
N-Acetylcysteine or Sodium Selenite Prevent the p38-Mediated Production of Proinflammatory Cytokines by Microglia during Exposure to Mercury (II)





Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability
2.3. Reactive Oxygen Species (ROS)
2.4. Preparation of Cell Lysates
2.5. Total Protein Determination
2.6. Total Glutathione Quantification
2.7. Thioredoxin Reductase Activity
2.8. Nuclear Expression of p38 and p50
2.9. Expression of Pro-Inflammatory Cytokines
2.10. Statistical Analysis
3. Results
3.1. Cell Viability
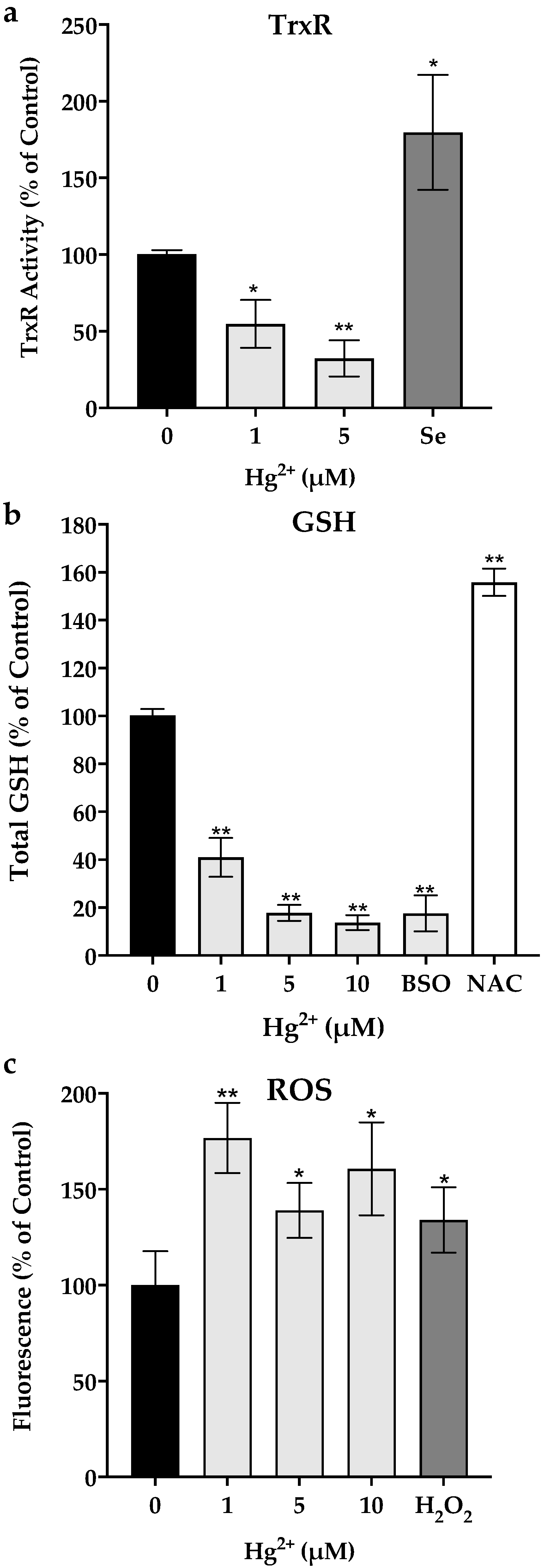
3.2. Thioredoxin Reductase Activity, Total Glutathione Levels and ROS Production
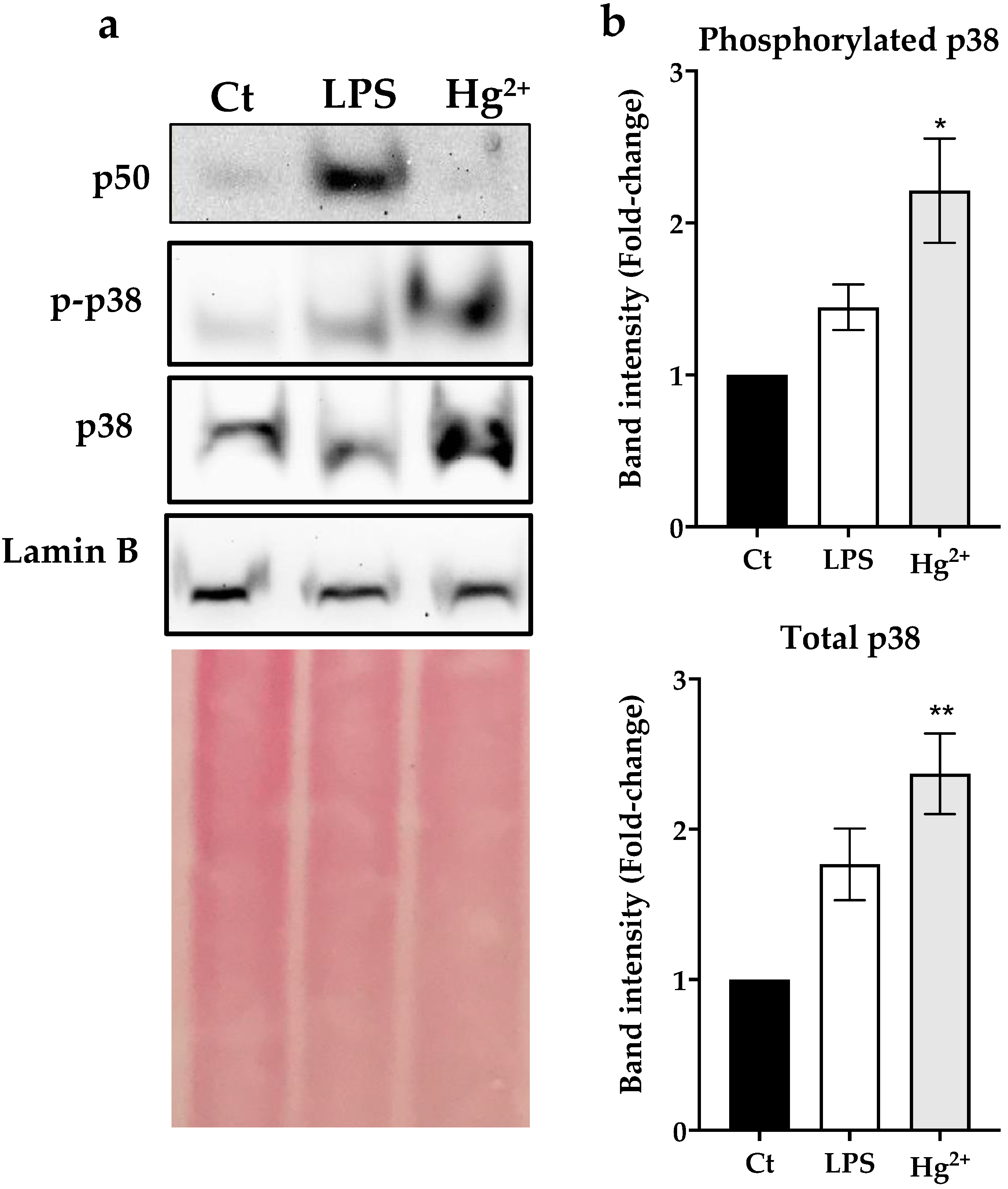
3.3. Effect of Hg2+ on p50 and p38 Nuclear Levels
3.4. mRNA Levels of IL1-ß, TNF-α and iNOS
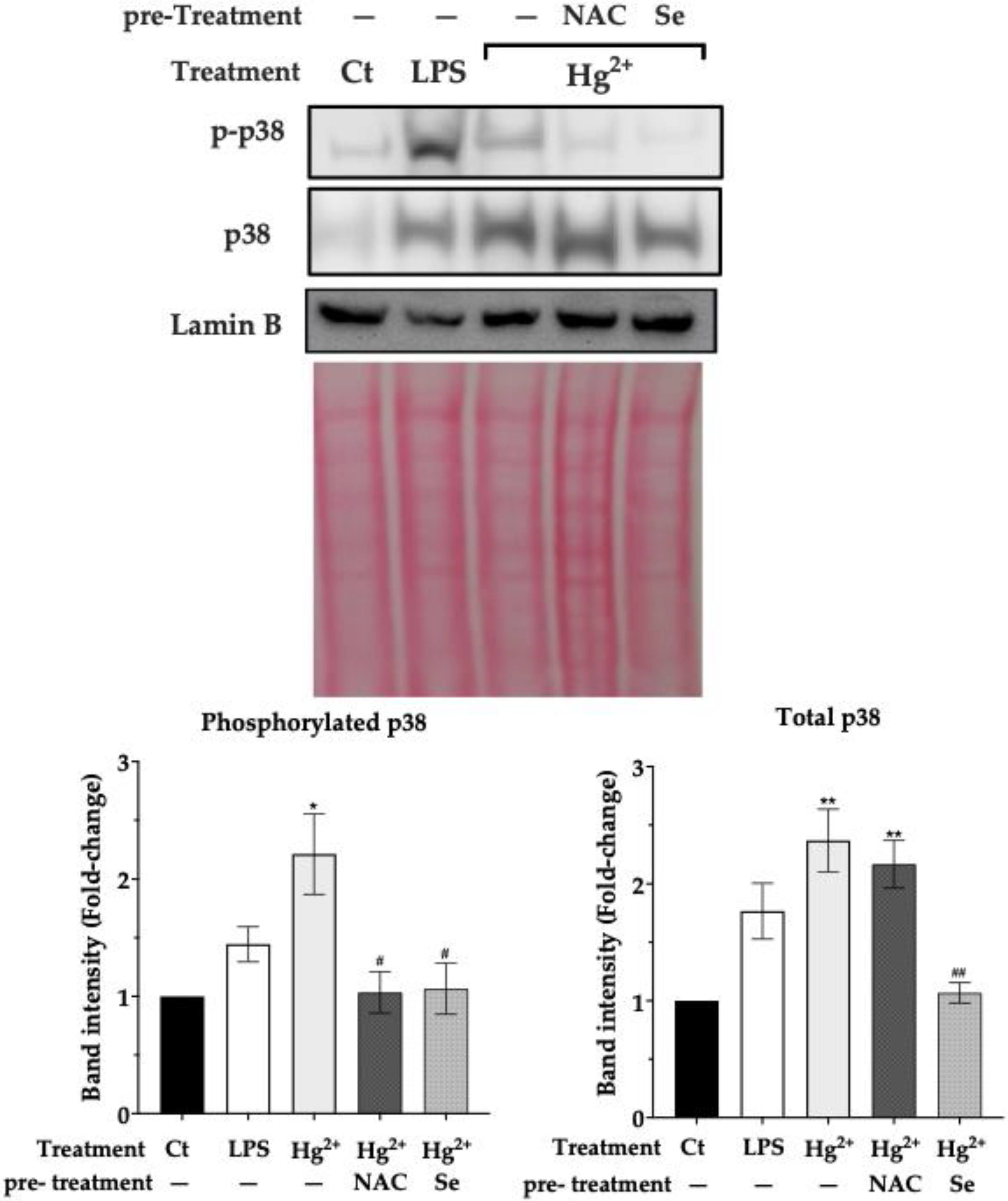
3.5. Effect of Pre-Exposure to NAC and Selenite over p38 Activation by Hg2+
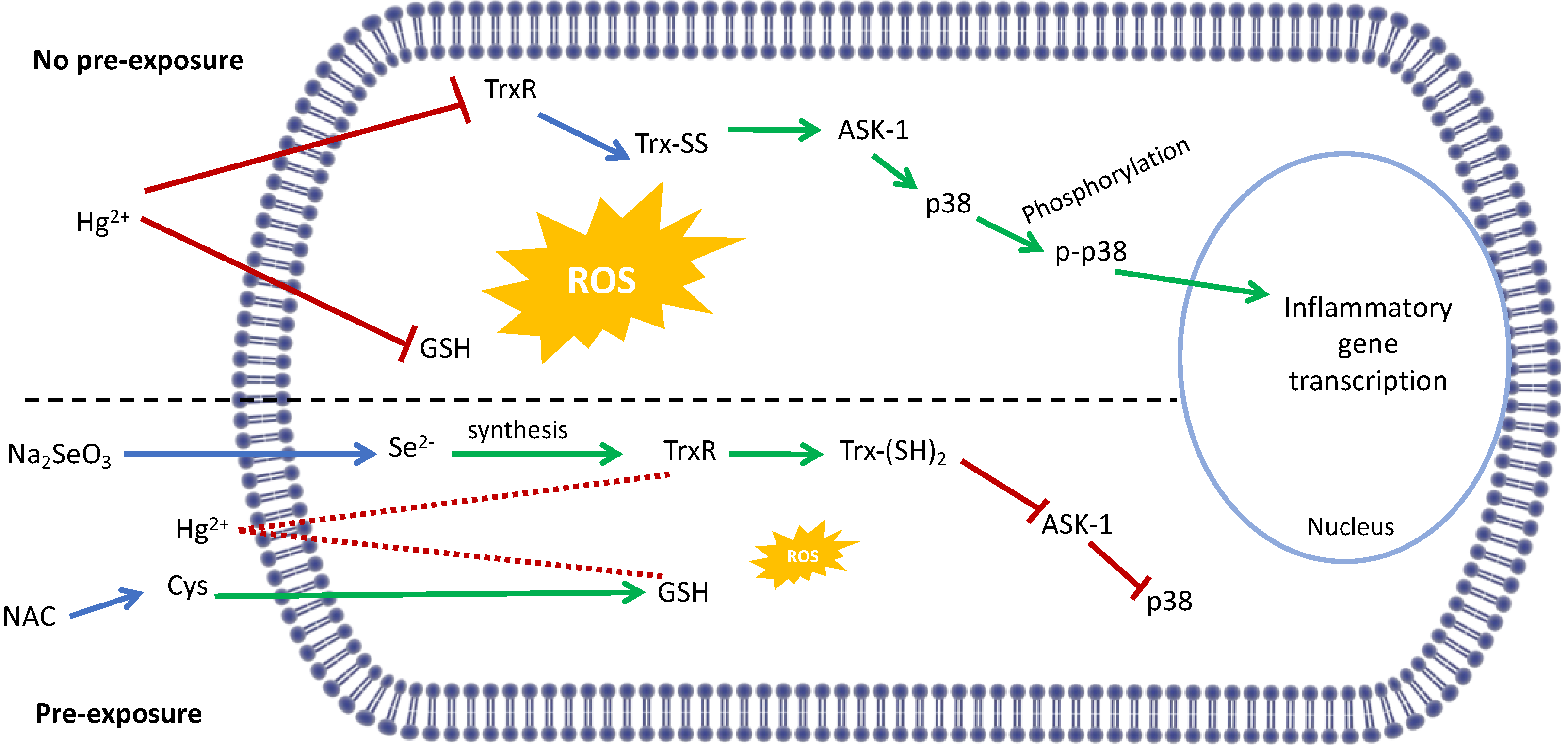
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Clarkson, T.W.; Magos, L. The toxicology of mercury and its chemical compounds. Crit. Rev. Toxicol. 2006, 36, 609–662. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, T.W. Mechanisms of Mercury Disposition in the Body. Am. J. Ind. Med. 2007, 50, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Branco, V.; Aschner, M.; Carvalho, C. Neurotoxicity of mercury: An old issue with contemporary significance. In Advances in Neurotoxicology; Aschner, M., Costa, L.G., Eds.; Academic Press: Amsterdam, The Netherlands, 2021; ISBN 2468-7480. [Google Scholar]
