
Exposure to the Dioxin-like Pollutant PCB 126 Afflicts Coronary Endothelial Cells via Increasing 4-Hydroxy-2 Nonenal: A Role for Aldehyde Dehydrogenase 2

 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
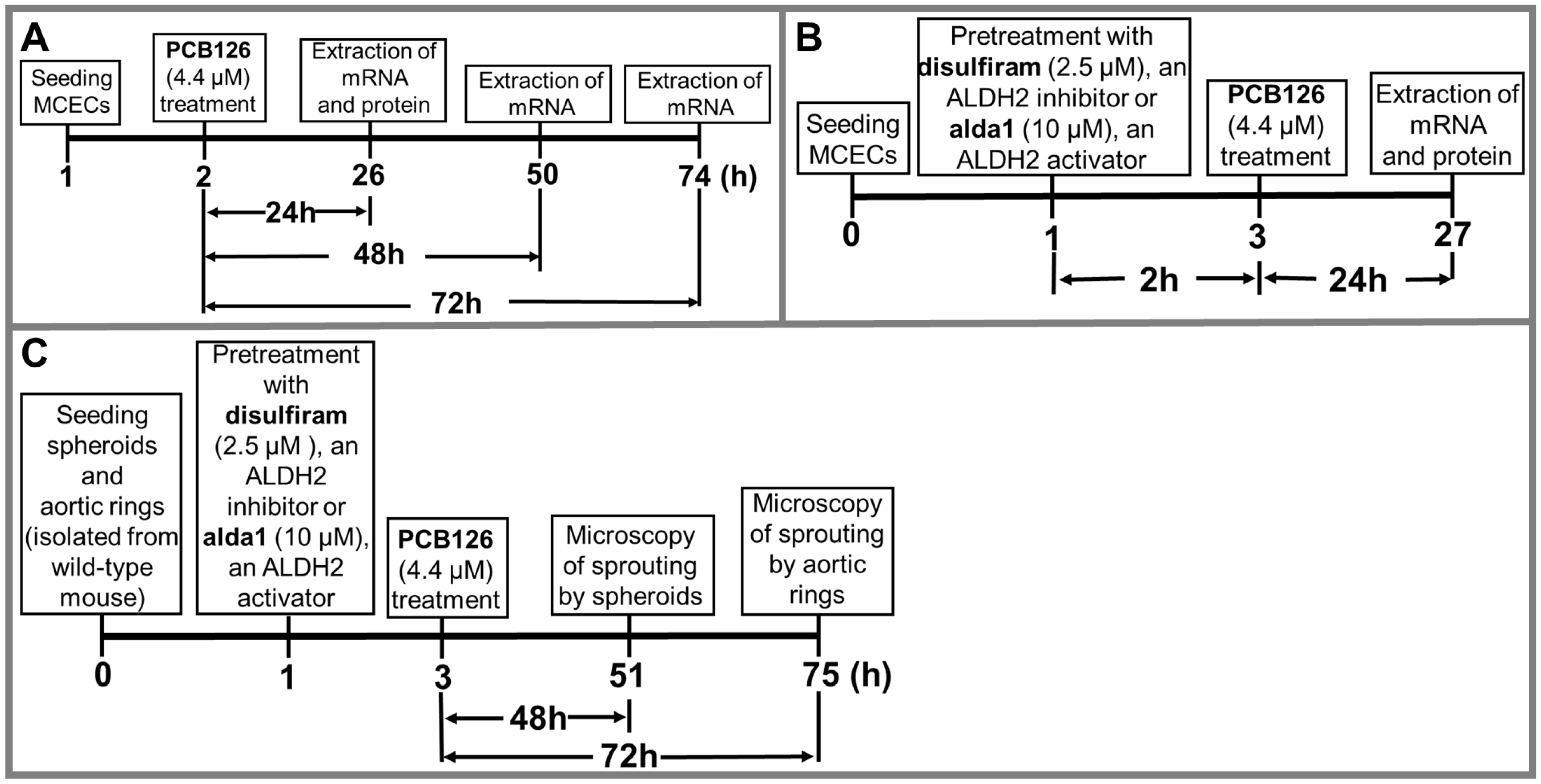
2. Materials and Methods
2.1. Experimental Animal
2.2. Cell Culture
2.3. Treatment Protocols
2.4. Spheroid Assay
2.5. Aortic Ring Assay
2.6. Real-Time qPCR
2.7. ALDH2 Activity Assay
2.8. Western Immunoblotting
2.9. Statistical Analysis
3. Results
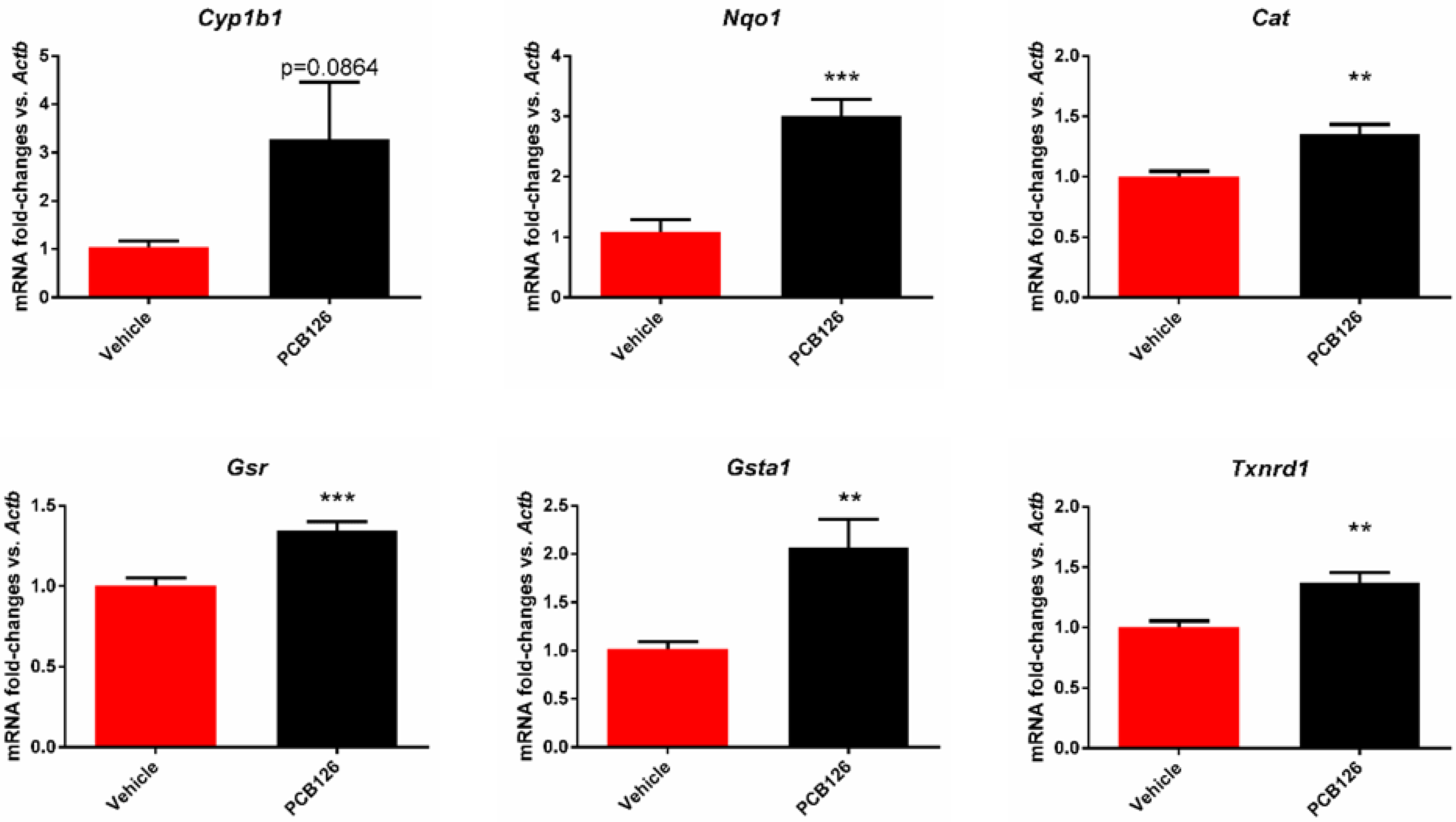
3.1. PCB 126 Activates the Aryl Hydrocarbon Receptor and Increases Oxidative Stress in Cultured Mouse Coronary Endothelial Cells
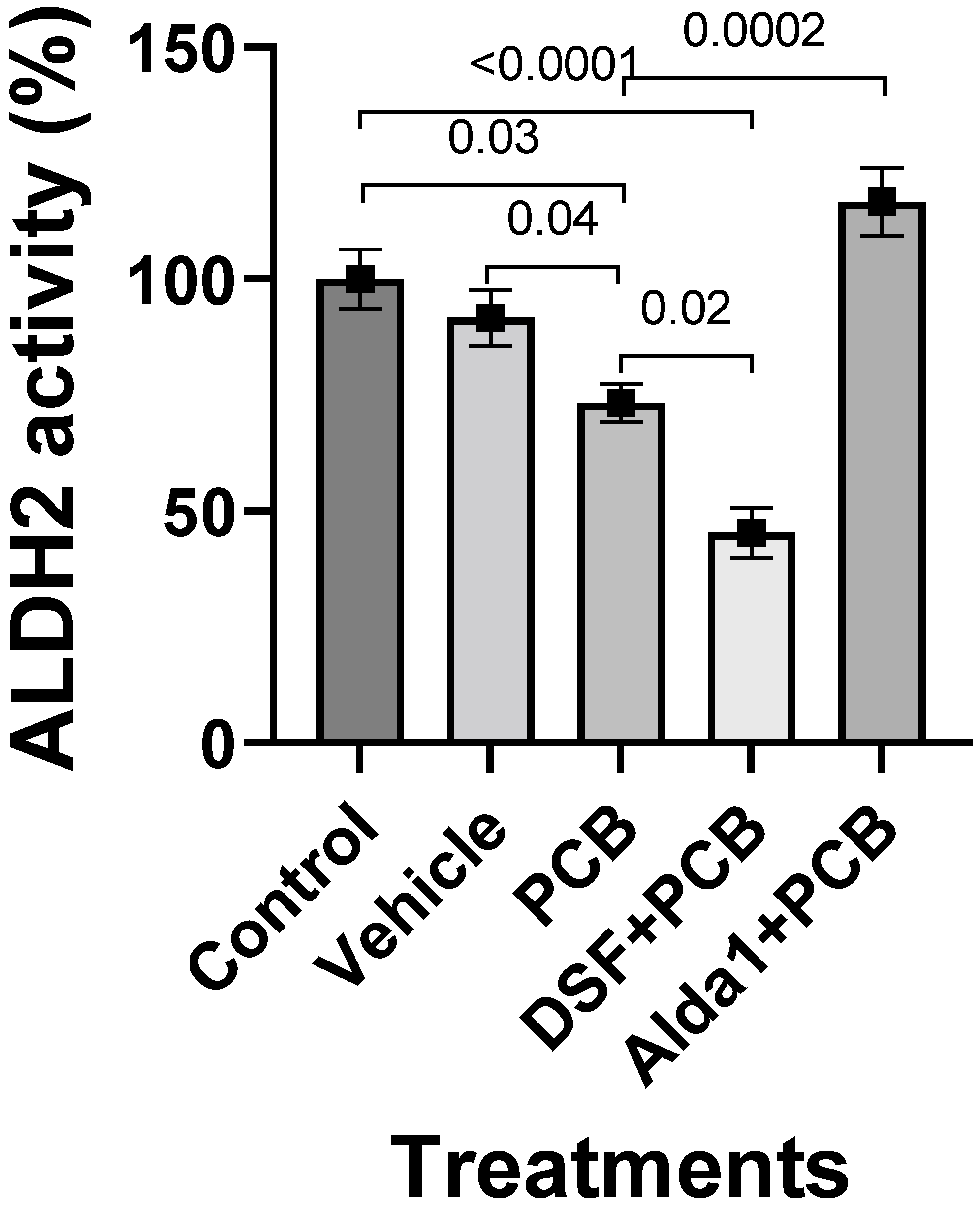
3.2. PCB 126 Decreases ALDH2 Activity, and Pharmacological Inhibition of ALDH2 Exacerbates the PCB-Mediated Effects, Whereas the Pharmacological Activation of ALDH2 Rescues the PCB-Mediated Effects in Cultured Mouse Coronary Endothelial Cells
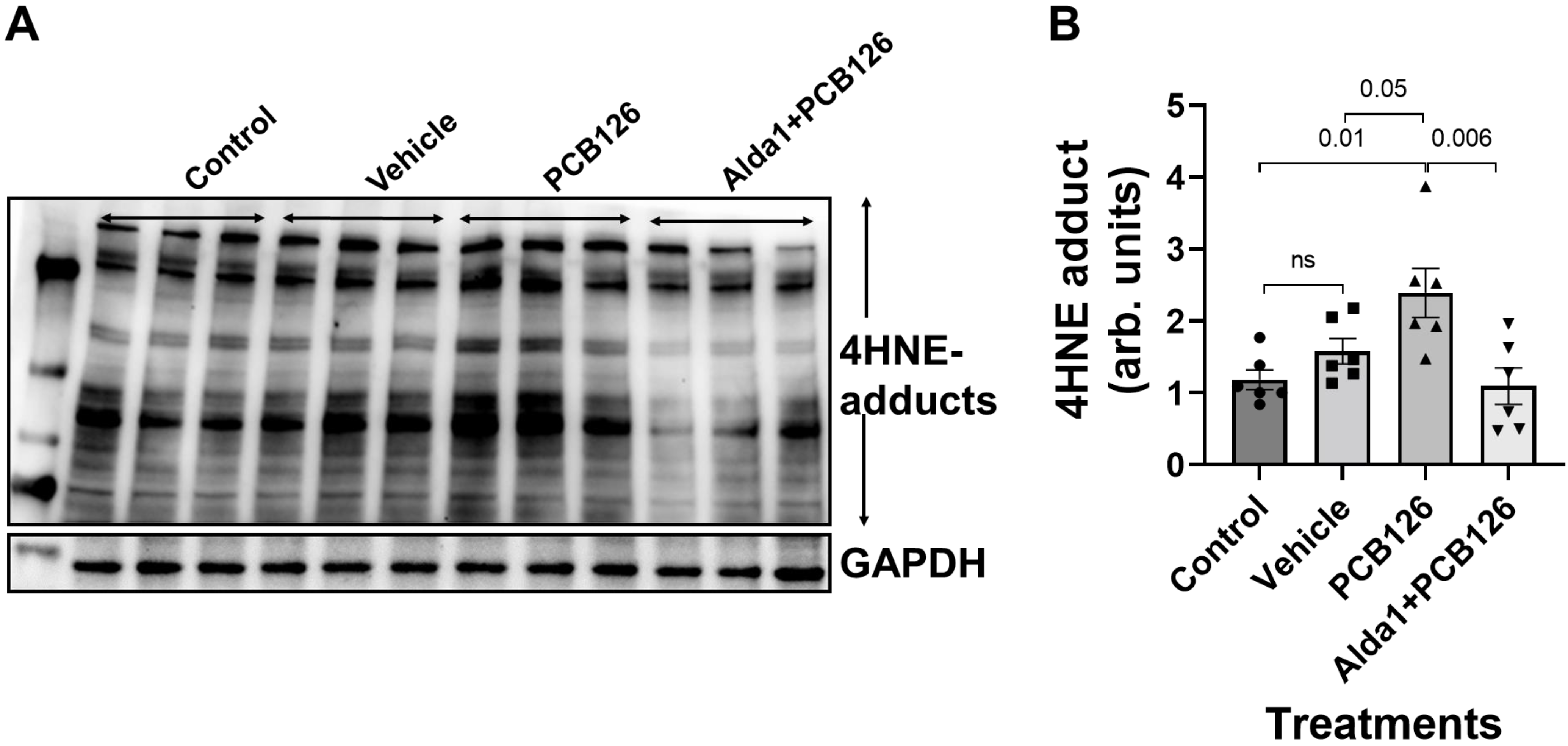
3.3. PCB 126 Increases the 4HNE Protein Adduct Levels, Whereas the Pharmacological Activation of ALDH2 Rescues the PCB-Mediated Effects in Cultured Mouse Coronary Endothelial Cells
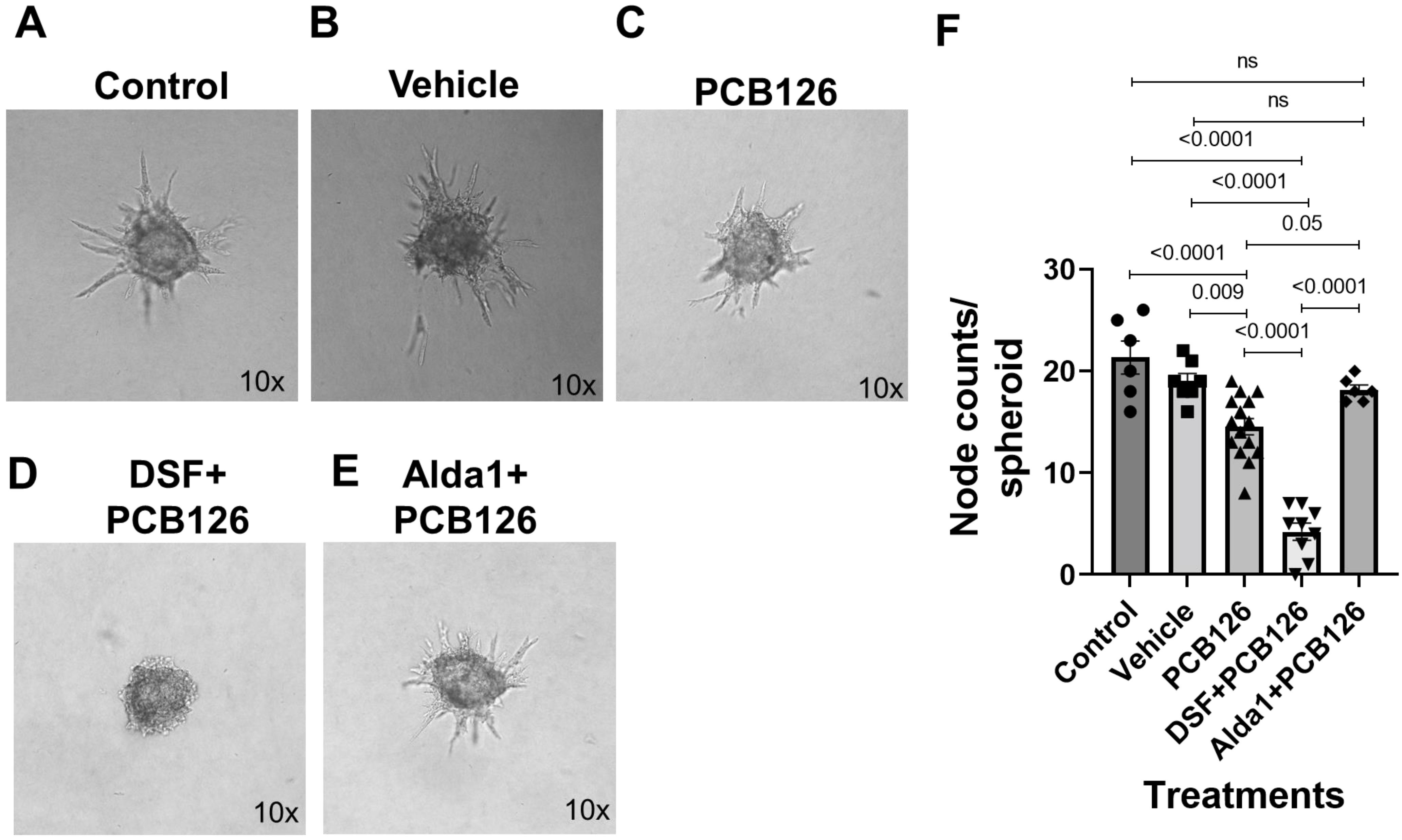
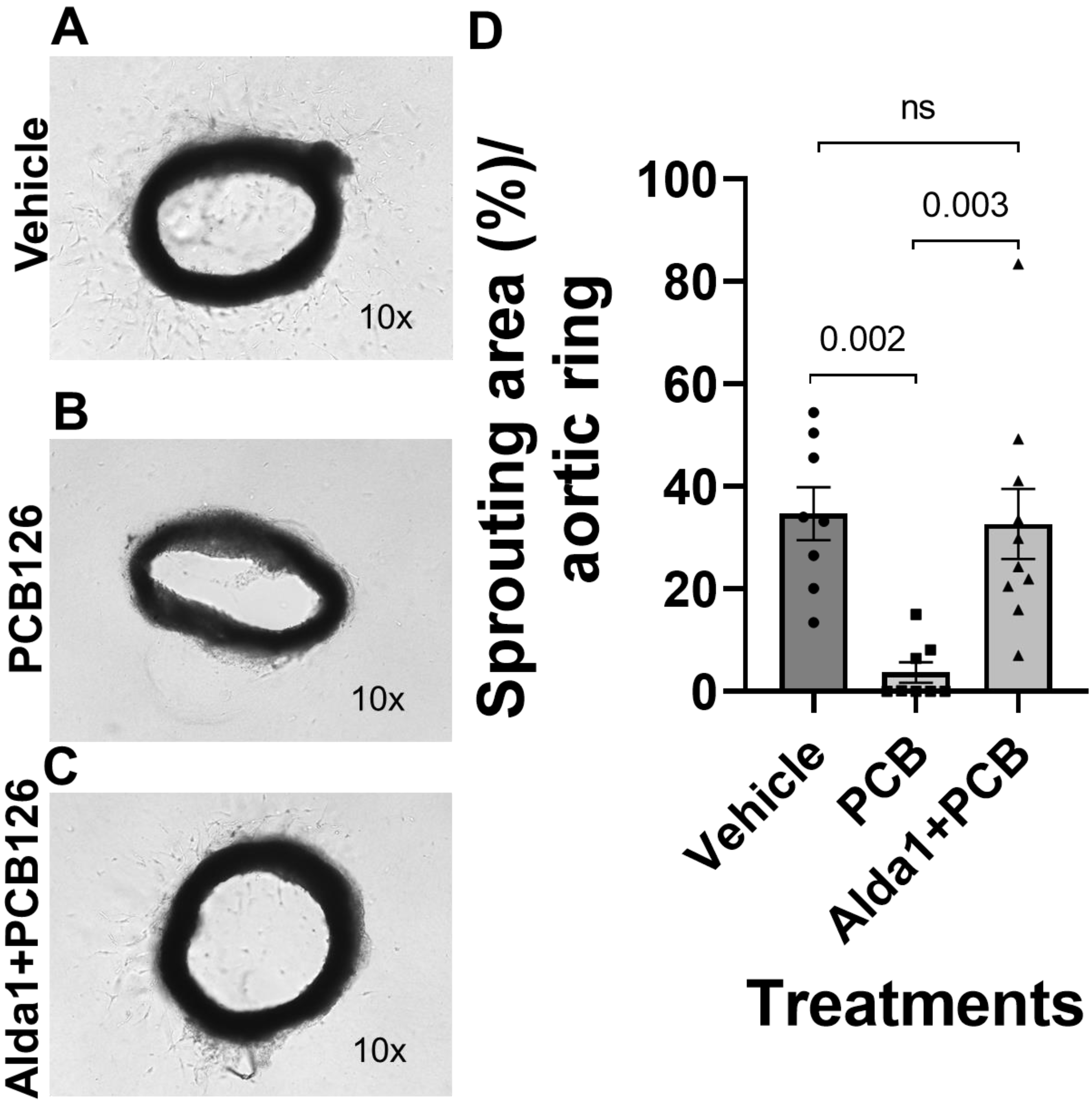
3.4. Pharmacological Inhibition of ALDH2 Exacerbates the PCB 126-Mediated Decrease in Angiogenesis, Whereas the Activation of ALDH2 Attenuates the PCB 126-Mediated Effect in Cultured Mouse Coronary Endothelial Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roy, B.; Palaniyandi, S.S. Aldehyde dehydrogenase 2 inhibition potentiates 4-hydroxy-2-nonenal induced decrease in angiogenesis of coronary endothelial cells. Cell Biochem. Funct. 