
Proteomics-Based Identification of Interaction Partners of the Xenobiotic Detoxification Enzyme FMO3 Reveals Involvement in Urea Cycle



Abstract
1. Introduction
2. Materials and Methods
2.1. Animal
2.2. Cell Culture
2.3. Co-IP
2.4. Mass Spectrometry Analysis
2.5. Immunoblotting
2.6. Urea/Ammonia Assays
2.7. RT-qPCR
2.8. Statistical Analysis
3. Results
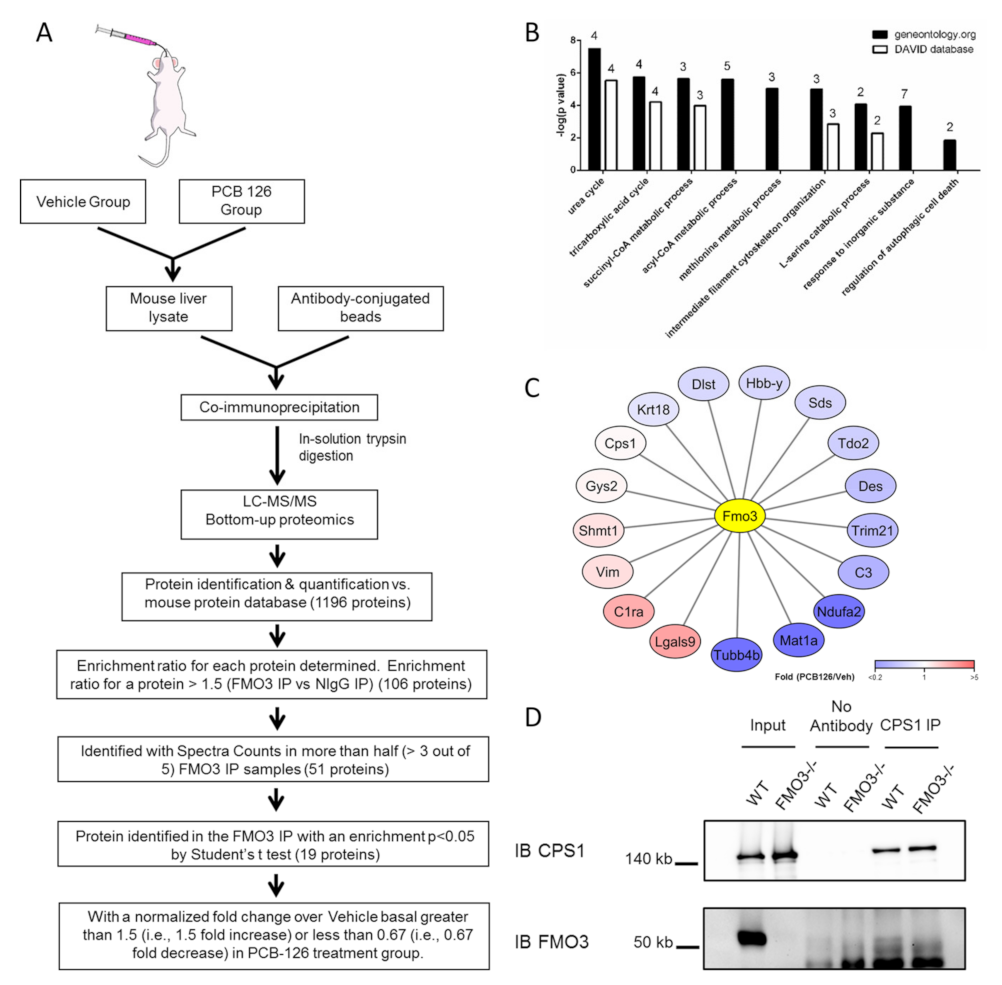
3.1. Hepatic FMO3 Interacts with Multiple Proteins Involved in Urea Cycle
3.2. FMO3 Interacts with CPS1 and Other Proteins of the Urea Cycle
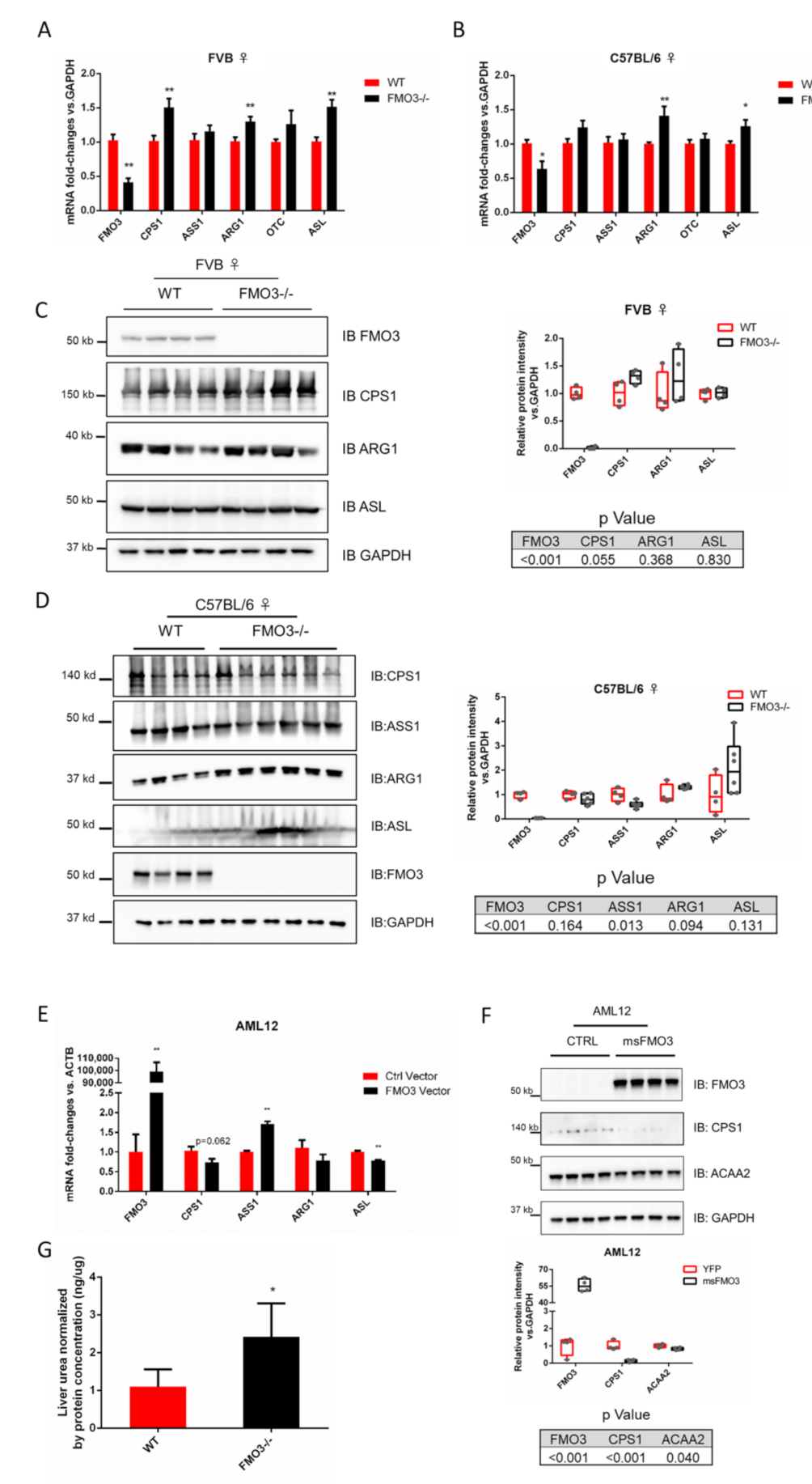
3.3. Modulation of FMO3 Expression Impacts Hepatic Urea Levels
4. Discussion
5. Conclusion and Future Direction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Phillips, I.R.; Shephard, E.A. Flavin-containing monooxygenase 3 (FMO3): Genetic variants and their consequences for drug metabolism and disease. Xenobiotica 2020, 50, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Scimone, C.; Alibrandi, S.; Donato, L.; Giofre, S.V.; Rao, G.; Sidoti, A.; D’Angelo, R. Antiretroviral treatment leading to secondary trimethylaminuria: Genetic associations and successful management with riboflavin. J. Clin. Pharm. Ther. 2021, 46, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Phillips, I.R.; Shephard, E.A. Flavin-containing monooxygenases: Mutations, disease and drug response. Trends Pharmacol. Sci. 2008, 29, 294–301. [Google Scholar] [CrossRef]
- Bennett, B.J.; de Aguiar Vallim, T.Q.; Wang, Z.; Shih, D.M.; Meng, Y.; Gregory, J.; Allayee, H.; Lee, R.; Graham, M.; Crooke, R.; et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013, 17, 49–60. [Google Scholar] [CrossRef]
- Longin-Sauvageon, C.; Lattard, V.; Lilaz-Michel, C.; Buronfosse, T.; Benoit, E. Expression of two different FMOs in sheep liver. Drug Metab. Dispos. 1998, 26, 284–287. [Google Scholar]
- Lattard, V.; Lachuer, J.; Buronfosse, T.; Garnier, F.; Benoit, E. Physiological factors affecting the expression of FMO1 and FMO3 in the rat liver and kidney. Biochem. Pharmacol. 2002, 63, 1453–1464. [Google Scholar] [CrossRef]
- Lattard, V.; Longin-Sauvageon, C.; Lachuer, J.; Delatour, P.; Benoit, E. Cloning, sequencing, and tissue-dependent expression of flavin-containing monooxygenase (FMO) 1 and FMO3 in the dog. Drug Metab. Dispos. 2002, 30, 119–128. [Google Scholar] [CrossRef]
- Ripp, S.L.; Itagaki, K.; Philpot, R.M.; Elfarra, A.A. Species and sex differences in expression of flavin-containing monooxygenase form 3 in liver and kidney microsomes. Drug Metab. Dispos. 1999, 27, 46–52. [Google Scholar]
- Petriello, M.C.; Hoffman, J.B.; Sunkara, M.; Wahlang, B.; Perkins, J.T.; Morris, A.J.; Hennig, B. Dioxin-like pollutants increase hepatic flavin containing monooxygenase (FMO3) expression to promote synthesis of the pro-atherogenic nutrient biomarker trimethylamine N-oxide from dietary precursors. J. Nutr. Biochem. 2016, 33, 145–153. [Google Scholar] [CrossRef]
- Howell, B.W.; Lagace, M.; Shore, G.C. Activity of the carbamyl phosphate synthetase I promoter in liver nuclear extracts is dependent on a cis-acting C/EBP recognition element. Mol. Cell. Biol. 1989, 9, 2928–2933. [Google Scholar] [CrossRef]
- Brown, J.M.; Hazen, S.L. The gut microbial endocrine organ: Bacterially derived signals driving cardiometabolic diseases. Annu. Rev. Med. 2015, 66, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.; Alibrandi, S.; Scimone, C.; Castagnetti, A.; Rao, G.; Sidoti, A.; D’Angelo, R. Gut-Brain Axis Cross-Talk and Limbic Disorders as Biological Basis of Secondary TMAU. J. Pers. Med. 2021, 11, 87. [Google Scholar] [CrossRef]