- Ni, M.; Li, X.; Yin, Z.; Sidoryk-Weogonekgrzynowicz, M.; Jiang, H.; Farina, M.; Rocha, J.B.T.; Syversen, T.; Aschner, M. Comparative study on the response of rat primary astrocytes and microglia to methylmercury toxicity. Glia 2011, 59, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Liu, Z.; Li, H.; Liu, Y.; Xia, Z.; Xiao, Y.; Usman, M.; Du, Y.; Bi, H.; Wei, L. Hormesis of mercuric chloride-human serum albumin adduct on N9 microglial cells via the ERK/MAPKs and JAK/STAT3 signaling pathways. Toxicology 2018, 408, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Johnson, V.J.; Sharma, R.P. Mercury inhibits nitric oxide production but activates proinflammatory cytokine expression in murine macrophage: Differential modulation of NF-κB and p38 MAPK signaling pathways. Nitric Oxide 2002, 7, 67–74. [Google Scholar] [CrossRef]
- Branco, V.; Carvalho, C. The thioredoxin system as a target for mercury compounds. Biochim. Biophys. Acta—Gen. Subj. 2019, 1863, 129255. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A.; Johansson, C.; Berndt, C.; Lönn, M.E.; Hudemann, C.; Lillig, C.H.; Laurent, T.C.; Moore, E.C.; Reichard, P.; Holmgren, A.; et al. Thiol redox control via thioredoxin and glutaredoxin systems. Biochem. Soc. Trans. 2005, 33, 1375–1377. [Google Scholar] [CrossRef] [Green Version]
- Lorenzen, I.; Mullen, L.; Bekeschus, S.; Hanschmann, E.-M. Redox Regulation of Inflammatory Processes Is Enzymatically Controlled. Oxid. Med. Cell. Longev. 2017, 2017, 8459402. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, C.M.L.; Chew, E.H.; Hashemy, S.I.; Lu, J.; Holmgren, A. Inhibition of the human thioredoxin system: A molecular mechanism of mercury toxicity. J. Biol. Chem. 2008, 283, 11913–11923. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, C.M.L.; Lu, J.; Zhang, X.; Arnér, E.S.J.; Holmgren, A. Effects of selenite and chelating agents on mammalian thioredoxin reductase inhibited by mercury: Implications for treatment of mercury poisoning. FASEB J. 2011, 25, 370–381. [Google Scholar] [CrossRef] [Green Version]
