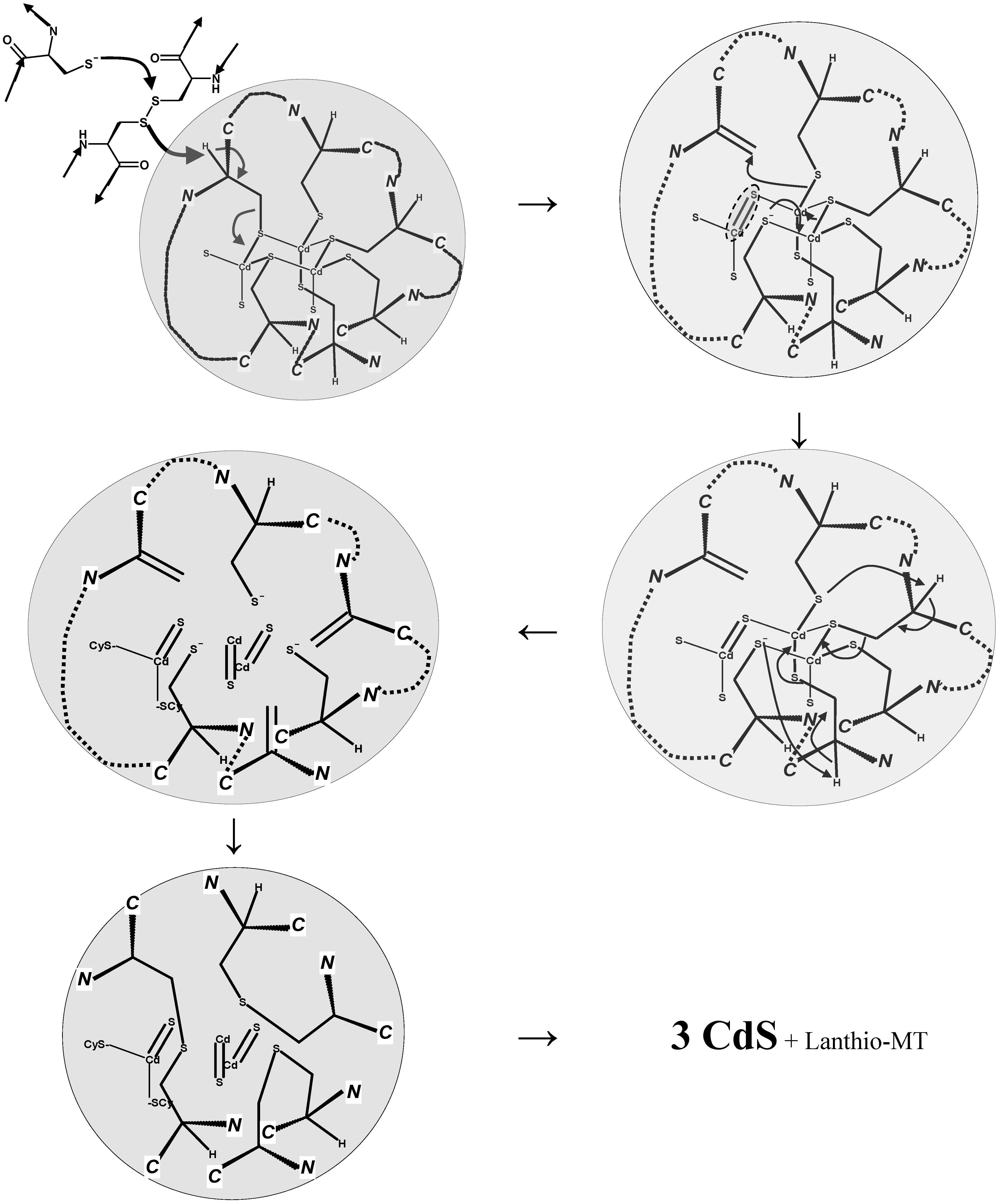
6. Metallothioneins and Their Conjugates with Arsenic, Cadmium, Mercury and Lead
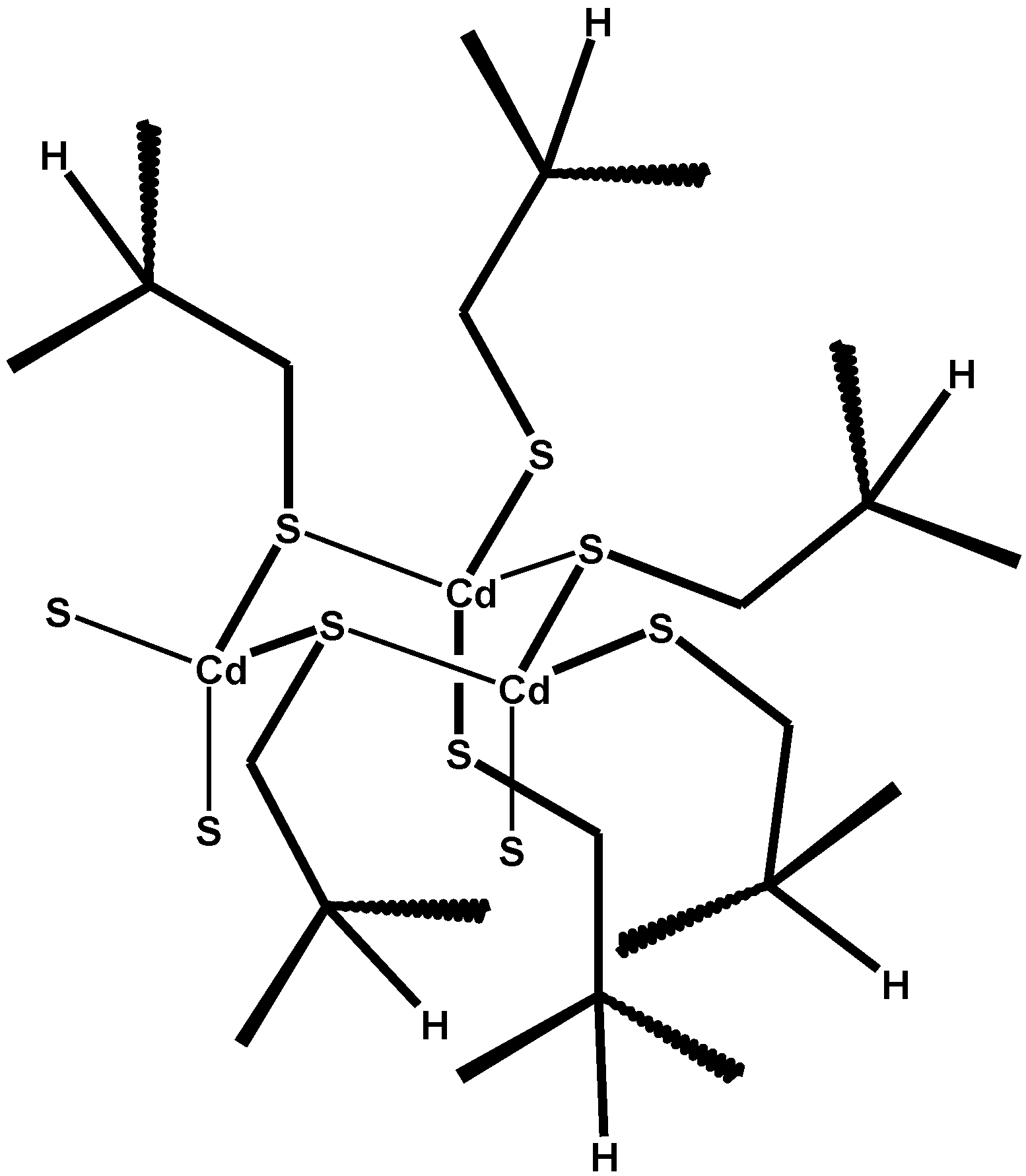
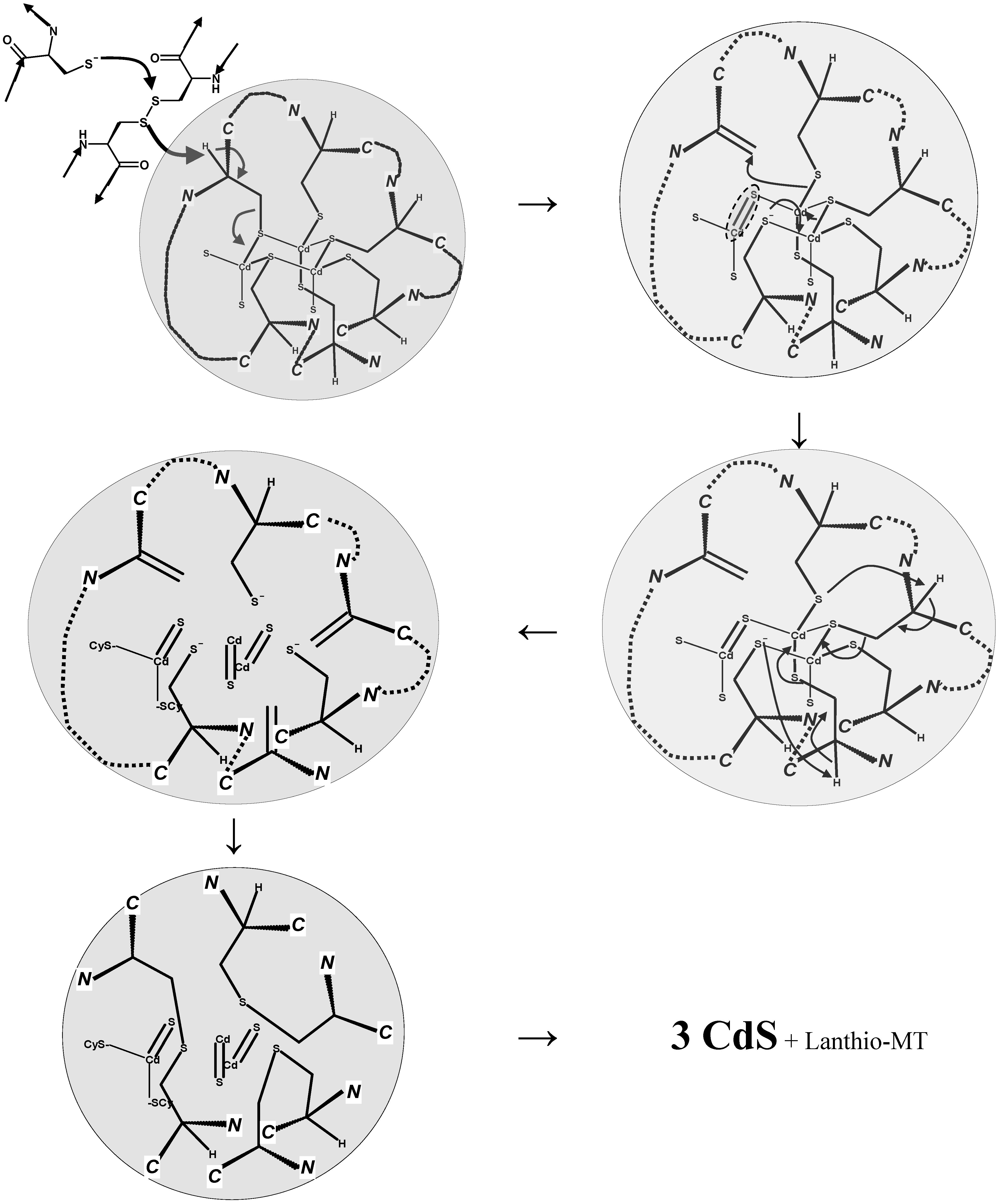
7. Conclusions and Perspectives
Acknowledgments
Abbreviations
| ATP | adenosine-triphosphate |
| BBB | blood-brain barrier |
| CAD | collision-activated decomposition (technique of mass spectrometry) |
| CAD-MS-MS | collision-activated decomposition tandem mass spectrometry (technique of mass spectrometry) |
| DMPO | 5,5-Dimethyl-1-pyrroline N-oxide (a spin-trap compound) |
| EdAG | γ-glutamyl-dehydroalanyl-glycine |
| ERMS | Energy-Resolved (tandem) Mass Spectrometry (technique of mass spectrometry) |
| EXAFS | extended X-ray absorption fine structure (spectroscopic technique) |
| FAD | flavin adenine dinucleotide, oxidized form |
| GCL | glutamate-cysteine-ligase (holo-enzyme) |
| GGT | γ-glutamyl-transpeptidase (enzyme) |
| GIBMS | Guided-Ion Beam Mass Spectrometry (technique of mass spectrometry) |
| GLCL | glutamate-cysteine-ligase (functional enzyme sub-unit) |
| GLCR | glutamate-cysteine-ligase (regulatory enzyme sub-unit) |
| Gpx | Glutathione peroxidase (enzyme/s) |
| GR | Glutathione reductase (enzyme/s) |
| Grx | Glutaredoxins (enzyme/s) |
| GSH | glutathione (γ-ECG), reduced form |
| GSSG | glutathione disulphide |
| GST | glutathione-S-transferase |
| GSTA1-1 | glutathione-S-transferase isoenzyme A-A1 |
| IARC | International Agency for Cancer Research |
| MerA | mercuric reductase A (enzyme) |
| MMA(V) | mono-methyl-arsonic acid (V) |
| MS-MS | tandem mass spectrometry |
| MTs | Metallothioneins |
| NAC | N-acetyl-cysteine |
| NADH | nicotinamide adenine dinucleotide, reduced form |
| NADPH | phosphorylated nicotinamide adenine dinucleotide, reduced form |
| NMR | Nuclear Magnetic Resonance (spectroscopic technique) |
| Prx | Peroxiredoxins (enzyme/s) |
| RBC | red blood cells (erythrocytes) |
| ROS | Reactive Oxygen Species |
| TBARS | thiobarbituric acid reactive substances (a class of organic electrophiles) |
| TEMPO | 2,2,6,6-Tetramethylpiperidine-1-Oxyl (a spin-trap compound) |
| XANES | X-ray absorption near edge structure (spectroscopic technique) |
Dedication
Conflicts of Interest
References
- Barregård, L.; Svalander, C.; Schütz, A.; Westberg, G.; Sällsten, G.; Blohmé, I.; Mölne, J.; Attman, P.O.; Haglind, P. Cadmium, mercury, and lead in kidney cortex of the general Swedish population: A study of biopsies from living kidney donors. Environ. Health Perspect. 1999, 107, 867–871. [Google Scholar] [CrossRef]
- Kinraide, T.B.; Yermiyahu, U. A scale of metal ion binding strengths correlating with ionic charge, Pauling electronegativity, toxicity, and other physiological effects. J. Inorg. Biochem. 2007, 101, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W. Manganese-induced parkinsonism and Parkinson’s disease. Ann. N. Y. Acad. Sci. 2004, 1012, 209–233. [Google Scholar] [CrossRef] [PubMed]
- Herrero Hernandez, E.; Discalzi, G.; Dessi, P.; Jarre, L.; Pira, E. Manganese intoxication: The cause of an inexplicable epileptic syndrome in a 3 year old child. Neurotoxicology 2003, 24, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Mascagni, P.; Consonni, D.; Bregante, G.; Chiappino, G.; Toffoletto, F. Olfactory function in workers exposed to moderate airborne cadmium levels. Neurotoxicology 2003, 24, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Vigliani, E.C.; Baldi, G. Unusual epidemics of mercury poisoning in a felt hat factory. Med. Lav. 1949, 92, 391–398. [Google Scholar]
- Wedeen, R.P. Were the hatters of New Jersey “mad”? Am. J. Ind. Med. 1989, 16, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Giavarini, C. Gli anni del piombo. In The Years of Lead; La Chimica e l’Industria: Milano, Italy, 1990; pp. 1027–1031. [Google Scholar]
- Lillig, C.H.; Berndt, C. Cellular functions of glutathione. Biochim. Biophys. Acta 2013, 1830, 3137–3138. [Google Scholar] [CrossRef] [PubMed]
- Stohs, S.J.; Bagchi, D. Oxidative mechanisms in the toxicity of metal ions. Free Radic. Biol. Med. 1995, 18, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B. Towards a unifying, systems biology understanding of large-scale cellular death and destruction caused by poorly liganded iron: Parkinson’s, Huntington’s, Alzheimer’s, prions, bactericides, chemical toxicology and others as examples. Arch. Toxicol. 2010, 84, 825–889. [Google Scholar] [CrossRef] [PubMed]
- Stohs, S.J.; Bagchi, D.; Hassoun, E.; Bagchi, M. Oxidative mechanisms in the toxicity of chromium and cadmium ions. J. Environ. Pathol. Toxicol. Oncol. 2000, 19, 201–213. [Google Scholar] [PubMed]
- International Agency for Cancer Research. Chromium, nickel and welding. IARC Monogr. Eval. Carcinog. Risks Hum. 1990, 49, 1–648. [Google Scholar]
- Stavrides, J.C. Lung carcinogenesis: Pivotal role of metals in tobacco smoke. Free Radic. Biol. Med. 2006, 41, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Burford, N.; Eelman, M.D.; Groom, K. Identification of complexes containing glutathione with As(III), Sb(III), Cd(II), Hg(II), Tl(I), Pb(II) or Bi(III) by electrospray ionization mass spectrometry. J. Inorg. Biochem. 2005, 99, 1992–1997. [Google Scholar] [CrossRef] [PubMed]