2020, 38, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Gogiraju, R.; Bochenek, M.L.; Schäfer, K. Angiogenic Endothelial Cell Signaling in Cardiac Hypertrophy and Heart Failure. Front. Cardiovasc. Med. 2019, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, A.; Neas, L.; Herbst, M.C.; Case, M.; Williams, R.W.; Cascio, W.; Hinderliter, A.; Holguin, F.; Buse, J.B.; Dungan, K.; et al. Endothelial Dysfunction: Associations with Exposure to Ambient Fine Particles in Diabetic Individuals. Environ. Health Perspect. 2008, 116, 1666–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Sargis, R.M.; Volden, P.A.; Carmean, C.M.; Sun, X.J.; Brady, M.J. PCB 126 and Other Dioxin-Like PCBs Specifically Suppress Hepatic PEPCK Expression via the Aryl Hydrocarbon Receptor. PLoS ONE 2012, 7, e37103. [Google Scholar] [CrossRef] [Green Version]
- Donat-Vargas, C.; Moreno-Franco, B.; Laclaustra, M.; Sandoval-Insausti, H.; Jarauta, E.; Guallar-Castillon, P. Exposure to dietary polychlorinated biphenyls and dioxins, and its relationship with subclinical coronary atherosclerosis: The Aragon Workers’ Health Study. Environ. Int. 2020, 136, 105433. [Google Scholar] [CrossRef]
- Lind, P.M.; Van Bavel, B.; Salihovic, S.; Lind, L. Circulating Levels of Persistent Organic Pollutants (POPs) and Carotid Atherosclerosis in the Elderly. Environ. Health Perspect. 2012, 120, 38–43. [Google Scholar] [CrossRef]
- Lind, Y.S.; Lind, L.; Salihovic, S.; van Bavel, B.; Lind, P.M. Persistent organic pollutants and abnormal geometry of the left ventricle in the elderly. J. Hypertens. 2013, 31, 1547–1553. [Google Scholar] [CrossRef] [Green Version]
- Åkesson, A.; Donat-Vargas, C.; Berglund, M.; Glynn, A.; Wolk, A.; Kippler, M. Dietary exposure to polychlorinated biphenyls and risk of heart failure—A population-based prospective cohort study. Environ. Int. 2019, 126, 1–6. [Google Scholar] [CrossRef]
- Loiola, R.A.; dos Anjos, F.M.; Shimada, A.L.; Cruz, W.S.; Drewes, C.C.; Rodrigues, S.F.; Cardozo, K.H.M.; Carvalho, V.M.; Pinto, E.; Farsky, S.H. Long-term in vivo polychlorinated biphenyl 126 exposure induces oxidative stress and alters proteomic profile on islets of Langerhans. Sci. Rep. 2016, 6, 27882. [Google Scholar] [CrossRef] [Green Version]