- Branco, V.; Canário, J.; Lu, J.; Holmgren, A.; Carvalho, C. Mercury and selenium interaction in vivo: Effects on thioredoxin reductase and glutathione peroxidase. Free Radic. Biol. Med. 2012, 52, 781–793. [Google Scholar] [CrossRef]
- Branco, V.; Godinho-Santos, A.; Gonçalves, J.; Lu, J.; Holmgren, A.; Carvalho, C. Mitochondrial thioredoxin reductase inhibition, selenium status, and Nrf-2 activation are determinant factors modulating the toxicity of mercury compounds. Free Radic. Biol. Med. 2014, 73, 95–105. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Lötzer, K.; Maurer, S.; Schultz, M.; Leist, M. Utilization of selenium from different chemical entities for selenoprotein biosynthesis by mammalian cell lines. Biofactors 1995, 5, 125–131. [Google Scholar]
- Xia, L.; Nordman, T.; Olsson, J.M.; Damdimopoulos, A.; Björkhem-Bergman, L.; Nalvarte, I.; Eriksson, L.C.; Arnér, E.S.J.; Spyrou, G.; Björnstedt, M. The Mammalian Cytosolic Selenoenzyme Thioredoxin Reductase Reduces Ubiquinone: A Novel Mechanism for Defense Against Oxidative Stress. J. Biol. Chem. 2003, 278, 2141–2146. [Google Scholar] [CrossRef] [Green Version]
- Rayman, M.P. Food-chain selenium and human health: Emphasis on intake. Br. J. Nutr. 2008, 100, 254–268. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Friberg, L.; Mottet, N.K. Accumulation of methylmercury and inorganic mercury in the brain. Biol. Trace Elem. Res. 1989, 21, 201–206. [Google Scholar] [CrossRef]
- Eto, K.; Takizawa, Y.; Akagi, H.; Haraguchi, K.; Asano, S.; Takahata, N.; Tokunaga, H. Differential Diagnosis between Organic and Inorganic Mercury Poisoning in Human Cases-The Pathologic Point of View. Toxicol. Pathol. 1999, 27, 664–671. [Google Scholar] [CrossRef]
- Rooney, J.P.K. The retention time of inorganic mercury in the brain—A systematic review of the evidence. Toxicol. Appl. Pharmacol. 2014, 274, 425–435. [Google Scholar] [CrossRef]