- Rubino, F.M.; Verduci, C.; Giampiccolo, R.; Pulvirenti, S.; Brambilla, G.; Colombi, A. Molecular characterization of homo- and heterodimeric mercury(II)-bis-thiolates of some biologically relevant thiols by electrospray ionization and triple quadrupole tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2004, 15, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Rubino, F.M.; Pitton, M.; Brambilla, G.; Colombi, A. Electrospray ionization and triple quadrupole tandem mass spectrometry study of some biologically relevant homo- and heterodimeric cadmium thiolate conjugates. J. Am. Soc. Mass Spectrom. 2006, 17, 1442–1455. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Ahmad, S. Molecular handling of cadmium in transporting epithelia. Toxicol. Appl. Pharmacol. 2003, 186, 163–188. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Cherian, M.G.; Barfuss, D.W. Mercury-metallothionein and the renal accumulation and handling of mercury. Toxicology 1993, 83, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K. Molecular interactions with mercury in the kidney. Pharmacol. Rev. 2000, 52, 113–143. [Google Scholar] [PubMed]
- Chan, H.M.; Satoh, M.; Zalups, R.K.; Cherian, M.G. Exogenous metallothionein and renal toxicity of cadmium and mercury in rats. Toxicology 1992, 76, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Diamond, G.L.; Zalups, R.K. Understanding renal toxicity of heavy metals. Toxicol. Pathol. 1998, 26, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.C.; Zalups, R.K. Molecular mimicry as a mechanism for the uptake of cysteine S-conjugates of methylmercury and inorganic mercury. Chem. Res. Toxicol. 2006, 19, 1117–1118, author reply 1118–1120. [Google Scholar] [CrossRef] [PubMed]
- Thorsen, M.; Lagniel, G.; Kristiansson, E.; Junot, C.; Nerman, O.; Labarre, J.; Tamas, M.J. Quantitative transcriptome, proteome, and sulfur metabolite profiling of the Saccharomyces cerevisiae response to arsenite. Physiol. Genomics 2007, 30, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Lafaye, A.; Junot, C.; Pereira, Y.; Lagniel, G.; Tabet, J.-C.; Ezan, E.; Labarre, J. Combined proteome and metabolite-profiling analyses reveal surprising insights into yeast sulfur metabolism. J. Biol. Chem. 2005, 280, 24723–24730. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.N.; Niedzwiecki, M.; Liu, X.; Harper, K.N.; Alam, S.; Slavkovich, V.; Ilievski, V.; Levy, D.; Siddique, A.B.; Parvez, F.; et al. Chronic arsenic exposure and blood glutathione and glutathione disulfide concentrations in Bangladeshi adults. Environ. Health Perspect. 2014, 121, 1068–1074. [Google Scholar]
- Garçon, G.; Leleu, B.; Zerimech, F.; Marez, T.; Haguenoer, J.M.; Furon, D.; Shirali, P. Biologic markers of oxidative stress and nephrotoxicity as studied in biomonitoring of adverse effects of occupational exposure to lead and cadmium. J. Occup. Environ. Med. 2004, 46, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Hambach, R.; Lison, D.; D’Haese, P.; Weyler, J.; François, G.; de Schryver, A.; Manuel-Y-Keenoy, B.; van Soom, U.; Caeyers, T.; van Sprundel, M.; et al. Adverse effects of low occupational cadmium exposure on renal and oxidative stress biomarkers in solderers. Occup. Environ. Med. 2013, 70, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Kobal, A.B.; Horvat, M.; Prezelj, M.; Briski, A.S.; Krsnik, M.; Dizdarevic, T.; Mazej, D.; Falnoga, I.; Stibilj, V.; Arneric, N.; et al. The impact of long-term past exposure to elemental mercury on antioxidative capacity and lipid peroxidation in mercury miners. J. Trace Elem. Med. Biol. 2004, 17, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Kobal, A.B.; Prezelj, M.; Horvat, M.; Krsnik, M.; Gibicar, D.; Osredkar, J. Glutathione level after long-term occupational elemental mercury exposure. Environ. Res. 2008, 107, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.; Igbaria, A.; D’Autreaux, B.; Planson, A.G.; Junot, C.; Godat, E.; Bachhawat, A.K.; Delaunay-Moisan, A.; Toledano, M.B. Glutathione revisited: A vital function in iron metabolism and ancillary role in thiol-redox control. EMBO J. 2011, 30, 2044–2056. [Google Scholar] [CrossRef]
- Qia, W.; Cowan, J.A. Mechanism of glutaredoxin-ISU [2Fe–2S] cluster exchange. Chem. Commun. 2011, 47, 4989–4991. [Google Scholar] [CrossRef]
- Rubino, F.M.; Pitton, M.; Brambilla, G.; Colombi, A. A study of the glutathione metaboloma peptides by energy-resolved mass spectrometry as a tool to investigate into the interference of toxic heavy metals with their metabolic processes. J. Mass Spectrom. 2006, 41, 1578–1593. [Google Scholar] [CrossRef] [PubMed]
- Carney, C.K.; Harry, S.R.; Sewell, S.L.; Wright, D.W. Detoxification biominerals. Biomineralization I Cryst. Self-Organ. Process Top. Curr. Chem. 2007, 270, 155–185. [Google Scholar]
- Perbellini, L.; Veronese, N.; Princivalle, A. Mercapturic acids in the biological monitoring of occupational exposure to chemicals. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2002, 781, 269–290. [Google Scholar] [CrossRef]