- Petriello, M.C.; Charnigo, R.; Sunkara, M.; Soman, S.; Pavuk, M.; Birnbaum, L.; Morris, A.J.; Hennig, B. Relationship between serum trimethylamine N-oxide and exposure to dioxin-like pollutants. Environ. Res. 2018, 162, 211–218. [Google Scholar] [CrossRef]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-kappaB. J. Am. Heart Assoc. 2016, 5, e002767. [Google Scholar] [CrossRef] [PubMed]
- Boini, K.M.; Hussain, T.; Li, P.L.; Koka, S. Trimethylamine-N-Oxide Instigates NLRP3 Inflammasome Activation and Endothelial Dysfunction. Cell. Physiol. Biochem. 2017, 44, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Geng, J.; Zhao, J.; Ni, Q.; Zhao, C.; Zheng, Y.; Chen, X.; Wang, L. Trimethylamine N-Oxide Exacerbates Cardiac Fibrosis via Activating the NLRP3 Inflammasome. Front. Physiol. 2019, 10, 866. [Google Scholar] [CrossRef]
- Hengeveld, L.M.; Praagman, J.; Beulens, J.W.J.; Brouwer, I.A.; van der Schouw, Y.T.; Sluijs, I. Fish consumption and risk of stroke, coronary heart disease, and cardiovascular mortality in a Dutch population with low fish intake. Eur. J. Clin. Nutr. 2018, 72, 942–950. [Google Scholar] [CrossRef]
- Zhong, V.W.; Van Horn, L.; Greenland, P.; Carnethon, M.R.; Ning, H.; Wilkins, J.T.; Lloyd-Jones, D.M.; Allen, N.B. Associations of Processed Meat, Unprocessed Red Meat, Poultry, or Fish Intake With Incident Cardiovascular Disease and All-Cause Mortality. JAMA Intern. Med. 2020, 180, 503–512. [Google Scholar] [CrossRef]
- Mohan, D.; Mente, A.; Dehghan, M.; Rangarajan, S.; O’Donnell, M.; Hu, W.; Dagenais, G.; Wielgosz, A.; Lear, S.; Wei, L.; et al. Associations of Fish Consumption With Risk of Cardiovascular Disease and Mortality Among Individuals with or without Vascular Disease from 58 Countries. JAMA Intern. Med. 2021, 181, 631–649. [Google Scholar] [CrossRef]
- Zhang, B.; Xiong, K.; Cai, J.; Ma, A. Fish Consumption and Coronary Heart Disease: A Meta-Analysis. Nutrients 2020, 12, 2278. [Google Scholar] [CrossRef] [PubMed]
- Erkkila, A.T.; Lichtenstein, A.H.; Mozaffarian, D.; Herrington, D.M. Fish intake is associated with a reduced progression of coronary artery atherosclerosis in postmenopausal women with coronary artery disease. Am. J. Clin. Nutr. 2004, 80, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Bennion, B.J.; Daggett, V.; Murphy, K.P. The molecular mechanism of stabilization of proteins by TMAO and its ability to counteract the effects of urea. J. Am. Chem. Soc. 2002, 124, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Zetterholm, S.G.; Verville, G.A.; Boutwell, L.; Boland, C.; Prather, J.C.; Bethea, J.; Cauley, J.; Warren, K.E.; Smith, S.A.; Magers, D.H.; et al. Noncovalent Interactions between Trimethylamine N-Oxide (TMAO), Urea, and Water. J. Phys. Chem. B 2018, 122, 8805–8811. [Google Scholar] [CrossRef]
- Hartiala, J.A.; Tang, W.H.; Wang, Z.; Crow, A.L.; Stewart, A.F.; Roberts, R.; McPherson, R.; Erdmann, J.; Willenborg, C.; Hazen, S.L.; et al. Genome-wide association study and targeted metabolomics identifies sex-specific association of CPS1 with coronary artery disease. Nat. Commun. 2016, 7, 10558. [Google Scholar] [CrossRef]
- Catucci, G.; Querio, G.; Sadeghi, S.J.; Gilardi, G.; Levi, R. Enzymatically Produced Trimethylamine N-Oxide: Conserving It or Eliminating It. Catalysts 2019, 9, 1028. [Google Scholar] [CrossRef]
- Alexovic, M.; Urban, P.L.; Tabani, H.; Sabo, J. Recent advances in robotic protein sample preparation for clinical analysis and other biomedical applications. Clin. Chim. Acta 2020, 507, 104–116. [Google Scholar] [CrossRef]
- Pourhaghighi, R.; Ash, P.E.A.; Phanse, S.; Goebels, F.; Hu, L.Z.M.; Chen, S.; Zhang, Y.; Wierbowski, S.D.; Boudeau, S.; Moutaoufik, M.T.; et al. BraInMap Elucidates the Macromolecular Connectivity Landscape of Mammalian Brain. Cell Syst. 2020, 10, 333–350.e14. [Google Scholar] [CrossRef]
- Zhou, Y.; Eid, T.; Hassel, B.; Danbolt, N.C. Novel aspects of glutamine synthetase in ammonia homeostasis. Neurochem. Int. 2020, 140, 104809. [Google Scholar] [CrossRef]