- Björkman, L.; Lundekvam, B.F.; Lægreid, T.; Bertelsen, B.I.; Morild, I.; Lilleng, P.; Lind, B.; Palm, B.; Vahter, M. Mercury in human brain, blood, muscle and toenails in relation to exposure: An autopsy study. Environ. Health 2007, 6, 30. [Google Scholar] [CrossRef] [Green Version]
- Branco, V.; Matos, B.; Mourato, C.; Diniz, M.; Carvalho, C.; Martins, M. Synthesis of glutathione as a central aspect of PAH toxicity in liver cells: A comparison between phenanthrene, Benzo[b]Fluoranthene and their mixtures. Ecotoxicol. Environ. Saf. 2021, 208, 111637. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2006, 1, 3159–3165. [Google Scholar] [CrossRef] [PubMed]
- Arnér, E.S.; Holmgren, A. Measurement of thioredoxin and thioredoxin reductase. Curr. Protoc. Toxicol. 2005, 24, 7-4. [Google Scholar]
- Underwood, D.C.; Osborn, R.R.; Kotzer, C.J.; Adams, J.L.; Lee, J.C.; Webb, E.F.; Carpenter, D.C.; Bochnowicz, S.; Thomas, H.C.; Hay, D.W.P.; et al. SB 239063, a Potent p38 MAP Kinase Inhibitor, Reduces Inflammatory Cytokine Production, Airways Eosinophil Infiltration, and Persistence. J. Pharmacol. Exp. Ther. 2000, 293, 281–288. [Google Scholar] [PubMed]
- Strassburger, M.; Braun, H.; Reymann, K.G. Anti-inflammatory treatment with the p38 mitogen-activated protein kinase inhibitor SB239063 is neuroprotective, decreases the number of activated microglia and facilitates neurogenesis in oxygen–glucose-deprived hippocampal slice cultures. Eur. J. Pharmacol. 2008, 592, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.; Gümbel, D.; Hanschmann, E.M.; Mandelkow, R.; Gelbrich, N.; Zimmermann, U.; Walther, R.; Ekkernkamp, A.; Sckell, A.; Kramer, A.; et al. Cold atmospheric plasma treatment induces anti-proliferative effects in prostate cancer cells by redox and apoptotic signaling pathways. PLoS ONE 2015, 10, e0130350. [Google Scholar] [CrossRef]
