
Application of Multi-Omics Techniques in Aquatic Ecotoxicology: A Review




Abstract
1. Introduction
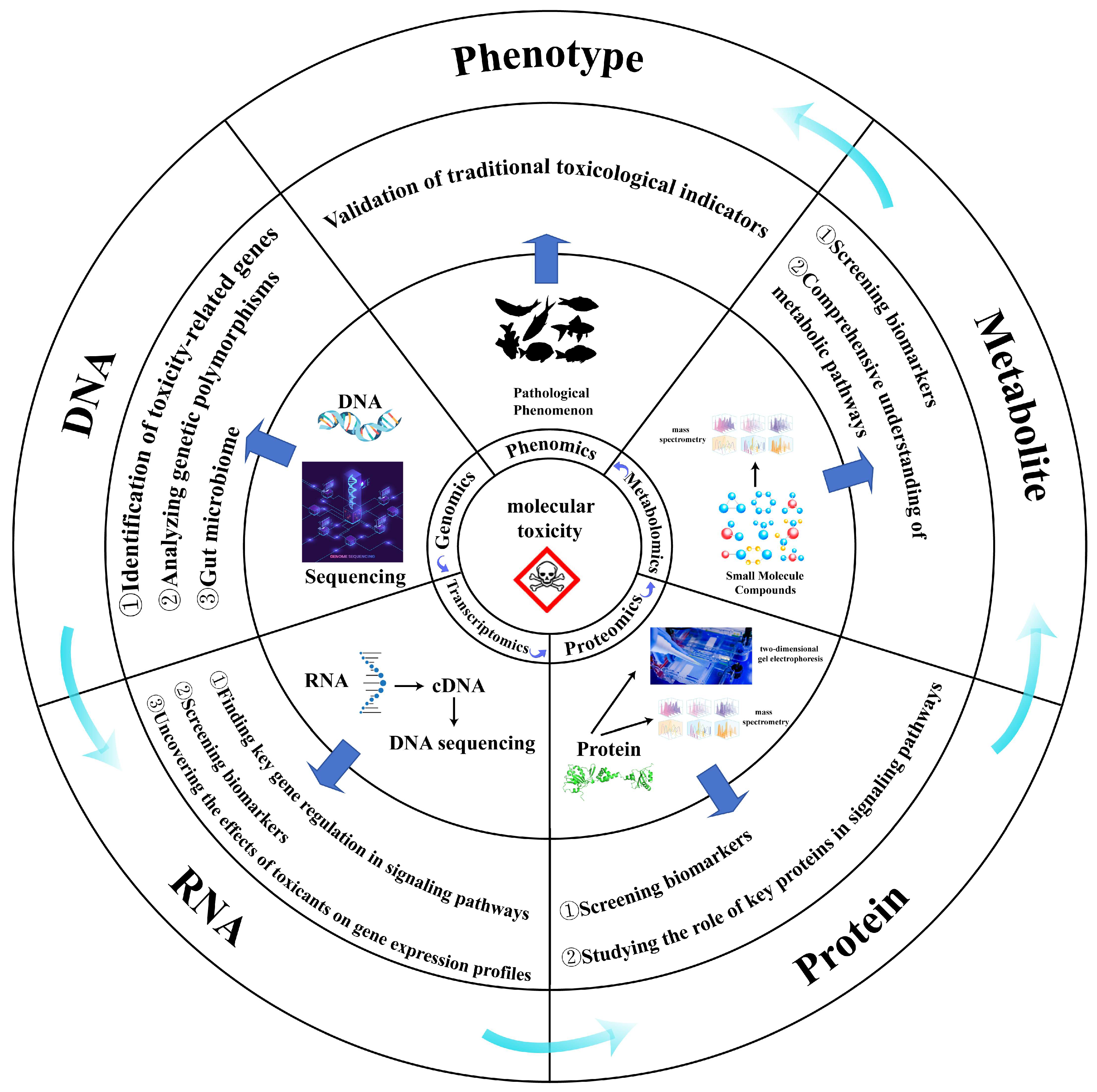
2. Common Single-Omics Method
2.1. Genomics
2.2. Transcriptomics
2.3. Proteomics
2.4. Metabolomics
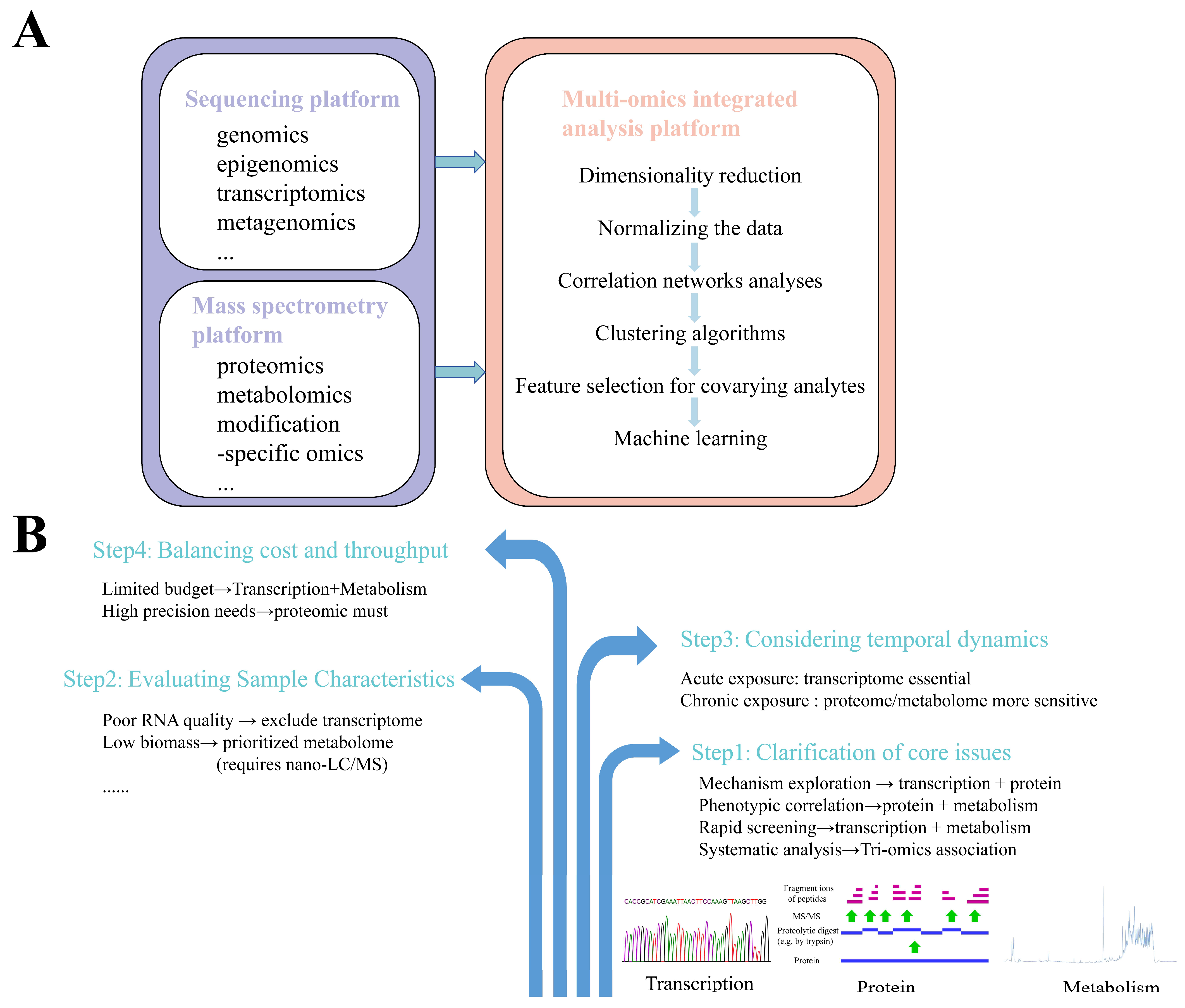
3. Multi-Omics Integrated Analysis Platform

{kind=link}
{kind=link}
{kind=link}
| Tool | Core Function | Accessibility | Aquatic Model Suitability | Reference |
|---|---|---|---|---|
| Data-Driven Approaches | ||||
| mixOmics | Multi-omics integration and dimensionality reduction | R package | Supports transcriptome–metabolome integration | [40] |
| MOFA | Multi-omics factor analysis | Python/R | Supports time-series exposure experiments | [41] |
| OmicsNotebook | Cloud-based multi-omics integration | Web platform | Dedicated aquatic toxicology module | NA |
| SNF | Clustering and classification Similarity network fusion | R package | Supports transcriptome–metabolome integration | [42] |
| Knowledge-Based Approaches | ||||
| clusterProfiler | KEGG/GO enrichment analysis | R package | Supports model organism pathways | [43] |
| ReactomePA | Reactome pathway analysis | R package | 30% improved coverage of fish signaling pathways | [44] |
| Cytoscape | Biological network visualization | Desktop | Supports microbe–host interaction networks | [45] |
| Pathview | Multi-omics pathway mapping | R/Bioconductor | Supports model fish and non-model species | [46] |
| Online Platforms | ||||
| Galaxy | Aquatic toxicology workflows | Cloud | Fish transcriptomics | [47] |
| OmicsNet | Multi-omics network analysis | Web platform | Supports aquatic toxicity biomarker networks | [48] |
| KNIME | Graphical workflow design | Desktop | Requires custom aquatic biology plugins | NA |
4. Application of Multi-Omics in Ecotoxicological Studies of Aquatic Organisms
4.1. Combination of Proteomics and Metabolomics Analysis
4.2. Combination of Gut Microbiome and Other Omics Analysis
4.3. Combination of Transcriptomics and Metabolomics Analysis
4.4. Combination of Transcriptomics and Proteomics Analysis
4.5. Combination of Transcriptomics, Proteomics, and Metabolomics Analysis
5. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
| Sequencing Platform | Reads | Accuracy |
|---|---|---|
| Single-Molecule Real-Time (SMRT) | 10~25 kp | 97% |
| Single-Molecule Nanopore | 1000~4500 bp | 87% (Read Mode), 99% (Accuracy Mode) |
| True Single Molecular Sequencing (tSMS) | 100 kb | 96% |
| Fluorescence Resonance Energy Transfer (FRET) | >1500 bp | >99% |
| Technology | Principle | Aquatic Application Advantages |
|---|---|---|
| Bulk RNA-Seq | ||
| Illumina Short-Read | NGS of fragmented RNA | Gold standard for DEG analysis High sensitivity for low-abundance transcripts in fish gills |
| PacBio Iso-Seq | Long-read full-length cDNA | Resolves complex splice variants in crustaceans No assembly needed |
| Oxford Nanopore | Direct RNA sequencing | Real-time mRNA surveillance Detects RNA modifications in live algae |
| Single-Cell | ||
| 10× Genomics | Barcoded droplet partitioning | Cell-type resolution in zebrafish embryos Immune cell profiling |
| Smart-seq2 | Full-length scRNA-seq | High sensitivity for rare cell types |
| Spatial | ||
| Visium (10×) | Spatial barcoding on slides | Maps pollutant gradients in fish liver lobules Correlates histopathology with gene expression |
| MERFISH | Multiplexed FISH imaging | Single-molecule resolution in biofilm communities Quantifies horizontal gene transfer |
| Technology | Advantages | Disadvantages |
|---|---|---|
| Mass Spectrometry | ||
| MALDI-TOF | Rapid analysis, tolerant to salt/washers | Limited resolution (<50,000 m/z), poor detection of low-abundance proteins |
| Orbitrap | Ultra-high resolution (>150,000 m/z), low detection limit | High instrument cost, strict desalination required for high-salt samples |
| Separation Techniques | ||
| Gel-based (2D-PAGE) | Intuitive separation of post-translationally modified proteins | Low recovery for hydrophobic/membrane proteins (<30%), fails in high-salt conditions |
| Non-gel-based (LC-MS) | Enhanced peak capacity, improved detection of low-abundance proteins | Surface-active agents may interfere with chromatographic separation |
| Quantitative Methods | ||
| Labeled (TMT) | 16-channel simultaneous quantification, relative error <10% | High reagent cost (USD 500/sample), limited channel number |
| Label-free (LFQ) | Unlimited sample throughput, cost reduction (60%) | Poor reproducibility (CV > 20%), high missing value rate (>15%) |
References
- Zhou, H.; Wu, H.; Liao, C.; Diao, X.; Zhen, J.; Chen, L.; Xue, Q. Toxicology mechanism of the persistent organic pollutants (POPs) in fish through AhR pathway. Toxicol. Mech. Methods 2010, 20, 279–286. [Google Scholar] [CrossRef]