- Fuhr, B.J.; Rabenstein, D.L. Nuclear magnetic resonance studies of the solution chemistry of metal complexes. IX. The binding of cadmium, zinc, lead, and mercury by glutathione. J. Am. Chem. Soc. 1973, 95, 6944–6950. [Google Scholar] [CrossRef] [PubMed]
- Rabenstein, D.L.; Fairhurst, M.T. Nuclear magnetic resonance studies of the solution chemistry of metal complexes. XI. The binding of methylmercury by sulfhydryl-containing amino acids and by glutathione. J. Am. Chem. Soc. 1975, 97, 2086–2092. [Google Scholar] [CrossRef] [PubMed]
- Kadima, W.; Rabenstein, D.L. A quantitative study of the complexation of cadmium in hemolyzed human erythrocytes by 1H NMR spectroscopy. J. Inorg. Biochem. 1990, 40, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Rabenstein, D.L.; Isab, A.A. A proton nuclear magnetic resonance study of the interaction of mercury with intact human erythrocytes. Biochim. Biophys. Acta 1982, 721, 374–384. [Google Scholar] [CrossRef]
- Rabenstein, D.L.; Isab, A.A.; Reid, R.S. A proton nuclear magnetic resonance study of the binding of methylmercury in human erythrocytes. Biochim. Biophys. Acta 1982, 720, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Rabenstein, D.L.; Isab, A.A.; Kadima, W.; Mohanakrishnan, P. A proton nuclear magnetic resonance study of the interaction of cadmium with human erythrocytes. Biochim. Biophys. Acta 1983, 762, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Osawa, M.; Magos, L. The chemical form of the methylmercury complex in the bile of the rat. Biochem. Pharmacol. 1974, 23, 1903–1905. [Google Scholar] [CrossRef] [PubMed]
- Refsvik, T.; Norseth, T. Methyl mercuric compounds in rat bile. Acta Pharmacol. Toxicol. 1975, 36, 67–78. [Google Scholar] [CrossRef]
- Naganuma, A.; Imura, N. Methylmercury binds to a low molecular weight substance in rabbit and human erythrocytes. Toxicol. Appl. Pharmacol. 1979, 47, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Omata, S.; Sakimura, K.; Ishii, T.; Sugano, H. Chemical nature of a methylmercury complex with a low molecular weight in the liver cytosol of rats exposed to methylmercury chloride. Biochem. Pharmacol. 1978, 27, 1700–1702. [Google Scholar] [CrossRef] [PubMed]
- Urano, T.; Naganuma, A.; Imura, N. Methylmercury-cysteinylglycine constitutes the main form of methylmercury in rat bile. Res. Commun. Chem. Pathol. Pharmacol. 1988, 6, 197–210. [Google Scholar]
- Ballatori, N.; Clarkson, T.W. Inorganic mercury secretion into bile as a low molecular weight complex. Biochem. Pharmacol. 1984, 33, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Mizutani, N.; Keino, H.; Kashiwamata, S. A low-molecular-weight cadmium-binding substance in human and rat livers and human blood. Toxicol. Appl. Pharmacol. 1984, 73, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Kadima, W.; Rabenstein, D.L. Nuclear magnetic resonance studies of the solution chemistry of metal complexes. 26. Mixed ligand complexes of cadmium, nitrilotriacetic acid, glutathione, and related ligands. J. Inorg. Biochem. 1990, 38, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Połeć-Pawlak, K.; Ruzik, R.; Lipiec, E. Investigation of Cd(II), Pb(II) and Cu(I) complexation by glutathione and its component amino acids by ESI-MS and size exclusion chromatography coupled to ICP-MS and ESI-MS. Talanta 2007, 72, 1564–1572. [Google Scholar] [CrossRef] [PubMed]
- Leslie, E.M.; Haimeur, A.; Waalkes, M.P. Arsenic transport by the human multidrug resistance protein 1 (MRP1/ABCC1). Evidence that a tri-glutathione conjugate is required. J. Biol. Chem. 2004, 279, 32700–32708. [Google Scholar] [CrossRef] [PubMed]
- Carew, M.W.; Naranmandura, H.; Shukalek, C.B.; Le, X.C.; Leslie, E.M. Monomethylarsenic diglutathione transport by the human multidrug resistance protein 1 (MRP1/ABCC1). Drug Metab. Dispos. 2011, 39, 2298–2304. [Google Scholar] [CrossRef]
- Leslie, E.M. Arsenic-glutathione conjugate transport by the human multidrug resistance proteins (MRPs/ABCCs). J. Inorg. Biochem. 2012, 108, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, D.; Cakir, Y. Arsenate V induced glutathione efflux from humanerythrocytes. J. Trace Elem. Med. Biol. 2012, 26, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, D.; Cakir, Y. Efflux of glutathione and glutathione complexes from human erythrocytes in response to inorganic arsenic exposure. Biol. Trace Elem. Res. 2012, 150, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Kojima, C.; Sakurai, T.; Waalkes, M.P.; Himeno, S. Cytolethality of glutathione conjugates with monomethylarsenic or dimethylarsenic compounds. Biol. Pharm. Bull. 2005, 28, 1827–1832. [Google Scholar] [CrossRef] [PubMed]
