Abnormal Copper Homeostasis: Mechanisms and Roles in Neurodegeneration

Abstract
:1. Introduction
2. Metabolism of Copper
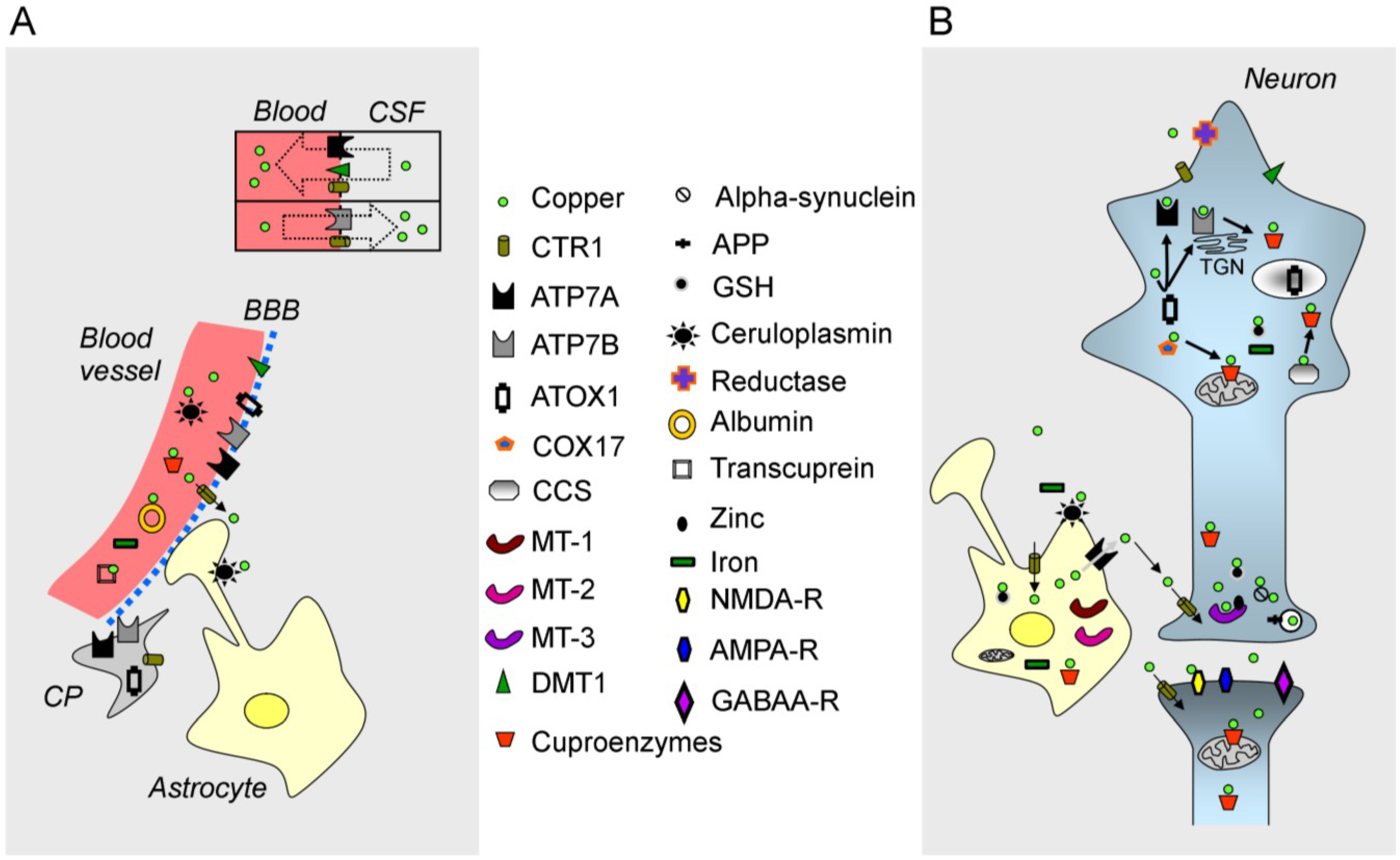
2.1. Free Copper
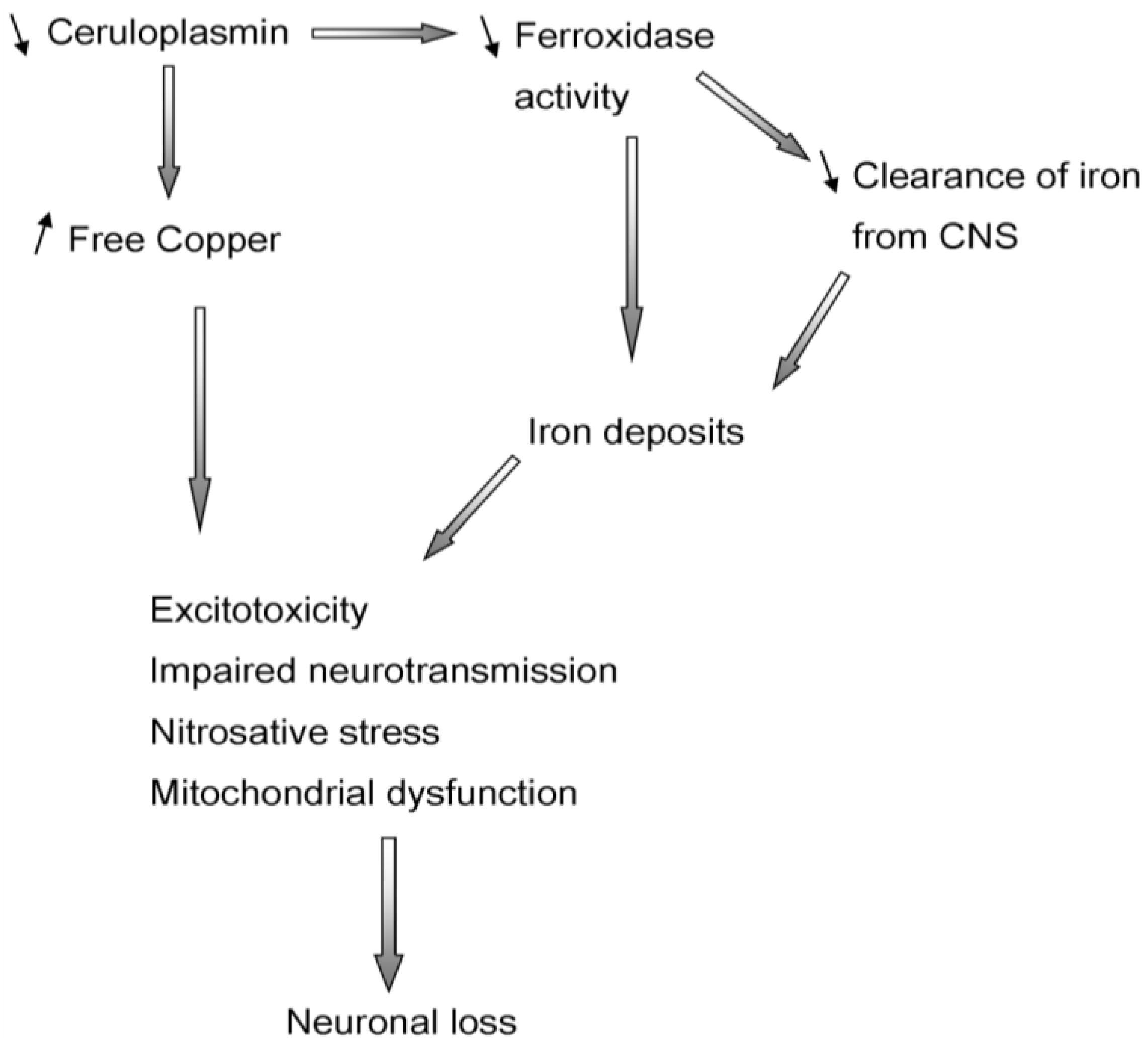
2.2. Ceruloplasmin
2.3. The ATPases ATP7A/ATP7B, CTR1 and Chaperones

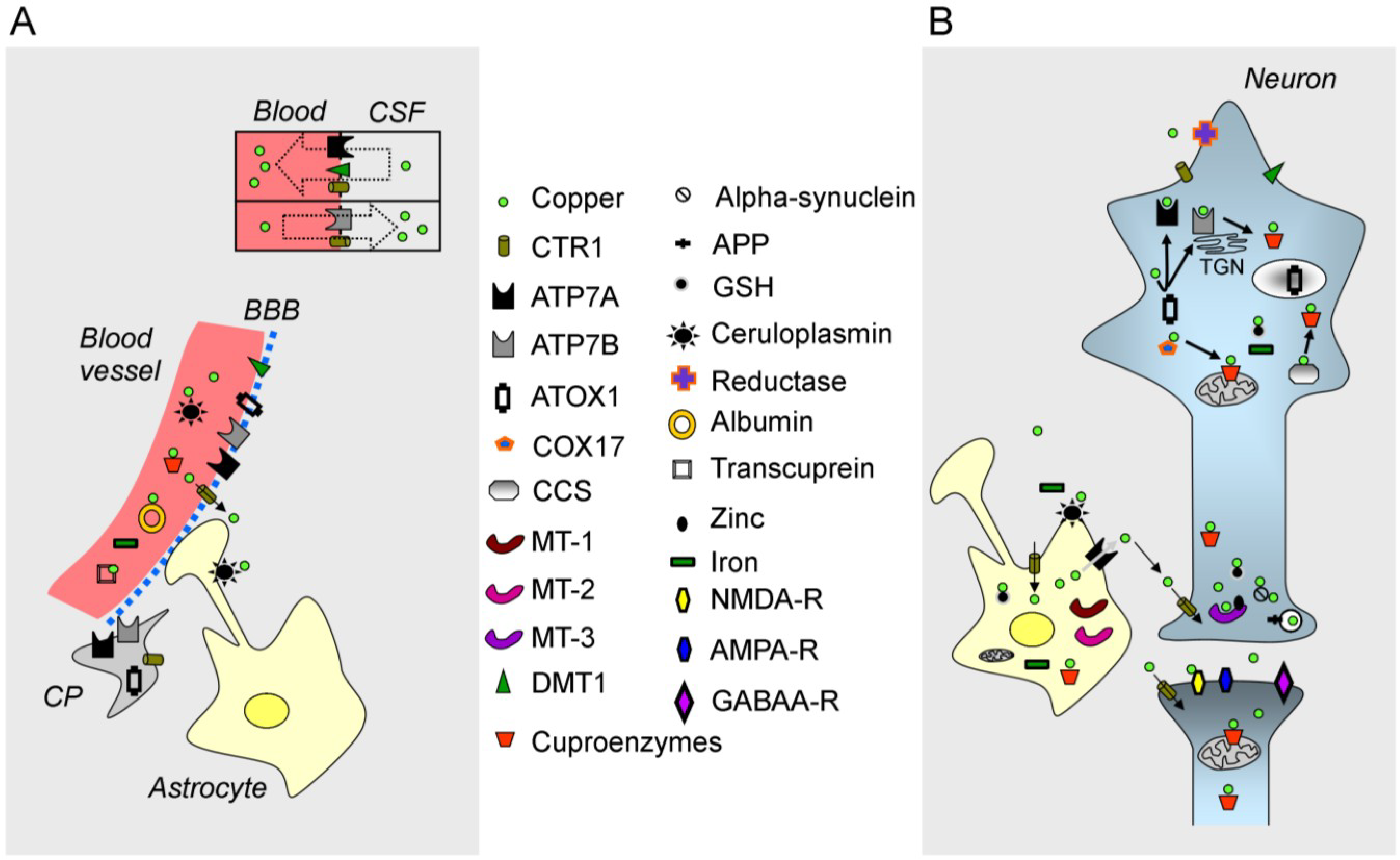{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Function | Physiological Roles |
|---|---|---|
| Superoxide dismutase 1 (SOD1) and 3 (SOD3) * | Converts superoxide to hydrogen peroxide | Anti-oxidative defense |
| Dopamine-beta-hydroxylase | Catecholamine production | Regulation of autonomic nervous system |
| Monoamine oxidase | Pigment and neurotransmitter metabolism | Oxidation of monoamines |
| Cytochrome C oxidase COX (COX, complex IV of the respiratory chain) | Converts molecular oxygen to water | Energy metabolism |
| Tyrosinase | Production of melanin; conversion of tyrosine to L-DOPA | Protection of skin |
| Catalase | Conversion of hydrogen peroxide to water and oxygen | Prevents oxidative-induced damage in the heart |
| Glutathione peroxidase | Converts hydroperoxide and hydrogen peroxide | Antioxidative defense |
| Hephaestin (homolog of ceruloplasmin) | Ferroxidase activity Involved in intestinal iron absorption | Control of iron efflux |
| Cartilage matrix glycoprotein (homolog of ceruloplasmin) | Ferroxidase activity Oxidase activity | Synthesis of the extracellular matrix |
| Lysyl oxidase | Cross-linking of elastin and oxygen | Stabilization of connective tissues |
2.4. Metallothioneins (MTs)
2.5. The Blood-Brain Barrier (BBB)
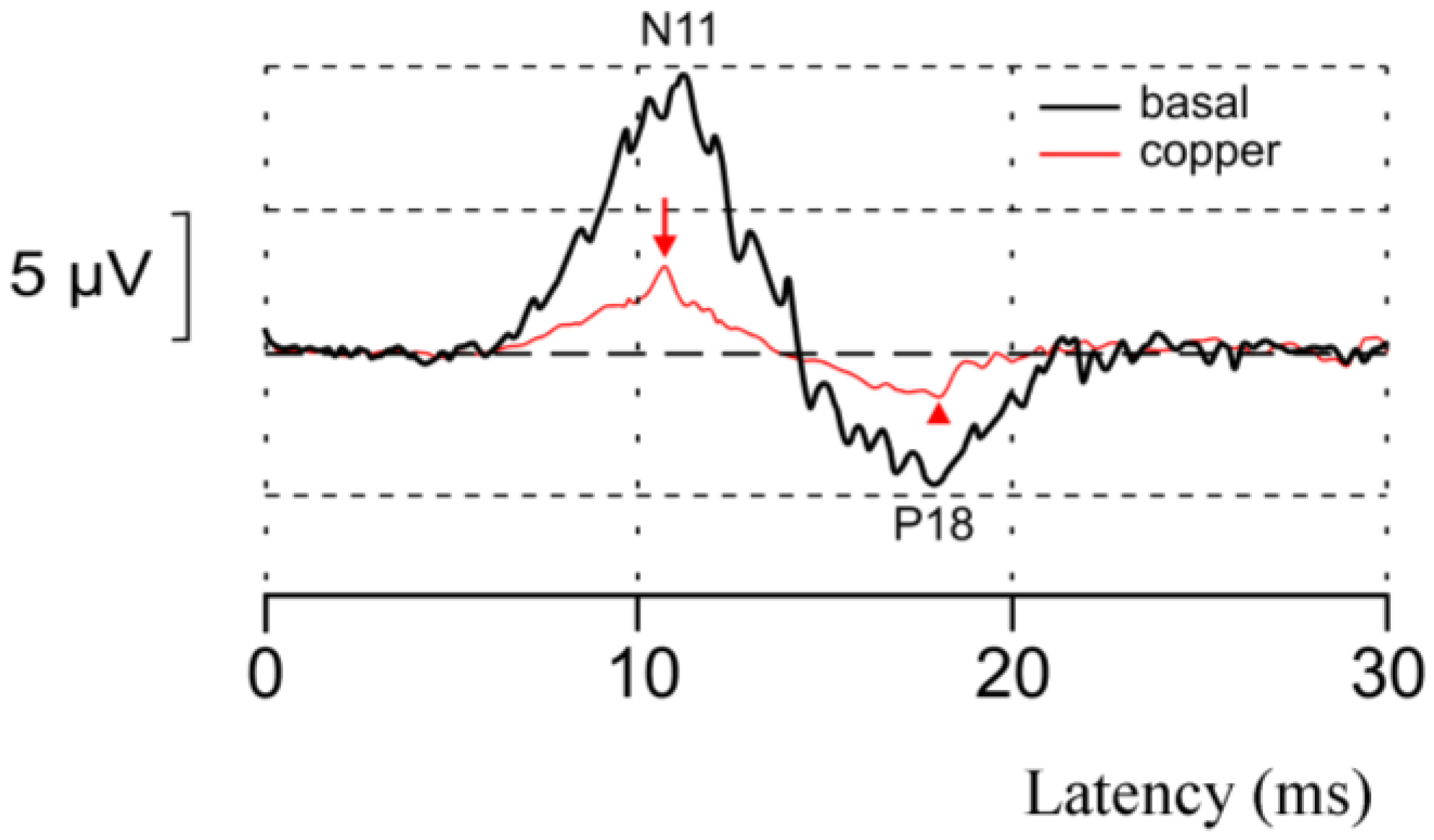
2.6. Copper and Synapses in the CNS
3. Copper Deficiency Syndromes (CDS): Menkes’ Disease (MD), Occipital Horn Syndrome, ATP7A-Related Isolated Distal Motor Neuropathy and Zinc-Induced Myeloneuropathy
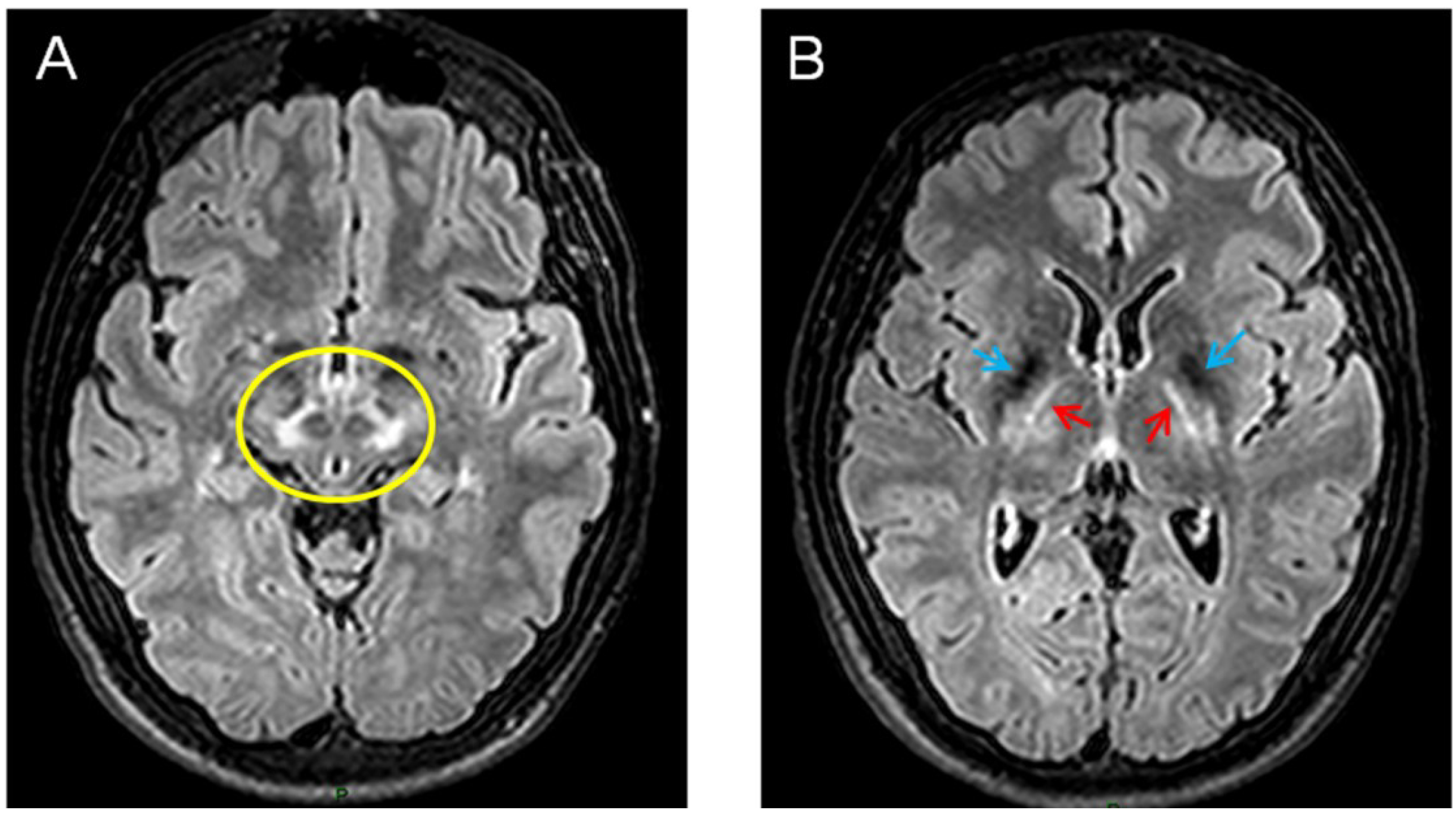
4. Wilson’s Disease (WD)
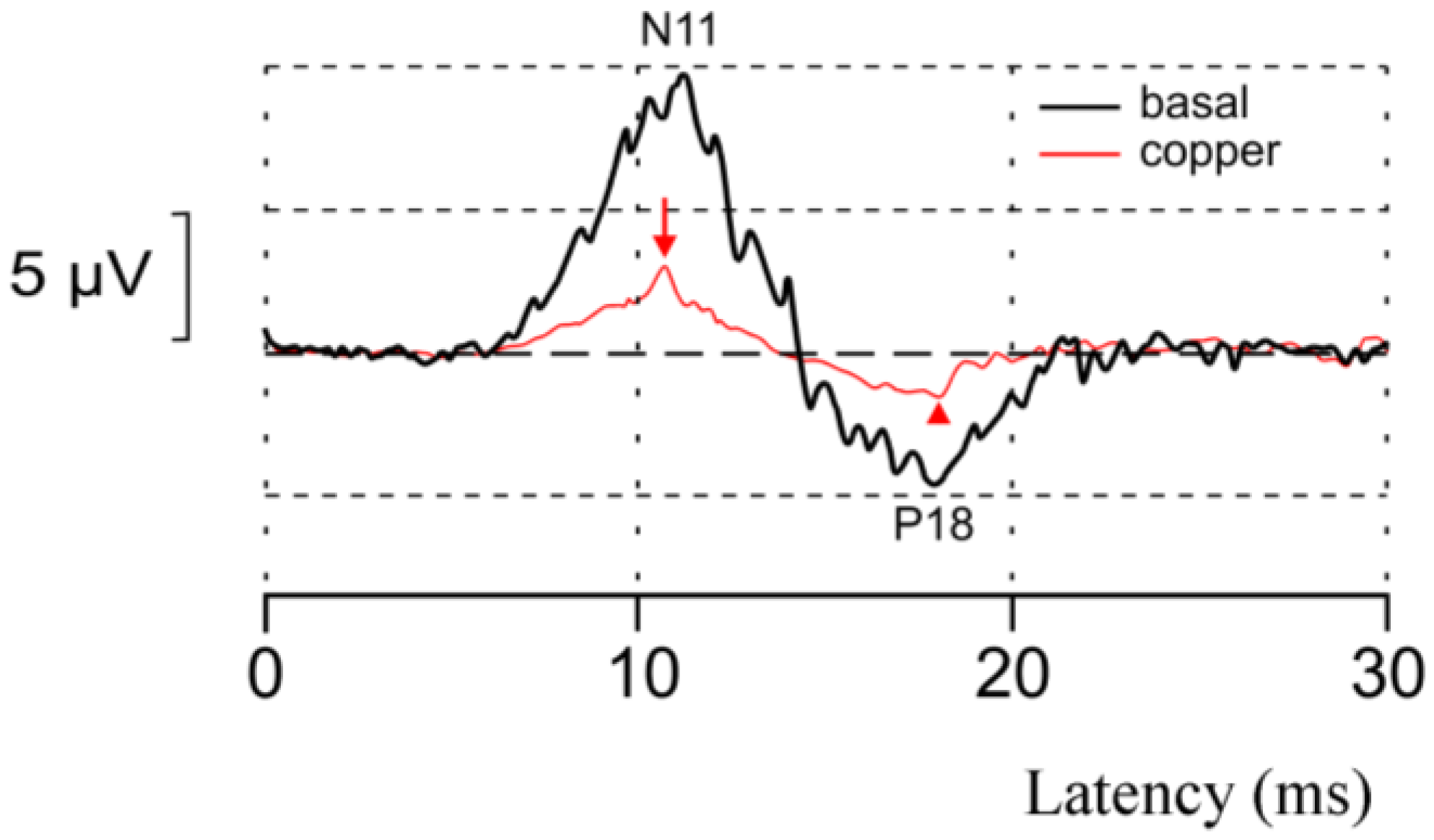
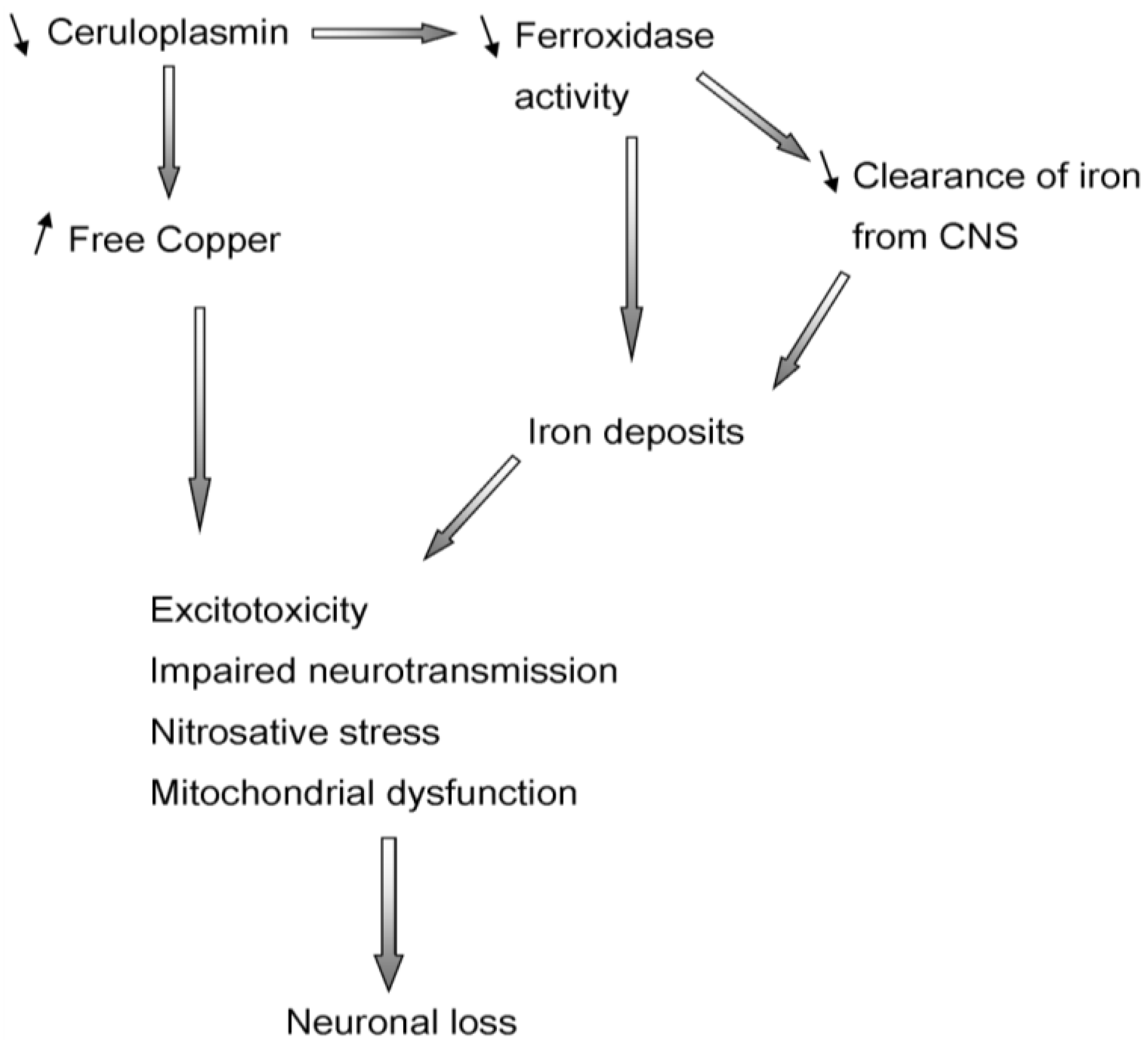
5. Aceruloplasminemia
6. Copper Toxicosis
7. Alzheimer’s Disease (AD)
| A. Alzheimer’s disease (AD) | B. Mechanisms of protein aggregation in Parkinson’s disease (PD) |
|---|---|
| Aggregation of β-Amyloid (Aβ) Peptide and Tau Proteins (Amyloid Cascade) * | Defect of the Ubiquitin-Proteasome System |
| Oxidative stress * | Overproduction of free radicals * |
| Inflammation * | Mitochondrial dysfunction * |
| Impaired energy metabolism * | Inflammation * |
| Impaired neurotransmission * | Impaired homeostasy of biometals * |
| Exposure to environmental pollutants * |
8. Parkinson’s Disease (PD)
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Squitti, R.; Polimanti, R. Copper phenotype in Alzheimer’s disease: Dissecting the pathway. Am. J. Neurodegener. Dis. 2013, 2, 46–56. [Google Scholar]
- Gaggelli, E.; Kozlowski, H.; Valensin, D.; Valensin, G. Copper homeostasis and neurodegenerative disorders (Alzheimer’s, prion, and Parkinson’s diseases and amyotrophic lateral sclerosis). Chem. Rev. 2006, 106, 1995–2044. [Google Scholar] [CrossRef]
