


A Physiologically Based Pharmacokinetic (PBPK) Modeling Framework for Mixtures of Dioxin-like Compounds



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Model Structure
2.2. Automated Vector Representation of Congener ODEs
2.3. Model Parameterization
2.4. Monte Carlo Simulation
2.5. Calculation of TEQ
2.6. Modeling Tools and Sharing
3. Results
3.1. Model Validation for TCDD Exposure
3.2. Simulations of Binary Mixtures
3.3. Simulation of High-Order Mixtures
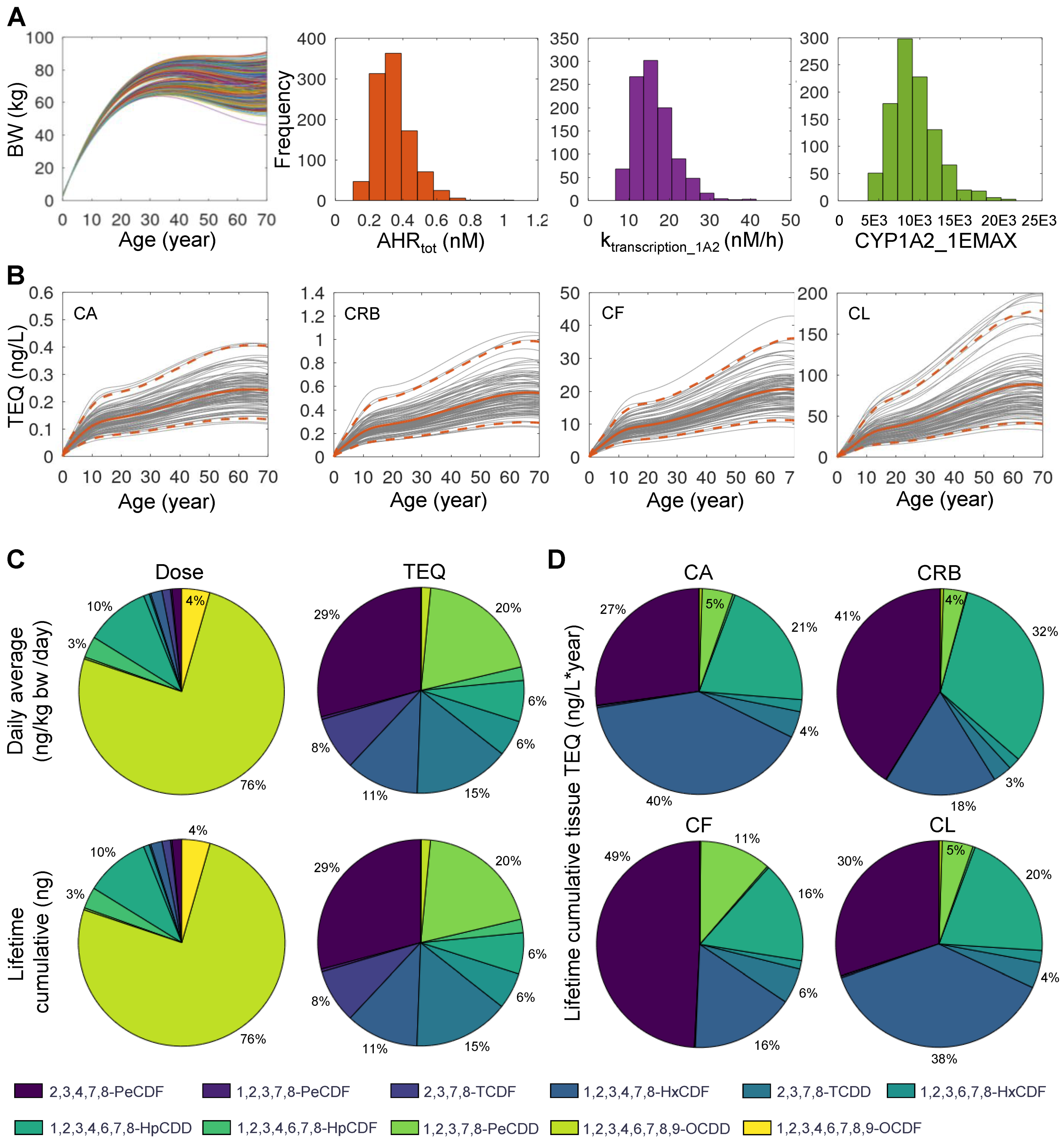
3.4. Monte Carlo Simulation of Population Variability
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- White, S.S.; Birnbaum, L.S. An overview of the effects of dioxins and dioxin-like compounds on vertebrates, as documented in human and ecological epidemiology. J. Environ. Sci. Health C. Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, M.; Birnbaum, L.S.; Denison, M.; De Vito, M.; Farland, W.; Feeley, M.; Fiedler, H.; Hakansson, H.; Hanberg, A.; Haws, L.; et al. The 2005 World Health Organization reevaluation of human and Mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicol. Sci. 2006, 93, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Dopico, M.; Gómez, A. Review of the current state and main sources of dioxins around the world. J. Air Waste Manag. Assoc. 2015, 65, 1033–1049. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.D.; Kaley, R.G., 2nd. Applications of polychlorinated biphenyls. Environ. Sci. Pollut. Res. Int. 2011, 18, 135–151. [Google Scholar] [CrossRef] [PubMed]
- ATSDR. Toxicological Profile for Chlorinated Dibenzo-p-Dioxins (CDDs); U.S. Department of Health and Human Services, P.H.S.: Atlanta, GA, USA, 1998. Available online: https://www.atsdr.cdc.gov/toxprofiles/tp104.pdf (accessed on 14 November 2022).