- Chapman, P.M. Integrating toxicology and ecology: Putting the “eco” into ecotoxicology. Mar. Pollut. Bull. 2002, 44, 7–15. [Google Scholar] [CrossRef]
- Zhu, J.; Liu, H.; Wang, W.; Dong, S.; Wang, H.; Jiao, X. Research progress on toxicity and ecological risk of microplastics and their combined pollution to aquatic organisms. Acta Sci. Circumstantiae 2023, 43, 125–136. [Google Scholar]
- Dayal, L.; Raj, D.; Kumari, P.; Sinha, S. Abundance of Microplastics in Marine and Freshwater Ecosystem and it’s Impact on Biotic and Abiotic Components. Water Air Soil Pollut. 2024, 235, 416. [Google Scholar] [CrossRef]
- Bertolatus, D.W.; Barber, L.B.; Martyniuk, C.J.; Zhen, H.; Collette, T.W.; Ekman, D.R.; Jastrow, A.; Rapp, J.L.; Vajda, A.M. Multi-omic responses of fish exposed to complex chemical mixtures in the Shenandoah River watershed. Sci. Total Environ. 2023, 902, 165975. [Google Scholar] [CrossRef]
- Liu, J.; Li, W.; Wang, L.; Li, J.; Li, E.; Luo, Y. Multi-omics technology and its applications to life sciences: A review. Chin. J. Biotechnol. 2022, 38, 3581–3593. [Google Scholar] [CrossRef]
- Shahrajabian, M.H.; Sun, W.L. Survey on Multi-omics, and Multi-omics Data Analysis, Integration and Application. Curr. Pharm. Anal. 2023, 19, 267–281. [Google Scholar] [CrossRef]
- Ge, F.; He, Q.-Y. Genomic and proteomic approaches for predicting toxicity and adverse drug reactions. Expert. Opin. Drug Metab. Toxicol. 2009, 5, 29–37. [Google Scholar] [CrossRef]
- Jiang, Y.; Tan, L.J.; Yin, Y.; Shen, Y.M.; Gong, B.; Shao, Z.F. Cleavable Linkers in DNA Sequencing by Synthesis. Prog. Chem. 2016, 28, 58–66. [Google Scholar] [CrossRef]
- Garcia-Reyero, N.; Perkins, E.J. Systems biology: Leading the revolution in ecotoxicology. Environ. Toxicol. Chem. 2011, 30, 265–273. [Google Scholar] [CrossRef]
- Zhang, D.; Ma, Q.; Yin, T.; Xia, T. The Third Generation Sequencing Technology and Its Application. J. Chin. Biotechnol. 2013, 33, 125–131. [Google Scholar]
- Tan, D.; Ou, T. Research progress and clinical application of the third- generation sequencing techniques. Sheng Wu Gong Cheng Xue Bao Chin. J. Biotechnol. 2022, 38, 3121–3130. [Google Scholar] [CrossRef]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef]
- Mutz, K.O.; Heilkenbrinker, A.; Lonne, M.; Walter, J.G.; Stahl, F. Transcriptome analysis using next-generation sequencing. Curr. Opin. Biotechnol. 2013, 24, 22–30. [Google Scholar] [CrossRef]
- Zhao, S.; Fung-Leung, W.P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Connell, M.L.; Meyer, D.N.; Haimbaugh, A.; Baker, T.R. Status of single-cell RNA sequencing for reproductive toxicology in zebrafish and the transcriptomic trade-off. Curr. Opin. Toxicol. 2024, 38, 100463. [Google Scholar] [CrossRef]
- Zhang, W.; Xia, S.Y.; Zhong, X.R.; Gao, G.Y.; Yang, J.; Wang, S.; Cao, M.; Liang, Z.; Yang, C.B.; Wang, J.G. Characterization of 2,2′,4,4′-tetrabromodiphenyl ether (BDE47)-induced testicular toxicity via single-cell RNA-sequencing. Precis. Clin. Med. 2022, 5, pbac016. [Google Scholar] [CrossRef]
- Du, J.; Yang, Y.C.; An, Z.J.; Zhang, M.H.; Fu, X.H.; Huang, Z.F.; Yuan, Y.; Hou, J. Advances in spatial transcriptomics and related data analysis strategies. J. Transl. Med. 2023, 21, 330. [Google Scholar] [CrossRef]
- Chilimoniuk, J.; Erol, A.; Rödiger, S.; Burdukiewicz, M. Challenges and opportunities in processing NanoString nCounter data. Comput. Struct. Biotechnol. J. 2024, 23, 1951–1958. [Google Scholar] [CrossRef]
- Goswami, P.; Cesare, J.; Rekowski, M.J.; Clark, Z.; Thornton, J.; Washburn, M.P. Analysis of FAIMS for the study of affinity-purified protein complexes using the orbitrap ascend tribrid mass spectrometer. Mol. Omics 2025, 21, 303–314. [Google Scholar] [CrossRef]
- Jin, Y.; Sun, Z.; Gu, A.Z.; Zhou, X. Proteomics technologies in toxicity screening: A review. Environ. Chem. Lett. 2025, 23, 67–80. [Google Scholar] [CrossRef]
- Alcaraz, A.J.G.; Baraniuk, S.; Mikulasek, K.; Park, B.; Lane, T.; Burbridge, C.; Ewald, J.; Potesil, D.; Xia, J.; Zdrahal, Z.; et al. Comparative analysis of transcriptomic points-of-departure (tPODs) and apical responses in embryo-larval fathead minnows exposed to fluoxetine. Environ. Pollut. 2022, 295, 118667. [Google Scholar] [CrossRef]
- Silvestre, F.; Gillardin, V.; Dorts, J. Proteomics to assess the role of phenotypic plasticity in aquatic organisms exposed to pollution and global warming. Integr. Comp. Biol. 2012, 52, 681–694. [Google Scholar] [CrossRef]
