
Perfluorooctane Sulfonate Induces Dysfunction of Human Umbilical Vein Endothelial Cells via Ferroptosis Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Assay
2.3. Transmission Electron Microscope
2.4. Lipid-ROS Assay by Flow Cytometry
2.5. Western Blot Analysis
2.6. Nitric Oxide (NO) Content Determination
2.7. Statistical Analysis
3. Results
3.1. PFOS Can Inhibit the Multiplication and Viability of HUVECs In Vitro
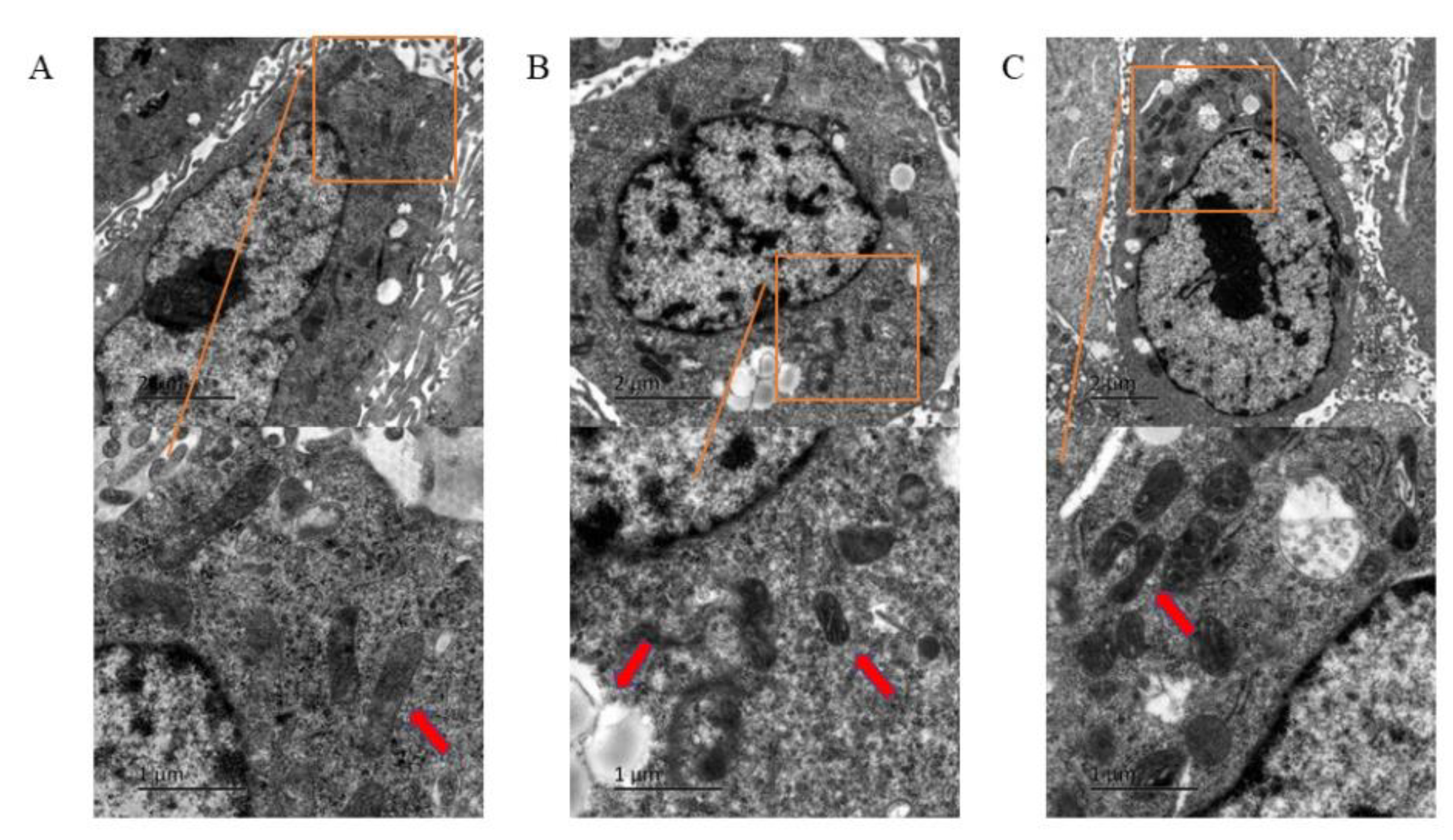
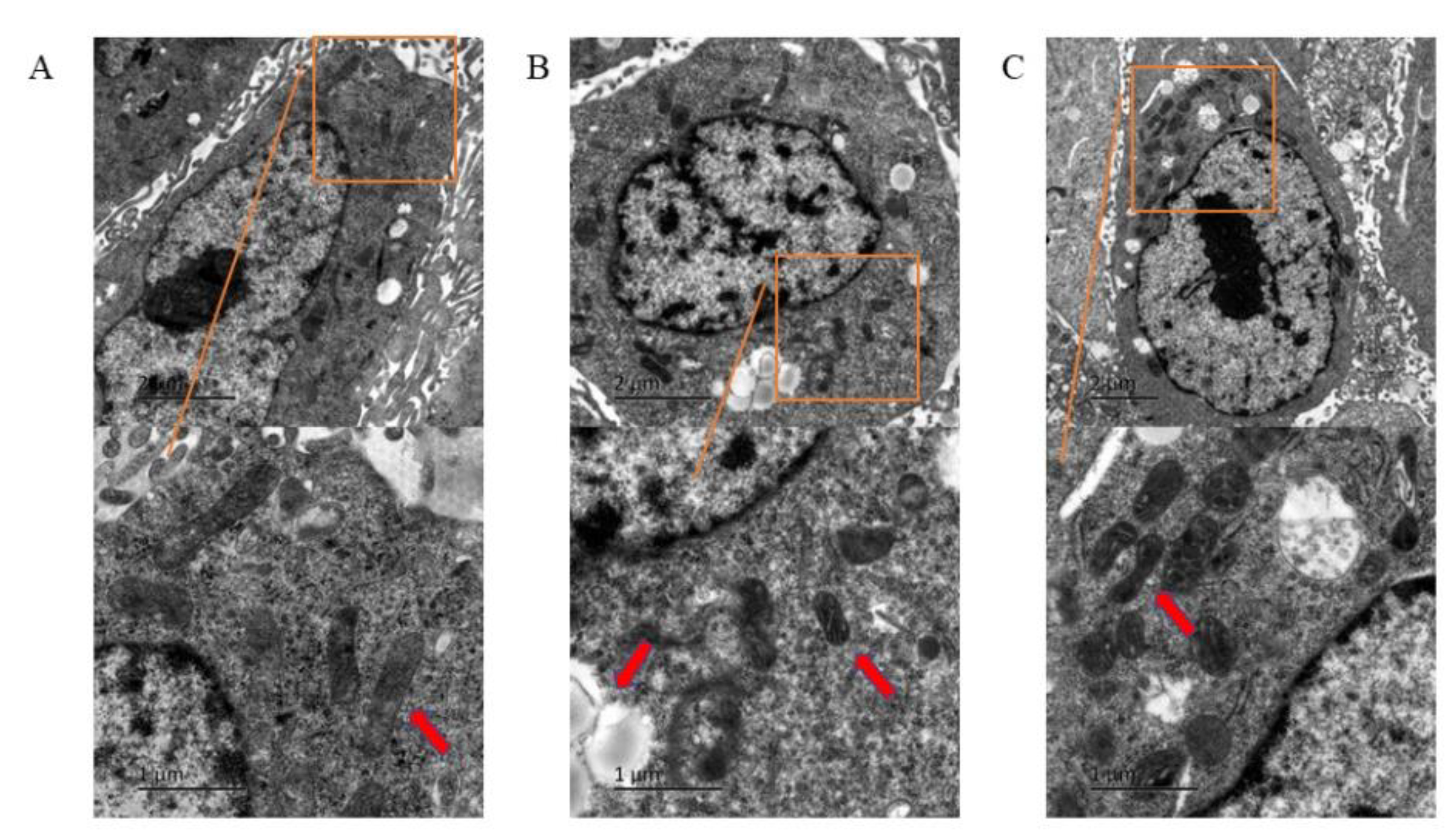
3.2. Ferroptosis of HUVECs Observed under Transmission Electron Microscope
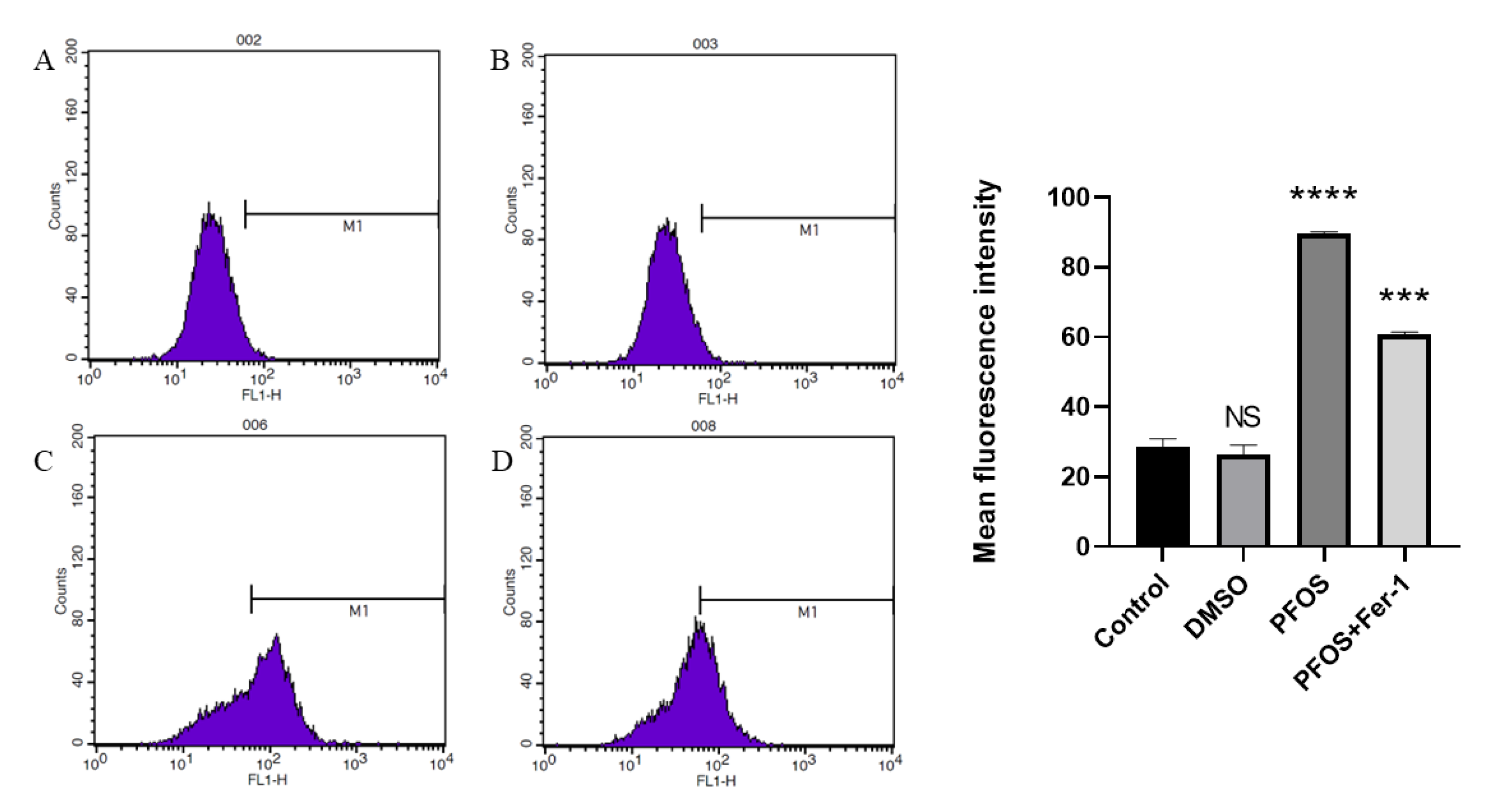
3.3. PFOS Can Increase the Production of ROS
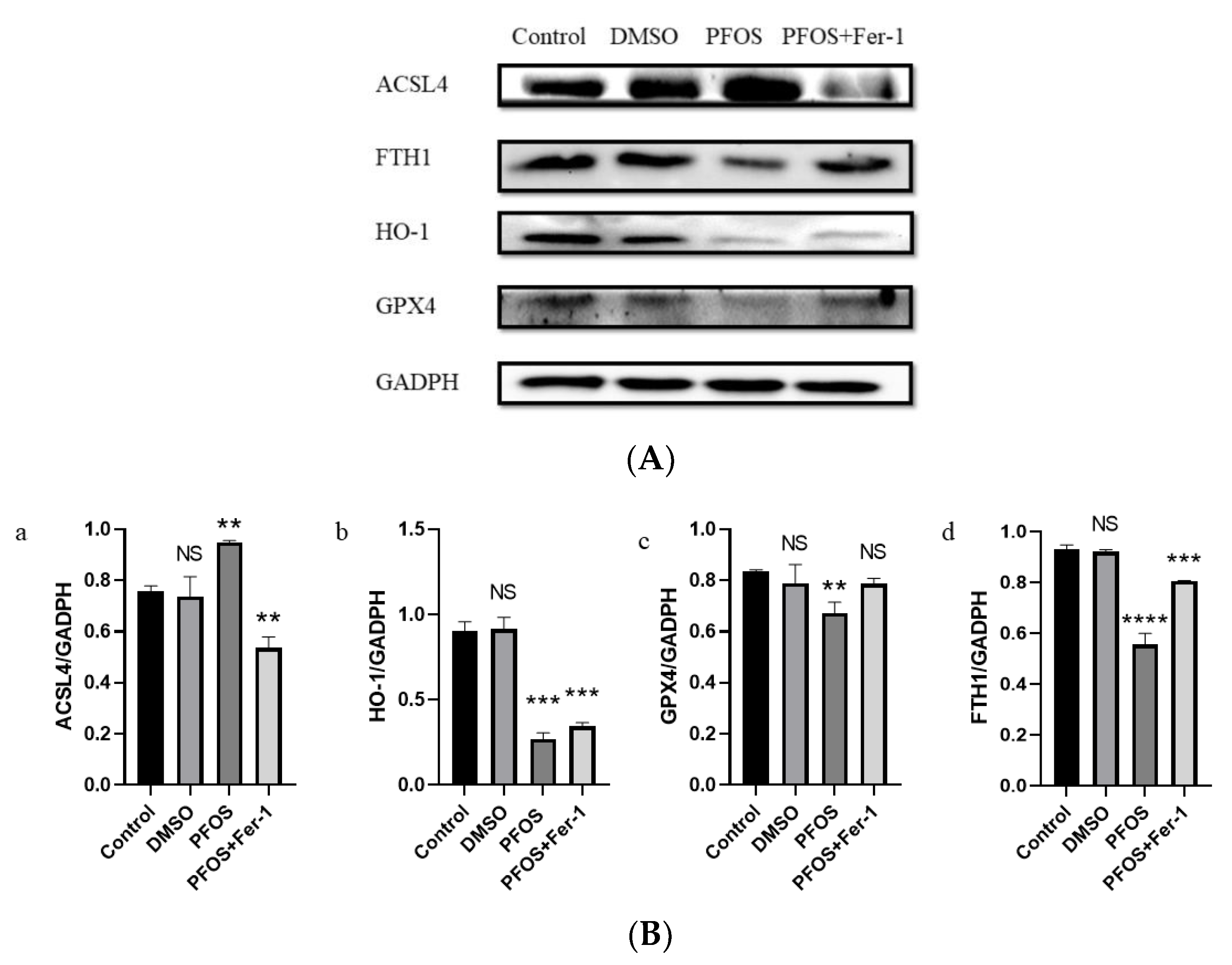
3.4. PFOS Can Induce Ferroptosis via Ferroptosis-Related Pathway
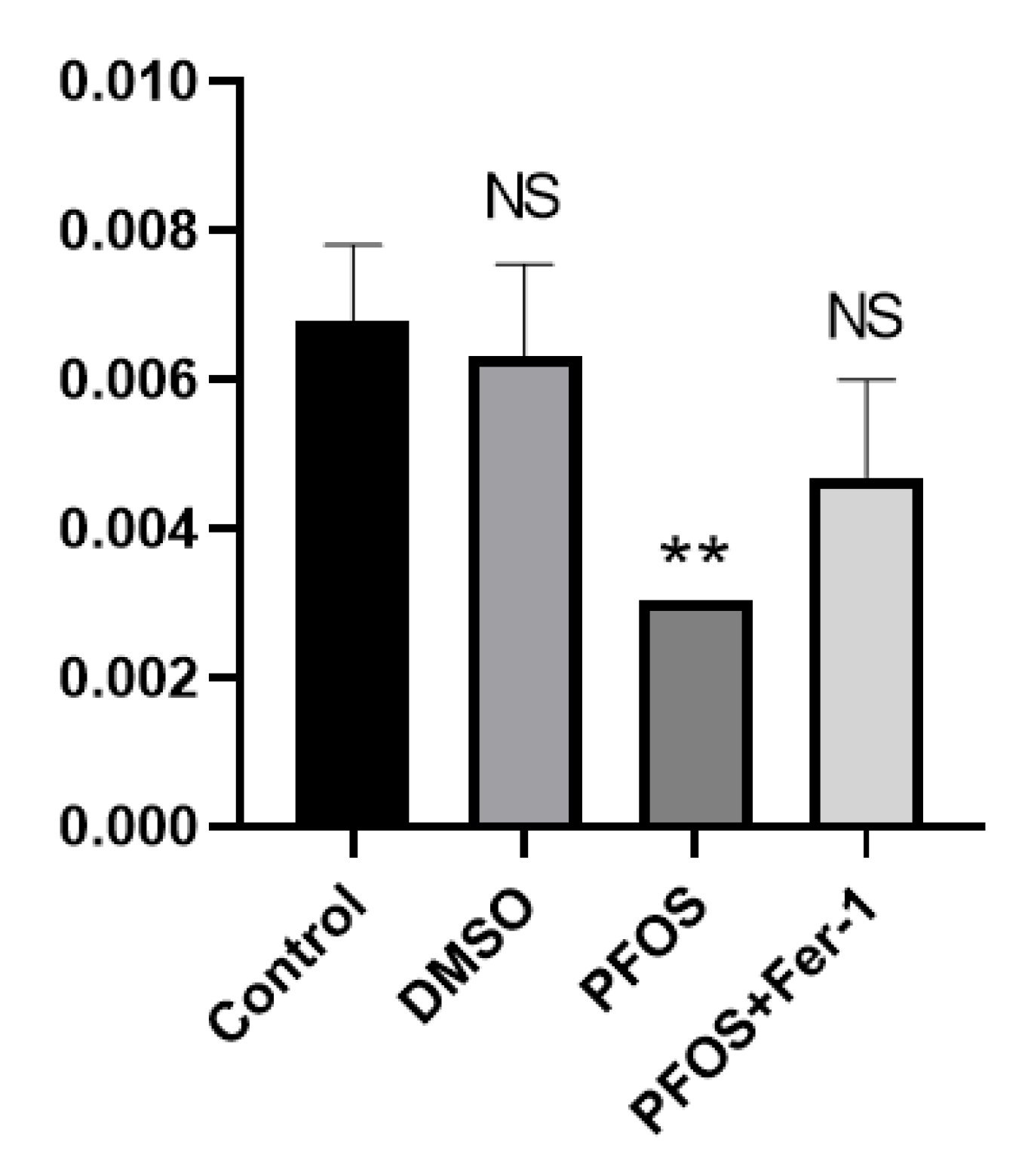
3.5. PFOS Can Decrease the Production of NO
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lindstrom, A.B.; Strynar, M.J.; Libelo, E.L. Polyfluorinated compounds: Past, present, and future. Environ. Sci. Technol. 2011, 45, 7954–7961. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.; Anitole, K.; Hodes, C.; Lai, D.; Pfahles-Hutchens, A.; Seed, J. Perfluoroalkyl acids: A review of monitoring and toxicological findings. Toxicol. Sci. 2007, 99, 366–394. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.G.; Wong, C.K.; Wong, M.H. Environmental contamination, human exposure and body loadings of perfluorooctane sulfonate (PFOS), focusing on Asian countries. Chemosphere 2012, 89, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Chain EpoCitF; Schrenk, D.; Bignami, M.; Bodin, L.; Chipman, J.K.; Del Mazo, J.; Grasl-Kraupp, B.; Hogstrand, C.; Hoogenboom, L.R.; Leblanc, J.C.; et al. Risk to human health related to the presence of perfluoroalkyl substances in food. EFSA J. 2020, 18, e06223. [Google Scholar] [PubMed]
- Gockener, B.; Weber, T.; Rudel, H.; Bucking, M.; Kolossa-Gehring, M. Human biomonitoring of per- and polyfluoroalkyl substances in German blood plasma samples from 1982 to 2019. Environ. Int. 2020, 145, 106123. [Google Scholar] [CrossRef]
- Tsuda, S. Differential toxicity between perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA). J. Toxicol. Sci. 2016, 41, SP27–SP36. [Google Scholar] [CrossRef]
- Saikat, S.; Kreis, I.; Davies, B.; Bridgman, S.; Kamanyire, R. The impact of PFOS on health in the general population: A review. Environ. Sci Process. Impacts 2013, 15, 329–335. [Google Scholar] [CrossRef]