- Brewer, G.J. Copper in medicine. Curr. Opin. Chem. Biol. 2003, 7, 207–212. [Google Scholar] [CrossRef]
- Eid, C.; Hémadi, M.; Ha-Duong, N.T.; El Hage Chahine, J.M. Iron uptake and transfer from ceruloplasmin to transferrin. Biochim. Biophys. Acta 2014, 1840, 1771–1781. [Google Scholar] [CrossRef]
- Scheiber, I.F.; Mercer, J.F.; Dringen, R. Metabolism and functions of copper in brain. Prog. Neurobiol. 2014, 116C, 33–57. [Google Scholar]
- Tapiero, H.; Townsend, D.M.; Tew, K.D. Trace elements in human physiology and pathology. Copper. Biomed. Pharmacother. 2003, 57, 386–398. [Google Scholar] [CrossRef]
- Chambers, A.; Krewski, D.; Birkett, N.; Plunkett, L.; Hertzberg, R.; Danzeisen, R.; Aggett, P.J.; Starr, T.B.; Baker, S.; Dourson, M.; et al. An exposure-response curve for copper excess and deficiency. J. Toxicol. Environ. Health B 2010, 13, 546–578. [Google Scholar] [CrossRef]
- Hordyjewska, A.; Popiołek, L.; Kocot, J. The many “faces” of copper in medicine and treatment. Biometals 2014. [Google Scholar] [CrossRef]
- Thiele, D.J. Integrating trace element metabolism from the cell to the whole organism. J. Nutr. 2003, 133, 1579S–1580S. [Google Scholar]
- Linder, M.C. Biochemistry of Copper; Plenum Press: New York, NY, USA, 1991. [Google Scholar]
- Davies, K.M.; Hare, D.J.; Cottam, V.; Chen, N.; Hilgers, L.; Halliday, G.; Mercer, J.F.; Double, K.L. Localization of copper and copper transporters in the human brain. Metallomics 2013, 5, 43–51. [Google Scholar] [CrossRef]
- Davies, K.M.; Bohic, S.; Carmona, A.; Ortega, R.; Cottam, V.; Hare, D.J.; Finberg, J.P.; Reyes, S.; Halliday, G.M.; Mercer, J.F.; et al. Copper pathology in vulnerable brain regions in Parkinson’s disease. Neurobiol. Aging 2014, 35, 858–866. [Google Scholar] [CrossRef]
- Hung, Y.H.; Bush, A.I.; La Fontaine, S. Links between copper and cholesterol in Alzheimer’s disease. Front. Physiol. 2013, 4, 111. [Google Scholar]
- Hopt, A.; Korte, S.; Fink, H.; Panne, U.; Niessner, R.; Jahn, R.; Kretzschmar, H.; Herms, J. Methods for studying synaptosomal copper release. J. Neurosci. Methods 2003, 128, 159–172. [Google Scholar] [CrossRef]
- Huffman, D.L.; O’Halloran, T.V. Function, structure, and mechanism of intracellular copper trafficking proteins. Annu. Rev. Biochem. 2001, 70, 677–701. [Google Scholar] [CrossRef]
- Rae, T.D.; Schmidt, P.J.; Pufahl, R.A.; Culotta, V.C.; O’Halloran, T.V. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science 1999, 284, 805–808. [Google Scholar] [CrossRef]
- Hellman, N.E.; Gitlin, J.D. Ceruloplasmin metabolism and function. Annu. Rev. Nutr. 2002, 22, 439–458. [Google Scholar] [CrossRef]
- Vashchenko, G.; MacGillivray, R.T. Multi-copper oxidases and human iron metabolism. Nutrients 2013, 5, 2289–2313. [Google Scholar] [CrossRef]