- Qiao, Q.; Le Manach, S.; Huet, H.; Duvernois-Berthet, E.; Chaouch, S.; Duval, C.; Sotton, B.; Ponger, L.; Marie, A.; Mathéron, L.; et al. An integrated omic analysis of hepatic alteration in medaka fish chronically exposed to cyanotoxins with possible mechanisms of reproductive toxicity. Environ. Pollut. 2016, 219, 119–131. [Google Scholar] [CrossRef]
- Liu, L.; Wu, Q.; Miao, X.; Fan, T.; Meng, Z.; Chen, X.; Zhu, W. Study on toxicity effects of environmental pollutants based on metabolomics: A review. Chemosphere 2022, 286, 131815. [Google Scholar] [CrossRef]
- Canuto, G.A.B.; da Costa, J.L.; da Cruz, P.L.R.; de Souza, A.R.L.; Faccio, A.T.; Klassen, A.; Rodrigues, K.T.; Tavares, M.F.M. Metabolomics: Definitions, state-of-the-art and representative applications. Quim. Nova 2018, 41, 75–91. [Google Scholar] [CrossRef]
- Chen, Y.; Li, E.M.; Xu, L.Y. Guide to Metabolomics Analysis: A Bioinformatics Workflow. Metabolites 2022, 12, 357. [Google Scholar] [CrossRef]
- Levy, A.J.; Oranzi, N.R.; Ahmadireskety, A.; Kemperman, R.H.J.; Wei, M.S.; Yost, R.A. Recent progress in metabolomics using ion mobility-mass spectrometry. Trac Trends Anal. Chem. 2019, 116, 274–281. [Google Scholar] [CrossRef]
- Ribay, V.; Praud, C.; Letertre, M.P.M.; Dumez, J.N.; Giraudeau, P. Hyperpolarized NMR metabolomics. Curr. Opin. Chem. Biol. 2023, 74, 102307. [Google Scholar] [CrossRef]
- Coleman, M.A.; Escobar, P.A.; Mahadevan, B. Omics-current applications in toxicology. Mutat. Res. 2011, 722, 93. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef]
- Chen, C.; Wang, J.; Pan, D.; Wang, X.; Xu, Y.; Yan, J.; Wang, L.; Yang, X.; Yang, M.; Liu, G.-P. Applications of multi-omics analysis in human diseases. Medcomm 2023, 4, e315. [Google Scholar] [CrossRef]
- Liew, A.W.-C.; Law, N.-F.; Yan, H. Missing value imputation for gene expression data: Computational techniques to recover missing data from available information. Brief. Bioinform. 2011, 12, 498–513. [Google Scholar] [CrossRef]
- Vivian, J.; Eizenga, J.; Morozova-Vaske, O.; Paten, B. A Bayesian Framework for Detecting Gene Expression Outliers in Individual Samples. JCO Clin. Cancer Inform. 2020, 4, 160–170. [Google Scholar] [CrossRef]
- Krassowski, M.; Das, V.; Sahu, S.K.; Misra, B.B. State of the Field in Multi-Omics Research: From Computational Needs to Data Mining and Sharing. Front. Genet. 2020, 11, 610798. [Google Scholar] [CrossRef]
- Tarazona, S.; Balzano-Nogueira, L.; Gomez-Cabrero, D.; Schmidt, A.; Imhof, A.; Hankemeier, T.; Tegner, J.; Westerhuis, J.A.; Conesa, A. Harmonization of quality metrics and power calculation in multi-omic studies. Nat. Commun. 2020, 11, 3092. [Google Scholar] [CrossRef]
- Tong, L.; Shi, W.Q.; Isgut, M.; Zhong, Y.S.; Lais, P.; Gloster, L.; Sun, J.M.; Swain, A.; Giuste, F.; Wang, M.D. Integrating Multi-Omics Data With EHR for Precision Medicine Using Advanced Artificial Intelligence. IEEE Rev. Biomed. Eng. 2024, 17, 80–97. [Google Scholar] [CrossRef]
- Tripathy, R.K.; Frohock, Z.; Wang, H.; Cary, G.A.; Keegan, S.; Carter, G.W.; Li, Y. Effective integration of multi-omics with prior knowledge to identify biomarkers via explainable graph neural networks. Npj Syst. Biol. Appl. 2025, 11, 43. [Google Scholar] [CrossRef]
- Terzin, M.; Robbins, S.J.; Bell, S.C.; Cao, K.-A.L.; Gruber, R.K.; Frade, P.R.; Webster, N.S.; Yeoh, Y.K.; Bourne, D.G.; Laffy, P.W. Gene content of seawater microbes is a strong predictor of water chemistry across the Great Barrier Reef. Microbiome 2025, 13, 11. [Google Scholar] [CrossRef]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis-a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 2018, 14, e8124. [Google Scholar] [CrossRef]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333–337. [Google Scholar] [CrossRef]
- Xu, S.; Hu, E.; Cai, Y.; Xie, Z.; Luo, X.; Zhan, L.; Tang, W.; Wang, Q.; Liu, B.; Wang, R.; et al. Using clusterProfiler to characterize multiomics data. Nat. Protoc. 2024, 19, 3292–3320. [Google Scholar] [CrossRef]
- Yu, G.; He, Q.-Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Otasek, D.; Morris, J.H.; Boucas, J.; Pico, A.R.; Demchak, B. Cytoscape Automation: Empowering workflow-based network analysis. Genome Biol. 2019, 20, 185. [Google Scholar] [CrossRef]
- Luo, W.; Pant, G.; Bhavnasi, Y.K.; Blanchard, S.G., Jr.; Brouwer, C. Pathview Web: User friendly pathway visualization and data integration. Nucleic Acids Res. 2017, 45, W501–W508. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Zhou, G.; Pang, Z.; Lu, Y.; Ewald, J.; Xia, J. OmicsNet 2.0: A web-based platform for multi-omics integration and network visual analytics. Nucleic Acids Res. 2022, 50, W527–W533. [Google Scholar] [CrossRef]
- ISO 23893-3:2013; Water Quality—Biochemical and Physiological Measurements on Fish—Part 3: Determination of Vitellogenin. International Organization for Standardization: Geneva, Switzerland, 2013.
