

The Role of Epigenetic Mechanisms in the Development of PM2.5-Induced Cognitive Impairment

 and
and
Abstract
1. Introduction
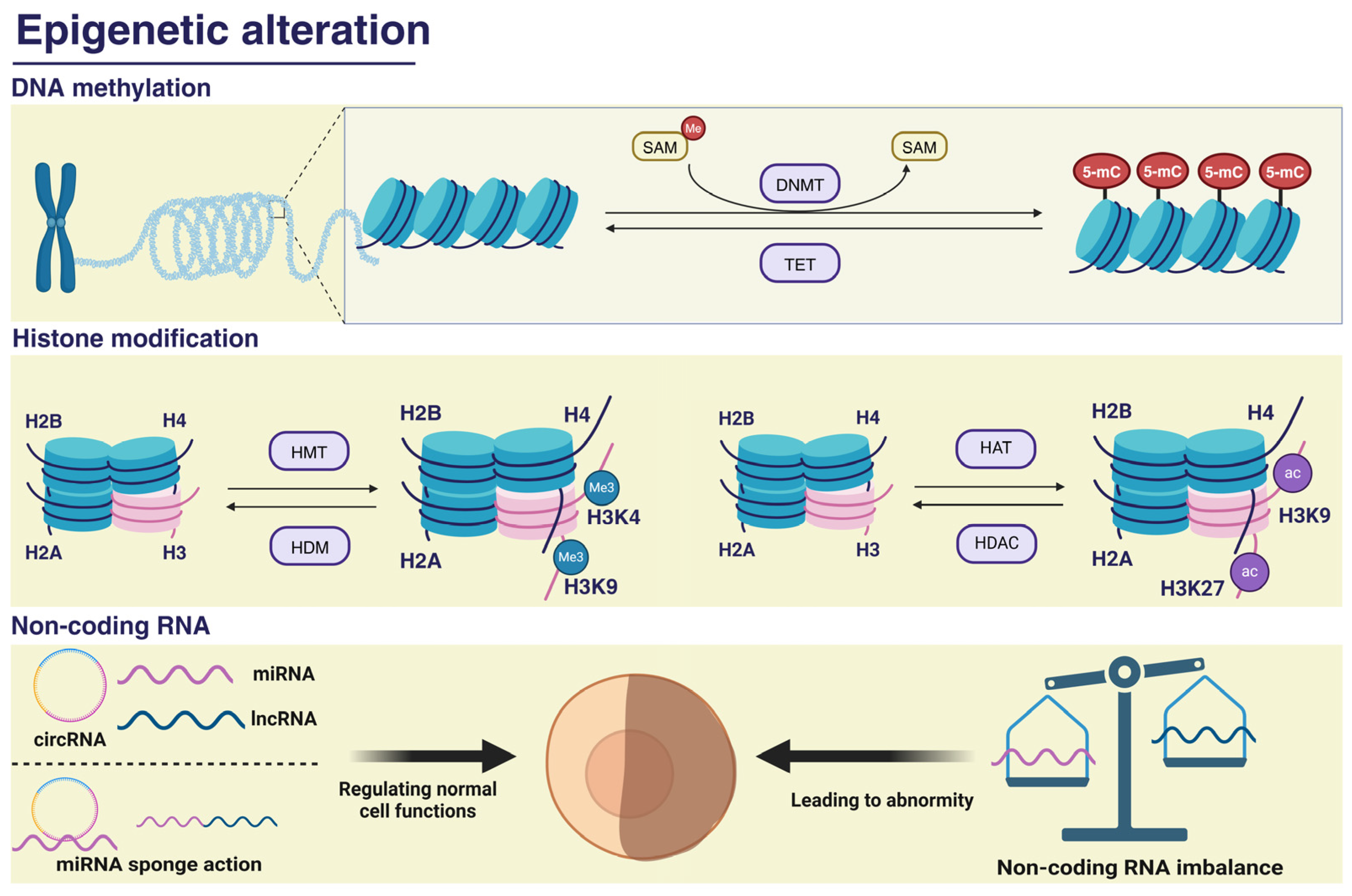
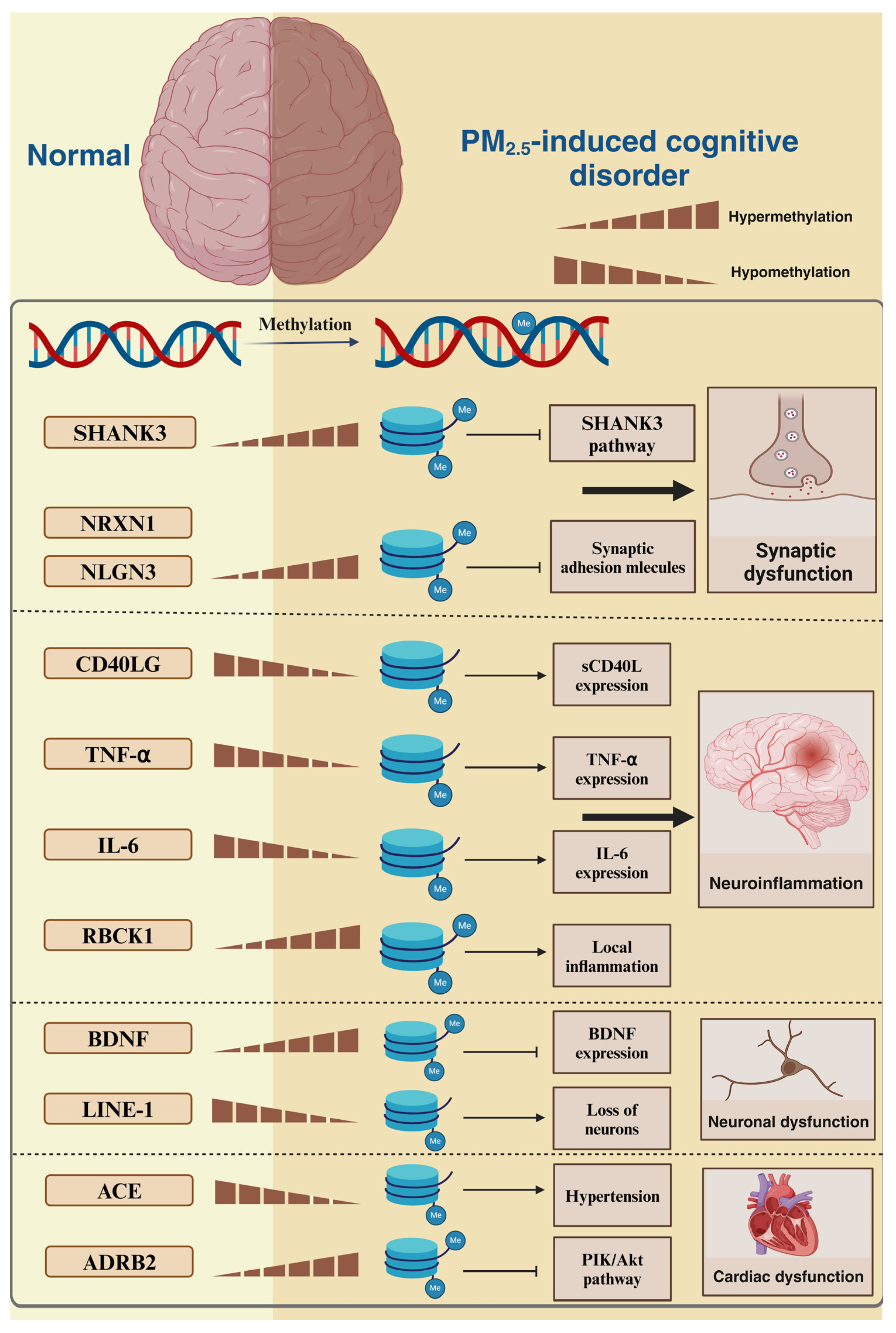
2. PM2.5-Induced DNA Methylation
2.1. PM2.5-Induced DNA Methylation Alterations and Synaptic Dysfunction
2.2. PM2.5-Induced DNA Methylation Alterations and Inflammation
2.3. PM2.5-Induced DNA Methylation Alterations and Neuronal Structural and Functional Abnormalities
2.4. PM2.5-Induced DNA Methylation Alterations and Cardiovascular Factors
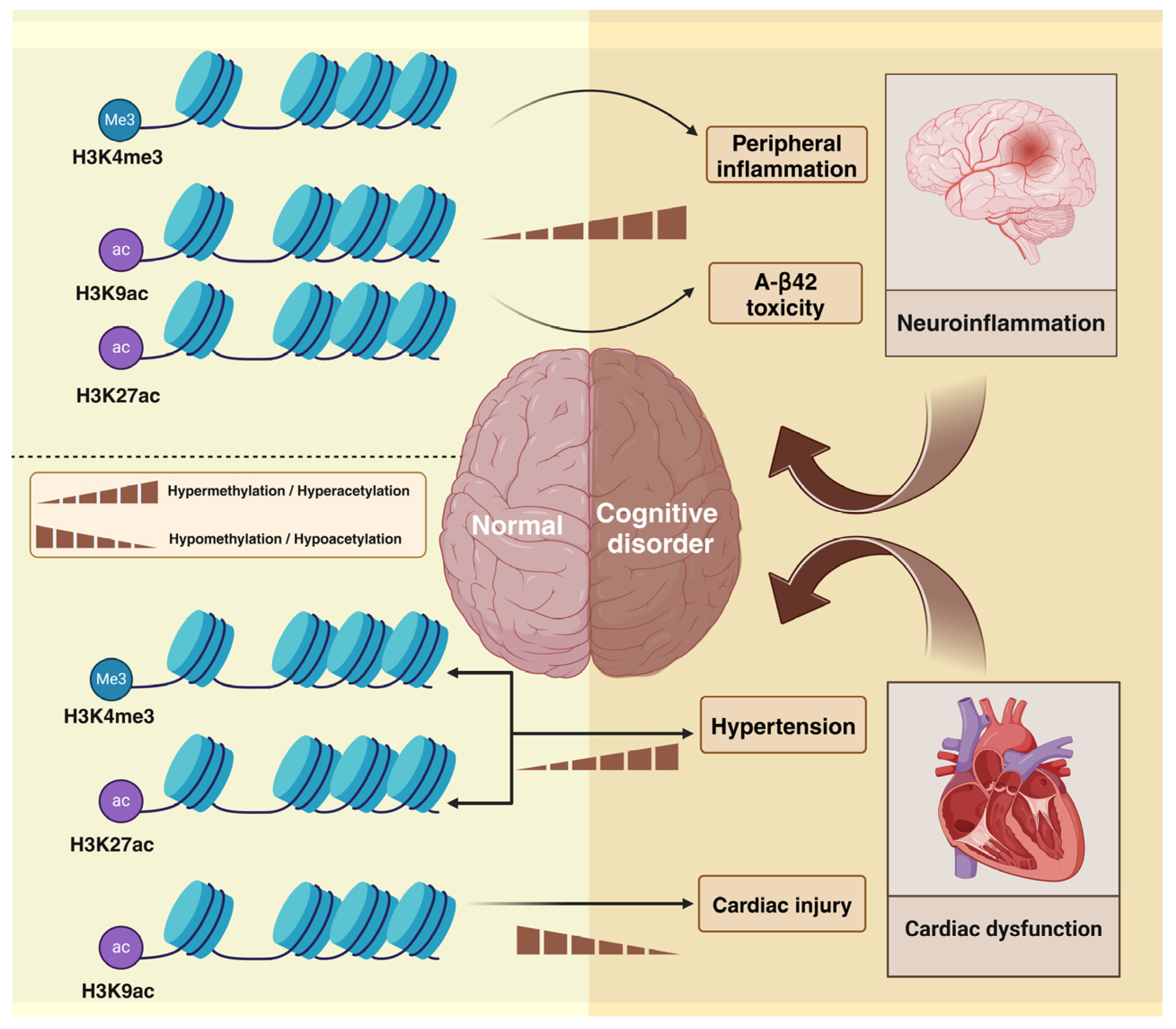
3. PM2.5 Alters Histone Modifications and Induces Cognitive Impairment
3.1. PM2.5-Induced Histone Methylation
3.1.1. PM2.5-Induced Histone Methylation and Inflammation
3.1.2. PM2.5-Induced Histone Methylation and Hypertension
3.2. PM2.5-Induced Histone Acetylation
3.2.1. PM2.5-Induced Histone Acetylation and Amyloid Toxicity
3.2.2. PM2.5-Induced Histone Acetylation and Cardiovascular Factors
4. PM2.5-Associated Non-Coding RNA and Cognitive Impairment
4.1. PM2.5-Associated Non-Coding RNA Alterations and Inflammation
4.2. PM2.5-Associated Non-Coding RNA Alterations and Synaptic Dysfunction
4.3. PM2.5-Associated Non-Coding RNA Alterations and Aβ Deposition and Tau Protein Hyperphosphorylation
4.4. PM2.5-Associated Non-Coding RNA Alterations and Oxidative Stress
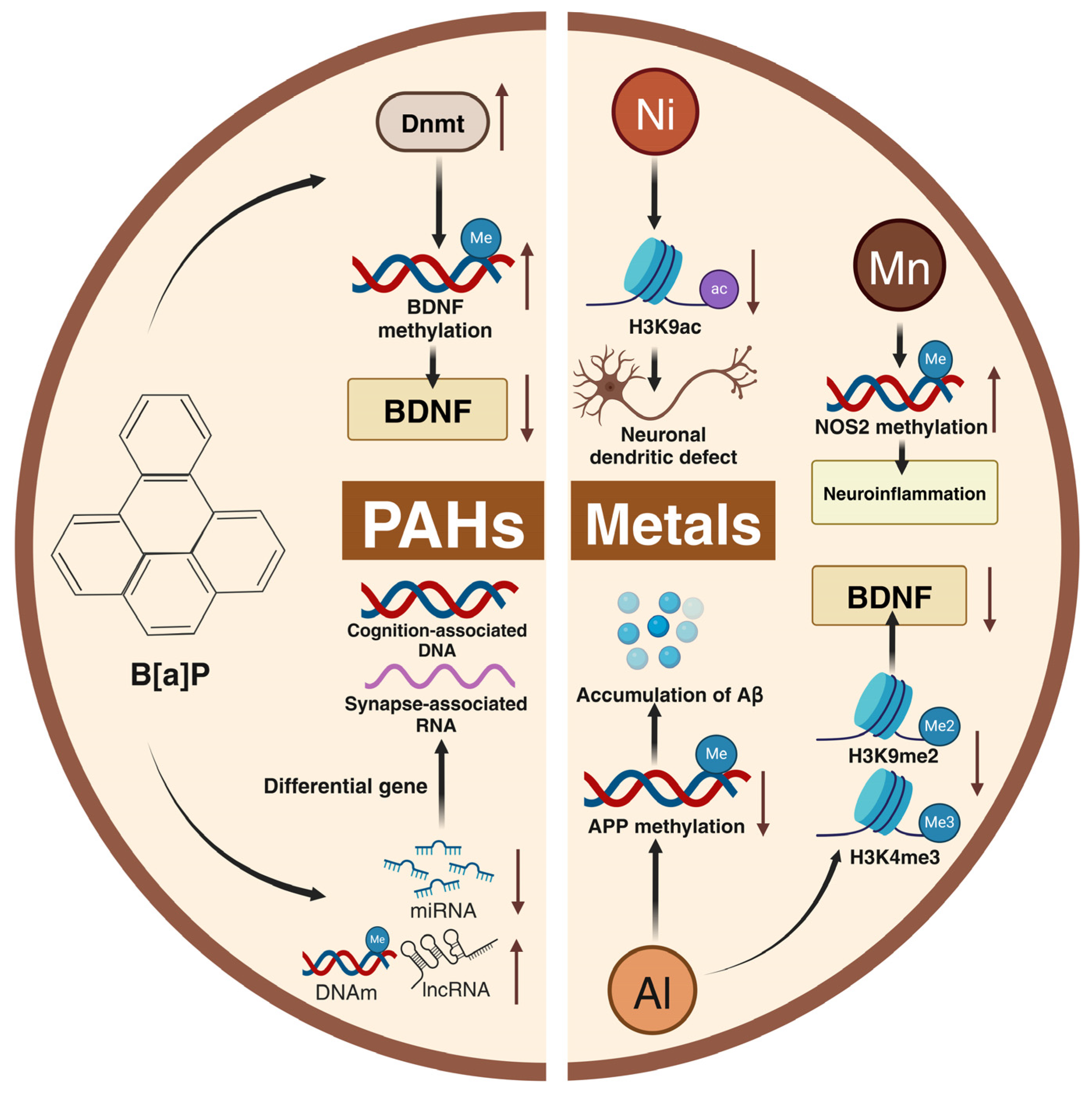
5. Role of Harmful Components of PM2.5 in Cognitive Impairment
5.1. Heavy Metals
5.2. PAHs
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, C.; Chen, R.; Sera, F.; Vicedo-Cabrera, A.M.; Guo, Y.; Tong, S.; Coelho, M.S.Z.S.; Saldiva, P.H.N.; Lavigne, E.; Matus, P.; et al. Ambient Particulate Air Pollution and Daily Mortality in 652 Cities. N. Engl. J. Med. 2019, 381, 705–715. [Google Scholar] [CrossRef]
- Li, J.; Tang, W.; Li, S.; He, C.; Dai, Y.; Feng, S.; Zeng, C.; Yang, T.; Meng, Q.; Meng, J.; et al. Ambient PM2.5 and its components associated with 10-year atherosclerotic cardiovascular disease risk in Chinese adults. Ecotoxicol. Environ. Saf. 2023, 263, 115371. [Google Scholar] [CrossRef]
- Feng, S.; Huang, F.; Zhang, Y.; Feng, Y.; Zhang, Y.; Cao, Y.; Wang, X. The pathophysiological and molecular mechanisms of atmospheric PM2.5 affecting cardiovascular health: A review. Ecotoxicol. Environ. Saf. 2023, 249, 114444. [Google Scholar] [CrossRef]
- He, B.; Xu, H.-M.; Liu, H.-W.; Zhang, Y.-F. Unique regulatory roles of ncRNAs changed by PM2.5 in human diseases. Ecotoxicol. Environ. Saf. 2023, 255, 114812. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ma, T.; Ma, D.; Li, H.; Hua, L.; He, Q.; Deng, X. The Impact of Air Pollution on Neurodegenerative Diseases. Ther. Drug Monit. 2021, 43, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fang, J.; Tang, S.; Du, H.; Zhao, L.; Wang, Y.; Deng, F.; Liu, Y.; Du, Y.; Cui, L.; et al. PM2.5 exposure associated with microbiota gut-brain axis: Multi-omics mechanistic implications from the BAPE study. Innovation 2022, 3, 100213. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Fang, J.; Tang, S.; Deng, F.; Liu, X.; Shen, Y.; Liu, Y.; Kong, F.; Du, Y.; Cui, L.; et al. PM2.5 and Serum Metabolome and Insulin Resistance, Potential Mediation by the Gut Microbiome: A Population-Based Panel Study of Older Adults in China. Environ. Health Perspect. 2022, 130, 027007. [Google Scholar] [CrossRef]
- Meo, S.A.; Salih, M.A.; Al-Hussain, F.; Alkhalifah, J.M.; Meo, A.S.; Akram, A. Akram Environmental pollutants PM2.5, PM10, carbon monoxide (CO), nitrogen dioxide (NO2), sulfur dioxide (SO2), and ozone (O3) impair human cognitive functions. Eur. Rev. Med. Pharmacol. Sci. 2024, 28, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Ke, L.; Zhang, Y.; Fu, Y.; Shen, X.; Zhang, Y.; Ma, X.; Di, Q. Short-term PM2.5 exposure and cognitive function: Association and neurophysiological mechanisms. Environ. Int. 2022, 170, 107593. [Google Scholar] [CrossRef] [PubMed]
- Ball, H.A.; McWhirter, L.; Ballard, C.; Bhome, R.; Blackburn, D.J.; Edwards, M.J.; Fleming, S.M.; Fox, N.C.; Howard, R.; Huntley, J.; et al. Functional cognitive disorder: Dementia’s blind spot. Brain 2020, 143, 2895–2903. [Google Scholar] [CrossRef] [PubMed]
- Rundek, T.; Tolea, M.; Ariko, T.; Fagerli, E.A.; Camargo, C.J. Vascular Cognitive Impairment (VCI). Neurotherapeutics 2021, 19, 68–88. [Google Scholar] [CrossRef]
- Velikonja, T.; Fett, A.-K.; Velthorst, E. Patterns of Nonsocial and Social Cognitive Functioning in Adults With Autism Spectrum Disorder. JAMA Psychiatry 2019, 76, 135. [Google Scholar] [CrossRef]