- Beaumont, C. Molecular mechanisms of iron homeostasis. Med. Sci. 2004, 20, 68–72. [Google Scholar]
- Kono, S. Aceruloplasminemia: An update. Int. Rev. Neurobiol. 2013, 110, 125–151. [Google Scholar] [CrossRef]
- Hare, D.; Ayton, S.; Bush, A.; Lei, P. A delicate balance: Iron metabolism and diseases of the brain. Front. Aging Neurosci. 2013, 5, 34. [Google Scholar]
- Vulpe, C.; Levinson, B.; Whitney, S.; Packman, S.; Gitschier, J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat. Genet. 1993, 3, 7–13. [Google Scholar] [CrossRef]
- Chelly, J.; Tümer, Z.; Tønnesen, T.; Petterson, A.; Ishikawa-Brush, Y.; Tommerup, N.; Horn, N.; Monaco, A.P. Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein. Nat. Genet. 1993, 3, 14–19. [Google Scholar] [CrossRef]
- Qian, Y.; Tiffany-Castiglioni, E.; Welsh, J.; Harris, E.D. Copper efflux from murine microvascular cells requires expression of the menkes disease Cu-ATPase. J. Nutr. 1998, 128, 1276–1282. [Google Scholar]
- Kennerson, M.L.; Nicholson, G.A.; Kaler, S.G.; Kowalski, B.; Mercer, J.F.; Tang, J.; Llanos, R.M.; Chu, S.; Takata, R.I.; Speck-Martins, C.E.; et al. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am. J. Hum. Genet. 2010, 86, 343–352. [Google Scholar] [CrossRef]
- Petris, M.J.; Mercer, J.F.; Culvenor, J.G.; Lockhart, P.; Gleeson, P.A.; Camakaris, J. Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: A novel mechanism of regulated trafficking. EMBO J. 1996, 15, 6084–6095. [Google Scholar]
- Wee, N.K.; Weinstein, D.C.; Fraser, S.T.; Assinder, S.J. The mammalian copper transporters CTR1 and CTR2 and their roles in development and disease. Int. J. Biochem. Cell Biol. 2013, 45, 960–963. [Google Scholar] [CrossRef]
- Choi, B.S.; Zheng, W. Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res. 2009, 1248, 14–21. [Google Scholar]
- Van den Berghe, P.V.; Klomp, L.W. Posttranslational regulation of copper transporters. J. Biol. Inorg. Chem. 2010, 15, 37–46. [Google Scholar] [CrossRef]
- Larson, C.A.; Adams, P.L.; Blair, B.G.; Safaei, R.; Howell, S.B. The role of the methionines and histidines in the transmembrane domain of mammalian copper transporter 1 in the cellular accumulation of cisplatin. Mol. Pharmacol. 2010, 78, 333–339. [Google Scholar] [CrossRef]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar]
- Bertinato, J.; Swist, E.; Plouffe, L.J.; Brooks, S.P.; L’abbé, M.R. Ctr2 is partially localized to the plasma membrane and stimulates copper uptake in COS-7 cells. Biochem. J. 2008, 409, 731–740. [Google Scholar] [CrossRef]
- Zheng, G.; Chen, J.; Zheng, W. Relative contribution of CTR1 and DMT1 in copper transport by the blood-CSF barrier: Implication in manganese-induced neurotoxicity. Toxicol. Appl. Pharmacol. 2012, 260, 285–293. [Google Scholar] [CrossRef]
- Lee, J.; Prohaska, J.R.; Thiele, D.J. Essential role for mammalian copper transporter Ctr1 in copper homeostasis and embryonic development. Proc. Natl. Acad. Sci. USA 2001, 98, 6842–6847. [Google Scholar] [CrossRef]