- Canzler, S.; Schor, J.; Busch, W.; Schubert, K.; Rolle-Kampczyk, U.E.; Seitz, H.; Kamp, H.; von Bergen, M.; Buesen, R.; Hackermüller, J. Prospects and challenges of multi-omics data integration in toxicology. Arch. Toxicol. 2020, 94, 371–388. [Google Scholar] [CrossRef]
- Wang, Y.W.; Zhao, J.H.; Xu, Y.P.; Tao, C.M.; Tong, J.; Luo, Y.J.; Chen, Y.; Liu, X.S.; Xu, T.F. Uncovering SOD3 and GPX4 as new targets of Benzo α pyrene-induced hepatotoxicity through Metabolomics and Chemical Proteomics. Redox Biol. 2023, 67, 102930. [Google Scholar] [CrossRef]
- Martyniuk, C.J.; Kroll, K.J.; Doperalski, N.J.; Barber, D.S.; Denslow, N.D. Genomic and Proteomic Responses to Environmentally Relevant Exposures to Dieldrin: Indicators of Neurodegeneration? Toxicol. Sci. 2010, 117, 190–199. [Google Scholar] [CrossRef]
- Wu, H.; Liu, X.; Zhang, X.; Ji, C.; Zhao, J.; Yu, J. Proteomic and metabolomic responses of clam Ruditapes philippinarum to arsenic exposure under different salinities. Aquat. Toxicol. 2013, 136–137, 91–100. [Google Scholar] [CrossRef]
- Ji, C.; Wu, H.; Wei, L.; Zhao, J.; Yu, J. Proteomic and metabolomic analysis reveal gender-specific responses of mussel Mytilus galloprovincialis to 2,2′,4,4′-tetrabromodiphenyl ether (BDE 47). Aquat. Toxicol. 2013, 140–141, 449–457. [Google Scholar] [CrossRef]
- Wu, H.; Xu, L.; Ji, C.; Yu, D. Proteomic and metabolomic responses in D-shape larval mussels Mytilus galloprovincialis exposed to cadmium and arsenic. Fish. Shellfish. Immunol. 2016, 58, 514–520. [Google Scholar] [CrossRef]
- Yu, D.; Ji, C.; Zhao, J.; Wu, H. Proteomic and metabolomic analysis on the toxicological effects of As (III) and As (V) in juvenile mussel Mytilus galloprovincialis. Chemosphere 2016, 150, 194–201. [Google Scholar] [CrossRef]
- Chen, H.; Diao, X.; Wang, H.; Zhou, H. An integrated metabolomic and proteomic study of toxic effects of Benzo[a]pyrene on gills of the pearl oyster Pinctada martensii. Ecotoxicol. Environ. Saf. 2018, 156, 330–336. [Google Scholar] [CrossRef]
- Dumas, T.; Courant, F.; Almunia, C.; Boccard, J.; Rosain, D.; Duporte, G.; Armengaud, J.; Fenet, H.; Gomez, E. An integrated metabolomics and proteogenomics approach reveals molecular alterations following carbamazepine exposure in the male mussel Mytilus galloprovincialis. Chemosphere 2022, 286, 131793. [Google Scholar] [CrossRef]
- Dumas, T.; Gomez, E.; Boccard, J.; Ramirez, G.; Armengaud, J.; Escande, A.; Mathieu, O.; Fenet, H.; Courant, F. Mixture effects of pharmaceuticals carbamazepine, diclofenac and venlafaxine on Mytilus galloprovincialis mussel probed by metabolomics and proteogenomics combined approach. Sci. Total Environ. 2024, 907, 168015. [Google Scholar] [CrossRef]
- Xu, L.; Lu, Z.; Ji, C.; Cong, M.; Li, F.; Shan, X.; Wu, H. Toxicological effects of As (V) in juvenile rockfish Sebastes schlegelii by a combined metabolomic and proteomic approach. Environ. Pollut. 2019, 255, 113333. [Google Scholar] [CrossRef]
- Ji, C.; Lu, Z.; Xu, L.; Li, F.; Cong, M.; Shan, X.; Wu, H. Global responses to tris(1-chloro-2-propyl)phosphate (TCPP) in rockfish Sebastes schlegeli using integrated proteomic and metabolomic approach. Sci. Total Environ. 2020, 724, 138307. [Google Scholar] [CrossRef]
- Ren, X.; Jia, S.; Gao, B.; Zhou, Q.; Xu, Y.; Liu, P.; Li, J. Application of proteomics and metabolomics to assess ammonia stress response and tolerance mechanisms of juvenile ornate rock lobster Panulirus ornatus. Sci. Total Environ. 2022, 837, 155751. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hu, Y.; He, J.; Chen, J.; Giesy, J.P.; Xie, P. Responses of the Proteome and Metabolome in Livers of Zebrafish Exposed Chronically to Environmentally Relevant Concentrations of Microcystin-LR. Environ. Sci. Technol. 2017, 51, 596–607. [Google Scholar] [CrossRef]
- Le Manach, S.; Sotton, B.; Huet, H.; Duval, C.; Paris, A.; Marie, A.; Yepremian, C.; Catherine, A.; Matheron, L.; Vinh, J.; et al. Physiological effects caused by microcystin-producing and non-microcystin producing Microcystis aeruginosa on medaka fish: A proteomic and metabolomic study on liver. Environ. Pollut. 2018, 234, 523–537. [Google Scholar] [CrossRef]
- Fernandez-Cisnal, R.; Garcia-Sevillano, M.A.; Garcia-Barrera, T.; Gomez-Ariza, J.L.; Abril, N. Metabolomic alterations and oxidative stress are associated with environmental pollution in Procambarus clarkii. Aquat. Toxicol. 2018, 205, 76–88. [Google Scholar] [CrossRef]
- Hu, C.; Liu, M.; Wan, T.; Tang, L.; Sun, B.; Zhou, B.; Lam, J.C.W.; Lam, P.K.S.; Chen, L. Disturbances in Microbial and Metabolic Communication across the Gut-Liver Axis Induced by a Dioxin-like Pollutant: An Integrated Metagenomics and Metabolomics Analysis. Environ. Sci. Technol. 2021, 55, 529–537. [Google Scholar] [CrossRef]
- Sun, X.; Tian, S.; Yan, S.; Sun, W.; Miao, J.; Yue, Y.; Han, S.; Huang, S.; Xu, N.; Diao, J.; et al. Bifidobacterium mediate gut microbiota-remedied intestinal barrier damage caused by cyproconazole in zebrafish (Danio rerio). Sci. Total Environ. 2024, 912, 169556. [Google Scholar] [CrossRef]
- Foucault, P.; Gallet, A.; Duval, C.; Marie, B.; Duperron, S. Gut microbiota and holobiont metabolome composition of the medaka fish (Oryzias latipes) are affected by a short exposure to the cyanobacterium Microcystis aeruginosa. Aquat. Toxicol. 2022, 253, 106329. [Google Scholar] [CrossRef]
- Chu, T.; Zhang, R.; Guo, F.; Zhu, M.; Zan, S.; Yang, R. The toxicity of polystyrene micro- and nano-plastics on rare minnow (Gobiocypris rarus) varies with the particle size and concentration. Aquat. Toxicol. 2024, 269, 106879. [Google Scholar] [CrossRef]