- Morozova, A.; Zorkina, Y.; Abramova, O.; Pavlova, O.; Pavlov, K.; Soloveva, K.; Volkova, M.; Alekseeva, P.; Andryshchenko, A.; Kostyuk, G.; et al. Neurobiological Highlights of Cognitive Impairment in Psychiatric Disorders. Int. J. Mol. Sci. 2022, 23, 1217. [Google Scholar] [CrossRef]
- Nunez, Y.; Boehme, A.K.; Weisskopf, M.G.; Re, D.B.; Navas-Acien, A.; van Donkelaar, A.; Martin, R.V.; Kioumourtzoglou, M.-A. Fine Particle Exposure and Clinical Aggravation in Neurodegenerative Diseases in New York State. Environ. Health Perspect. 2021, 129, 27003. [Google Scholar] [CrossRef] [PubMed]
- Cory-Slechta, D.A.; Merrill, A.; Sobolewski, M. Air Pollution–Related Neurotoxicity Across the Life Span. Annu. Rev. Pharmacol. 2023, 63, 143–163. [Google Scholar] [CrossRef]
- Rump, K.; Adamzik, M. Epigenetic Mechanisms of Postoperative Cognitive Impairment Induced by Anesthesia and Neuroinflammation. Cells 2022, 11, 2954. [Google Scholar] [CrossRef]
- Harman, M.F.; Martín, M.G. Epigenetic mechanisms related to cognitive decline during aging. J. Neurosci. Res. 2019, 98, 234–246. [Google Scholar] [CrossRef]
- Maity, S.; Farrell, K.; Navabpour, S.; Narayanan, S.N.; Jarome, T.J. Epigenetic Mechanisms in Memory and Cognitive Decline Associated with Aging and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 12280. [Google Scholar] [CrossRef]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications—Cause and consequence of genome function. Nat. Rev. Genet. 2022, 23, 563–580. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Q.; Su, Z.; Liu, Y.; Yan, T.; Zhang, Y.; Wang, T.; Wei, X.; Chen, Z.; Hu, G.; et al. Genetic damage and potential mechanism exploration under different air pollution patterns by multi-omics. Environ. Int. 2022, 170, 107636. [Google Scholar] [CrossRef] [PubMed]
- Younesian, S.; Yousefi, A.-M.; Momeny, M.; Ghaffari, S.H.; Bashash, D. The DNA Methylation in Neurological Diseases. Cells 2022, 11, 3439. [Google Scholar] [CrossRef]
- Alcalà-Vida, R.; Awada, A.; Boutillier, A.-L.; Merienne, K. Epigenetic mechanisms underlying enhancer modulation of neuronal identity, neuronal activity and neurodegeneration. Neurobiol. Dis. 2021, 147, 105155. [Google Scholar] [CrossRef] [PubMed]
- Xylaki, M.; Atzler, B.; Outeiro, T.F. Epigenetics of the Synapse in Neurodegeneration. Curr. Neurol. Neurosci. 2019, 19, 72. [Google Scholar] [CrossRef] [PubMed]
- Prasher, D.; Greenway, S.C.; Singh, R.B. The impact of epigenetics on cardiovascular disease. Biochem. Cell Biol. 2020, 98, 12–22. [Google Scholar] [CrossRef]
- Giallongo, S.; Longhitano, L.; Denaro, S.; D’Aprile, S.; Torrisi, F.; La Spina, E.; Giallongo, C.; Mannino, G.; Lo Furno, D.; Zappalà, A.; et al. The Role of Epigenetics in Neuroinflammatory-Driven Diseases. Int. J. Mol. Sci. 2022, 23, 15218. [Google Scholar] [CrossRef]
- Li, K.; Liang, X.; Xie, X.; Tian, L.; Yan, J.; Lin, B.; Liu, H.; Lai, W.; Liu, X.; Xi, Z. Role of SHANK3 in concentrated ambient PM2. 5 exposure induced autism-like phenotype. Heliyon 2023, 9, e14328. [Google Scholar] [CrossRef]
- Moutin, E.; Sakkaki, S.; Compan, V.; Bouquier, N.; Giona, F.; Areias, J.; Goyet, E.; Hemonnot-Girard, A.-L.; Seube, V.; Glasson, B.; et al. Restoring glutamate receptosome dynamics at synapses rescues autism-like deficits in Shank3-deficient mice. Mol. Psychiatry 2021, 26, 7596–7609. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Liang, F.; Meng, G.; Nie, Z.; Zhou, R.; Cheng, W.; Wu, X.; Feng, Y.; Wang, Y. Redox/methylation mediated abnormal DNA methylation as regulators of ambient fine particulate matter-induced neurodevelopment related impairment in human neuronal cells. Sci. Rep. 2016, 6, 33402. [Google Scholar] [CrossRef] [PubMed]
- Tononi, G.; Cirelli, C. Sleep and synaptic down-selection. Eur. J. Neurosci. 2019, 51, 413–421. [Google Scholar] [CrossRef]
- Khlghatyan, J.; Evstratova, A.; Bozoyan, L.; Chamberland, S.; Chatterjee, D.; Marakhovskaia, A.; Soares Silva, T.; Toth, K.; Mongrain, V.; Beaulieu, J.M. Fxr1 regulates sleep and synaptic homeostasis. EMBO J. 2020, 39, e103864. [Google Scholar] [CrossRef]
- Hepsomali, P.; Coxon, C. Inflammation and diet: Focus on mental and cognitive health. Adv. Clin. Exp. Med. 2022, 31, 821–825. [Google Scholar] [CrossRef]