- Naeve, G.S.; Vana, A.M.; Eggold, J.R.; Kelner, G.S.; Maki, R.; Desouza, E.B.; Foster, A.C. Expression profile of the copper homeostasis gene, rAtox1, in the rat brain. Neuroscience 1999, 93, 1179–1187. [Google Scholar] [CrossRef]
- Penkowa, M.; Tio, L.; Giralt, M.; Quintana, A.; Molinero, A.; Atrian, S.; Vasák, M.; Hidalgo, J. Specificity and divergence in the neurobiologic effects of different metallothioneins after brain injury. J. Neurosci. Res. 2006, 83, 974–984. [Google Scholar] [CrossRef]
- Michael, G.J.; Esmailzadeh, S.; Moran, L.B.; Christian, L.; Pearce, R.K.; Graeber, M.B. Up-regulation of metallothionein gene expression in parkinsonian astrocytes. Neurogenetics 2011, 12, 295–305. [Google Scholar] [CrossRef]
- Haywood, S.; Vaillant, C. Overexpression of copper transporter CTR1 in the brain barrier of North Ronaldsay sheep: Implications for the study of neurodegenerative diseases. J. Comp. Pathol. 2014, 150, 216–224. [Google Scholar] [CrossRef]
- Squitti, R. Copper dysfunction in Alzheimer’s disease: From meta-analysis of biochemical studies to new insight into genetics. J. Trace Elem. Med. Biol. 2012, 26, 93–96. [Google Scholar]
- Gaier, E.D.; Eipper, B.A.; Mains, R.E. Copper signaling in the mammalian nervous system: Synaptic effects. J. Neurosci. Res. 2013, 91, 2–19. [Google Scholar]
- Schlief, M.L.; Craig, A.M.; Gitlin, J.D. NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J. Neurosci. 2005, 25, 239–246. [Google Scholar] [CrossRef]
- Vlachová, V.; Zemková, H.; Vyklický, L., Jr. Copper modulation of NMDA responses in mouse and rat cultured hippocampal neurons. Eur. J. Neurosci. 1996, 8, 2257–2264. [Google Scholar] [CrossRef]
- Schlief, M.L.; Gitlin, J.D. Copper homeostasis in the CNS: A novel link between the NMDA receptor and copper homeostasis in the hippocampus. Mol. Neurobiol. 2006, 33, 81–90. [Google Scholar] [CrossRef]
- Weiser, T.; Wienrich, M. The effects of copper ions on glutamate receptors in cultured rat cortical neurons. Brain Res. 1996, 742, 211–218. [Google Scholar] [CrossRef]
- Kardos, J.; Kovács, I.; Hajós, F.; Kálmán, M.; Simonyi, M. Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci. Lett. 1989, 103, 139–144. [Google Scholar] [CrossRef]
- McGee, T.P.; Houston, C.M.; Brickley, S.G. Copper block of extrasynaptic GABAA receptors in the mature cerebellum and striatum. J. Neurosci. 2013, 33, 13431–13435. [Google Scholar] [CrossRef]
- Kaler, S.G. Translational research investigations on ATP7A: An important human copper ATPase. Ann. N. Y. Acad. Sci. 2014, 1314, 64–68. [Google Scholar] [CrossRef]
- Shibata, N.; Hirano, A.; Kobayashi, M.; Umahara, T.; Kawanami, T.; Asayama, K. Cerebellar superoxide dismutase expression in Menkes’ kinky hair disease: An immunohistochemical investigation. Acta Neuropathol. 1995, 90, 198–202. [Google Scholar] [CrossRef]