- Xu, Y.; Fletcher, T.; Pineda, D.; Lindh, C.H.; Nilsson, C.; Glynn, A.; Vogs, C.; Norstrom, K.; Lilja, K.; Jakobsson, K.; et al. Serum Half-Lives for Short- and Long-Chain Perfluoroalkyl Acids after Ceasing Exposure from Drinking Water Contaminated by Firefighting Foam. Environ. Health Perspect. 2020, 128, 77004. [Google Scholar] [CrossRef]
- Haug, L.S.; Huber, S.; Becher, G.; Thomsen, C. Characterisation of human exposure pathways to perfluorinated compounds--comparing exposure estimates with biomarkers of exposure. Environ. Int. 2011, 37, 687–693. [Google Scholar] [CrossRef]
- Olsen, G.W.; Burris, J.M.; Ehresman, D.J.; Froehlich, J.W.; Seacat, A.M.; Butenhoff, J.L.; Zobel, L.R. Half-life of serum elimination of perfluorooctanesulfonate, perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers. Environ. Health Perspect. 2007, 115, 1298–1305. [Google Scholar] [CrossRef]
- Kummu, M.; Sieppi, E.; Koponen, J.; Laatio, L.; Vahakangas, K.; Kiviranta, H.; Rautio, A.; Myllynen, P. Organic anion transporter 4 (OAT 4) modifies placental transfer of perfluorinated alkyl acids PFOS and PFOA in human placental ex vivo perfusion system. Placenta 2015, 36, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Olsen, G.W.; Butenhoff, J.L.; Zobel, L.R. Perfluoroalkyl chemicals and human fetal development: An epidemiologic review with clinical and toxicological perspectives. Reprod. Toxicol. 