- Dolphin, C.T.; Janmohamed, A.; Smith, R.L.; Shephard, E.A.; Phillips, I.R. Missense mutation in flavin-containing mono-oxygenase 3 gene, FMO3, underlies fish-odour syndrome. Nat. Genet. 1997, 17, 491–494. [Google Scholar] [CrossRef]
- Yeung, C.K.; Adman, E.T.; Rettie, A.E. Functional characterization of genetic variants of human FMO3 associated with trimethylaminuria. Arch. Biochem. Biophys. 2007, 464, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Persaud, S.J.; Jones, P.M.; Howell, S.L. Activation of protein kinase C is not required for glyceraldehyde-stimulated insulin secretion from rat islets. Biochim. Biophys. Acta 1991, 1095, 183–185. [Google Scholar] [CrossRef]
- Cashman, J.R.; Motika, M.S. Monoamine Oxidases and Flavin-Containing Monooxygenases. In Comprehensive Toxicology, 2nd ed.; McQueen, C.A., Ed.; Elsevier: Oxford, UK, 2010; pp. 77–110. [Google Scholar]
- Gao, C.; Catucci, G.; Castrignano, S.; Gilardi, G.; Sadeghi, S.J. Inactivation mechanism of N61S mutant of human FMO3 towards trimethylamine. Sci. Rep. 2017, 7, 14668. [Google Scholar] [CrossRef] [PubMed]
- Alibrandi, S.; Nicita, F.; Donato, L.; Scimone, C.; Rinaldi, C.; D’Angelo, R.; Sidoti, A. Adaptive Modelling of Mutated FMO3 Enzyme Could Unveil Unexplored Scenarios Linking Variant Haplotypes to TMAU Phenotypes. Molecules 2021, 26, 7045. [Google Scholar] [CrossRef] [PubMed]
- Catucci, G.; Occhipinti, A.; Maffei, M.; Gilardi, G.; Sadeghi, S.J. Effect of human flavin-containing monooxygenase 3 polymorphism on the metabolism of aurora kinase inhibitors. Int. J. Mol. Sci. 2013, 14, 2707–2716. [Google Scholar] [CrossRef] [PubMed]
- Catucci, G.; Bortolussi, S.; Rampolla, G.; Cusumano, D.; Gilardi, G.; Sadeghi, S.J. Flavin-Containing Monooxygenase 3 Polymorphic Variants Significantly Affect Clearance of Tamoxifen and Clomiphene. Basic Clin. Pharmacol. Toxicol. 2018, 123, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Catucci, G.; Aramini, D.; Sadeghi, S.J.; Gilardi, G. Ligand stabilization and effect on unfolding by polymorphism in human flavin-containing monooxygenase 3. Int. J. Biol. Macromol. 2020, 162, 1484–1493. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Hazen, S.L. Microbial modulation of cardiovascular disease. Nat. Rev. Microbiol. 2018, 16, 171–181. [Google Scholar] [CrossRef]
- Guasti, L.; Galliazzo, S.; Molaro, M.; Visconti, E.; Pennella, B.; Gaudio, G.V.; Lupi, A.; Grandi, A.M.; Squizzato, A. TMAO as a biomarker of cardiovascular events: A systematic review and meta-analysis. Intern. Emerg. Med. 2021, 16, 201–207. [Google Scholar] [CrossRef]
- Motika, M.S.; Zhang, J.; Cashman, J.R. Flavin-containing monooxygenase 3 and human disease. Expert Opin. Drug Metab. Toxicol. 2007, 3, 831–845. [Google Scholar] [CrossRef]
- Schugar, R.C.; Shih, D.M.; Warrier, M.; Helsley, R.N.; Burrows, A.; Ferguson, D.; Brown, A.L.; Gromovsky, A.D.; Heine, M.; Chatterjee, A.; et al. The TMAO-Producing Enzyme Flavin-Containing Monooxygenase 3 Regulates Obesity and the Beiging of White Adipose Tissue. Cell Rep. 2017, 19, 2451–2461. [Google Scholar] [CrossRef] [PubMed]
- Wahlang, B.; Barney, J.; Thompson, B.; Wang, C.; Hamad, O.M.; Hoffman, J.B.; Petriello, M.C.; Morris, A.J.; Hennig, B. Editor’s Highlight: PCB126 Exposure Increases Risk for Peripheral Vascular Diseases in a Liver Injury Mouse Model. Toxicol. Sci. 2017, 160, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Gronenborn, A.M. Weak protein complexes: Challenging to study but essential for life. FEBS J. 2014, 281, 1948–1949. [Google Scholar] [CrossRef] [PubMed]