- Tümer, Z. An overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndrome. Hum. Mutat. 2013, 34, 417–429. [Google Scholar] [CrossRef]
- Møller, L.B.; Lenartowicz, M.; Zabot, M.T.; Josiane, A.; Burglen, L.; Bennett, C.; Riconda, D.; Fisher, R.; Janssens, S.; Mohammed, S.; et al. Clinical expression of Menkes disease in females with normal karyotype. Orphanet J. Rare Dis. 2012, 7, 6. [Google Scholar] [CrossRef]
- Haddad, M.R.; Choi, E.Y.; Kaler, S.G. AAVrh10 ATP7A administration to the cerebrospinal fluid, in combination with subcutaneous copper, normalizes neurological outcomes in a mouse model of Menkes disease. Mol. Ther. 2014, 22 (Suppl.). in press. [Google Scholar]
- Kaler, S.G. ATP7A-related copper transport diseases-emerging concepts and future trends. Nat. Rev. Neurol. 2011, 7, 15–29. [Google Scholar] [CrossRef]
- Yi, L.; Kaler, S. ATP7A trafficking and mechanisms underlying the distal motor neuropathy induced by mutations in ATP7A. Ann. N.Y. Acad. Sci. 2014, 1314, 49–54. [Google Scholar] [CrossRef]
- Schleper, B.; Stuerenburg, H.J. Copper deficiency-associated myelopathy in a 46-year-old woman. J. Neurol. 2001, 248, 705–706. [Google Scholar] [CrossRef]
- Bennetts, H.W.; Chapman, F.E. Copper deficiency in sheep in Western Australia: A preliminary account of the aetiology of enzootic ataxia of lambs and anaemia of ewes. Aust. Vet. J. 1937, 13, 138–149. [Google Scholar] [CrossRef]
- Lanska, D.J.; Remler, B. Myelopathy among zinc-smelter workers in Upper Silesia during the late 19th century. Neurology 2014, 82, 1175–1179. [Google Scholar] [CrossRef]
- Gabreyes, A.A.; Abbasi, H.N.; Forbes, K.P.; McQuaker, G.; Duncan, A.; Morrison, I. Hypocupremia associated cytopenia and myelopathy: A national retrospective review. Eur. J. Haematol. 2013, 90, 1–9. [Google Scholar]
- Choi, E.H.; Strum, W. Hypocupremia-related myeloneuropathy following gastrojejunal bypass surgery. Ann. Nutr. MeTable 2010, 57, 190–192. [Google Scholar] [CrossRef]
- Jaiser, S.R.; Winston, G.P. Copper deficiency myelopathy. J. Neurol. 2010, 257, 869–881. [Google Scholar] [CrossRef]
- Lorincz, M.T. Neurologic Wilson’s disease. Ann. N. Y. Acad. Sci. 2010, 1184, 173–187. [Google Scholar] [CrossRef]
- Faa, G.; Lisci, M.; Caria, M.P.; Ambu, R.; Sciot, R.; Nurchi, V.M.; Silvagni, R.; Diaz, A.; Crisponi, G. Brain copper, iron, magnesium, zinc, calcium, sulfur and phosphorus storage in Wilson’s disease. J. Trace Elem. Med. Biol. 2001, 15, 155–160. [Google Scholar] [CrossRef]
- Marmolino, D.; Manto, M. Pregabalin antagonizes copper-induced toxicity in the brain: In vitro and in vivo studies. Neurosignals 2010, 18, 210–222. [Google Scholar] [CrossRef]
- Sokol, R.J.; Devereaux, M.W.; O’Brien, K.; Khandwala, R.A.; Loehr, J.P. Abnormal hepatic mitochondrial respiration and cytochrome C oxidase activity in rats with long-term copper overload. Gastroenterology 1993, 105, 178–187. [Google Scholar]