- Watson, M.; van Leer, L.; Vanderlelie, J.J.; Perkins, A.V. Selenium supplementation protects trophoblast cells from oxidative stress. Placenta 2012, 33, 1012–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Franco, J.L.; Braga, H.C.; Stringari, J.; Missau, F.C.; Posser, T.; Mendes, B.G.; Leal, R.B.; Santos, A.R.S.; Dafre, A.L.; Pizzolatti, M.G.; et al. Mercurial-Induced Hydrogen Peroxide Generation in Mouse Brain Mitochondria: Protective Effects of Quercetin. Chem. Res. Toxicol. 2007, 20, 1919–1926. [Google Scholar] [CrossRef]
- Piccoli, C.; D’Aprile, A.; Scrima, R.; Ambrosi, L.; Zefferino, R.; Capitanio, N. Subcytotoxic mercury chloride inhibits gap junction intercellular communication by a redox-and phosphorylation-mediated mechanism. Free Radic. Biol. Med. 2012, 52, 916–927. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, H.; Yan, F.; Zhou, J. Oscillatory dynamics of p38 activity with transcriptional and translational time delays. Sci. Rep. 2017, 7, 11495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, S.; Gon, Y.; Matsumoto, K.; Takeshita, I.; Machinoand, T.; Horie, T. Intracellular glutathione regulates tumour necrosis factor-α-induced p38 MAP kinase activation and RANTES production by human bronchial epithelial cells. Clin. Exp. Allergy 2001, 31, 144–151. [Google Scholar] [PubMed]
- Kawai, T.; Akira, S. Signaling to NF-kB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Rachmawati, D.; Alsalem, I.W.A.; Bontkes, H.J.; Verstege, M.I.; Gibbs, S.; von Blomberg, B.M.E.; Scheper, R.J.; van Hoogstraten, I.M.W. Innate stimulatory capacity of high molecular weight transition metals Au (gold) and Hg (mercury). Toxicol. Vitr. 2015, 29, 363–369. [Google Scholar] [CrossRef]