- Chen, R.; Meng, X.; Zhao, A.; Wang, C.; Yang, C.; Li, H.; Cai, J.; Zhao, Z.; Kan, H. DNA hypomethylation and its mediation in the effects of fine particulate air pollution on cardiovascular biomarkers: A randomized crossover trial. Environ. Int. 2016, 94, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Zorkina, Y.; Abramova, O.; Ushakova, V.; Andreyuk, D.; Andriushchenko, N.; Pavlov, K.; Savilov, V.; Soloveva, K.; Kurmishev, M.; Syunyakov, T.; et al. Inflammatory biomarkers and lipid metabolism parameters in women with mild cognitive impairment and dementia. Women Health 2023, 63, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Lecca, D.; Jung, Y.J.; Scerba, M.T.; Hwang, I.; Kim, Y.K.; Kim, S.; Modrow, S.; Tweedie, D.; Hsueh, S.C.; Liu, D.; et al. Role of chronic neuroinflammation in neuroplasticity and cognitive function: A hypothesis. Alzheimer’s Dement. 2022, 18, 2327–2340. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; O’Brien, K.M.; Xu, Z.; Sandler, D.P.; Taylor, J.A.; Weinberg, C.R. Long-term ambient fine particulate matter and DNA methylation in inflammation pathways: Results from the Sister Study. Epigenetics 2019, 15, 524–535. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liang, D.; Ebelt, S.; Gearing, M.; Kobor, M.S.; Konwar, C.; Maclsaac, J.L.; Dever, K.; Wingo, A.; Levey, A.; et al. Differential DNA Methylation in the Brain as Potential Mediator of the Association between Traffic-related PM2.5 and Neuropathology Markers of Alzheimer’s Disease. Alzheimer’s Dement. 2024, 20, 2538–2551. [Google Scholar] [CrossRef] [PubMed]
- Sordillo, J.E.; Cardenas, A.; Qi, C.; Rifas-Shiman, S.L.; Coull, B.; Luttmann-Gibson, H.; Schwartz, J.; Kloog, I.; Hivert, M.-F.; DeMeo, D.L.; et al. Residential PM2.5 exposure and the nasal methylome in children. Environ. Int. 2021, 153, 106505. [Google Scholar] [CrossRef]
- Hasegawa-Ishii, S.; Shimada, A.; Imamura, F. Neuroplastic changes in the olfactory bulb associated with nasal inflammation in mice. J. Allergy Clin. Immunol. 2019, 143, 978–989.e3. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Namkung, H.; Smith, A.; Sakamoto, S.; Zhu, X.; Ishizuka, K.; Lane, A.P.; Sawa, A.; Kamiya, A. Causal impact of local inflammation in the nasal cavity on higher brain function and cognition. Neurosci. Res. 2021, 172, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, T.; Zhou, J.; Cao, Z.; Liao, Z.; Zhao, Y.; Su, X.; He, J.; Hua, J. Prenatal exposure to concentrated ambient PM2.5 results in spatial memory defects regulated by DNA methylation in male mice offspring. Environ. Sci. Pollut. Res. 2022, 30, 35142–35152. [Google Scholar] [CrossRef] [PubMed]
- Shou, Y.; Huang, Y.; Zhu, X.; Liu, C.; Hu, Y.; Wang, H. A review of the possible associations between ambient PM2.5 exposures and the development of Alzheimer’s disease. Ecotoxicol. Environ. Saf. 2019, 174, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Louis, S.; Carlson, A.K.; Suresh, A.; Rim, J.; Mays, M.; Ontaneda, D.; Dhawan, A. Impacts of Climate Change and Air Pollution on Neurologic Health, Disease, and Practice. Neurology 2023, 100, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.G.; Cole, T.B.; Dao, K.; Chang, Y.-C.; Coburn, J.; Garrick, J.M. Effects of air pollution on the nervous system and its possible role in neurodevelopmental and neurodegenerative disorders. Pharmacol. Therapeut 2020, 210, 107523. [Google Scholar] [CrossRef]
- Saleh, A.; Macia, A.; Muotri, A.R. Transposable Elements, Inflammation, and Neurological Disease. Front. Neurol. 2019, 10, 894. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Tangsuwansri, C.; Saeliw, T.; Thongkorn, S.; Chonchaiya, W.; Suphapeetiporn, K.; Mutirangura, A.; Tencomnao, T.; Hu, V.W.; Sarachana, T. Investigation of epigenetic regulatory networks associated with autism spectrum disorder (ASD) by integrated global LINE-1 methylation and gene expression profiling analyses. PLoS ONE 2018, 13, e0201071. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, M.; Liu, W.; Li, Y.; Qin, Y.; Xu, Y. Transgenerational transmission of neurodevelopmental disorders induced by maternal exposure to PM2.5. Chemosphere 2020, 255, 126920. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, Y.; Sterling, K.; Song, W. Brain-derived neurotrophic factor in Alzheimer’s disease and its pharmaceutical potential. Transl. Neurodegener. 2022, 11, 4. [Google Scholar] [CrossRef]
- Weaver, J.R.; Susiarjo, M.; Bartolomei, M.S. Imprinting and epigenetic changes in the early embryo. Mamm. Genome 2009, 20, 532–543. [Google Scholar] [CrossRef]
- Johnson, N.M.; Hoffmann, A.R.; Behlen, J.C.; Lau, C.; Pendleton, D.; Harvey, N.; Shore, R.; Li, Y.; Chen, J.; Tian, Y.; et al. Air pollution and children’s health—A review of adverse effects associated with prenatal exposure from fine to ultrafine particulate matter. Environ. Health Prev. Med. 2021, 26, 72. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Zhang, X.; Tian, F.; Pan, B.; Peng, R.; Wang, Y.; Xia, M.; Yang, M.; Hu, J.; Kan, H.; et al. Maternal exposure to ambient PM2.5 causes fetal growth restriction via the inhibition of spiral artery remodeling in mice. Ecotoxicol. Environ. Saf. 2022, 237, 113512. [Google Scholar] [CrossRef]