- Sheline, C.T.; Choi, D.W. Cu2+ toxicity inhibition of mitochondrial dehydrogenases in vitro and in vivo. Ann. Neurol. 2004, 55, 645–653. [Google Scholar] [CrossRef]
- White, A.R.; Multhaup, G.; Maher, F.; Bellingham, S.; Camakaris, J.; Zheng, H.; Bush, A.I.; Beyreuther, K.; Masters, C.L.; Cappai, R. The Alzheimer’s disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. J. Neurosci. 1999, 19, 9170–9179. [Google Scholar]
- Bertrand, E.; Lewandowska, E.; Szpak, G.M.; Hoogenraad, T.; Blaauwgers, H.G.; Czlonkowska, A.; Dymecki, J. Neuropathological analysis of pathological forms of astroglia in Wilson’s disease. Folia Neuropathol. 2001, 39, 73–79. [Google Scholar]
- Kono, S. Aceruloplasminemia. Curr. Drug Targets 2012, 13, 1190–1199. [Google Scholar] [CrossRef]
- Pan, P.L.; Tang, H.H.; Chen, Q.; Song, W.; Shang, H.F. Desferrioxamine treatment of aceruloplasminemia: Long-term follow-up. Mov. Disord. 2011, 26, 2142–2144. [Google Scholar] [CrossRef]
- Miyajima, H. Aceruloplasminemia. In GeneReviews; Pagon, R.A., Adam, M.P., Ardinger, H.H., Bird, T.D., Dolan, C.R., Fong, C.T., Smith, R.J.H., Stephens, K., Eds.; University of Washington: Seattle, WA, USA, 2003; pp. 1993–2014. [Google Scholar]
- Puig, S.; Thiele, D. Molecular mechanism of copper uptake and distribution. Curr. Opin. Chem. Biol. 2002, 6, 171–180. [Google Scholar] [CrossRef]
- Araya, M.; Olivares, M.; Pizarro, F.; Méndez, M.A.; González, M.; Uauy, R. Supplementing copper at the upper level of the adult dietary recommended intake induces detectable but transient changes in healthy adults. J. Nutr. 2005, 135, 2367–2371. [Google Scholar]
- Schramel, P.; Müller-Höcker, J.; Meyer, U.; Weiss, M.; Eife, R. Nutritional copper intoxication in three German infants with severe liver cell damage (features of Indian childhood cirrhosis). J. Trace Elem. Electrolytes Health Dis. 1988, 2, 85–89. [Google Scholar]
- Baker, A.; Gormally, S.; Saxena, R.; Baldwin, D.; Drumm, B.; Bonham, J.; Portmann, B.; Mowat, A.P. Copper-associated liver disease in childhood. J. Hepatol. 1995, 23, 538–543. [Google Scholar] [CrossRef]
- Ozcelik, D.; Uzun, H. Copper intoxication, antioxidant defenses and oxidative damage in rat brain. Biol. Trace Elem. Res. 2009, 127, 45–52. [Google Scholar] [CrossRef]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef]
- Bettens, K.; Sleegers, K.; van Broeckhoven, C. Genetic insights in Alzheimer’s disease. Lancet Neurol. 2013, 12, 92–104. [Google Scholar] [CrossRef]
- Noda, Y.; Asada, M.; Kubota, M.; Maesako, M.; Watanabe, K.; Uemura, M.; Kihara, T.; Shimohama, S.; Takahashi, R.; Kinoshita, A.; et al. Copper enhances APP dimerization and promotes Ab production. Neurosci. Lett. 2013, 547, 10–15. [Google Scholar] [CrossRef]
- Arnal, N.; Morel, G.R.; de Alaniz, M.J.T.; Castillo, O.; Marra, C.A. Role of copper and cholesterol association in the neurodegenerative process. Int. J. Alzheimer’s Dis. 2013, 10, 1–15. [Google Scholar]