- Lin, Y.-S.; Caffrey, J.L.; Hsu, P.-C.; Chang, M.-H.; Faramawi, M.F.; Lin, J.-W. Environmental exposure to dioxin-like compounds and the mortality risk in the U.S. population. Int. J. Hygiene Environ. Health 2012, 215, 541–546. [Google Scholar] [CrossRef]
- Turyk, M.E.; Anderson, H.A.; Persky, V.W. Relationships of Thyroid Hormones with Polychlorinated Biphenyls, Dioxins, Furans, and DDE in Adults. Environ. Health Perspect. 2007, 115, 1197–1203. [Google Scholar] [CrossRef]
- Mínguez-Alarcón, L.; Sergeyev, O.; Burns, J.S.; Williams, P.L.; Lee, M.M.; Korrick, S.A.; Smigulina, L.; Revich, B.; Hauser, R. A Longitudinal Study of Peripubertal Serum Organochlorine Concentrations and Semen Parameters in Young Men: The Russian Children’s Study. Environ. Health Perspect. 2017, 125, 460–466. [Google Scholar] [CrossRef]
- Tsai, Y.-A.; Mao, I.F.; Chi, K.H.; Chang, M.B.; Feng, C.-C.; Lin, C.-H.; Hung, P.C.; Chen, M.L. Health Risk from Exposure to PCDD/Fs from a Waelz Plant in Central Taiwan. Aerosol. Air Qual. Res. 2014, 14, 1310–1319. [Google Scholar] [CrossRef]
- Hilscherova, K.; Kannan, K.; Nakata, H.; Hanari, N.; Yamashita, N.; Bradley, P.W.; McCabe, J.M.; Taylor, A.B.; Giesy, J.P. Polychlorinated dibenzo-p-dioxin and dibenzofuran concentration profiles in sediments and flood-plain soils of the Tittabawassee River. Environ. Sci. Technol. 2003, 37, 468–474. [Google Scholar] [CrossRef]
- Taylor, A.B.; McCabe, J.M. Baseline Chemical Characterization of Saginaw Bay Watershed Sediments: A Report to the Office of the Great Lakes Michigan Department of Environmental Quality. 2002. Available online: https://semspub.epa.gov/work/05/276625.pdf (accessed on 14 November 2022).