- Sukenik, S.; Ren, P.; Gruebele, M. Weak protein-protein interactions in live cells are quantified by cell-volume modulation. Proc. Natl. Acad. Sci. USA 2017, 114, 6776–6781. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, B.; Walter, P. ERMES-mediated ER-mitochondria contacts: Molecular hubs for the regulation of mitochondrial biology. J. Cell Sci. 2010, 123, 1389–1393. [Google Scholar] [CrossRef]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef]
- Rieusset, J. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: An update. Cell Death Dis. 2018, 9, 388. [Google Scholar] [CrossRef]
- Doghman-Bouguerra, M.; Lalli, E. ER-mitochondria interactions: Both strength and weakness within cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 650–662. [Google Scholar] [CrossRef]
- Xu, L.; Wang, X.; Tong, C. Endoplasmic Reticulum-Mitochondria Contact Sites and Neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 428. [Google Scholar] [CrossRef]
- Xu, Z. Flavin-Containing Monooxygenase-3 and 5: Tissue Distribution, Age-Related Expression and Regulation by Endoplasmic Reticulum Stress. Master’s Thesis, University of Rhode Island, Ann Arbor, MI, USA, 2017. [Google Scholar]
- Zhang, J.; Cashman, J.R. Quantitative analysis of FMO gene mRNA levels in human tissues. Drug Metab. Dispos. 2006, 34, 19–26. [Google Scholar] [CrossRef]
- Gearing, M.E. Novel Mediators of Diabetes-Associated Cardiovascular Disease; Harvard University: Cambridge, MA, USA, 2017. [Google Scholar]
- Klick, D.E.; Shadley, J.D.; Hines, R.N. Differential regulation of human hepatic flavin containing monooxygenase 3 (FMO3) by CCAAT/enhancer-binding protein beta (C/EBPbeta) liver inhibitory and liver activating proteins. Biochem. Pharmacol. 2008, 76, 268–278. [Google Scholar] [CrossRef]
- Celius, T.; Roblin, S.; Harper, P.A.; Matthews, J.; Boutros, P.C.; Pohjanvirta, R.; Okey, A.B. Aryl hydrocarbon receptor-dependent induction of flavin-containing monooxygenase mRNAs in mouse liver. Drug Metab. Dispos. 2008, 36, 2499–2505. [Google Scholar] [CrossRef] [PubMed]
- Tijet, N.; Boutros, P.C.; Moffat, I.D.; Okey, A.B.; Tuomisto, J.; Pohjanvirta, R. Aryl hydrocarbon receptor regulates distinct dioxin-dependent and dioxin-independent gene batteries. Mol. Pharmacol. 2006, 69, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Sadeque, A.J.; Thummel, K.E.; Rettie, A.E. Purification of macaque liver flavin-containing monooxygenase: A form of the enzyme related immunochemically to an isozyme expressed selectively in adult human liver. Biochim. Biophys. Acta 1993, 1162, 127–134. [Google Scholar] [CrossRef]
- Janmohamed, A.; Hernandez, D.; Phillips, I.R.; Shephard, E.A. Cell-, tissue-, sex- and developmental stage-specific expression of mouse flavin-containing monooxygenases (Fmos). Biochem. Pharmacol. 2004, 68, 73–83. [Google Scholar] [CrossRef]
- Miao, J.; Ling, A.V.; Manthena, P.V.; Gearing, M.E.; Graham, M.J.; Crooke, R.M.; Croce, K.J.; Esquejo, R.M.; Clish, C.B.; Morbid Obesity Study Group; et al. Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis. Nat. Commun. 2015, 6, 6498. [Google Scholar] [CrossRef]
- Falls, J.G.; Ryu, D.Y.; Cao, Y.; Levi, P.E.; Hodgson, E. Regulation of mouse liver flavin-containing monooxygenases 1 and 3 by sex steroids. Arch. Biochem. Biophys. 1997, 342, 212–223. [Google Scholar] [CrossRef]
- Wu, J.C.; Merlino, G.; Fausto, N. Establishment and characterization of differentiated, nontransformed hepatocyte cell lines derived from mice transgenic for transforming growth factor alpha. Proc. Natl. Acad. Sci. USA 1994, 91, 674–678. [Google Scholar] [CrossRef]
- Sefried, S.; Haring, H.U.; Weigert, C.; Eckstein, S.S. Suitability of hepatocyte cell lines HepG2, AML12 and THLE-2 for investigation of insulin signalling and hepatokine gene expression. Open Biol. 2018, 8, 180147. [Google Scholar] [CrossRef]