- Daubeuf, S.; Leroy, P.; Paolicchi, A.; Pompella, A.; Wellman, M.; Galteau, M.M.; Visvikis, A. Enhanced resistance of HeLa cells to cisplatin by overexpression of γ-glutamyltransferase. Biochem. Pharmacol. 2002, 64, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Daubeuf, S.; Balin, D.; Leroy, P.; Visvikis, A. Different mechanisms for γ-glutamyltransferase-dependent resistance to carboplatin and cisplatin. Biochem. Pharmacol. 2003, 66, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Miao, R.; Yang, G.; Miao, Y.; Mei, Y.; Hong, J.; Zhao, C.; Zhu, L. Interactions of platinum(II) complexes with sulfur-containing peptides studied by electrospray ionization mass spectrometry and tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Y.; Mandal, R.; Li, X.F. Intact human holo-transferrin interaction with oxaliplatin. Rapid Commun. Mass Spectrom. 2005, 19, 1956–1962. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lyons, B.; Truscott, R.J.; Schey, K.L. Human protein aging: Modification and crosslinking through dehydroalanine and dehydrobutyrine intermediates. Aging Cell 2014, 13, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Schierl, R.; Rohrer, B.; Hohnloser, J. Long-term platinum excretion in patients treated with cisplatin. Cancer Chemother. Pharmacol. 1995, 36, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Hohnloser, J.H.; Schierl, R.; Hasford, B.; Emmerich, B. Cisplatin based chemotherapy in testicular cancer patients: Long term platinum excretion and clinical effects. Eur. J. Med. Res. 1996, 1, 509–514. [Google Scholar] [PubMed]
- Gerl, A.; Schierl, R. Urinary excretion of platinum in chemotherapy-treated long-term survivors of testicular cancer. Acta Oncol. 2000, 39, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Wagner, F.; Bernhauer, K. New aspects of the structure of corrinoid coenzymes. Ann. N. Y. Acad. Sci. 1964, 112, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Cregan, A.G.; Berben, L.A.; Brasch, N.E. Studies on the formation of glutathionylcobalamin: Any free intracellular aquacobalamin is likely to be rapidly and irreversibly converted to glutathionylcobalamin. Inorg. Chem. 2004, 43, 6848–6857. [Google Scholar] [CrossRef] [PubMed]
- Pezacka, E.; Green, R.; Jacobsen, D.W. Glutathionylcobalamin as an intermediate in the formation of cobalamin coenzymes. Biochem. Biophys. Res. Commun. 1990, 169, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Guttmann, D.; Poage, G.; Johnston, T.; Zhitkovich, A. Reduction with glutathione is a weakly mutagenic pathway in chromium(VI) metabolism. Chem. Res. Toxicol. 2008, 21, 2188–2194. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.; Armknecht, S.; Zhitkovich, A. Metabolism of Cr(VI) by ascorbate but not glutathione is a low oxidant-generating process. J. Trace Elem. Med. Biol. 2012, 26, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.J.; Shi, X.; Dalal, N.S. Synthesis of Cr(IV)-GSH, Its identification and its free hydroxyl radical generation: A model compound for Cr(VI) carcinogenicity. Biochem. Biophys. Res. Commun. 1997, 235, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.; Zhang, L.; Lay, P.A. Structure and reactivity of a Chromium(V) glutathione complex. Inorg. Chem. 2003, 42, 767–784. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.S.; Vallyathan, V.; Castranova, V.; Shi, X. Generation of reactive oxygen species in the enzymatic reduction of PbCrO4 and related DNA Damage. Mol. Cell. Biochem. 2002, 234/235, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Zhitkovich, A.; Song, Y.; Quievryn, G.; Voitkun, V. Non-oxidative mechanisms are responsible for the induction of mutagenesis by reduction of Cr(VI) with cysteine: Role of ternary DNA adducts in Cr(III)-dependent mutagenesis. Biochemistry 2001, 40, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Macfie, A.; Hagan, E.; Zhitkovich, A. Mechanism of DNA-protein crosslinking by chromium. Chem. Res. Toxicol. 2010, 23, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Qu, L.; Li, B.; Xing, L.; Jia, G.; Wang, T.; Gao, Y.; Zhang, P.; Li, M.; Chen, W.; et al. Increased oxidative DNA damage, as assessed by urinary 8-hydroxy-2'-deoxyguanosine concentrations, and serum redox status in persons exposed to mercury. Clin. Chem. 2005, 51, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Bando, I.; Sanchez Reus, M.I.; Andres, D.; Cascales, M. Endogenous antioxidant defence system in rat liver following mercury chloride oral intoxication. J. Biochem. Mol. Toxicol. 2005, 19, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.A.; Reddy, A.G.; Kumar, P.R.; Reddy, Y.R.; Rao, T.M.; Haritha, C. Protective role of N-Acetyl L-Cysteine against reproductive toxicity due to interaction of lead and cadmium in male Wistar rats. J. Nat. Sci. Biol. Med. 2013, 4, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2011, 30, 1191–1212. [Google Scholar] [CrossRef]
- Flohe, L. The fairytale of the GSSG/GSH redox potential. Biochim. Biophys. Acta 2013, 1830, 3139–3142. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Angele-Martinez, C.; Goodman, C.; Brumaghim, J. Metal-mediated DNA damage and cell death: Mechanisms, detection methods, and cellular consequences. Metallomics 2014, 6, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Rubino, F.M.; Pitton, M.; di Fabio, D.; Colombi, A. Toward an “omic” physiopathology of reactive chemicals: Thirty years of mass spectrometric study of the protein adducts with endogenous and xenobiotic compounds. Mass Spectrom. Rev. 2009, 28, 725–784. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Tak Yee, A.W. Glutathione and apoptosis. Free Radic. Res. 2008, 42, 689–706. [Google Scholar] [CrossRef] [PubMed]
- United Nations Environmental Program. Minamata Convenion on Mercury. Available online: http://www.mercuryconvention.org/ (accessed on 7 November 2014).