- Shimada, A.L.B.; Cruz, W.S.; Loiola, R.A.; Drewes, C.C.; Dorr, F.; Figueiredo, N.G.; Pinto, E.; Farsky, S.H.P. Absorption of PCB126 by upper airways impairs G protein-coupled receptor-mediated immune response. Sci. Rep. 2015, 5, 14917. [Google Scholar] [CrossRef] [Green Version]
- Wahlang, B.; Barney, J.; Thompson, B.; Wang, C.; Hamad, O.M.; Hoffman, J.B.; Petriello, M.C.; Morris, A.J.; Hennig, B. Editor’s Highlight: PCB126 Exposure Increases Risk for Peripheral Vascular Diseases in a Liver Injury Mouse Model. Toxicol. Sci. 2017, 160, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Perkins, J.T.; Hennig, B. EGCG prevents PCB-126-induced endothelial cell inflammation via epigenetic modifications of NF-kappaB target genes in human endothelial cells. J. Nutr. Biochem. 2016, 28, 164–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petriello, M.C.; Brandon, J.A.; Hoffman, J.; Wang, C.; Tripathi, H.; Abdel-Latif, A.; Ye, X.; Li, X.; Yang, L.; Lee, E.; et al. Dioxin-like PCB 126 Increases Systemic Inflammation and Accelerates Atherosclerosis in Lean LDL Re-ceptor-Deficient Mice. Toxicol. Sci. 2018, 162, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Kalkunte, S.; Huang, Z.; Lippe, E.; Kumar, S.; Robertson, L.W.; Sharma, S. Polychlorinated biphenyls target Notch/Dll and VEGF R2 in the mouse placenta and human trophoblast cell lines for their anti-angiogenic effects. Sci. Rep. 2017, 7, 39885. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Roy, B.; Palaniyandi, S.S. Diabetic Aldehyde Dehydrogenase 2 Mutant (ALDH2*2) Mice Are More Susceptible to Cardiac Ischemic-Reperfusion Injury Due to 4-Hydroxy-2-Nonenal Induced Coronary Endothelial Cell Damage. J. Am. Heart Assoc. 2021, 10, e021140. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-H.; Ferreira, J.C.B.; Gross, E.R.; Mochly-Rosen, D. Targeting Aldehyde Dehydrogenase 2: New Therapeutic Opportunities. Physiol. Rev. 2014, 94, 1–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, C.-L.; Chu, P.; Liu, Y.-X.; Qu, X.-L.; Gao, X.-F.; Wang, Z.-M.; Dong, J.; Chen, S.-L.; Zhang, J.-X. ALDH2 rs671 polymorphism and the risk of heart failure with preserved ejection fraction (HFpEF) in patients with cardiovascular diseases. J. Hum. Hypertens. 2020, 34, 16–23. [Google Scholar] [CrossRef]
- Roy, B.; Sundar, K.; Palaniyandi, S.S. 4-hydroxy-2-nonenal decreases coronary endothelial cell migration: Potentiation by aldehyde dehydrogenase 2 inhibition. Vasc. Pharmacol. 2020, 131, 106762. [Google Scholar] [CrossRef]
- Wang, C.; Petriello, M.C.; Zhu, B.; Hennig, B. PCB 126 induces monocyte/macrophage polarization and inflammation through AhR and NF-kappaB pathways. Toxicol. Appl. Pharmacol. 2019, 367, 71–81. [Google Scholar] [CrossRef]
- Stevens, E.A.; Mezrich, J.D.; Bradfield, C.A. The aryl hydrocarbon receptor: A perspective on potential roles in the immune system. Immunology 2009, 127, 299–311. [Google Scholar] [CrossRef]
- Jung, K.-A.; Kwak, M.-K. The Nrf2 System as a Potential Target for the Development of Indirect Antioxidants. Molecules 2010, 15, 7266–7291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schecter, A.; Birnbaum, L.; Ryan, J.J.; Constable, J.D. Dioxins: An overview. Environ. Res. 2006, 101, 419–428. [Google Scholar] [CrossRef] [PubMed]
- White, S.S.; Birnbaum, L.S. An Overview of the Effects of Dioxins and Dioxin-Like Compounds on Vertebrates, as Documented in Human and Ecological Epidemiology. J. Environ. Sci. Health Part C 2009, 27, 197–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowitz, G. From Industrial Toxins to Worldwide Pollutants: A Brief History of Polychlorinated Biphenyls. Public