- Shih, D.M.; Wang, Z.; Lee, R.; Meng, Y.; Che, N.; Charugundla, S.; Qi, H.; Wu, J.; Pan, C.; Brown, J.M.; et al. Flavin containing monooxygenase 3 exerts broad effects on glucose and lipid metabolism and atherosclerosis. J. Lipid Res. 2015, 56, 22–37. [Google Scholar] [CrossRef]
- Guo, D.; Shen, Y.; Li, W.; Li, Q.; Miao, Y.; Zhong, Y. Upregulation of flavin-containing monooxygenase 3 mimics calorie restriction to retard liver aging by inducing autophagy. Aging 2020, 12, 931–944. [Google Scholar] [CrossRef] [PubMed]
- Warrier, M.; Shih, D.M.; Burrows, A.C.; Ferguson, D.; Gromovsky, A.D.; Brown, A.L.; Marshall, S.; McDaniel, A.; Schugar, R.C.; Wang, Z.; et al. The TMAO-Generating Enzyme Flavin Monooxygenase 3 Is a Central Regulator of Cholesterol Balance. Cell Rep. 2015, 10, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Yang, P.; Liu, X.; Lu, L.; Chen, Y.; Zhong, X.; Li, Z.; Liu, H.; Ou, C.; et al. Trimethylamine-N-Oxide Promotes Vascular Calcification through Activation of NLRP3 (Nucleotide-Binding Domain, Leucine-Rich-Containing Family, Pyrin Domain-Containing-3) Inflammasome and NF-kappaB (Nuclear Factor kappaB) Signals. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Henderson, A.; Petriello, M.C.; Romano, K.A.; Gearing, M.; Miao, J.; Schell, M.; Sandoval-Espinola, W.J.; Tao, J.; Sha, B.; et al. Trimethylamine N-Oxide Binds and Activates PERK to Promote Metabolic Dysfunction. Cell Metab. 2019, 30, 1141–1151.e5. [Google Scholar] [CrossRef]
- Thorp, E.; Li, G.; Seimon, T.A.; Kuriakose, G.; Ron, D.; Tabas, I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe-/- and Ldlr-/- mice lacking CHOP. Cell Metab. 2009, 9, 474–481. [Google Scholar] [CrossRef]
- Tam, A.B.; Mercado, E.L.; Hoffmann, A.; Niwa, M. ER stress activates NF-kappaB by integrating functions of basal IKK activity, IRE1 and PERK. PLoS ONE 2012, 7, e45078. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef]
- De Chiara, F.; Heeboll, S.; Marrone, G.; Montoliu, C.; Hamilton-Dutoit, S.; Ferrandez, A.; Andreola, F.; Rombouts, K.; Gronbaek, H.; Felipo, V.; et al. Urea cycle dysregulation in non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 905–915. [Google Scholar] [CrossRef]
- Shih, D.M.; Zhu, W.; Schugar, R.C.; Meng, Y.; Jia, X.; Miikeda, A.; Wang, Z.; Zieger, M.; Lee, R.; Graham, M.; et al. Genetic Deficiency of Flavin-Containing Monooxygenase 3 (Fmo3) Protects against Thrombosis but Has Only a Minor Effect on Plasma Lipid Levels-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1045–1054. [Google Scholar] [CrossRef]
- Balmer, L.A.; Whiting, R.; Rudnicka, C.; Gallo, L.A.; Jandeleit, K.A.; Chow, Y.; Chow, Z.; Richardson, K.L.; Forbes, J.M.; Morahan, G. Genetic characterization of early renal changes in a novel mouse model of diabetic kidney disease. Kidney Int. 2019, 96, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Zaidan, M.; Burtin, M.; Zhang, J.D.; Blanc, T.; Barre, P.; Garbay, S.; Nguyen, C.; Vasseur, F.; Yammine, L.; Germano, S.; et al. Signaling pathways predisposing to chronic kidney disease progression. JCI Insight 2020, 5, e126183. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Sekine, K.; Nakamura, K.; Yanai, H.; Tanaka, M.; Miyajima, A. Y-box binding protein-1 down-regulates expression of carbamoyl phosphate synthetase-I by suppressing CCAAT enhancer-binding protein-alpha function in mice. Gastroenterology 2009, 137, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Chowdhury, S.; Tanaka, T.; Shimizu, A.; Iwase, K.; Oyadomari, S.; Gotoh, T.; Matsuzaki, H.; Mori, M.; Akira, S.; et al. CCAAT/enhancer-binding protein beta is required for activation of genes for ornithine cycle enzymes by glucocorticoids and glucagon in primary-cultured hepatocytes. FEBS Lett. 2001, 494, 105–111. [Google Scholar] [CrossRef]