- Yue, H.; Ji, X.; Li, G.; Hu, M.; Sang, N. Maternal Exposure to PM2.5 Affects Fetal Lung Development at Sensitive Windows. Environ. Sci. Technol. 2019, 54, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Wang, F.; Li, C. Effect of Fine Particulate Matter (PM2.5) on Rat Placenta Pathology and Perinatal Outcomes. Med. Sci. Monit. 2016, 22, 3274–3280. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, P.; Zhou, Y.; Xia, B.; Zhu, Q.; Ge, W.; Li, J.; Shi, H.; Xiao, X.; Zhang, Y. Prenatal fine particulate matter exposure, placental DNA methylation changes, and fetal growth. Environ. Int. 2021, 147, 106313. [Google Scholar] [CrossRef]
- Xu, C.; Tao, X.; Ma, X.; Zhao, R.; Cao, Z.; Morishita, R. Cognitive Dysfunction after Heart Disease: A Manifestation of the Heart-Brain Axis. Oxidative Med. Cell. Longev. 2021, 2021, 4899688. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Gottesman, R.F. Neurovascular and Cognitive Dysfunction in Hypertension. Circ. Res. 2019, 124, 1025–1044. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, R.; Cai, J.; Shi, J.; Yang, C.; Tse, L.A.; Li, H.; Lin, Z.; Meng, X.; Liu, C.; et al. Personal exposure to fine particulate matter and blood pressure: A role of angiotensin converting enzyme and its DNA methylation. Environ. Int. 2016, 94, 661–666. [Google Scholar] [CrossRef]
- van Nieuwkerk, A.C.; Delewi, R.; Wolters, F.J.; Muller, M.; Daemen, M.; Biessels, G.J. Cognitive Impairment in Patients With Cardiac Disease: Implications for Clinical Practice. Stroke 2023, 54, 2181–2191. [Google Scholar] [CrossRef]
- Faulkner, K.M.; Dickson, V.V.; Fletcher, J.; Katz, S.D.; Chang, P.P.; Gottesman, R.F.; Witt, L.S.; Shah, A.M.; D’Eramo Melkus, G. Factors Associated With Cognitive Impairment in Heart Failure With Preserved Ejection Fraction. J. Cardiovasc. Nurs. 2022, 37, 17–30. [Google Scholar] [CrossRef]
- Yang, X.; Feng, L.; Zhang, Y.; Hu, H.; Shi, Y.; Liang, S.; Zhao, T.; Fu, Y.; Duan, J.; Sun, Z. Cytotoxicity induced by fine particulate matter (PM2.5) via mitochondria-mediated apoptosis pathway in human cardiomyocytes. Ecotoxicol. Environ. Saf. 2018, 161, 198–207. [Google Scholar] [CrossRef]
- Yang, X.; Zhao, T.; Feng, L.; Shi, Y.; Jiang, J.; Liang, S.; Sun, B.; Xu, Q.; Duan, J.; Sun, Z. PM2.5-induced ADRB2 hypermethylation contributed to cardiac dysfunction through cardiomyocytes apoptosis via PI3K/Akt pathway. Environ. Int. 2019, 127, 601–614. [Google Scholar] [CrossRef]
- Grande, G.; Ljungman, P.L.S.; Eneroth, K.; Bellander, T.; Rizzuto, D. Association Between Cardiovascular Disease and Long-term Exposure to Air Pollution With the Risk of Dementia. JAMA Neurol. 2020, 77, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.E.; Greer, C.B.; Coleman, B.C.; Sweatt, J.D. Histone H3 lysine K4 methylation and its role in learning and memory. Epigenetics & Chromatin 2019, 12, 7. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of Histone Modification. In Histone Mutations and Cancer; Advances in Experimental Medicine and Biology Series; Springer: Singapore, 2021; Volume 1283, pp. 1–16. [Google Scholar] [CrossRef]
- Vrijens, K.; Trippas, A.-J.; Lefebvre, W.; Vanpoucke, C.; Penders, J.; Janssen, B.G.; Nawrot, T.S. Association of Prenatal Exposure to Ambient Air Pollution With Circulating Histone Levels in Maternal Cord Blood. JAMA Netw. Open 2020, 3, e205156. [Google Scholar] [CrossRef]
- Abrams, S.T.; Zhang, N.; Manson, J.; Liu, T.; Dart, C.; Baluwa, F.; Wang, S.S.; Brohi, K.; Kipar, A.; Yu, W.; et al. Circulating Histones Are Mediators of Trauma-associated Lung Injury. Am. J. Respir. Crit. Care Med. 2013, 187, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Kresovich, J.K.; Zhang, Z.; Fang, F.; Zheng, Y.; Sanchez-Guerra, M.; Joyce, B.T.; Zhong, J.; Chervona, Y.; Wang, S.; Chang, D.; et al. Histone 3 modifications and blood pressure in the Beijing Truck Driver Air Pollution Study. Biomarkers 2017, 22, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Toth, P.; Tarantini, S.; Prodan, C.I.; Sorond, F.; Merkely, B.; Csiszar, A. Hypertension-induced cognitive impairment: From pathophysiology to public health. Nat. Rev. Nephrol. 2021, 17, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Nativio, R.; Lan, Y.; Donahue, G.; Sidoli, S.; Berson, A.; Srinivasan, A.R.; Shcherbakova, O.; Amlie-Wolf, A.; Nie, J.; Cui, X.; et al. An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer’s disease. Nat. Genet. 2020, 52, 1024–1035. [Google Scholar] [CrossRef]
- Thiankhaw, K.; Chattipakorn, N.; Chattipakorn, S.C. PM2.5 exposure in association with AD-related neuropathology and cognitive outcomes. Environ. Pollut. 2022, 292, 118320. [Google Scholar] [CrossRef]