Health Rep. 2018, 133, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.; Herold, C.; Hollert, H.; Kamphues, J.; Ungemach, L.; Blepp, M.; Ballschmiter, K. Life cycle of PCBs and contamination of the environment and of food products from animal origin. Environ. Sci. Pollut. Res. 2018, 25, 16325–16343. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.H. Polychlorinated Biphenyls (PCBs): Environmental Impact, Biochemical and Toxic Responses, and Implications for Risk Assessment. Crit. Rev. Toxicol. 1994, 24, 87–149. [Google Scholar] [CrossRef]
- Risebrough, R.W.; Rieche, P.; Peakall, D.B.; Herman, S.G.; Kirven, M.N. Polychlorinated Biphenyls in the Global Ecosystem. Nature 1968, 220, 1098–1102. [Google Scholar] [CrossRef]
- Xu, W.; Wang, X.; Cai, Z. Analytical chemistry of the persistent organic pollutants identified in the Stockholm Convention: A review. Anal. Chim. Acta 2013, 790, 1–13. [Google Scholar] [CrossRef]
- Fernández-González, R.; Yebra-Pimentel, I.; Martínez-Carballo, E.; Simal-Gándara, J. A Critical Review about Human Exposure to Polychlorinated Dibenzo-p-Dioxins (PCDDs), Polychlorinated Dibenzofurans (PCDFs) and Polychlorinated Biphenyls (PCBs) through Foods. Crit. Rev. Food Sci. Nutr. 2015, 55, 1590–1617. [Google Scholar] [CrossRef]
- Schecter, A.; Cramer, P.; Boggess, K.; Stanley, J.; Päpke, O.; Olson, J.; Silver, A.; Schmitz, M. Intake of Dioxins and Related Compounds from Food in the U.S. Population. J. Toxicol. Environ. Health Part A 2001, 63, 1–18. [Google Scholar] [CrossRef]
- Xue, J.; Liu, S.V.; Zartarian, V.G.; Geller, A.M.; Schultz, B.D. Analysis of NHANES measured blood PCBs in the general US population and application of SHEDS model to identify key exposure factors. J. Expo. Sci. Environ. Epidemiol. 2014, 24, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Lim, J.-E.; Park, H.; Jee, S.H. Body burden of persistent organic pollutants on hypertension: A meta-analysis. Environ. Sci. Pollut. Res. Int. 2016, 23, 14284–14293. [Google Scholar] [CrossRef] [PubMed]