- Ventriglia, M.; Bucossi, S.; Panetta, V.; Squitti, R. Copper in Alzheimer’s disease: A meta-analysis of serum, plasma, and cerebrospinal fluid studies. J. Alzheimer’s Dis. 2012, 30, 981–984. [Google Scholar]
- Squitti, R.; Pasqualetti, P.; dal Forno, G.; Moffa, F.; Cassetta, E.; Lupoi, D.; Vernieri, F.; Rossi, L.; Baldassini, M.; Rossini, P.M. Excess of serum copper not related to ceruloplasmin in Alzheimer disease. Neurology 2005, 64, 1040–1046. [Google Scholar] [CrossRef]
- James, S.A.; Volitakis, I.; Adlard, P.A.; Duce, J.A.; Masters, C.L.; Cherny, R.A.; Bush, A.I. Elevated labile Cu is associated with oxidative pathology in Alzheimer disease. Free Radic. Biol. Med. 2012, 52, 298–302. [Google Scholar] [CrossRef]
- Brewer, G.J. Copper toxicity in Alzheimer’s disease: Cognitive loss from ingestion of inorganic copper. J. Trace Elem. Med. Biol. 2012, 26, 89–92. [Google Scholar] [CrossRef]
- Campbell, A. The role of aluminum and copper on neuroinflammation and Alzheimer’s disease. J. Alzheimers Dis. 2006, 10, 165–172. [Google Scholar]
- Becaria, A.; Lahiri, D.K.; Bondy, S.C.; Chen, D.; Hamadeh, A.; Li, H.; Taylor, R.; Campbell, A. Aluminum and copper in drinking water enhance inflammatory or oxidative events specifically in the brain. J. Neuroimmunol. 2006, 176, 16–23. [Google Scholar] [CrossRef]
- Spisni, E.; Valerii, M.C.; Manerba, M.; Strillacci, A.; Polazzi, E.; Mattia, T.; Griffoni, C.; Tomasi, V. Effect of copper on extracellular levels of key pro-inflammatory molecules in hypothalamic GN11 and primary neurons. Neurotoxicology 2009, 30, 605–612. [Google Scholar] [CrossRef]
- Crouch, P.J.; Barnham, K.J. Therapeutic redistribution of metal ions to treat Alzheimer’s disease. Acc. Chem. Res. 2012, 45, 1604–1611. [Google Scholar] [CrossRef]
- Ceccom, J.; Cosledan, F.; Halley, H.; Frances, B.; Lassalle, J.M.; Meunier, B. Copper chelator induced efficient episodic memory recovery in a non-transgenic Alzheimer’s mouse model. PLoS One 2012, 7, e43105. [Google Scholar]
- Rasia, R.M.; Bertoncini, C.W.; Marsh, D.; Hoyer, W.; Cherny, D.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernández, C.O. Insights into the bioinorganic chemistry of Parkinson’s disease. Structural characterization of copper(II) binding to alpha-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 4294–4299. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Riederer, P.; Sofic, E.; Rausch, W.D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J. Neurochem. 1989, 52, 515–520. [Google Scholar] [CrossRef]
- Berg, D.; Siefker, C.; Becker, G. Echogenicity of the substantia nigra in Parkinson’s disease and its relation to clinical findings. J. Neurol. 2001, 248, 684–689. [Google Scholar] [CrossRef]
- Martínez-Hernández, R.; Montes, S.; Higuera-Calleja, J.; Yescas, P.; Boll, M.C.; Diaz-Ruiz, A.; Rios, C. Plasma ceruloplasmin ferroxidase activity correlates with the nigral sonographic area in Parkinson’s disease patients: A pilot study. Neurochem. Res. 2011, 36, 2111–2115. [Google Scholar] [CrossRef]