- Ding, R.; Jin, Y.; Liu, X.; Zhu, Z.; Zhang, Y.; Wang, T.; Xu, Y. H3K9 acetylation change patterns in rats after exposure to traffic-related air pollution. Environ. Toxicol. Pharmacol. 2016, 42, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xu, J.; Chen, Y.; Guo, X.; Zheng, Y.; Wang, Q.; Chen, Y.; Ni, Y.; Zhu, Y.; Joyce, B.T.; et al. Characterization of genome-wide H3K27ac profiles reveals a distinct PM2.5-associated histone modification signature. Environ. Health 2015, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhao, Y.; Shi, J.; Zhao, C.; Xie, P.; Huang, W.; Yong, T.; Cai, Z. Effects of PM2.5 exposure in utero on heart injury, histone acetylation and GATA4 expression in offspring mice. Chemosphere 2020, 256, 127133. [Google Scholar] [CrossRef] [PubMed]
- Afouda, B.A. Towards Understanding the Gene-Specific Roles of GATA Factors in Heart Development: Does GATA4 Lead the Way? Int. J. Mol. Sci. 2022, 23, 5255. [Google Scholar] [CrossRef]
- Chen, Z.; Wu, H.; Zhang, M. Long non-coding RNA: An underlying bridge linking neuroinflammation and central nervous system diseases. Neurochem. Int. 2021, 148, 105101. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.; Lee, S.; Lee, H.; Min, J.-W.; Iwatsubo, T.; Teunissen, C.; Cho, H.-J.; Ryu, J.-H. Targeting MicroRNA-485-3p Blocks Alzheimer’s Disease Progression. Int. J. Mol. Sci. 2021, 22, 13136. [Google Scholar] [CrossRef] [PubMed]
- Balusu, S.; Horré, K.; Thrupp, N.; Craessaerts, K.; Snellinx, A.; Serneels, L.; T’Syen, D.; Chrysidou, I.; Arranz, A.M.; Sierksma, A.; et al. MEG3 activates necroptosis in human neuron xenografts modeling Alzheimer’s disease. Science 2023, 381, 1176–1182. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Herron, S.; Silveira, S.; Kleemann, K.; Gauthier, C.; Mallah, D.; Cheng, Y.; Margeta, M.A.; Pitts, K.M.; Barry, J.-L.; et al. Identification of a protective microglial state mediated by miR-155 and interferon-γ signaling in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2023, 26, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-S.; He, S.-L.; Chen, W.-C.; Wang, C.-M.; Huang, Q.-M.; Shi, Y.-C.; Lin, S.; He, H.-f. Recent progress on the role of non-coding RNA in postoperative cognitive dysfunction. Front. Cell. Neurosci. 2022, 16, 1024475. [Google Scholar] [CrossRef]
- Soutschek, M.; Schratt, G. Non-coding RNA in the wiring and remodeling of neural circuits. Neuron 2023, 111, 2140–2154. [Google Scholar] [CrossRef] [PubMed]
- Olufunmilayo, E.O.; Holsinger, R.M.D. Roles of Non-Coding RNA in Alzheimer’s Disease Pathophysiology. Int. J. Mol. Sci. 2023, 24, 12498. [Google Scholar] [CrossRef]
- Aghaei-Zarch, S.M.; Alipourfard, I.; Rasoulzadeh, H.; Najafi, S.; Aghaei-Zarch, F.; Partov, S.; Movafagh, A.; Jahanara, A.; Toolabi, A.; Sheikhmohammadi, A.; et al. Non-coding RNAs: An emerging player in particulate matter 2.5-mediated toxicity. Int. J. Biol. Macromol. 2023, 235, 123790. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, S.; Xu, C.; Zhao, J.; Hou, L.; Jiang, F.; Zhu, Z.; Wang, Y.; Tian, L. microRNA-149-5p mediates the PM2.5-induced inflammatory response by targeting TAB2 via MAPK and NF-κB signaling pathways in vivo and in vitro. Cell Biol. Toxicol. 2021, 39, 703–717. [Google Scholar] [CrossRef]
- Vassiliou, A.; Vitsas, V.; Kardara, M.; Keskinidou, C.; Michalopoulou, P.; Rovina, N.; Dimopoulou, I.; Orfanos, S.E.; Tsoukalas, G.; Koutsoukou, A.; et al. Study of Inflammatory Biomarkers in COPD and Asthma Exacerbations. Adv. Respir. Med. 2020, 88, 558–566. [Google Scholar] [CrossRef]
- Muñoz-Delgado, L.; Labrador-Espinosa, M.Á.; Macías-García, D.; Jesús, S.; Benítez Zamora, B.; Fernández-Rodríguez, P.; Adarmes-Gómez, A.D.; Reina Castillo, M.I.; Castro-Labrador, S.; Silva-Rodríguez, J.; et al. Peripheral Inflammation Is Associated with Dopaminergic Degeneration in Parkinson’s Disease. Mov. Disord. 2023, 38, 755–763. [Google Scholar] [CrossRef]
- Costas, C.; Faro, L.R.F. Do Naturally Occurring Antioxidants Protect Against Neurodegeneration of the Dopaminergic System? A Systematic Revision in Animal Models of Parkinson’s Disease. Curr. Neuropharmacol. 2022, 20, 432–459. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Li, Y.; Zhou, J.; Xu, J.; Yang, B. PM2.5 deregulated microRNA and inflammatory microenvironment in lung injury. Environ. Toxicol. Pharmacol. 2022, 91, 103832. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wong, J.Y.Y.; Dai, Y.; Ren, D.; Blechter, B.; Duan, H.; Niu, Y.; Xu, J.; Fu, W.; Meliefste, K.; et al. Occupational exposure to diesel engine exhaust and serum levels of microRNAs in a cross-sectional molecular epidemiology study in China. Environ. Mol. Mutagen. 2023, 64, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Wei, J.; Liang, S.; Sun, Z.; Duan, J. miR-205/IRAK2 signaling pathway is associated with urban airborne PM2.5-induced myocardial toxicity. Nanotoxicology 2020, 14, 1198–1212. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jia, Y.; Nan, A.; Zhang, N.; Zhou, H.; Chen, L.; Pan, X.; Qiu, M.; Zhu, J.; Zhang, H.; et al. CircRNA104250 and lncRNAuc001.dgp.1 promote the PM2.5-induced inflammatory response by co-targeting miR-3607-5p in BEAS-2B cells. Environ. Pollut. 2020, 258, 113749. [Google Scholar] [CrossRef] [PubMed]