- Christoffels, V.M.; Grange, T.; Kaestner, K.H.; Cole, T.J.; Darlington, G.J.; Croniger, C.M.; Lamers, W.H. Glucocorticoid receptor, C/EBP, HNF3, and protein kinase A coordinately activate the glucocorticoid response unit of the carbamoylphosphate synthetase I gene. Mol. Cell. Biol. 1998, 18, 6305–6315. [Google Scholar] [CrossRef][Green Version]
- Hoogenkamp, M.; Stallen, J.M.; Lamers, W.H.; Gaemers, I.C. In vivo footprinting of the carbamoylphosphate synthetase I cAMP-response unit indicates important roles for FoxA and PKA in formation of the enhanceosome. Biochimie 2006, 88, 1357–1366. [Google Scholar] [CrossRef]
- Hoogenkamp, M.; Gaemers, I.C.; Schoneveld, O.J.; Das, A.T.; Grange, T.; Lamers, W.H. Hepatocyte-specific interplay of transcription factors at the far-upstream enhancer of the carbamoylphosphate synthetase gene upon glucocorticoid induction. FEBS J. 2007, 274, 37–45. [Google Scholar] [CrossRef]
- Singh, C.; Benos, A.; Grenell, A.; Tran, V.; Hanna, D.; Anand-Apte, B.; Brunengraber, H.; Sears, J.E. The urea cycle is transcriptionally controlled by hypoxia-inducible factors. bioRxiv 2021. [Google Scholar] [CrossRef]
- Saheki, T.; Li, M.X.; Kobayashi, K. Antagonizing effect of AP-1 on glucocorticoid induction of urea cycle enzymes: A study of hyperammonemia in carnitine-deficient, juvenile visceral steatosis mice. Mol. Genet. Metab. 2000, 71, 545–551. [Google Scholar] [CrossRef]
- Li, L.; Mao, Y.; Zhao, L.; Li, L.; Wu, J.; Zhao, M.; Du, W.; Yu, L.; Jiang, P. p53 regulation of ammonia metabolism through urea cycle controls polyamine biosynthesis. Nature 2019, 567, 253–256. [Google Scholar] [CrossRef]
- Liu, H.; Dong, H.; Robertson, K.; Liu, C. DNA methylation suppresses expression of the urea cycle enzyme carbamoyl phosphate synthetase 1 (CPS1) in human hepatocellular carcinoma. Am. J. Pathol. 2011, 178, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Corvi, M.M.; Soltys, C.L.; Berthiaume, L.G. Regulation of mitochondrial carbamoyl-phosphate synthetase 1 activity by active site fatty acylation. J. Biol. Chem. 2001, 276, 45704–45712. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, A.A.; Choudhury, M.; Rahman, S.M.; McCurdy, C.E.; Friederich, M.; Van Hove, J.L.; Watson, P.A.; Birdsey, N.; Bao, J.; Gius, D.; et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem. J. 2011, 433, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Peng, C.; Anderson, K.A.; Chhoy, P.; Xie, Z.; Dai, L.; Park, J.; Chen, Y.; Huang, H.; Zhang, Y.; et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014, 19, 605–617. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, X.; Bi, J.; Li, Z.; Zhang, Z.; Kong, C. Caspase recruitment domain family member 10 regulates carbamoyl phosphate synthase 1 and promotes cancer growth in bladder cancer cells. J. Cell. Mol. Med. 2019, 23, 8128–8138. [Google Scholar] [CrossRef]
- Lu, M.; Bai, J.; Xu, B.; Sun, Q.Y.; Wei, F.X.; Tang, X.F.; Zhang, H.F.; Li, J.; Wang, G.L.; Yin, Q.Q.; et al. Effect of alpha-lipoic acid on relieving ammonia stress and hepatic proteomic analyses of broilers. Poult. Sci. 2017, 96, 88–97. [Google Scholar] [CrossRef]
- Saito, H.; Kaneko, Y.; Hashimoto, Y.; Shirai, T.; Kudo, H.; Otsuka, K.; Mitsubayashi, K. Ammonia bio-sniffer with flavin-containing monooxygenase. Sens. Actuat. B Chem. 2007, 123, 877–881. [Google Scholar] [CrossRef]
- Davie, S.A.; Maglione, J.E.; Manner, C.K.; Young, D.; Cardiff, R.D.; MacLeod, C.L.; Ellies, L.G. Effects of FVB/NJ and C57Bl/6J strain backgrounds on mammary tumor phenotype in inducible nitric oxide synthase deficient mice. Transgenic Res. 2007, 16, 193–201. [Google Scholar] [CrossRef]
- Szade, A.; Nowak, W.N.; Szade, K.; Gese, A.; Czypicki, R.; Was, H.; Dulak, J.; Jozkowicz, A. Effect of crossing C57BL/6 and FVB mouse strains on basal cytokine expression. Mediat. Inflamm. 2015, 2015, 762419. [Google Scholar] [CrossRef]