- Zhang, C.; Bao, F.; Wang, F.; Xue, Z.; Lin, D. Toxic effects of nanoplastics and microcystin-LR coexposure on the liver-gut axis of Hypophthalmichthys molitrix. Sci. Total Environ. 2024, 916, 170011. [Google Scholar] [CrossRef]
- Wei, X.Y.; Jia, P.P.; Hu, H.; Liu, L.; Li, T.Y.; Li, Y.Z.; Pei, D.S. Multi-omics reveal mechanisms underlying chronic kidney disease of unknown etiology (CKDu) pathogenesis using zebrafish. Environ. Pollut. 2023, 337, 122524. [Google Scholar] [CrossRef]
- Jia, P.P.; Li, Y.; Zhang, L.C.; Wu, M.F.; Li, T.Y.; Pei, D.S. Metabolome evidence of CKDu risks after chronic exposure to simulated Sri Lanka drinking water in zebrafish. Ecotoxicol. Environ. Saf. 2024, 273, 116149. [Google Scholar] [CrossRef]
- Yu, K.; Song, Y.; Wang, N.; Yu, X.; Sun, T.; Yu, H.; Ruan, Z.; Qiu, Y. Exposure of Danio rerio to environmental sulfamethoxazole may contribute to neurobehavioral abnormalities via gut microbiome disturbance. Sci. Total Environ. 2024, 918, 170546. [Google Scholar] [CrossRef]
- Ma, S.; Shu, X.; Wang, W.X. Multi-omics reveals the regulatory mechanisms of zinc exposure on the intestine-liver axis of golden pompano Trachinotus ovatus. Sci. Total Environ. 2022, 816, 151497. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, X.; Jiang, J.; Dong, Y.; Pan, Y.; Guan, X.; Wang, B.; Gao, S.; Chen, Z.; Zhou, Z. Adverse effects of polystyrene nanoplastics on sea cucumber Apostichopus japonicus and their association with gut microbiota dysbiosis. Chemosphere 2023, 330, 138568. [Google Scholar] [CrossRef]
- Fu, Z.; Han, F.; Huang, K.; Zhang, J.; Qin, J.G.; Chen, L.; Li, E. Combined toxic effects of thiamethoxam on intestinal flora, transcriptome and physiology of Pacific white shrimp Litopenaeus vannamei. Sci. Total Environ. 2022, 830, 154799. [Google Scholar] [CrossRef]
- Spaink, H.P.; Jansen, H.J.; Dirks, R.P. Advances in genomics of bony fish. Brief. Funct. Genom. 2014, 13, 144–156. [Google Scholar] [CrossRef]
- Alestrom, P.; Holter, J.L.; Nourizadeh-Lillabadi, R. Zebrafish in functional genomics and aquatic biomedicine. Trends Biotechnol. 2006, 24, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.D.; Wu, H.; Santos, E.M.; Ball, J.; Katsiadaki, I.; Brown, M.M.; Baker, P.; Ortega, F.; Falciani, F.; Craft, J.A.; et al. Hepatic transcriptomic and metabolomic responses in the stickleback (Gasterosteus aculeatus) exposed to environmentally relevant concentrations of dibenzanthracene. Environ. Sci. Technol. 2009, 43, 6341–6348. [Google Scholar] [CrossRef]
- Ortiz-Villanueva, E.; Navarro-Martin, L.; Jaumot, J.; Benavente, F.; Sanz-Nebot, V.; Pina, B.; Tauler, R. Metabolic disruption of zebrafish (Danio rerio) embryos by bisphenol A. An integrated metabolomic and transcriptomic approach. Environ. Pollut. 2017, 231, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Weng, Y.; Zhao, Y.; Wang, D.; Luo, T.; Jin, Y. Transcriptomic and targeted metabolomic analysis revealed the toxic effects of prochloraz on larval zebrafish. Sci. Total Environ. 2022, 822, 153625. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhang, C.; Wang, L.; Wang, X.; He, H.; Wu, S.; Zhao, X. Insights into the combined effects of environmental concentration of difenoconazole and tebuconazole on zebrafish early life stage. Sci. Total Environ. 2022, 830, 154687. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.; Zhu, W.; Wang, D.; Qi, S.; Wang, Y.; Yan, J.; Dong, K.; Zheng, M.; Wang, C. Metabolomics and transcriptomics reveal the toxicity of difenoconazole to the early life stages of zebrafish (Danio rerio). Aquat. Toxicol. 2018, 194, 112–120. [Google Scholar] [CrossRef]
- Li, Y.; Liang, H.; Ren, B.; Zhao, T.; Chen, H.; Zhao, Y.; Liang, H. Enantioselective toxic effects of mefentrifluconazole in the liver of adult zebrafish (Danio rerio) based on transcription level and metabolomic profile. Toxicology 2022, 467, 153095. [Google Scholar] [CrossRef]
- Zhang, S.; Wu, L.; Zhang, J.; Wang, X.; Yang, X.; Xin, Y.; Chen, L.; Li, J.; Niu, P. Multi-omics analysis reveals Mn exposure affects ferroptosis pathway in zebrafish brain. Ecotoxicol. Environ. Saf. 2023, 253, 114616. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Yan, S.; Meng, Z.; Huang, S.; Sun, W.; Jia, M.; Teng, M.; Zhou, Z.; Zhu, W. New insights into bisphenols induced obesity in zebrafish (Danio rerio): Activation of cannabinoid receptor CB1. J. Hazard. Mater. 2021, 418, 126100. [Google Scholar] [CrossRef]
- Zhang, Y.; Zeng, H.; Zhou, L.; Wang, C.; Yang, X.; Liu, S. Integrated histopathology and transcriptome metabolome profiling reveal the toxicity mechanism of phenazine-1-carboxylic acid in zebrafish. Environ. Pollut. 2024, 344, 123402. [Google Scholar] [CrossRef]
- Wang, T.; Wang, Y.; Liu, X.; Gao, X.; Hu, K. Combined Transcriptomics and Metabolomics Analyses in Grass Carp Under Anesthetic Stress. Front. Cell Infect. Microbiol. 2022, 12, 931696. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.Q.; Yan, H.; Luo, X.W.; Kang, Y.H.; Hu, J.M.; Chen, L.Q. Integration of transcriptomics and metabolomics reveals damage and recovery mechanisms of fish gills in response to nanosilver exposure. Aquat. Toxicol. 2021, 237, 105895. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Z.; Li, S.; Ding, L.; Wei, X.; Han, S.; Wang, P.; Sun, Y. Multi-omics profiling and biochemical assays reveal the acute toxicity of environmental related concentrations of Di-(2-ethylhexyl) phthalate (DEHP) on the gill of crucian carp (Carassius auratus). Chemosphere 2022, 307, 135814. [Google Scholar] [CrossRef]