- Emami, M.H.; Sereshki, N.; Malakoutikhah, Z.; Dehkordi, S.A.E.; Fahim, A.; Mohammadzadeh, S.; Maghool, F. Nrf2 signaling pathway in trace metal carcinogenesis: A cross-talk between oxidative stress and angiogenesis. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2022, 254, 109266. [Google Scholar] [CrossRef]
- Yue, W.; Tong, L.; Liu, X.; Weng, X.; Chen, X.; Wang, D.; Dudley, S.C.; Weir, E.K.; Ding, W.; Lu, Z.; et al. Short term Pm2.5 exposure caused a robust lung inflammation, vascular remodeling, and exacerbated transition from left ventricular failure to right ventricular hypertrophy. Redox Biol. 2019, 22, 101161. [Google Scholar] [CrossRef]
- Xu, J.; Xu, H.; Ma, K.; Wang, Y.; Niu, B.; Zhang, L.; Li, F. lncRNA Gm16410 Mediates PM2.5-Induced Macrophage Activation via PI3K/AKT Pathway. Front. Cell Dev. Biol. 2021, 9, 18045. [Google Scholar] [CrossRef]
- Liao, F.; Tan, Y.; Wang, Y.; Zhou, C.; Wang, Q.; Li, J.; He, L.; Peng, X. lncRNA AABR07005593.1 potentiates PM2.5-induced interleukin-6 expression by targeting MCCC1. Ecotoxicol. Environ. Saf. 2021, 226, 112834. [Google Scholar] [CrossRef]
- Xie, J.; Li, S.; Ma, X.; Li, R.; Zhang, H.; Li, J.; Yan, X. MiR-217-5p inhibits smog (PM2.5)-induced inflammation and oxidative stress response of mouse lung tissues and macrophages through targeting STAT1. Aging 2022, 14, 6796–6808. [Google Scholar] [CrossRef]
- Peng, Y.-H.; Wu, B.-R.; Su, C.-H.; Liao, W.-C.; Muo, C.-H.; Hsia, T.-C.; Kao, C.-H. Adult asthma increases dementia risk: A nationwide cohort study. J. Epidemiol. Community Health 2015, 69, 123–128. [Google Scholar] [CrossRef]
- Firoozi, Z.; Shahi, A.; Mohammadisoleimani, E.; Afzali, S.; Mansoori, B.; Bahmanyar, M.; Mohaghegh, P.; Dastsooz, H.; Pezeshki, B.; Nikfar, G.; et al. CircRNA-associated ceRNA networks (circCeNETs) in chronic obstructive pulmonary disease (COPD). Life Sciences 2024, 349, 122715. [Google Scholar] [CrossRef]
- Huang, J.; Hu, Y.; Wang, Y.; Jin, Z. Activation of Notch1-GATA3 pathway in asthma bronchial epithelial cells induced by acute PM2.5 exposure and the potential protective role of microRNA-139-5p. J. Asthma 2024, 61, 959–969. [Google Scholar] [CrossRef]
- Wang, L.; Xu, J.; Liu, H.; Li, J.; Hao, H. PM2.5 inhibits SOD1 expression by up-regulating microRNA-206 and promotes ROS accumulation and disease progression in asthmatic mice. Int. Immunopharm. 2019, 76, 105871. [Google Scholar] [CrossRef]
- Bairamian, D.; Sha, S.; Rolhion, N.; Sokol, H.; Dorothée, G.; Lemere, C.A.; Krantic, S. Microbiota in neuroinflammation and synaptic dysfunction: A focus on Alzheimer’s disease. Mol. Neurodegener. 2022, 17, 19. [Google Scholar] [CrossRef]
- Liu, J.; Liu, B.; Yuan, P.; Cheng, L.; Sun, H.; Gui, J.; Pan, Y.; Huang, D.; Chen, H.; Jiang, L. Role of PKA/CREB/BDNF signaling in PM2.5-induced neurodevelopmental damage to the hippocampal neurons of rats. Ecotoxicol. Environ. Saf. 2021, 214, 112005. [Google Scholar] [CrossRef]
- Liu, F.; Liu, C.; Liu, Y.; Wang, J.; Wang, Y.; Yan, B. Neurotoxicity of the air-borne particles: From molecular events to human diseases. J. Hazard. Mater. 2023, 457, 131827. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Steenland, K.; Liu, P.; van Donkelaar, A.; Martin, R.V.; Chang, H.H.; Caudle, W.M.; Schwartz, J.; Koutrakis, P.; et al. Long-term effects of PM2.5 components on incident dementia in the northeastern United States. Innovation 2022, 3, 100208. [Google Scholar] [CrossRef]
- Liu, X.-q.; Huang, J.; Song, C.; Zhang, T.-l.; Liu, Y.-p.; Yu, L. Neurodevelopmental toxicity induced by PM2.5 Exposure and its possible role in Neurodegenerative and mental disorders. Hum. Exp. Toxicol. 2023, 42, 9603271231191436. [Google Scholar] [CrossRef]
- Chopra, N.; Wang, R.; Maloney, B.; Nho, K.; Beck, J.S.; Pourshafie, N.; Niculescu, A.; Saykin, A.J.; Rinaldi, C.; Counts, S.E.; et al. MicroRNA-298 reduces levels of human amyloid-β precursor protein (APP), β-site APP-converting enzyme 1 (BACE1) and specific tau protein moieties. Mol. Psychiatry 2020, 26, 5636–5657. [Google Scholar] [CrossRef]
- Ku, T.; Li, B.; Gao, R.; Zhang, Y.; Yan, W.; Ji, X.; Li, G.; Sang, N. NF-κB-regulated microRNA-574-5p underlies synaptic and cognitive impairment in response to atmospheric PM2.5 aspiration. Part. Fibre Toxicol. 2017, 14, 34. [Google Scholar] [CrossRef]