- Ostergren, C.; Shim, J.; Larsen, J.V.; Nielsen, L.B.; Bentzon, J.F. Genetic analysis of ligation-induced neointima formation in an F2 intercross of C57BL/6 and FVB/N inbred mouse strains. PLoS ONE 2015, 10, e0121899. [Google Scholar] [CrossRef]
- Sato, Y.; Seo, N.; Kobayashi, E. Genetic background differences between FVB and C57BL/6 mice affect hypnotic susceptibility to pentobarbital, ketamine and nitrous oxide, but not isoflurane. Acta Anaesthesiol. Scand. 2006, 50, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Missailidis, C.; Hallqvist, J.; Qureshi, A.R.; Barany, P.; Heimburger, O.; Lindholm, B.; Stenvinkel, P.; Bergman, P. Serum Trimethylamine-N-Oxide Is Strongly Related to Renal Function and Predicts Outcome in Chronic Kidney Disease. PLoS ONE 2016, 11, e0141738. [Google Scholar] [CrossRef] [PubMed]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ruan, J.; Li, D.; Wang, M.; Han, Z.; Qiu, W.; Wu, G. The Role of Intestinal Bacteria and Gut-Brain Axis in Hepatic Encephalopathy. Front. Cell. Infect. Microbiol. 2020, 10, 595759. [Google Scholar] [CrossRef]
- Rai, R.; Saraswat, V.A.; Dhiman, R.K. Gut microbiota: Its role in hepatic encephalopathy. J. Clin. Exp. Hepatol. 2015, 5, S29–S36. [Google Scholar] [CrossRef]
- Maura, D.; Elmekki, N.; Goddard, C.A. The ammonia oxidizing bacterium Nitrosomonas eutropha blocks T helper 2 cell polarization via the anti-inflammatory cytokine IL-10. Sci. Rep. 2021, 11, 14162. [Google Scholar] [CrossRef]
- Vince, A.; Dawson, A.M.; Park, N.; O’Grady, F. Ammonia production by intestinal bacteria. Gut 1973, 14, 171–177. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene Name | FMO3 IP vs. NIgG IP | Fold Change Norm. by NIgG IP (PCB Treated/Vehicle) | Half Sample Size Criteria | |||||
|---|---|---|---|---|---|---|---|---|
| General (n = 9) | Vehicle (n = 5) | PCB-126 Treated (n = 4) | ||||||
| Enrichment Fold | Enrichment p-Value | Enrichment Fold | Enrichment p-Value | Enrichment Fold | Enrichment p-Value | |||
| CPS1 | 1.7 | 0.014 | 1.7 | 0.186 | 1.7 | 0.037 | 1.2 | Passed in both groups |
| ASS1 | 0.8 | 0.786 | 0.5 | 0.392 | 1.7 | 0.424 | 4.0 | |
| ARG1 | 1.8 | 0.170 | 2.5 | 0.308 | 1.4 | 0.373 | 0.7 | |
| ACAA2 | 1.5 | 0.240 | 1.5 | 0.471 | 1.5 | 0.422 | 1.2 | |
| C3 | 13.8 | 0.000 | 19.0 | 0.000 | 9.1 | 0.016 | 0.6 | |
| CYP1A2 | 1.4 | 0.553 | 7.3 | 0.138 | 1.2 | 0.568 | 0.2 | |
| DES | 3.1 | 0.022 | 4.1 | 0.064 | 2.2 | 0.264 | 0.7 | |
| DLST | 9.5 | 0.000 | 11.0 | 0.001 | 7.0 | 0.015 | 0.8 | |
| FMO5 | 1.8 | 0.330 | 2.4 | 0.394 | 1.0 | 0.804 | 0.5 | |
| GSTM1 | 1.3 | 0.497 | 1.7 | 0.479 | 1.1 | 0.654 | 0.8 | |
| GYS2 | 1.8 | 0.000 | 1.6 | 0.014 | 2.0 | 0.015 | 1.3 | |
| HBB-Y | 3.4 | 0.000 | 3.8 | 0.007 | 3.0 | 0.016 | 0.8 | |
| KRT18 | 5.2 | 0.020 | 5.9 | 0.126 | 4.1 | 0.037 | 0.9 | |
| KRT8 | 2.1 | 0.102 | 1.6 | 0.386 | 2.7 | 0.204 | 2.1 | |
| MRI1 | 6.5 | 0.002 | 34.0 | 0.041 | 3.4 | 0.042 | 0.1 | |
| MVP | 1.5 | 0.358 | 1.3 | 0.716 | 1.7 | 0.414 | 1.6 | |
| RPL13 | 0.9 | 0.984 | 3.0 | 0.282 | 0.3 | 0.085 | 0.1 | |
| SHMT1 | 2.9 | 0.049 | 2.4 | 0.276 | 3.4 | 0.134 | 1.8 | |
| SLC25A3 | 1.4 | 0.392 | 0.9 | 0.880 | 2.0 | 0.278 | 2.8 | |
| TDO2 | 13.0 | 0.004 | 15.0 | 0.019 | 9.0 | 0.079 | 0.8 | |
| TUBB4B | 4.4 | 0.036 | 12.0 | 0.100 | 2.5 | 0.259 | 0.3 | |
| VIM | 5.8 | 0.005 | 5.0 | 0.137 | 7.5 | 0.000 | 1.9 | |
| ATP5PD | 1.5 | 0.427 | 3.3 | 0.123 | 0.7 | 0.926 | 0.3 | Passed in the Vehicle group |
| C1QB | 1.9 | 0.156 | 2.0 | 0.351 | 1.8 | 0.368 | 1.1 | |
| C1RA | 12.0 | 0.021 | 1.6 | 0.014 | 4.0 | 0.407 | 3.1 | |
| ETFA | 1.0 | 0.825 | 1.5 | 0.562 | 0.5 | 0.543 | 0.4 | |
| FTCD | 1.4 | 0.640 | 2.9 | 0.443 | 0.3 | 0.382 | 0.1 | |
| GLUL | 2.9 | 0.106 | 6.0 | 0.185 | 1.5 | 0.481 | 0.3 | |
| GNMT | 1.8 | 0.277 | 1.9 | 0.490 | 1.6 | 0.468 | 1.1 | |