2009, 27, 212–230. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Huang, Q.S.; Fang, C.; Kiyama, R.; Shen, H.; Dong, S. Evaluation of cellular response to perfluorooctane sulfonate in human umbilical vein endothelial cells. Toxicol. In Vitro 2012, 26, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Ye, T.; Yang, L.; Shen, Y.; Li, H. Ferroptosis Signaling and Regulators in Atherosclerosis. Front. Cell Dev. Biol. 2021, 9, 809457. [Google Scholar] [CrossRef]
- Zhao, Y.; Lu, J.; Mao, A.; Zhang, R.; Guan, S. Autophagy Inhibition Plays a Protective Role in Ferroptosis Induced by Alcohol via the p62-Keap1-Nrf2 Pathway. J. Agric. Food Chem. 2021, 69, 9671–9683. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr. Top. Microbiol. Immunol. 2017, 403, 143–170. [Google Scholar] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Shi, Y.; Vanhoutte, P.M. Macro- and microvascular endothelial dysfunction in diabetes. J. Diabetes 2017, 9, 434–449. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Kelaini, S.; Caines, R.; Margariti, A. RBPs Play Important Roles in Vascular Endothelial Dysfunction Under Diabetic Conditions. Front. Physiol. 2018, 9, 1310. [Google Scholar] [CrossRef] [PubMed]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef] [PubMed]