- Yang, J.; Jia, L.; Li, Y.; Qiu, Q.; Quan, M.; Jia, J. Fluid Biomarkers in Clinical Trials for Alzheimer’s Disease: Current and Future Application. J. Alzheimers Dis. 2021, 81, 19–32. [Google Scholar] [CrossRef]
- Watson, C.M.; Dammer, E.B.; Ping, L.; Duong, D.M.; Modeste, E.; Carter, E.K.; Johnson, E.C.B.; Levey, A.I.; Lah, J.J.; Roberts, B.R.; et al. Quantitative Mass Spectrometry Analysis of Cerebrospinal Fluid Protein Biomarkers in Alzheimer’s Disease. Sci. Data 2023, 10, 261. [Google Scholar] [CrossRef]
- Chauhan, A.; Chauhan, V. Beneficial Effects of Walnuts on Cognition and Brain Health. Nutrients 2020, 12, 550. [Google Scholar] [CrossRef]
- Imbimbo, B.P.; Ippati, S.; Watling, M.; Imbimbo, C. Role of monomeric amyloid-β in cognitive performance in Alzheimer’s disease: Insights from clinical trials with secretase inhibitors and monoclonal antibodies. Pharmacol. Res. 2023, 187, 106631. [Google Scholar] [CrossRef]
- Hou, T.; Liao, J.; Zhang, C.; Sun, C.; Li, X.; Wang, G. Elevated expression of miR-146, miR-139 and miR-340 involved in regulating Th1/Th2 balance with acute exposure of fine particulate matter in mice. Int. Immunopharm. 2018, 54, 68–77. [Google Scholar] [CrossRef]
- Tan, X.; Luo, Y.; Pi, D.; Xia, L.; Li, Z.; Tu, Q. MiR-340 Reduces the Accumulation of Amyloid-β Through Targeting BACE1 (β-site Amyloid Precursor Protein Cleaving Enzyme 1) in Alzheimer’s Disease. Curr. Neurovasc. Res. 2020, 17, 86–92. [Google Scholar] [CrossRef]
- Liu, C.-g.; Wang, J.-l.; Li, L.; Xue, L.-x.; Zhang, Y.-q.; Wang, P.-c. MicroRNA-135a and -200b, potential Biomarkers for Alzheimer׳s disease, regulate β secretase and amyloid precursor protein. Brain Res. 2014, 1583, 55–64. [Google Scholar] [CrossRef]
- Lee, H.-W. Elevated microRNA-135a is associated with pulmonary arterial hypertension in experimental mouse model. Oncotarget 2017, 8, 22. [Google Scholar] [CrossRef]
- Wegmann, S.; Biernat, J.; Mandelkow, E. A current view on Tau protein phosphorylation in Alzheimer’s disease. Curr. Opin. Neurobiol. 2021, 69, 131–138. [Google Scholar] [CrossRef]
- Henneghan, A.; Haley, A.P.; Kesler, S. Exploring Relationships Among Peripheral Amyloid Beta, Tau, Cytokines, Cognitive Function, and Psychosomatic Symptoms in Breast Cancer Survivors. Biol. Res. Nurs. 2019, 22, 126–138. [Google Scholar] [CrossRef]
- Opland, C.K.; Bryan, M.R.; Harris, B.; McGillion-Moore, J.; Tian, X.; Chen, Y.; Itano, M.S.; Diering, G.H.; Meeker, R.B.; Cohen, T.J. Activity-dependent tau cleavage by caspase-3 promotes neuronal dysfunction and synaptotoxicity. iScience 2023, 26, 106905. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Aman, Y.; Ng, C.T.; Chau, W.-H.; Zhang, Z.; Yue, M.; Bohm, C.; Jia, Y.; Li, S.; et al. Amyloid-β toxicity modulates tau phosphorylation through the PAX6 signalling pathway. Brain 2021, 144, 2759–2770. [Google Scholar] [CrossRef]
- Ionescu-Tucker, A.; Cotman, C.W. Emerging roles of oxidative stress in brain aging and Alzheimer’s disease. Neurobiol. Aging 2021, 107, 86–95. [Google Scholar] [CrossRef]
- Feng, S.; Gao, D.; Liao, F.; Zhou, F.; Wang, X. The health effects of ambient PM2.5 and potential mechanisms. Ecotoxicol. Environ. Saf. 2016, 128, 67–74. [Google Scholar] [CrossRef]
- Moufarrej, L.; Verdin, A.; Cazier, F.; Ledoux, F.; Courcot, D. Oxidative stress response in pulmonary cells exposed to different fractions of PM2.5-0.3 from urban, traffic and industrial sites. Environ. Res. 2023, 216, 114572. [Google Scholar] [CrossRef]
- Kumari, S.; Prakash, S.; Gupta, S.K. NRF2: A key regulator of endothelial microRNA transcription. Cardiovasc. Res. 2021, 117, 1241–1242. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, H.; Sun, G.; Zhou, M.; Zhang, H.; Liu, H.; Wang, M.; Zhang, Z.; Chu, H. linc01515 regulates PM2.5-induced oxidative stress via targeting NRF2 in airway epithelial cells. Environ. Pollut. 2023, 331, 121798. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Huang, N.; Wei, S.; Xv, J.; Meng, Q.; Aschner, M.; Li, X.; Chen, R. lncRNA TUG1 as a ceRNA promotes PM exposure-induced airway hyper-reactivity. J. Hazard. Mater. 2021, 416, 125878. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Qu, R.; Sun, B.; Chen, R.; Kan, H.; An, Z.; Jiang, J.; Li, J.; Zhang, Y.; Wu, W. Associations of Short-Term Exposure to Fine Particulate Matter with Neural Damage Biomarkers: A Panel Study of Healthy Retired Adults. Environ. Sci. Technol. 2021, 56, 7203–7213. [Google Scholar] [CrossRef] [PubMed]