- Barkay, T.; Miller, S.M.; Summers, A.O. Bacterial mercury resistance from atoms to ecosystems. FEMS Microbiol. Rev. 2003, 27, 355–384. [Google Scholar] [CrossRef] [PubMed]
- Wardwell, L.H.; Jude, B.A.; Moody, J.P.; Olcerst, A.I.; Gyure, R.A.; Nelson, R.E.; Fekete, F.A. Co-selection of mercury and antibiotic resistance in sphagnum core samples dating back 2000 years. Geomicrobiol. J. 2009, 26, 238–247. [Google Scholar] [CrossRef]
- Summers, A.O.; Sugarman, L.I. Cell-free mercury(II)-reducing activity in a plasmid-bearing strain of Escherichia coli. J. Bacteriol. 1974, 119, 242–249. [Google Scholar] [PubMed]
- Schottel, J.L. The mercuric and organomercurial detoxifying enzymes from a plasmid-bearing strain of Escherichia coli. J. Biol. Chem. 1978, 253, 4341–4349. [Google Scholar] [PubMed]
- Benison, G.C.; di Lello, P.; Shokes, J.E.; Cosper, N.J.; Scott, R.A.; Legault, P.; Omichinski, J.G. A stable mercury-containing complex of the organomercurial lyase MerB: Catalysis, product release, and direct transfer to MerA. Biochemistry 2004, 43, 8333–8345. [Google Scholar] [CrossRef] [PubMed]
- Ledwidge, R.; Patel, B.; Dong, A.; Fiedler, D.; Falkowski, M.; Zelikova, J.; Summers, A.O.; Pai, E.F.; Miller, S.M. NmerA, the metal binding domain of mercuric ion reductase, removes Hg2+ from proteins, delivers it to the catalytic core, and protects cells under glutathione-depleted conditions. Biochemistry 2005, 44, 11402–11416. [Google Scholar] [CrossRef] [PubMed]
- Ledwidge, R.; Hong, B.; Dötsch, V.; Miller, S.M. NmerA of Tn501 mercuric ion reductase: Structural modulation of the pKa values of the metal binding cysteine thiols. Biochemistry 2010, 49, 8988–8998. [Google Scholar] [CrossRef] [PubMed]
- Schiering, N.; Kabsch, W.; Moore, M.J.; Distefano, M.D.; Walsh, C.T.; Pai, E.F. Structure of the detoxification catalyst mercuric ion reductase from Bacillus sp. strain RC607. Nature 1991, 352, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Cummings, R.T.; Walsh, C.T. Interaction of Tn501 mercuric reductase and dihydroflavin adenine dinucleotide anion with metal ions: Implications for the mechanism of mercuric reductase mediated Hg(II) reduction. Biochemistry 1992, 31, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Siedler, F.; Rudolph-Böhner, S.; Doi, M.; Musiol, H.J.; Moroder, L. Redox potentials of active-site bis(cysteinyl) fragments of thiol-protein oxidoreductases. Biochemistry 1993, 32, 7488–7495. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeyer, R.E.; Singh, S.P.; Doonan, C.J.; Ross, A.R.; Hughes, R.J.; Pickering, I.J.; George, G.N. Molecular mimicry in mercury toxicology. Chem. Res. Toxicol. 2006, 19, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Qiu, L.; Divine, K.K.; Ashbaugh, M.D.; McIntyre, L.C., Jr.; Fernando, Q.; Gandolfi, A.J. Toxicity and transport of three synthesized mercury-thiol-complexes in isolated rabbit renal proximal tubule suspensions. Drug Chem. Toxicol. 1999, 22, 323–341. [Google Scholar] [CrossRef] [PubMed]
- Busch, K.; Glish, G.; Mcluckey, S. Mass Spectrometry/Mass Spectrometry: Techniques and Applications of Tandem Mass Spectrometry; VCH Publishing: New York, NY, USA, 1988. [Google Scholar]
- Rubino, F.M.; Verduci, C.; Giampiccolo, R.; Pulvirenti, S.; Brambilla, G.; Colombi, A. Characterization of the disulfides of bio-thiols by electrospray ionization and triple-quadrupole tandem mass spectrometry. J. Mass Spectrom. 2004, 39, 1408–1416. [Google Scholar] [CrossRef] [PubMed]
- Rubino, F.M.; Pitton, M.; Caneva, E.; Pappini, M.; Colombi, A. Thiol-disulfide redox equilibria of glutathione metaboloma compounds investigated by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 3935–3948. [Google Scholar] [CrossRef] [PubMed]
- Harrison, A.G. The gas-phase basicities and proton affinities of amino acids and peptides. Mass Spectrom. Rev. 1997, 16, 201–217. [Google Scholar] [CrossRef]
- Klassen, J.S.; Kebarle, P. Collison-induced dissociation threshold energies of protonated glycine, glycinamide, and some related small peptides and peptide amino amides. J. Am. Chem. Soc. 1997, 119, 6552–6563. [Google Scholar] [CrossRef]
- Price, W.D.; Schnier, P.D.; Williams, E.R. Binding energies of the proton-bound amino acid dimers Gly·Gly, Ala·Ala, Gly·Ala, and Lys·Lys measured by blackbody infrared radiative dissociation. J. Phys. Chem. B 1997, 101, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Bleiholder, C.; Suhai, S.; Paizs, B. Revising the proton affinity scale of the naturally occurring α-amino acids. J. Am. Soc. Mass Spectrom. 2006, 17, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Greco, F.; Liguori, A.; Sindona, G.; Uccella, N. Gas-phase proton affinity of deoxyribonucleosides and related nucleobases by fast atom bombardment tandem mass spectrometry. J. Am. Chem. Soc. 1990, 112, 9092–9096. [Google Scholar] [CrossRef]
- Liguori, A.; Napoli, A.; Sindona, G. Survey of the proton affinities of adenine, cytosine, thymine and uracil dideoxyribonucleosides, deoxyribonucleosides and ribonucleosides. J. Mass Spectrom. 2000, 35, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Di Donna, L.; Napoli, A.; Sindona, G.; Athanassopoulos, C. A comprehensive evaluation of the kinetic method applied in the determination of the proton affinity of the nucleic acid molecules. J. Am. Soc. Mass Spectrom. 2004, 15, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Armentrout, P.B. Mass spectrometry—Not just a structural tool: The use of guided ion beam tandem mass spectrometry to determine thermochemistry. J. Am. Soc. Mass Spectrom. 2002, 13, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, M.T.; Armentrout, P.B. Noncovalent metal-ligand bond energies as studied by threshold collision-induced dissociation. Mass Spectrom. Rev. 2000, 19, 215–247. [Google Scholar] [CrossRef] [PubMed]
- Enoiu, M.; Aberkane, H.; Salazar, J.F.; Leroy, P.; Groffen, J.; Siest, G.; Wellman, M. Evidence for the pro-oxidant effect of γ-glutamyltranspeptidase-related enzyme. Free Radic. Biol. Med. 2000, 29, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W.; Tate, S.S. The apparent glutathione oxidase activity of γ-glutamyl transpeptidase. Chemical mechanism. J. Biol. Chem. 1980, 255, 5011–5014. [Google Scholar] [PubMed]
- Divine, K.K.; Ayala-Fierro, F.; Barber, D.S.; Carter, D.E. Glutathione, albumin, cysteine, and cys-gly effects on toxicity and accumulation of mercuric chloride in LLC-PK1 cells. J. Toxicol. Environ. Health A 1999, 57, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Brandão, R.; Santos, F.W.; Zeni, G.; Rocha, J.B.; Nogueira, C.W. DMPS and N-acetylcysteine induced renal toxicity in mice exposed to mercury. Biometals 2006, 19, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Bulaj, G.; Kortemme, T.; Goldenberg, D.P. Ionization-reactivity relationships for cysteine thiols in polypeptides. Biochemistry 1998, 37, 8965–8972. [Google Scholar] [CrossRef] [PubMed]
- Noszál, B.; Visky, D.; Kraszni, M. Population, acid-base, and redox properties of N-acetylcysteine conformers. J. Med. Chem. 2000, 43, 2176–2182. [Google Scholar] [CrossRef] [PubMed]