- Liu, L.; Fu, J.; Tang, Q.; Wang, H.; Lin, C.; Wei, L. Combined transcriptomics and metabolomics analysis reveals lipid metabolic disruption in swamp eel (Monopterus albus) under chronic waterborne copper exposure. Aquat. Toxicol. 2023, 259, 106520. [Google Scholar] [CrossRef]
- Li, Y.; Yang, H.; Fu, B.; Kaneko, G.; Li, H.; Tian, J.; Wang, G.; Wei, M.; Xie, J.; Yu, E. Integration of Multi-Omics, Histological, and Biochemical Analysis Reveals the Toxic Responses of Nile Tilapia Liver to Chronic Microcystin-LR Exposure. Toxins 2024, 16, 149. [Google Scholar] [CrossRef]
- Bi, X.; Qiu, M.; Li, D.; Zhang, Y.; Zhan, W.; Wang, Z.; Lv, Z.; Li, H.; Chen, G. Transcriptomic and metabolomic analysis of the mechanisms underlying stress responses of the freshwater snail, Pomacea canaliculata, exposed to different levels of arsenic. Aquat. Toxicol. 2024, 267, 106835. [Google Scholar] [CrossRef]
- Yang, H.; Shen, M.; Zhang, Q.; Li, Y.; Tan, X.; Li, X.; Chen, H.; Wu, L.; He, S.; Zhu, X. Transcriptome and metabolomics analysis of adaptive mechanism of Chinese mitten crab (Eriocheir sinensis) to aflatoxin B1. PLoS ONE 2023, 18, e0295291. [Google Scholar] [CrossRef]
- Jamers, A.; Blust, R.; De Coen, W.; Griffin, J.L.; Jones, O.A. An omics based assessment of cadmium toxicity in the green alga Chlamydomonas reinhardtii. Aquat. Toxicol. 2013, 126, 355–364. [Google Scholar] [CrossRef]
- Li, Q.; Lu, D.; Sun, H.; Guo, J.; Mo, J. Tylosin toxicity in the alga Raphidocelis subcapitata revealed by integrated analyses of transcriptome and metabolome: Photosynthesis and DNA replication-coupled repair. Aquat. Toxicol. 2021, 239, 105964. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.H.; Altin, D.; Booth, A.; Vang, S.H.; Frenzel, M.; Sorheim, K.R.; Brakstad, O.G.; Storseth, T.R. Molecular effects of diethanolamine exposure on Calanus finmarchicus (Crustacea: Copepoda). Aquat. Toxicol. 2010, 99, 212–222. [Google Scholar] [CrossRef]
- Vandenbrouck, T.; Jones, O.A.; Dom, N.; Griffin, J.L.; De Coen, W. Mixtures of similarly acting compounds in Daphnia magna: From gene to metabolite and beyond. Environ. Int. 2010, 36, 254–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Ng, Q.X.; Zhang, B.; Wei, Z.; Hassan, M.; He, Y.; Ong, C.N. Employing multi-omics to elucidate the hormetic response against oxidative stress exerted by nC(60) on Daphnia pulex. Environ. Pollut. 2019, 251, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Poynton, H.C.; Taylor, N.S.; Hicks, J.; Colson, K.; Chan, S.; Clark, C.; Scanlan, L.; Loguinov, A.V.; Vulpe, C.; Viant, M.R. Metabolomics of microliter hemolymph samples enables an improved understanding of the combined metabolic and transcriptional responses of Daphnia magna to cadmium. Environ. Sci. Technol. 2011, 45, 3710–3717. [Google Scholar] [CrossRef]
- Guo, J.; Mo, J.; Qi, Q.; Peng, J.; Qi, G.; Kanerva, M.; Iwata, H.; Li, Q. Prediction of adverse effects of effluents containing phenolic compounds in the Ba River on the ovary of fish (Hemiculter leucisculus) using transcriptomic and metabolomic analyses. Sci. Total Environ. 2021, 801, 149554. [Google Scholar] [CrossRef]
- Xiao, J.; Li, Q.Y.; Tu, J.P.; Chen, X.L.; Chen, X.H.; Liu, Q.Y.; Liu, H.; Zhou, X.Y.; Zhao, Y.Z.; Wang, H.L. Stress response and tolerance mechanisms of ammonia exposure based on transcriptomics and metabolomics in Litopenaeus vannamei. Ecotoxicol. Environ. Saf. 2019, 180, 491–500. [Google Scholar] [CrossRef]
- Katsiadaki, I.; Williams, T.D.; Ball, J.S.; Bean, T.P.; Sanders, M.B.; Wu, H.; Santos, E.M.; Brown, M.M.; Baker, P.; Ortega, F.; et al. Hepatic transcriptomic and metabolomic responses in the Stickleback (Gasterosteus aculeatus) exposed to ethinyl-estradiol. Aquat. Toxicol. 2010, 97, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Colas-Ruiz, N.R.; Courant, F.; Gomez, E.; Lara-Martin, P.A.; Hampel, M. Transcriptomic and metabolomic integration to assess the response of gilthead sea bream (Sparus aurata) exposed to the most used insect repellent: DEET. Environ. Pollut. 2023, 316, 120678. [Google Scholar] [CrossRef] [PubMed]
- Colas-Ruiz, N.R.; Ramirez, G.; Courant, F.; Gomez, E.; Hampel, M.; Lara-Martin, P.A. Multi-omic approach to evaluate the response of gilt-head sea bream (Sparus aurata) exposed to the UV filter sulisobenzone. Sci. Total Environ. 2022, 803, 150080. [Google Scholar] [CrossRef]
- Olsvik, P.A.; Berntssen, M.H.; Softeland, L. Modifying effects of vitamin E on chlorpyrifos toxicity in atlantic salmon. PLoS ONE 2015, 10, e0119250. [Google Scholar] [CrossRef]
- Liu, F.; Li, S.; Yu, Y.; Sun, M.; Xiang, J.; Li, F. Effects of ammonia stress on the hemocytes of the Pacific white shrimp Litopenaeus vannamei. Chemosphere 2020, 239, 124759. [Google Scholar] [CrossRef]
- Shi, Q.Q.; Zhang, X.Q.; Zhang, Z.M.; Wang, N.B.; Liu, H.; Zhang, R.R.; Sun, A.L.; Chen, J.; Shi, X.Z. Transcriptome sequencing and metabolite analysis reveal the single and combined effects of microplastics and di-(2-ethylhexyl) phthalate on Peneaus vannamei. Sci. Total Environ. 2023, 867, 161549. [Google Scholar] [CrossRef]
- Li, F.; Meng, X.; Wang, X.; Ji, C.; Wu, H. Graphene-triphenyl phosphate (TPP) co-exposure in the marine environment: Interference with metabolism and immune regulation in mussel Mytilus galloprovincialis. Ecotoxicol. Environ. Saf. 2021, 227, 112904. [Google Scholar] [CrossRef]