- Adameova, A.; Goncalvesova, E.; Szobi, A.; Dhalla, N.S. Necroptotic cell death in failing heart: Relevance and proposed mechanisms. Heart Fail. Rev. 2016, 21, 213–221. [Google Scholar] [CrossRef]
- Dangudubiyyam, S.V.; Mishra, J.S.; Zhao, H.; Kumar, S. Perfluorooctane sulfonic acid (PFOS) exposure during pregnancy increases blood pressure and impairs vascular relaxation mechanisms in the adult offspring. Reprod. Toxicol. 2020, 98, 165–173. [Google Scholar] [CrossRef]
- Zeng, Z.; Song, B.; Xiao, R.; Zeng, G.; Gong, J.; Chen, M.; Xu, P.; Zhang, P.; Shen, M.; Yi, H. Assessing the human health risks of perfluorooctane sulfonate by in vivo and in vitro studies. Environ. Int. 2019, 126, 598–610. [Google Scholar] [CrossRef]
- Li, J.; Zhou, Y.; Wang, H.; Lou, J.; Lenahan, C.; Gao, S.; Wang, X.; Deng, Y.; Chen, H.; Shao, A. Oxidative Stress-Induced Ferroptosis in Cardiovascular Diseases and Epigenetic Mechanisms. Front. Cell Dev. Biol 2021, 9, 685775. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef]
- Gao, M.; Jiang, X. To eat or not to eat-the metabolic flavor of ferroptosis. Curr. Opin. Cell Biol. 2018, 51, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, P.; Chen, W.; Chen, G. Ferroptosis