- Woods, J.S.; Calas, C.A.; Aicher, L.D. Stimulation of porphyrinogen oxidation by mercuric ion. I. Evidence of free radical formation in the presence of thiols and hydrogen peroxide. Mol. Pharmacol. 1990, 38, 253–260. [Google Scholar] [PubMed]
- Woods, J.S.; Calas, C.A.; Aicher, L.D. Stimulation of porphyrinogen oxidation by mercuric ion. II. Promotion of oxidation from the interaction of mercuric ion, glutathione, and mitochondria-generated hydrogen peroxide. Mol. Pharmacol. 1990, 38, 261–266. [Google Scholar] [PubMed]
- Miller, D.M.; Woods, J.S. Redox activities of mercury-thiol complexes: Implications for mercury-induced porphyria and toxicity. Chem. Biol. Interact. 1993, 88, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Halbach, S.; Clarkson, T.W. Enzymatic oxidation of mercury vapor by erythrocytes. Biochim. Biophys. Acta 1978, 523, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Wigfield, D.C.; Tse, S. The mechanism of biooxidation of mercury. J. Appl. Toxicol. 1986, 6, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Banu, L.; Blagojevic, V.; Bohme, D.K. Locating Pb2+ and Zn2+ in zinc finger-like peptides using mass spectrometry. J. Am. Soc. Mass Spectrom. 2013, 24, 1534–1542. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Scholz, K.; Donegan, M.; Burton, L.; Wingate, J.; Völkel, W. Metabonomics and biomarker discovery: LC-MS metabolic profiling and constant neutral loss scanning combined with multivariate data analysis for mercapturic acid analysis. Anal. Chem. 2006, 78, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Scholz, K.; Sieber, M.; Kellert, M.; Voelkel, W. Tools in metabonomics: An integrated validation approach for LC-MS metabolic profiling of mercapturic acids in human urine. Anal. Chem. 2007, 79, 2918–2926. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.; Krasnikov, B.F.; Niatsetskaya, Z.V.; Pinto, J.T.; Callery, P.S.; Villar, M.T.; Artigues, A.; Bruschi, S.A. Cysteine S-conjugate β-lyases: Important roles in the metabolism of naturally occurring sulfur and selenium-containing compounds, xenobiotics and anticancer agents. Amino Acids 2011, 41, 7–27. [Google Scholar] [CrossRef] [PubMed]
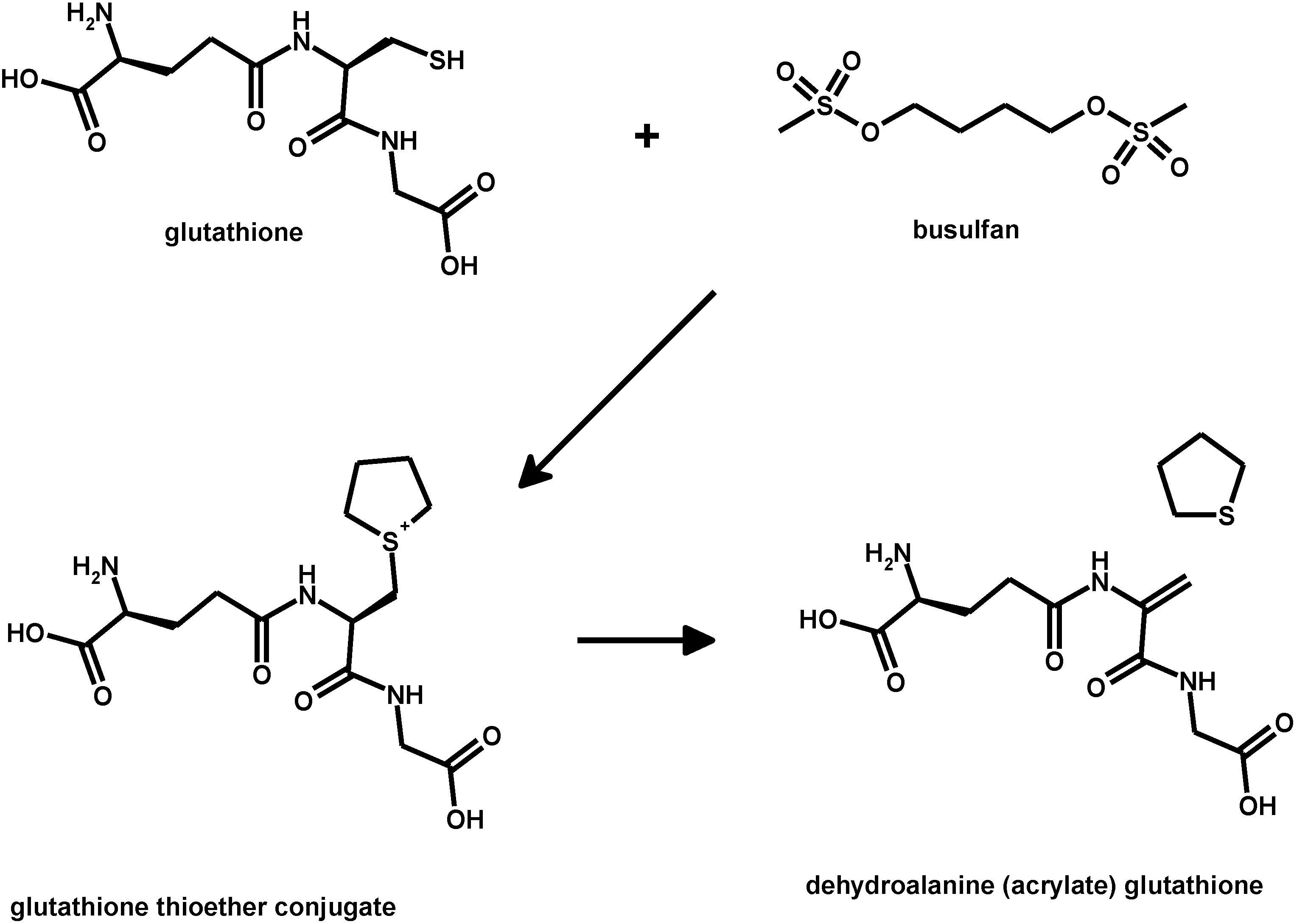
- Younis, I.R.; Elliott, M.; Peer, C.J.; Cooper, A.J.; Pinto, J.T.; Konat, G.W.; Kraszpulski, M.; Petros, W.P.; Callery, P.S. Dehydroalanine analog of glutathione: An electrophilic busulfan metabolite that binds to human glutathione S-transferase A1-1. J. Pharmacol. Exp. Ther. 2008, 327, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Nath, C.E.; Shaw, P.J. Busulphan in blood and marrow transplantation: Dose, route, frequency and role of therapeutic drug monitoring. Curr. Clin. Pharmacol. 2007, 2, 75–91. [Google Scholar] [CrossRef] [PubMed]
- McCune, J.S.; Holmberg, L.A. Busulfan in hematopoietic stem cell transplant setting. Expert Opin. Drug Metab. Toxicol. 2009, 5, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [PubMed]
- Sinning, I.; Kleywegt, G.J.; Cowan, S.W.; Reinemer, P.; Dirr, H.W.; Huber, R.; Gilliland, G.L.; Armstrong, R.N.; Ji, X.; Board, P.G.; et al. Structure determination and refinement of human α class glutathione transferase A1-1, and a comparison with the Mu and Pi class enzymes. J. Mol. Biol. 1993, 232, 192–212. [Google Scholar] [CrossRef] [PubMed]
- Oakley, A.J.; Lo Bello, M.; Battistoni, A.; Ricci, G.; Rossjohn, J.; Villar, H.O.; Parker, M.W. The structures of human glutathione transferase P1-1 in complex with glutathione and various inhibitors at high resolution. J. Mol. Biol. 1997, 274, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Oakley, A.J.; Lo Bello, M.; Mazzetti, A.P.; Federici, G.; Parker, M.W. The glutathione conjugate of ethacrynic acid can bind to human pi class glutathione transferase P1-1 in two different modes. FEBS Lett. 1997, 419, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-J.; Boehlert, C.C.; Rider, K.; Armstrong, R.N. Synthesis and characterization of the oxygen and des-thioanalogue of glutathione as dead-end inhibitors of glutathione-S-transferase. Biochem. Biophys. Res. Commun. 1985, 128, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.; Pinto, J.T.; Callery, P.S. Reversible and irreversible protein glutathionylation: Biological and clinical aspects. Expert Opin. Drug Metab. Toxicol 2011, 7, 891–910. [Google Scholar] [CrossRef] [PubMed]
- Seelig, G.F.; Meister, A. Cystamine-Sepharose. A probe for the active site of γ-glutamylcysteine synthetase. J. Biol. Chem. 1982, 257, 5092–5096. [Google Scholar] [PubMed]
- Seelig, G.F.; Meister, A. γ-glutamylcysteine synthetase. Interactions of an essential sulfhydryl group. J. Biol. Chem. 1984, 259, 3534–3538. [Google Scholar] [PubMed]
- Lebo, R.V.; Kredich, N.M. Inactivation of human γ-glutamylcysteine synthetase by cystamine. Demonstration and quantification of enzyme-ligand complexes. J. Biol. Chem. 1978, 253, 2615–2623. [Google Scholar] [PubMed]
- Huang, C.S.; Moore, W.R.; Meister, A. On the active site thiol of γ-glutamylcysteine synthetase: Relationships to catalysis, inhibition, and regulation. Proc. Natl. Acad. Sci. USA 1988, 85, 2464–2468. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Anders, M.W. Identification of an important cysteine residue in human glutamate-cysteine ligase catalytic subunit by site-directed mutagenesis. Biochem. J. 1998, 336, 675–680. [Google Scholar] [PubMed]
- Ghaemmaghami, S.; Huh, W.K.; Bower, K.; Howson, R.W.; Belle, A.; Dephoure, N.; O’Shea, E.K.; Weissman, J.S. Global analysis of protein expression in yeast. Nature 2003, 425, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Kil, I.S.; Park, J.W. Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J. Biol. Chem. 2005, 280, 10846–10854. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Martinez-Garcia, E.; Nguyen, H.; Mullen, A.R.; Dufour, E.; Sudarshan, S.; Licht, J.D.; Deberardinis, R.J.; Chandel, N.S. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol. Cell 2013, 51, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Cho, C.S. Blot-based detection of dehydroalanine-containing glutathione peroxidase with the use of biotin-conjugated cysteamine. Methods Enzymol. 2010, 474, 23–34. [Google Scholar] [PubMed]
- Chen, J.; Shiyanov, P.; Schlager, J.J.; Green, K.B. A pseudo MS3 approach for identification of disulfide-bonded proteins: Uncommon product ions and database search. J. Am. Soc. Mass Spectrom. 2012, 23, 225–243. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, A. Zinc finger proteins as potential targets for toxic metal ions: Differential effects on structure and function. Antioxid. Redox Signal. 2001, 3, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz-Kucharczyk, A.; Bal, W. Damage of zinc fingers in DNA repair proteins, a novel molecular mechanism in carcinogenesis. Toxicol. Lett. 2006, 162, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.M.; Ciriolo, M.R.; Marcocci, L.; Rotilio, G. Copper(I) transfer into metallothionein mediated by glutathione. Biochem. J. 1993, 292, 673–676. [Google Scholar] [PubMed]