- Valera, B.; Jørgensen, M.E.; Jeppesen, C.; Bjerregaard, P. Exposure to persistent organic pollutants and risk of hypertension among Inuit from Greenland. Environ. Res. 2013, 122, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.-H.; Lee, D.-H.; Son, H.-K.; Park, S.-K.; Jacobs, D.R. Association between serum concentrations of persistent organic pollutants and prevalence of newly diagnosed hypertension: Results from the National Health and Nutrition Examination Survey 1999–2002. J. Hum. Hypertens. 2009, 23, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Lind, Y.S.; Lind, P.M.; Salihovic, S.; van Bavel, B.; Lind, L. Circulating levels of persistent organic pollutants (POPs) are associated with left ventricular systolic and diastolic dysfunction in the elderly. Environ. Res. 2013, 123, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.G.; Gerstenblith, G.; Bottomley, P.A. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc. Natl. Acad. Sci. USA 2005, 102, 808–813. [Google Scholar] [CrossRef] [Green Version]
- Dham, D.; Roy, B.; Gowda, A.; Pan, G.; Sridhar, A.; Zeng, X.; Thandavarayan, R.A.; Palaniyandi, S.S. 4-Hydroxy-2-nonenal, a lipid peroxidation product, as a biomarker in diabetes and its complications: Challenges and opportunities. Free Radic. Res. 2021, 55, 510–524. [Google Scholar] [CrossRef]
- Xiao, M.; Zhong, H.; Xia, L.; Tao, Y.; Yin, H. Pathophysiology of mitochondrial lipid oxidation: Role of 4-hydroxynonenal (4-HNE) and other bioactive lipids in mitochondria. Free Radic. Biol. Med. 2017, 111, 316–327. [Google Scholar] [CrossRef]