- Ikram, M.F.; Farhat, S.M.; Mahboob, A.; Baig, S.; Yaqinuddin, A.; Ahmed, T. Expression of DnMTs and MBDs in AlCl3-Induced Neurotoxicity Mouse Model. Biol. Trace Elem. Res. 2020, 199, 3433–3444. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Zhou, Y.; Li, H.; Li, Y.; Xue, X.; Li, L.; Liu, Q.; Zhao, X.; Niu, Q. Relationship between occupational aluminium exposure and histone lysine modification through methylation. J. Trace Elem. Med. Biol. 2020, 61, 126551. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Liu, M.; Mei, X.; Li, Q.; Zhang, W.; Deng, P.; He, Z.; Xi, Y.; Tong, T.; Pi, H.; et al. Histone hypoacetylation contributes to neurotoxicity induced by chronic nickel exposure in vivo and in vitro. Sci. Total Environ. 2021, 783, 147014. [Google Scholar] [CrossRef] [PubMed]
- Searles Nielsen, S.; Checkoway, H.; Criswell, S.R.; Farin, F.M.; Stapleton, P.L.; Sheppard, L.; Racette, B.A. Inducible nitric oxide synthase gene methylation and parkinsonism in manganese-exposed welders. Park. Relat. D 2015, 21, 355–360. [Google Scholar] [CrossRef]
- Olasehinde, T.A.; Olaniran, A.O. Neurotoxicity of Polycyclic Aromatic Hydrocarbons: A Systematic Mapping and Review of Neuropathological Mechanisms. Toxics 2022, 10, 417. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cao, J.; Hao, Z.; Liu, A.; Li, X.; Li, H.; Xia, N.; Wang, Z.; Zhang, Z.; Bai, J.; et al. Aspirin ameliorates the cognition impairment in mice following benzo[a]pyrene treatment via down-regulating BDNF IV methylation. Neurotoxicology 2022, 89, 20–30. [Google Scholar] [CrossRef]
- Wang, J.; Li, C.-L.; Tu, B.-J.; Yang, K.; Mo, T.-T.; Zhang, R.-Y.; Cheng, S.-Q.; Chen, C.-Z.; Jiang, X.-J.; Han, T.-L.; et al. Integrated epigenetics, transcriptomics, and metabolomics to analyze the mechanisms of Benzo[a]pyrene neurotoxicity in the hippocampus. Toxicol. Sci. 2018, 166, 65–81. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Regulate | Output | Ref. |
|---|---|---|---|
| miR-149-5p | ↓ | miR-149-5p plays a crucial role in PM2.5-induced inflammation. | [76] |
| miR-217-5p | ↓ | miR-217-5p mimics can reduce the level of ROS in lung tissues of mice treated with PM2.5. | [87] |
| miR-298 | ↓ | It can inhibit the expression of neurotoxic peptides. | [92] |
| miR-574-5p | ↓ | PM2.5-induced down-regulation of mir-574-5p causes neurotoxicity and further disrupts the integrity of synaptic function. | [93] |
| miR-340 | ↑ | PM2.5-induced up-regulation of miR-340 can reduce the accumulation of amyloid-β and reduce cell apoptosis by targeting BACE1. | [99] |
| miR-135a | ↑ | PM2.5-induced up-regulation of miR-135a can inhibit the expression and activity of BACE-1. | [101] |
| miR-21-5p miR-126-3p miR-100-5p | \ \ \ | They are the miRNAs most regulated by Nrf2. | [109] |
| miR-206 | ↑ | PM2.5-induced up-regulation of miRNA-206 inhibits the translation of superoxide dismutase 1 (SOD1), leading to ROS accumulation and inflammation. | [111] |
| miR-205 | ↓ | PM2.5-induced down-regulation of miR-205 activates the IRAK2/TRAF6/NF-kB signaling pathway, causing cardiomyocyte apoptosis and myocardial inflammation. | [81] |
| uc001.dgp.1 | ↑ | PM2.5 will up-regulate lncRNAuc001.dgp.1 to inhibit the expression of miR-3607-5p. | [82] |
| linc01515 | ↑ | PM2.5-induced up-regulated linc01515 regulates NrF2-induced oxidative damage in airway epithelial cells, ultimately leading to inflammation. | [110] |
| lncgm16410 | ↓ | PM2.5-induced down-regulation of lncgm16410 enhances the activation of macrophages and promotes lung inflammation. | [85] |
| AABR07005593.1 | ↑ | PM2.5-induced up-regulation of AABR07005593.1 is involved in the activation of the κ-B signaling pathway, which ultimately promotes the expression of IL-6 and causes inflammation. | [86] |
| circ104250 | ↑ | PM2.5 will up-regulate circ104250 to inhibit the expression of miR-3607-5p. | [82] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, L.; Shao, M.; Song, C.; Zhou, L.; Nie, W.; Yu, H.; Wang, S.; Liu, Y.; Yu, L. The Role of Epigenetic Mechanisms in the Development of PM2.5-Induced Cognitive Impairment. Toxics 2025, 13, 119. https://doi.org/10.3390/toxics13020119
Jiang L, Shao M, Song C, Zhou L, Nie W, Yu H, Wang S, Liu Y, Yu L. The Role of Epigenetic Mechanisms in the Development of PM2.5-Induced Cognitive Impairment. Toxics. 2025; 13(2):119. https://doi.org/10.3390/toxics13020119
Chicago/Turabian StyleJiang, Lishan, Mingxia Shao, Chao Song, Li Zhou, Wenke Nie, Hang Yu, Siqi Wang, Yongping Liu, and Li Yu. 2025. "The Role of Epigenetic Mechanisms in the Development of PM2.5-Induced Cognitive Impairment" Toxics 13, no. 2: 119. https://doi.org/10.3390/toxics13020119
APA StyleJiang, L., Shao, M., Song, C., Zhou, L., Nie, W., Yu, H., Wang, S., Liu, Y., & Yu, L. (2025). The Role of Epigenetic Mechanisms in the Development of PM2.5-Induced Cognitive Impairment. Toxics, 13(2), 119. https://doi.org/10.3390/toxics13020119