- Tallkvist, J.; Persson, E.; Henriksson, J.; Tjälve, H. Cadmium-metallothionein interactions in the olfactory pathways of rats and pikes. Toxicol. Sci. 2002, 67, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Afonso, C.; Hathout, Y.; Fenselau, C. Qualitative characterization of biomolecular zinc complexes by collisionally induced dissociation. J. Mass Spectrom. 2002, 37, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Ngu, T.T.; Stillman, M.J. Metalation of metallothioneins. IUBMB Life 2009, 61, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Otvos, J.D.; Armitage, I.M. Structure of the metal clusters in rabbit liver metallothionein. Proc. Natl. Acad. Sci. USA 1980, 77, 7094–7098. [Google Scholar] [CrossRef] [PubMed]
- Greisen, P.; Jespersen, J.B.; Kepp, K.P. Metallothionein Zn2+- and Cu2+-clusters from first-principles calculations. Dalton Trans. 2012, 41, 2247–2256. [Google Scholar] [CrossRef] [PubMed]
- Henkel, G.; Krebs, B. Metallothioneins: Zinc, cadmium, mercury, and copper thiolates and selenolates mimicking protein active site features—Structural aspects and biological implications. Chem. Rev. 2004, 104, 801–824. [Google Scholar] [CrossRef] [PubMed]
- Kepp, K.P. Full quantum-mechanical structure of the human protein Metallothionein-2. J. Inorg. Biochem. 2012, 107, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Tjalve, H.; Henriksson, J. Uptake of metals in the brain via olfactory pathways. Neurotoxicology 1999, 20, 181–195. [Google Scholar] [PubMed]
- Persson, E.; Henriksson, J.; Tallkvist, J.; Rouleau, C.; Tjälve, H. Transport and subcellular distribution of intranasally administered zinc in the olfactory system of rats and pikes. Toxicology 2003, 191, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Gottofrey, J.; Tjalve, H. Axonal transport of cadmium in the olfactory nerve of the pike. Pharmacol. Toxicol. 1991, 69, 242–252. [Google Scholar] [PubMed]
- Tjälve, H.; Henriksson, J.; Tallkvist, J.; Larsson, B.S.; Lindquist, N.G. Uptake of manganese and cadmium from the nasal mucosa into the central nervous system via olfactory pathways in rats. Pharmacol. Toxicol. 1996, 79, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Borg-Neczak, K.; Tjalve, H. Uptake of 203Hg2+ in the olfactory system in pike. Toxicol. Lett. 1996, 84, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, J.; Tjalve, H. Uptake of inorganic mercury in the olfactory bulbs via olfactory pathways in rats. Environ. Res. 1998, 77, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Tjälve, H.; Mejàre, C.; Borg-Neczak, K. Uptake and transport of manganese in primary and secondary olfactory neurones in pike. Pharmacol. Toxicol. 1995, 77, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, J.; Tallkvist, J.; Tjälve, H. Transport of manganese via the olfactory pathway in rats: Dosage dependency of the uptake and subcellular distribution of the metal in the olfactory epithelium and the brain. Toxicol. Appl. Pharmacol. 1999, 156, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, J.; Tjalve, H. Manganese taken up into the CNS via the olfactory pathway in rats affects astrocytes. Toxicol. Sci. 2000, 55, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Persson, E.; Henriksson, J.; Tjälve, H. Uptake of cobalt from the nasal mucosa into the brain via olfactory pathways in rats. Toxicol. Lett. 2003, 145, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, J.; Tallkvist, J.; Tjälve, H. Uptake of nickel into the brain via olfactory neurons in rats. Toxicol. Lett. 1997, 91, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Azzouz, I.; Trabelsi, H.; Hanini, A.; Ferchichi, S.; Tebourbi, O.; Sakly, M.; Abdelmelek, H. Interaction between nanoparticles generated by zinc chloride treatment and oxidative responses in rat liver. Int. J. Nanomed. 2014, 9, 223–229. [Google Scholar]
- Trabelsi, H.; Azzouz, I.; Sakly, M.; Abdelmelek, H. Subacute toxicity of cadmium on hepatocytes and nephrocytes in the rat could be considered as a green biosynthesis of nanoparticles. Int. J. Nanomed. 2013, 8, 1121–1128. [Google Scholar] [CrossRef]
- Trabelsi, H.; Azzouz, I.; Ferchichi, S.; Tebourbi, O.; Sakly, M.; Abdelmelek, H. Nanotoxicological evaluation of oxidative responses in rat nephrocytes induced by cadmium. Int. J. Nanomed. 2013, 8, 3447–3453. [Google Scholar] [CrossRef]
- Reese, R.N.; White, C.A.; Winge, D.R. Cadmium-sulfide crystallites in Cd-('YEC)nG peptide complexes from tomato. Plant Physiol. 1992, 98, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Kowshik, M.; Vogel, W.; Urban, J.; Kulkarni, S.K.; Paknikar, K.M. Microbial synthesis of semiconductor PbS nanocrystallites. Adv. Mater. 2002, 14, 815–818. [Google Scholar] [CrossRef]
- Flenniken, M.; Allen, M.; Douglas, T. Microbe manufacturers of semiconductors. Chem. Biol. 2004, 11, 1478–1480. [Google Scholar] [CrossRef] [PubMed]
- Mandal, D.; Bolander, M.E.; Mukhopadhyay, D.; Sarkar, G.; Mukherjee, P. The use of microorganisms for the formation of metal nanoparticles and their application. Appl. Microbiol. Biotechnol. 2006, 69, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, R.Y.; Mao, C.; Gao, X.; Burt, J.L.; Belcher, A.M.; Georgiou, G.; Iverson, B.L. Bacterial biosynthesis of cadmium sulfide nanocrystals. Chem. Biol. 2004, 11, 1553–1559. [Google Scholar] [CrossRef] [PubMed]
- Diacomanolis, V.; Ng, J.C.; Sadler, R.; Nomura, M.; Noller, B.N.; Harris, H.H. Consistent chemical form of Cd in liver and kidney tissues in rats dosed with a range of Cd treatments: XAS of intact tissues. Chem. Res. Toxicol. 2010, 23, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Elinder, C.G.; Nordberg, M.; Palm, B. Is cadmium released from metallothionein in rejected human kidneys? Biol. Met. 1990, 2, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.B.; Barregard, L.; Sallsten, G.; Broberg, K. Cadmium, mercury, and lead in kidney cortex are not associated with urinary 8-oxo-7,8-dihydro-2'-deoxyguanosine (8-oxodG) in living kidney donors. Int. Arch. Occup. Environ. Health 2014, 87, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Cerulli, N.; Campanella, L.; Grossi, R.; Politi, L.; Scandurra, R.; Soda, G.; Gallo, F.; Damiani, S.; Alimonti, A.; Petrucci, F.; et al. Determination of Cd, Cu, Pb and Zn in neoplastic kidneys and in renal tissue of fetuses, newborns and corpses. J. Trace Elem. Med. Biol. 2006, 20, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Danscher, G. Applications of autometallography to heavy metal toxicology. Pharmacol. Toxicol. 1991, 68, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Danscher, G.; Stoltenberg, M.; Juhl, S. How to detect gold, silver and mercury in human brain and other tissues by autometallographic silver amplification. Neuropathol. Appl. Neurobiol. 1994, 20, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Stoltenberg, M.; Juhl, S.; Danscher, G. Bismuth ions are metabolized into autometallographic traceable bismuth-sulphur quantum dots. Eur. J. Histochem. 2007, 51, 53–57. [Google Scholar] [PubMed]