| GOLGA1 | 1.7 | 0.463 | 4.8 | 0.159 | 0.3 | 0.595 | 0.1 | |
| GOLGA3 | 12.0 | 0.154 | 2.4 | 0.133 | 0 | 0.407 | 0.0 | |
| GSTZ1 | 14.0 | 0.032 | 1.2 | 0.040 | 8.0 | 0.197 | 8.3 | |
| HINT1 | 1.4 | 0.335 | 3.3 | 0.079 | 0.8 | 0.958 | 0.3 | |
| MAT1A | 4.6 | 0.028 | 8.8 | 0.040 | 1.8 | 0.475 | 0.3 | |
| NDUFA2 | 3.2 | 0.045 | 6.0 | 0.046 | 1.3 | 0.619 | 0.3 | |
| OGDH | 2.0 | 0.355 | 2.0 | 0.545 | 2.0 | 0.407 | 1.3 | |
| PHB2 | 1.0 | 0.817 | 0.7 | 0.461 | 1.6 | 0.495 | 2.8 | |
| PKLR | 3.5 | 0.069 | 2.3 | 0.373 | 7.0 | 0.141 | 3.8 | |
| RPL7A | 2.2 | 0.093 | 2.8 | 0.130 | 1.5 | 0.534 | 0.7 | |
| SDS | 9.0 | 0.026 | 1.6 | 0.014 | 1.0 | 0.879 | 0.8 | |
| SERBP1 | 4.5 | 0.068 | 7.0 | 0.094 | 2.0 | 0.563 | 0.4 | |
| SUCLG2 | 2.6 | 0.092 | 3.2 | 0.153 | 1.8 | 0.483 | 0.7 | |
| TLN1 | 1.8 | 0.357 | 2.5 | 0.420 | 1.0 | 0.814 | 0.5 | |
| VWA8 | 3.0 | 0.081 | 7.0 | 0.128 | 1.7 | 0.443 | 0.3 | |
| OTC | 2.1 | 0.319 | 2.3 | 0.606 | 2.0 | 0.334 | 1.1 | Passed in the PCB-126-treated group |
| KRT10 | 1.0 | 0.876 | 0.4 | 0.371 | 3.3 | 0.272 | 9.3 | |
| LGALS9 | 2.4 | 0.133 | 1.3 | 0.862 | 3.4 | 0.048 | 3.4 | |
| PCCA | 3.8 | 0.084 | 0.8 | 0.207 | 2.8 | 0.142 | 4.3 | |
| PRDX1 | 2.2 | 0.182 | 2.8 | 0.391 | 1.8 | 0.353 | 0.8 | |
| SUCLG1 | 2.0 | 0.179 | 1.6 | 0.633 | 2.7 | 0.111 | 2.1 | |
| TRIM21 | 4.3 | 0.062 | 2.3 | 0.471 | 1.5 | 0.011 | 0.6 | |
| Gene Name | Function/ Compartment | FMO3−/− | Ms FMO3 Overexpression | ||||
|---|---|---|---|---|---|---|---|
| C57BL/6 ♀ | FVB ♀ | AML12 | |||||
| Fold | p-Value | Fold | p-Value | Fold | p-Value | ||
| Cps1 | Urea cycle | 1.23 | 0.070 | 1.48 | 0.005 | 0.72 | 0.062 |
| Ass1 | Urea cycle | 1.05 | 0.695 | 1.12 | 0.347 | 1.70 | <0.001 |
| Arg1 | Urea cycle | 1.41 | 0.008 | 1.28 | 0.008 | 0.71 | 0.221 |
| Otc | Urea cycle | 1.07 | 0.455 | 1.26 | 0.199 | Not detected | |
| Asl | Urea cycle | 1.26 | 0.018 | 1.50 | 0.001 | 0.78 | <0.001 |
| Glul | Ammonia metabolism | 1.05 | 0.594 | 1.55 | 0.028 | 0.71 | <0.001 |
| Vim | Intermediate filament | 1.52 | 0.027 | 1.53 | 0.034 | 1.26 | 0.001 |
| Krt18 | Intermediate filament | 0.74 | 0.014 | 0.97 | 0.833 | 1.04 | 0.186 |
| Mvp | Autophagic cell death | 1.41 | 0.003 | 1.05 | 0.687 | 0.92 | 0.038 |
| Phb2 | Autophagic cell death | 1.31 | 0.009 | 1.05 | 0.361 | 1.04 | 0.408 |
| C3 | Complement | 0.99 | 0.910 | 1.19 | 0.350 | 0.75 | 0.036 |
| C1qb | Complement | 1.23 | 0.069 | 1.16 | 0.362 | Not detected | |
| Slc25a3 | Mitochondrial | 1.41 | 0.030 | 1.19 | 0.187 | 0.80 | <0.001 |
| Acaa2 | Mitochondrial | 1.24 | 0.088 | 1.15 | 0.334 | 0.69 | <0.001 |
| Dlst | Mitochondrial | 1.34 | 0.035 | 1.15 | 0.119 | 0.90 | 0.062 |
| Shmt1 | Mitochondrial | 1.25 | 0.087 | 1.02 | 0.891 | 0.94 | 0.193 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Stemmer, P.M.; Petriello, M.C. Proteomics-Based Identification of Interaction Partners of the Xenobiotic Detoxification Enzyme FMO3 Reveals Involvement in Urea Cycle. Toxics 2022, 10, 60. https://doi.org/10.3390/toxics10020060
Yang Z, Stemmer PM, Petriello MC. Proteomics-Based Identification of Interaction Partners of the Xenobiotic Detoxification Enzyme FMO3 Reveals Involvement in Urea Cycle. Toxics. 2022; 10(2):60. https://doi.org/10.3390/toxics10020060
Chicago/Turabian StyleYang, Zhao, Paul M. Stemmer, and Michael C. Petriello. 2022. "Proteomics-Based Identification of Interaction Partners of the Xenobiotic Detoxification Enzyme FMO3 Reveals Involvement in Urea Cycle" Toxics 10, no. 2: 60. https://doi.org/10.3390/toxics10020060
APA StyleYang, Z., Stemmer, P. M., & Petriello, M. C. (2022). Proteomics-Based Identification of Interaction Partners of the Xenobiotic Detoxification Enzyme FMO3 Reveals Involvement in Urea Cycle. Toxics, 10(2), 60. https://doi.org/10.3390/toxics10020060