mediated DSS-induced ulcerative colitis associated with Nrf2/HO-1 signaling pathway. Immunol. Lett. 2020, 225, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.J.; Zhang, D.; Wu, Y.; Jia, Q.H.; Zhang, L.; Li, Y.X.; Yang, Y.F.; Wang, H.; Wu, C.T.; Wang, L.S. miRNA-17–92 protects endothelial cells from erastin-induced ferroptosis through targeting the A20-ACSL4 axis. Biochem. Biophys. Res. Commun. 2019, 515, 448–454. [Google Scholar] [CrossRef]
- Konukoglu, D.; Uzun, H. Endothelial Dysfunction and Hypertension. Adv. Exp. Med. Biol. 2017, 956, 511–540. [Google Scholar]
- Verma, S.; Anderson, T.J. Fundamentals of endothelial function for the clinical cardiologist. Circulation 2002, 105, 546–549. [Google Scholar] [CrossRef]
- Caserta, D.; Mantovani, A.; Marci, R.; Fazi, A.; Ciardo, F.; La Rocca, C.; Maranghi, F.; Moscarini, M. Environment and women’s reproductive health. Hum. Reprod. Update 2011, 17, 418–433. [Google Scholar] [CrossRef]
- Palomba, S.; de Wilde, M.A.; Falbo, A.; Koster, M.P.; La Sala, G.B.; Fauser, B.C. Pregnancy complications in women with polycystic ovary syndrome. Hum. Reprod. Update 2015, 21, 575–592. [Google Scholar] [CrossRef]
- Palomba, S.; Piltonen, T.T.; Giudice, L.C. Endometrial function in women with polycystic ovary syndrome: A comprehensive review. Hum. Reprod. Update 2021, 27, 584–618. [Google Scholar] [CrossRef] [PubMed]