- Danscher, G. In vivo liberation of gold ions from gold implants. Autometallographic tracing of gold in cells adjacent to metallic gold. Histochem. Cell Biol. 2002, 117, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Danscher, G.; Rungby, J. Differentiation of histochemically visualized mercury and silver. Histochem. J. 1986, 18, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Baatrup, E.; Nielsen, M.G.; Danscher, G. Histochemical demonstration of two mercury pools in trout tissues: Mercury in kidney and liver after mercuric chloride exposure. Ecotoxicol. Environ. Saf. 1986, 12, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.C.; Reske-Nielsen, E.; Thorlacius-Ussing, O.; Rungby, J.; Danscher, G. Distribution of dietary mercury in a dog. Quantitation and localization of total mercury in organs and central nervous system. Sci. Total Environ. 1989, 78, 23–43. [Google Scholar] [CrossRef] [PubMed]
- Bolewska, J.; Holmstrup, P.; Møller-Madsen, B.; Kenrad, B.; Danscher, G. Amalgam associated mercury accumulations in normal oral mucosa, oral mucosal lesions of lichen planus and contact lesions associated with amalgam. J. Oral Pathol. Med. 1990, 19, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Danscher, G.; Hørsted-Bindslev, P.; Rungby, J. Traces of mercury in organs from primates with amalgam fillings. Exp. Mol. Pathol. 1990, 52, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.B.; Hansen, J.C.; Mulvad, G.; Pedersen, H.S.; Gregersen, M.; Danscher, G. Mercury accumulations in brains from populations exposed to high and low dietary levels of methyl mercury. Concentration, chemical form and distribution of mercury in brain samples from autopsies. Int. J. Circumpolar Health 1999, 58, 96–107. [Google Scholar]
- Loumbourdis, N.S.; Danscher, G. Autometallographic tracing of Hg-S quantum dots in frogs exposed to inorganic mercury. Biometals 2008, 21, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Stoltenberg, M.; Hogenhuis, J.A.; Hauw, J.J.; Danscher, G. Autometallographic tracing of bismuth in human brain autopsies. J. Neuropathol. Exp. Neurol. 2001, 60, 705–710. [Google Scholar] [PubMed]
- Hardman, R. A toxicologic review of quantum dots: Toxicity depends on physicochemical and environmental factors. Environ. Health. Perspect. 2006, 114, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, K.M.; Dai, Q.; Alman, B.A.; Chan, W.C. Are quantum dots toxic? Exploring the discrepancy between cell culture and animal studies. Acc. Chem. Res. 2013, 46, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.P.; Leong, K.W. Quantum dot-based theranostics. Nanoscale 2010, 2, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Shao, Y.; Liu, J.; Chen, G.; Ho, P.C. The medicinal use of realgar (As4S4) and its recent development as an anticancer agent. J. Ethnopharmacol. 2011, 135, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-Y.; Shen, J.-C.; Zhuang, Z.-X.; Wang, X.-R.; Lee Frank, S.C. Investigation of metal-binding metallothioneins in the tissues of rats after oral intake of cinnabar. Anal. Bioanal. Chem. 2004, 379, 427–432. [Google Scholar] [CrossRef] [PubMed]
- L’Azou, B.; Passagne, I.; Mounicou, S.; Tréguer-Delapierre, M.; Puljalté, I.; Szpunar, J.; Lobinskib, R.; Ohayon-Courtès, C. Comparative cytotoxicity of cadmium forms (CdCl2, CdO, CdS micro- and nano-particles) in renal cells. Toxicol. Res. 2014, 3, 32–41. [Google Scholar] [CrossRef]
- Green, M.; Howman, E. Semiconductor quantum dots and free radical induced DNA nicking. Chem. Commun. 2005, 7, 121–123. [Google Scholar] [CrossRef]
- Ipe, B.I.; Lehnig, M.; Niemeyer, C.M. On the generation of free radical species from quantum dots. Small 2005, 1, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Billone, P.S.; Maretti, L.; Maurel, V.; Scaiano, J.C. Dynamics of the dissociation of a disulfide biradical on a CdSe nanoparticle surface. J. Am. Chem. Soc. 2007, 129, 14150–14151. [Google Scholar] [CrossRef] [PubMed]
- Aryal, B.P.; Neupane, K.P.; Sandros, M.G.; Benson, D.E. Metallothioneins initiate semiconducting nanoparticle cellular toxicity. Small 2006, 2, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Holmes, J.D.; Richardson, D.J.; Saed, S.; Evans-Gowing, R.; Russell, D.A.; Sodeau, J.R. Cadmium-specific formation of metal sulfide 'Q-particles' by Klebsiella pneumoniae. Microbiology 1997, 143, 2521–2530. [Google Scholar] [CrossRef] [PubMed]
- Zangger, K.; Shen, G.; Oz, G.; Otvos, J.D.; Armitage, I.M. Oxidative dimerization in metallothionein is a result of intermolecular disulphide bonds between cysteines in the α-domain. Biochem. J. 2001, 359, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Krezel, A.; Maret, W. Different redox states of metallothionein/thionein in biological tissue. Biochem. J. 2007, 402, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Ogra, Y.; Suzuki, K.T. Nuclear trafficking of metallothionein: Possible mechanisms and current knowledge. Cell. Mol. Biol. 2000, 46, 357–365. [Google Scholar] [PubMed]
- Woo, E.S.; Dellapiazza, D.; Wang, A.S.; Lazo, J.S. Energy-dependent nuclear binding dictates metallothionein localization. J. Cell. Physiol. 2000, 182, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Ogra, Y.; Suzuki, K.T. Nuclear trafficking of metallothionein requires oxidation of a cytosolic partner. J. Cell. Physiol. 2005, 202, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Dowd, P.; Hershline, R.; Ham, S.W.; Naganathan, S. Vitamin K and energy transduction: A base strength amplification mechanism. Science 1995, 269, 1684–1691. [Google Scholar] [CrossRef] [PubMed]
- Linetsky, M.; LeGrand, R.D. Glutathionylation of lens proteins through the formation of thioether bond. Mol. Cell. Biochem. 2005, 272, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.S.; Lee, S.; Lee, G.T.; Woo, H.A.; Choi, E.J.; Rhee, S.G. Irreversible inactivation of glutathione peroxidase 1 and reversible inactivation of peroxiredoxin II by H2O2 in red blood cells. Antioxid. Redox Signal. 2010, 12, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.K.; Weaver, J.D.; Zhang, S.; Lei, X.G. Knockout of SOD1 promotes conversion of selenocysteine to dehydroalanine in murine hepatic GPX1 protein. Free Radic. Biol. Med. 2011, 51, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Kim, Y.; Kyung Seong, J.; Lee, K.J. Comprehensive identification of novel post-translational modifications in cellular peroxiredoxin 6. Proteomics 2012, 12, 1452–1462. [Google Scholar] [CrossRef] [PubMed]
- García-Sevillano, M.A.; García-Barrera, T.; Gómez-Ariza, J.L. Application of metallomic and metabolomic approaches in exposure experiments on laboratory mice for environmental metal toxicity assessment. Metallomics 2014, 6, 237–248. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubino, F.M. Toxicity of Glutathione-Binding Metals: A Review of Targets and Mechanisms. Toxics 2015, 3, 20-62. https://doi.org/10.3390/toxics3010020
Rubino FM. Toxicity of Glutathione-Binding Metals: A Review of Targets and Mechanisms. Toxics. 2015; 3(1):20-62. https://doi.org/10.3390/toxics3010020
Chicago/Turabian StyleRubino, Federico Maria. 2015. "Toxicity of Glutathione-Binding Metals: A Review of Targets and Mechanisms" Toxics 3, no. 1: 20-62. https://doi.org/10.3390/toxics3010020
APA StyleRubino, F. M. (2015). Toxicity of Glutathione-Binding Metals: A Review of Targets and Mechanisms. Toxics, 3(1), 20-62. https://doi.org/10.3390/toxics3010020