- Li, F.; Yu, Y.; Guo, M.; Lin, Y.; Jiang, Y.; Qu, M.; Sun, X.; Li, Z.; Zhai, Y.; Tan, Z. Integrated analysis of physiological, transcriptomics and metabolomics provides insights into detoxication disruption of PFOA exposure in Mytilus edulis. Ecotoxicol. Environ. Saf. 2021, 214, 112081. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tian, X.; Gong, X.; Han, D.; Ren, L.; Cui, Y.; Jiang, F.; Zhao, J.; Chen, J.; Jiang, L.; et al. Analyzing toxicological effects of AsIII and AsV to Chlamys farreri by integrating transcriptomic and metabolomic approaches. Mar. Pollut. Bull. 2023, 186, 114385. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, Q.; Li, X.; Xu, H.; Ren, C.; Yang, Y.; Xu, S.; Wei, G.; Duan, Y.; Tan, Z.; et al. Norgestrel causes digestive gland injury in the clam Mactra veneriformis: An integrated histological, transcriptomics, and metabolomics study. Sci. Total Environ. 2023, 871, 162110. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Fang, J.; Du, M.; Gao, Y.; Fang, J.; Jiang, Z. Integrated transcriptomics and metabolomics analyses reveal benzo[a]pyrene enhances the toxicity of mercury to the Manila clam, Ruditapes philippinarum. Ecotoxicol. Environ. Saf. 2021, 213, 112038. [Google Scholar] [CrossRef] [PubMed]
- Paganos, P.; Ullmann, C.V.; Gaglio, D.; Bonanomi, M.; Salmistraro, N.; Arnone, M.I.; Jimenez-Guri, E. Plastic leachate-induced toxicity during sea urchin embryonic development: Insights into the molecular pathways affected by PVC. Sci. Total Environ. 2023, 864, 160901. [Google Scholar] [CrossRef]
- Ying, Z.; Xie, X.; Li, Y.; Bao, Y.; Ye, G.; Chen, X.; Zhang, W.; Gu, Y.G. A novel cadmium detoxification pathway in Tri-spine horseshoe crab (Tachypleus tridentatus): A 430-million-years-ago organism. Ecotoxicol. Environ. Saf. 2023, 252, 114585. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhang, M.; Bao, J.; Liu, J. Physiological, metabolomic, and transcriptomic analyses reveal the dynamic redox homeostasis upon extended exposure of Dunaliella salina GY-H13 cells to Cd. Ecotoxicol. Environ. Saf. 2021, 223, 112593. [Google Scholar] [CrossRef]
- Lyu, L.; Tao, Y.; Wu, S.; Abaakil, K.; Zhong, G.; Gu, Y.; Hu, Y.; Zhang, Y. Tissue-specific accumulation of DEHP and involvement of endogenous arachidonic acid in DEHP-induced spleen information and injury. Sci. Total Environ. 2023, 904, 166841. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, Y.; Wang, J.; Peng, C.; Wang, Z.; Qin, H.; Li, G.; Li, D. Geosmin disrupts energy metabolism and locomotor behavior of zebrafish in early life stages. Sci. Total Environ. 2023, 859, 160222. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Chen, L.; Liu, X.; Wang, L.; Wu, S.; Zhao, X. Histology and multi-omic profiling reveal the mixture toxicity of tebuconazole and difenoconazole in adult zebrafish. Sci. Total Environ. 2021, 795, 148777. [Google Scholar] [CrossRef]
- Huang, S.S.Y.; Benskin, J.P.; Veldhoen, N.; Chandramouli, B.; Butler, H.; Helbing, C.C.; Cosgrove, J.R. A multi-omic approach to elucidate low-dose effects of xenobiotics in zebrafish (Danio rerio) larvae. Aquat. Toxicol. 2017, 182, 102–112. [Google Scholar] [CrossRef]
- Fu, J.; Tan, Y.X.R.; Gong, Z.; Bae, S. The toxic effect of triclosan and methyl-triclosan on biological pathways revealed by metabolomics and gene expression in zebrafish embryos. Ecotoxicol. Environ. Saf. 2020, 189, 110039. [Google Scholar] [CrossRef]
- Chang, J.; Jiao, M.; Zhang, Z.; Liu, W.; Li, W.; Xu, P.; Wan, B. Mechanistic insight into the adverse outcome of tire wear and road particle leachate exposure in zebrafish (Danio rerio) larvae. Environ. Int. 2023, 178, 108053. [Google Scholar] [CrossRef] [PubMed]
- De Wit, M.; Keil, D.; Remmerie, N.; van der Ven, K.; van den Brandhof, E.J.; Knapen, D.; Witters, E.; De Coen, W. Molecular targets of TBBPA in zebrafish analysed through integration of genomic and proteomic approaches. Chemosphere 2008, 74, 96–105. [Google Scholar] [CrossRef]
- Xu, Y.; Yang, L.; Teng, Y.; Li, J.; Li, N. Exploring the underlying molecular mechanism of tri(1,3-dichloropropyl) phosphate-induced neurodevelopmental toxicity via thyroid hormone disruption in zebrafish by multi-omics analysis. Aquat. Toxicol. 2023, 258, 106510. [Google Scholar] [CrossRef]
- Singh, S.K.; Aravamudhan, S.; Armant, O.; Krüger, M.; Grabher, C. Proteome dynamics in neutrophils of adult zebrafish upon chemically-induced inflammation. Fish. Shellfish. Immun. 2014, 40, 217–224. [Google Scholar] [CrossRef]
- Lavelle, C.; Smith, L.C.; Bisesi, J.H.; Vu, F.H.; Silva-Sanchez, C.; Moraga-Amador, D.; Buerger, A.N.; Garcia-Reyero, N.; Sabo-Attwood, T.; Denslow, N.D. Tissue-Based Mapping of the Fathead Minnow (Pimephales promelas) Transcriptome and Proteome. Front. Endocrinol. 2018, 9, 611. [Google Scholar] [CrossRef]
- Yuan, J.; Zheng, Y.; Gu, Z. Effects of cypermethrin on the hepatic transcriptome and proteome of the red claw crayfish Cherax quadricarinatus. Chemosphere 2021, 263, 128060. [Google Scholar] [CrossRef]
- Esperanza, M.; Seoane, M.; Rioboo, C.; Herrero, C.; Cid, A. Early alterations on photosynthesis-related parameters in Chlamydomonas reinhardtii cells exposed to atrazine: A multiple approach study. Sci. Total Environ. 2016, 554–555, 237–245. [Google Scholar] [CrossRef]
- Mielecki, D.; Grzesiuk, E.; Bednarska, A.; Garbicz, D.; Swiderska, B.; Grzesiuk, M. Contamination of aquatic environment with anticancer reagents influences Daphnia magna—Ecotoxicogenomics approach. Ecotoxicol. Environ. Saf. 2023, 249, 114372. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tian, X.; Jiang, L.; Han, D.; Hu, S.; Cui, Y.; Jiang, F.; Liu, Y.; Xu, Y.; Li, H. Sources, bioaccumulation, and toxicity mechanisms of cadmium in Chlamys farreri. J. Hazard. Mater. 2023, 453, 131395. [Google Scholar] [CrossRef]