- Palomba, S. Is fertility reduced in ovulatory women with polycystic ovary syndrome? An opinion paper. Hum. Reprod. 2021, 36, 2421–2428. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, J.; Wang, P.; Yan, S.; Liang, Y.; Liu, D.; Ren, S. Perfluorooctane Sulfonate Induces Dysfunction of Human Umbilical Vein Endothelial Cells via Ferroptosis Pathway. Toxics 2022, 10, 503. https://doi.org/10.3390/toxics10090503
Cui J, Wang P, Yan S, Liang Y, Liu D, Ren S. Perfluorooctane Sulfonate Induces Dysfunction of Human Umbilical Vein Endothelial Cells via Ferroptosis Pathway. Toxics. 2022; 10(9):503. https://doi.org/10.3390/toxics10090503
Chicago/Turabian StyleCui, Jiajing, Pingwei Wang, Shuqi Yan, Yujun Liang, Dongge Liu, and Shuping Ren. 2022. "Perfluorooctane Sulfonate Induces Dysfunction of Human Umbilical Vein Endothelial Cells via Ferroptosis Pathway" Toxics 10, no. 9: 503. https://doi.org/10.3390/toxics10090503
APA StyleCui, J., Wang, P., Yan, S., Liang, Y., Liu, D., & Ren, S. (2022). Perfluorooctane Sulfonate Induces Dysfunction of Human Umbilical Vein Endothelial Cells via Ferroptosis Pathway. Toxics, 10(9), 503. https://doi.org/10.3390/toxics10090503