- Bharucha, K.J.; Friedman, J.K.; Vincent, A.S.; Ross, E.D. Lower serum ceruloplasmin levels correlate with younger age of onset in Parkinson’s disease. J. Neurol. 2008, 255, 1957–1962. [Google Scholar] [CrossRef]
- Boll, M.C.; Alcaraz-Zubeldia, M.; Montes, S.; Rios, C. Free copper, ferroxidase and SOD1 activities, lipid peroxidation and NO(x) content in the CSF. A different marker profile in four neurodegenerative diseases. Neurochem. Res. 2008, 33, 1717–1723. [Google Scholar] [CrossRef]
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Kortsha, G.X.; Brown, G.G.; Richardson, R.J. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson’s disease. Neurotoxicology 1999, 20, 239–247. [Google Scholar]
- Ayton, S.; Lei, P.; Duce, J.A.; Wong, B.X.; Sedjahtera, A.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I. Ceruloplasmin dysfunction and therapeutic potential for Parkinson disease. Ann. Neurol. 2013, 73, 554–559. [Google Scholar] [CrossRef]
- Spencer, W.A.; Jeyabalan, J.; Kichambre, S.; Gupta, R.C. Oxidatively generated DNA damage after Cu(II) catalysis of dopamine and related catecholamine neurotransmitters and neurotoxins: Role of reactive oxygen species. Free Radic. Biol. Med. 2011, 50, 139–147. [Google Scholar]
- Yu, W.R.; Jiang, H.; Wang, J.; Xie, J.X. Copper (Cu2+) induces degeneration of dopaminergic neurons in the nigrostriatal system of rats. Neurosci. Bull. 2008, 24, 73–78. [Google Scholar] [CrossRef]
- Przedborski, S.; Kostic, V.; Jackson-Lewis, V.; Naini, A.B.; Simonetti, S.; Fahn, S.; Carlson, E.; Epstein, C.J.; Cadet, J.L. Transgenic mice with increased Cu/Zn-superoxide dismutase activity are resistant to N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. J. Neurosci. 1992, 12, 1658–1667. [Google Scholar]
- Ebadi, M.; Sharma, S. Metallothioneins 1 and 2 attenuate peroxynitrite-induced oxidative stress in Parkinson disease. Exp. Biol. Med. 2006, 231, 1576–1583. [Google Scholar]
- Ayton, S.; Zhang, M.; Roberts, B.R.; Lam, L.Q.; Lind, M.; McLean, C.; Bush, A.I.; Frugier, T.; Crack, P.J.; Duce, J.A. Ceruloplasmin and β-amyloid precursor protein confer neuroprotection in traumatic brain injury and lower neuronal iron. Free Radic. Biol. Med. 2014, 69, 331–337. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Manto, M. Abnormal Copper Homeostasis: Mechanisms and Roles in Neurodegeneration. Toxics 2014, 2, 327-345. https://doi.org/10.3390/toxics2020327
Manto M. Abnormal Copper Homeostasis: Mechanisms and Roles in Neurodegeneration. Toxics. 2014; 2(2):327-345. https://doi.org/10.3390/toxics2020327
Chicago/Turabian StyleManto, Mario. 2014. "Abnormal Copper Homeostasis: Mechanisms and Roles in Neurodegeneration" Toxics 2, no. 2: 327-345. https://doi.org/10.3390/toxics2020327
APA StyleManto, M. (2014). Abnormal Copper Homeostasis: Mechanisms and Roles in Neurodegeneration. Toxics, 2(2), 327-345. https://doi.org/10.3390/toxics2020327