- Sadowska, A.M.; Manuel-y-Keenoy, B.; De Backer, W.A. Antioxidant and anti-inflammatory efficacy of NAC in the treatment of COPD: Discordant in vitro and in vivo dose-effects: A review. Pulm. Pharmacol. Ther. 2007, 20, 9–22. [Google Scholar] [CrossRef]
- Wuyts, W.A.; Vanaudenaerde, B.M.; Dupont, L.J.; Demedts, M.G.; Verleden, G.M. N-acetylcysteine reduces chemokine release via inhibition of p38 MAPK in human airway smooth muscle cells. Eur. Respir. J. 2003, 22, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Holenya, P.; Can, S.; Alborzinia, H.; Rubbiani, R.; Ott, I.; Wölfl, S. A TrxR inhibiting gold(I) NHC complex induces apoptosis through ASK1-p38-MAPK signaling in pancreatic cancer cells. Mol. Cancer 2014, 13, 221. [Google Scholar] [CrossRef] [Green Version]
- Branco, V.; Coppo, L.; Solá, S.; Lu, J.; Rodrigues, C.M.P.; Holmgren, A.; Carvalho, C. Impaired cross-talk between the thioredoxin and glutathione systems is related to ASK-1 mediated apoptosis in neuronal cells exposed to mercury. Redox Biol. 2017, 13, 278–287. [Google Scholar] [CrossRef]
- Park, H.-S.; Park, E.; Kim, M.-S.; Ahn, K.; Kim, I.Y.; Choi, E.-J. Selenite Inhibits the c-Jun N-terminal Kinase/Stress-activated Protein Kinase (JNK/SAPK) through a Thiol Redox Mechanism*. J. Biol. Chem. 2000, 275, 2527–2531. [Google Scholar] [CrossRef] [Green Version]
- Rudolf, E.; Rudolf, K.; Červinka, M. Selenium activates p53 and p38 pathways and induces caspase-independent cell death in cervical cancer cells. Cell Biol. Toxicol. 2008, 24, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Fairweather-Tait, S.J.; Collings, R.; Hurst, R. Selenium bioavailability: Current knowledge and future research requirements. Am. J. Clin. Nutr. 2010, 91, 1484S–1491S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burk, R.F.; Hill, K.E.; Motley, A.K.; Winfrey, V.P.; Kurokawa, S.; Mitchell, S.L.; Zhang, W. Selenoprotein P and apolipoprotein E receptor-2 interact at the blood-brain barrier and also within the brain to maintain an essential selenium pool that protects against neurodegeneration. FASEB J. 2014, 28, 3579–3588. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mRNA Target | Forward Primer (5-3′) | Reverse Primer (5-3′) |
|---|---|---|
| Interleukin 1 (IL-1β) | TGCCACCTTTTGACAGTGATG | ATGTGCTGCTGCGAGATTTG |
| Nitric oxide synthase 2 (iNOS) | GTTCTCAGCCCAACAATACAAGA | GTGGACGGGTCGATGTCAC |
| Tumor Necrosis Factor (TNF-α) | TAGCCCACGTCGTAGCAAAC | GCAGCCTTGTCCCTTGAAGA |
| Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | GGAGAGTGTTTCCTCGTCCC | ATGAAGGGGTCGTTGATGGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Branco, V.; Coppo, L.; Aschner, M.; Carvalho, C. N-Acetylcysteine or Sodium Selenite Prevent the p38-Mediated Production of Proinflammatory Cytokines by Microglia during Exposure to Mercury (II). Toxics 2022, 10, 433. https://doi.org/10.3390/toxics10080433
Branco V, Coppo L, Aschner M, Carvalho C. N-Acetylcysteine or Sodium Selenite Prevent the p38-Mediated Production of Proinflammatory Cytokines by Microglia during Exposure to Mercury (II). Toxics. 2022; 10(8):433. https://doi.org/10.3390/toxics10080433
Chicago/Turabian StyleBranco, Vasco, Lucia Coppo, Michael Aschner, and Cristina Carvalho. 2022. "N-Acetylcysteine or Sodium Selenite Prevent the p38-Mediated Production of Proinflammatory Cytokines by Microglia during Exposure to Mercury (II)" Toxics 10, no. 8: 433. https://doi.org/10.3390/toxics10080433
APA StyleBranco, V., Coppo, L., Aschner, M., & Carvalho, C. (2022). N-Acetylcysteine or Sodium Selenite Prevent the p38-Mediated Production of Proinflammatory Cytokines by Microglia during Exposure to Mercury (II). Toxics, 10(8), 433. https://doi.org/10.3390/toxics10080433