- Barrera, G.; Pizzimenti, S.; Daga, M.; Dianzani, C.; Arcaro, A.; Cetrangolo, G.P.; Giordano, G.; Cucci, M.A.; Graf, M.; Gentile, F. Lipid Peroxidation-Derived Aldehydes, 4-Hydroxynonenal and Malondialdehyde in Aging-Related Disorders. Antioxidants 2018, 7, 102. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-H.; Budas, G.R.; Churchill, E.N.; Disatnik, M.-H.; Hurley, T.D.; Mochly-Rosen, D. Activation of Aldehyde Dehydrogenase-2 Reduces Ischemic Damage to the Heart. Science 2008, 321, 1493–1495. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, M.; Mali, V.R.; Pan, G.; Xu, J.; Yang, X.-P.; Thandavarayan, R.A.; Palaniyandi, S.S. Increased 4-hydroxy-2-nonenal-induced proteasome dysfunction is correlated with cardiac damage in streptozotocin-injected rats with isoproterenol infusion. Cell Biochem. Funct. 2016, 34, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Mali, V.R.; Pan, G.; Deshpande, M.; Thandavarayan, R.A.; Xu, J.; Yang, X.-P.; Palaniyandi, S.S. Cardiac Mitochondrial Respiratory Dysfunction and Tissue Damage in Chronic Hyperglycemia Correlate with Reduced Aldehyde Dehydrogenase-2 Activity. PLoS ONE 2016, 11, e0163158. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Fan, F.; Cao, Q.; Shen, C.; Zhu, H.; Wang, P.; Zhao, X.; Sun, X.; Dong, Z.; Ma, X.; et al. Mitochondrial aldehyde dehydrogenase 2 deficiency aggravates energy metabolism disturbance and diastolic dysfunction in diabetic mice. J. Mol. Med. 2016, 94, 1229–1240. [Google Scholar] [CrossRef]
- Pan, G.; Munukutla, S.; Kar, A.; Gardinier, J.; Thandavarayan, R.A.; Palaniyandi, S.S. Type-2 diabetic aldehyde dehydrogenase 2 mutant mice (ALDH 22) exhibiting heart failure with preserved ejection fraction phenotype can be determined by exercise stress echocar-diography. PLoS ONE 2018, E13, e0195796. [Google Scholar]
- Roy, B.; Palaniyandi, S.S. A role for aldehyde dehydrogenase (ALDH) 2 in angiotensin II-mediated decrease in angiogenesis of coronary endothelial cells. Microvasc. Res. 2021, 135, 104133. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| NQO1 | GGCATCCAGTCCTCCATCAA | GTTAGTCCCTCGGCCATTGTT |