- Meng, J.; Wang, W.; Li, L.; Yin, Q.; Zhang, G. Cadmium effects on DNA and protein metabolism in oyster (Crassostrea gigas) revealed by proteomic analyses. Sci. Rep. 2017, 7, 11716. [Google Scholar] [CrossRef] [PubMed]
- Dondero, F.; Negri, A.; Boatti, L.; Marsano, F.; Mignone, F.; Viarengo, A. Transcriptomic and proteomic effects of a neonicotinoid insecticide mixture in the marine mussel (Mytilus galloprovincialis, Lam.). Sci. Total Environ. 2010, 408, 3775–3786. [Google Scholar] [CrossRef]
- Lee, H.; Sung, E.J.; Seo, S.; Min, E.K.; Lee, J.Y.; Shim, I.; Kim, P.; Kim, T.Y.; Lee, S.; Kim, K.T. Integrated multi-omics analysis reveals the underlying molecular mechanism for developmental neurotoxicity of perfluorooctanesulfonic acid in zebrafish. Environ. Int. 2021, 157, 106802. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Gao, Y.; Ko, E.; Lee, J.; Lee, H.K.; Lee, S.; Choi, M.; Shin, S.; Park, Y.H.; Moon, H.B.; et al. Nonmonotonic response of type 2 diabetes by low concentration organochlorine pesticide mixture: Findings from multi-omics in zebrafish. J. Hazard. Mater. 2021, 416, 125956. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Xie, C.; Dong, M.; Zhang, Y.; Huang, H.; Han, Y.; Liu, Y.; Wei, L.; Wang, X. Effects of ambient UVB light on Pacific oyster Crassostrea gigas mantle tissue based on multivariate data. Ecotoxicol. Environ. Saf. 2024, 274, 116236. [Google Scholar] [CrossRef] [PubMed]
- Uppal, K.; Ma, C.; Go, Y.M.; Jones, D.P.; Wren, J. xMWAS: A data-driven integration and differential network analysis tool. Bioinformatics 2018, 34, 701–702. [Google Scholar] [CrossRef]
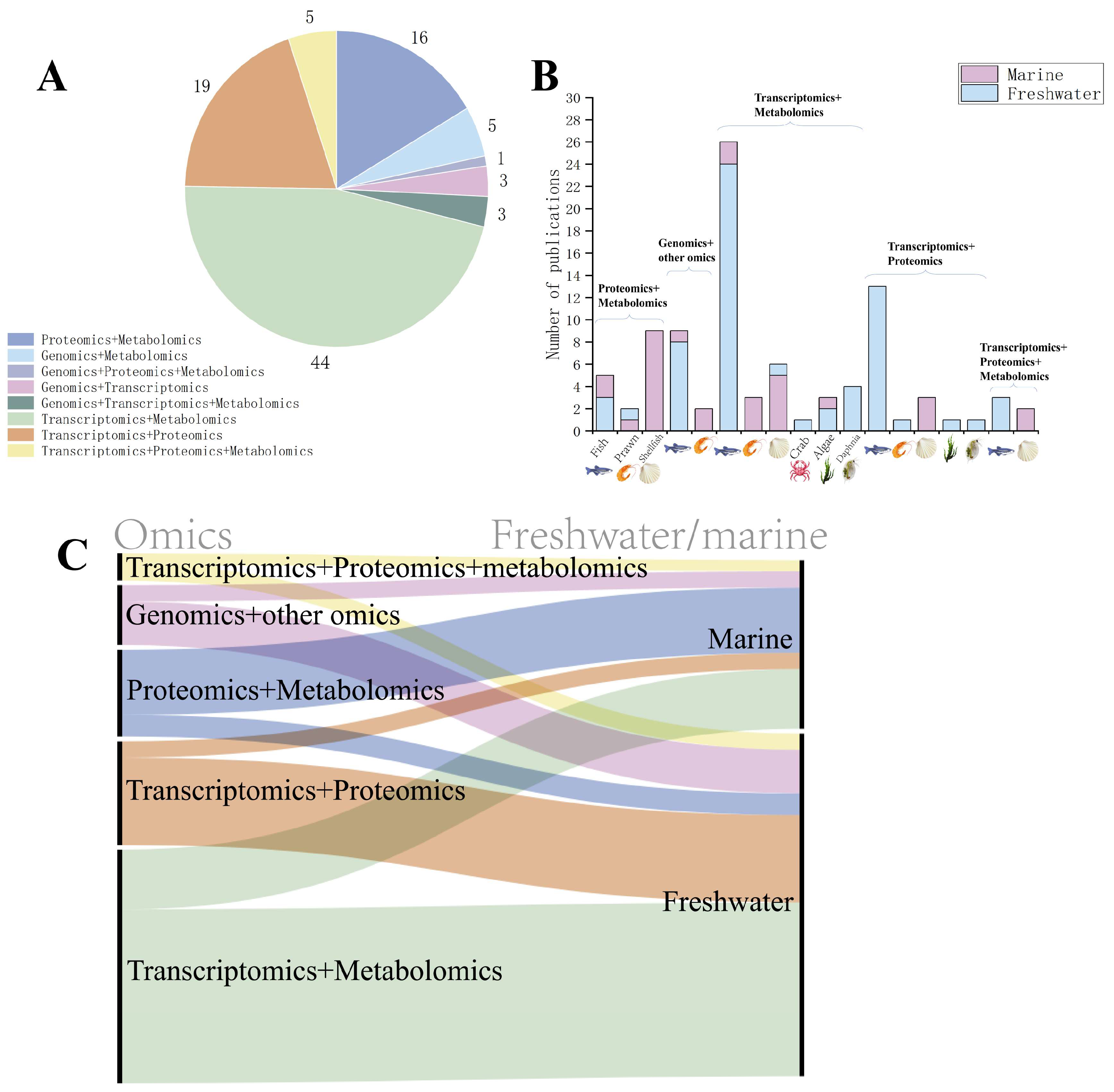
| Number | Biological Group | Species | Contaminant | Reference |
|---|---|---|---|---|
| Freshwater organisms | ||||
| 1 | Fish | Danio rerio | Di-(2-ethylhexyl) phthalate | [117] |
| 2 | Fish | Danio rerio | Geosmin | [118] |
| 3 | Fish | Danio rerio | Tebuconazole, difenoconazole | [119] |
| 4 | Fish | Danio rerio | Acetaminophen, diphenhydramine, carbamazepine, and fluoxetine | [120] |
| 5 | Fish | Danio rerio | Triclosan and its derivative, methyl-triclosan | [121] |
| 6 | Fish | Danio rerio | Bisphenol A | [80] |
| 7 | Fish | Danio rerio | Mefentrifluconazole | [84] |
| 8 | Fish | Danio rerio | Tire wear particles, road particles | [122] |
| 9 | Fish | Danio rerio | Manganese | [85] |
| 10 | Fish | Danio rerio | Prochloraz | [81] |
| 11 | Fish | Danio rerio | Difenoconazole Tebuconazole | [82] |
| 12 | Fish | Danio rerio | Difenoconazole | [83] |
| 13 | Fish | Danio rerio | Phenazine-1-carboxylic acid | [87] |
| 14 | Fish | Danio rerio | Bisphenol A Tetrabromobisphenol A | [86] |
| 15 | Fish | Cyprinus carpio | MS-222 and 2-PE | [88] |
| 16 | Fish | Cyprinus carpio | Silver nanoparticles | [89] |
| 17 | Fish | Carassius auratus | Di-(2-ethylhexyl) phthalate | [90] |
| 18 | Fish | Hemiculter leucisculus | Phenolic compounds | [101] |
| 19 | Fish | Monopterus albus | Copper | [91] |
| 20 | Fish | Nile tilapia | Microcystin-LR | [92] |
| 21 | Shellfish | Pomacea canaliculata | Arsenic | [93] |
| 22 | Crab | Eriocheir sinensis | Aflatoxin B1 | [94] |
| 23 | Algae | Chlamydomonas reinhardtii | Cadmium | [95] |
| 24 | Algae | Raphidocelis subcapitata | Tylosin | [96] |
| 25 | Daphnia | Calanus finmarchicus | Alkanolamines | [97] |
| 26 | Daphnia | Daphnia magna | Pyrene, fluoranthene | [98] |
| 27 | Daphnia | Daphnia pulex | Fullerene crystals (nC(60)) | [99] |
| 28 | Daphnia | Daphnia magna | Cadmium | [100] |
| Marine organisms | ||||
| 29 | Fish | Gasterosteus aculeatus | 1,2:5,6-Dibenzanthracene | [79] |
| 30 | Fish | Gasterosteus aculeatus | Ethinyl-estradiol | [103] |
| 31 | Fish | Sparus aurata | N,N-Diethyl-3-methyl benzoyl amide | [104] |
| 32 | Fish | Sparus aurata | Sulisobenzone | [105] |
| 33 | Fish | Salmo salar | Vitamin E, chlorpyrifos | [106] |
| 34 | Prawn | Litopenaeus vannamei | Ammonia | [102] |
| 35 | Prawn | Litopenaeus vannamei | Ammonia | [107] |
| 36 | Prawn | Litopenaeus vannamei | Microplastics, di-(2-ethylhexyl) phthalate | [108] |
| 37 | Shellfish | Mytilus galloprovincialis | Graphene nanomaterials, triphenyl phosphate | [109] |
| 38 | Shellfish | Mytilus edulis | Perfluorooctanoic acid | [110] |
| 39 | Shellfish | Chlamys farreri | Inorganic arsenic | [111] |
| 40 | Shellfish | Mactra veneriformis | Progestins | [112] |
| 41 | Shellfish | Uditapes philippinarum | Mercury, benzo(a)pyrene | [113] |
| 42 | Shellfish | Strongylocentrotus purpuratus | Polyvinyl chloride microplastics | [114] |
| 43 | Crab | Tachypleus tridentatus | Cadmium | [115] |
| 44 | Algae | Dunaliella salina | Cadmium | [116] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Zhang, Y.; Du, J.; Liu, C.; Zhou, G.; Li, M.; Yan, Z. Application of Multi-Omics Techniques in Aquatic Ecotoxicology: A Review. Toxics 2025, 13, 653. https://doi.org/10.3390/toxics13080653
Li B, Zhang Y, Du J, Liu C, Zhou G, Li M, Yan Z. Application of Multi-Omics Techniques in Aquatic Ecotoxicology: A Review. Toxics. 2025; 13(8):653. https://doi.org/10.3390/toxics13080653
Chicago/Turabian StyleLi, Boyang, Yizhang Zhang, Jinzhe Du, Chen Liu, Guorui Zhou, Mingrui Li, and Zhenguang Yan. 2025. "Application of Multi-Omics Techniques in Aquatic Ecotoxicology: A Review" Toxics 13, no. 8: 653. https://doi.org/10.3390/toxics13080653
APA StyleLi, B., Zhang, Y., Du, J., Liu, C., Zhou, G., Li, M., & Yan, Z. (2025). Application of Multi-Omics Techniques in Aquatic Ecotoxicology: A Review. Toxics, 13(8), 653. https://doi.org/10.3390/toxics13080653