| CAT | CAGAGAGCGGATTCCTGAGAGA | CTTTGCCTTGGAGTATGTGGTGAT |
| GSTM1 | ATACTGGGATACTGGAACGTCC | AGTCAGGGTTGTAACAGAGCAT |
| GSR | TCGGAATTCATGCACGATCA | GGCTCACATAGGCATCCCTTT |
| TXNRD1 | GGCCAAAATCGGTGAACACATGGAAG | CGCCAGCAACACTGTGTTAAATTCGCCC |
| GSTA1 | AAGCCCGTGCTTCACTACTTC | GGGCACTTGGTCAAACATCAAA |
| GPX2 | GTGGCGTCACTCTGAGGAACA | CAGTTCTCCTGATGTCCGAACTG |
| CYP1B1 | AATGAGGAGTTCGGGCGCACA | GGCGTGTGGAATGGTGACAGG |
| ACTB | GCCACTGTCGAGTCGCGT | GATACCTCTCTTGCTCTGGGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, B.; Yang, Z.; Pan, G.; Roth, K.; Agarwal, M.; Sharma, R.; Petriello, M.C.; Palaniyandi, S.S. Exposure to the Dioxin-like Pollutant PCB 126 Afflicts Coronary Endothelial Cells via Increasing 4-Hydroxy-2 Nonenal: A Role for Aldehyde Dehydrogenase 2. Toxics 2022, 10, 328. https://doi.org/10.3390/toxics10060328
Roy B, Yang Z, Pan G, Roth K, Agarwal M, Sharma R, Petriello MC, Palaniyandi SS. Exposure to the Dioxin-like Pollutant PCB 126 Afflicts Coronary Endothelial Cells via Increasing 4-Hydroxy-2 Nonenal: A Role for Aldehyde Dehydrogenase 2. Toxics. 2022; 10(6):328. https://doi.org/10.3390/toxics10060328
Chicago/Turabian StyleRoy, Bipradas, Zhao Yang, Guodong Pan, Katherine Roth, Manisha Agarwal, Rahul Sharma, Michael C. Petriello, and Suresh Selvaraj Palaniyandi. 2022. "Exposure to the Dioxin-like Pollutant PCB 126 Afflicts Coronary Endothelial Cells via Increasing 4-Hydroxy-2 Nonenal: A Role for Aldehyde Dehydrogenase 2" Toxics 10, no. 6: 328. https://doi.org/10.3390/toxics10060328
APA StyleRoy, B., Yang, Z., Pan, G., Roth, K., Agarwal, M., Sharma, R., Petriello, M. C., & Palaniyandi, S. S. (2022). Exposure to the Dioxin-like Pollutant PCB 126 Afflicts Coronary Endothelial Cells via Increasing 4-Hydroxy-2 Nonenal: A Role for Aldehyde Dehydrogenase 2. Toxics, 10(6), 328. https://doi.org/10.3390/toxics10060328