- Chan, J.K.Y.; Man, Y.B.; Xing, G.H.; Wu, S.C.; Murphy, M.B.; Xu, Y.; Wong, M.H. Dietary exposure to polychlorinated dibenzo-p-dioxins and dibenzofurans via fish consumption and dioxin-like activity in fish determined by H4IIE-luc bioassay. Sci. Total Environ. 2013, 463–464, 1192–1200. [Google Scholar] [CrossRef]
- Coady, K.K.; Jones, P.D.; Giesy, J.P. 2,3,7,8-tetrachlorodibenzo-p-dioxin equivalents in tissue samples from three species in the Denver, Colorado, USA, metropolitan area. Environ. Toxicol. Chem. 2001, 20, 2433–2442. [Google Scholar] [CrossRef]
- Liem, A.K.; Fürst, P.; Rappe, C. Exposure of populations to dioxins and related compounds. Food Addit. Contam. 2000, 17, 241–259. [Google Scholar] [CrossRef]
- Maruyama, W.; Yoshida, K.; Tanaka, T.; Nakanishi, J. Possible range of dioxin concentration in human tissues: Simulation with a physiologically based model. J. Toxicol. Environ. Health A 2002, 65, 2053–2073. [Google Scholar] [CrossRef]
- Shih, Y.-H.; Kasaon, S.J.e.; Tseng, C.-H.; Wang, H.-C.; Chen, L.-L.; Chang, Y.-M. Health risks and economic costs of exposure to PCDD/Fs from open burning: A case study in Nairobi, Kenya. Air Qual. Atmos. Health 2016, 9, 201–211. [Google Scholar] [CrossRef]
- Mari, M.; Nadal, M.; Schuhmacher, M.; Domingo, J.L. Body burden monitoring of dioxins and other organic substances in workers at a hazardous waste incinerator. Int. J. Hyg. Environ. Health 2013, 216, 728–734. [Google Scholar] [CrossRef]
- Iida, T.; Hirakawa, H.; Matsueda, T.; Takenaka, S.; Yu, M.L.; Guo, Y.L. Recent trend of polychlorinated dibenzo-p-dioxins and their related compounds in the blood and sebum of Yusho and Yu Cheng patients. Chemosphere 1999, 38, 981–993. [Google Scholar] [CrossRef]
- Iida, T.; Hirakawa, H.; Matsueda, T.; Nagayama, J.; Nagata, T. Polychlorinated dibenzo-p-dioxins and related compounds: Correlations of levels in human tissues and in blood. Chemosphere 1999, 38, 2767–2774. [Google Scholar] [CrossRef]
- Leung, H.W.; Poland, A.; Paustenbach, D.J.; Murray, F.J.; Andersen, M.E. Pharmacokinetics of [125I]-2-iodo-3,7,8-trichlorodibenzo-p-dioxin in mice: Analysis with a physiological modeling approach. Toxicol. Appl. Pharmacol. 1990, 103, 411–419. [Google Scholar] [CrossRef]
- Wang, X.; Santostefano, M.J.; Evans, M.V.; Richardson, V.M.; Diliberto, J.J.; Birnbaum, L.S. Determination of parameters responsible for pharmacokinetic behavior of TCDD in female Sprague-Dawley rats. Toxicol. Appl. Pharmacol. 1997, 147, 151–168. [Google Scholar] [CrossRef]
- Wang, X.; Santostefano, M.J.; DeVito, M.J.; Birnbaum, L.S. Extrapolation of a PBPK model for dioxins across dosage regimen, gender, strain, and species. Toxicol. Sci. 2000, 56, 49–60. [Google Scholar] [CrossRef][Green Version]
- Emond, C.; Michalek, J.E.; Birnbaum, L.S.; DeVito, M.J. Comparison of the use of a physiologically based pharmacokinetic model and a classical pharmacokinetic model for dioxin exposure assessments. Environ. Health Perspect. 2005, 113, 1666–1668. [Google Scholar] [CrossRef] [PubMed]
- Emond, C.; Birnbaum, L.S.; DeVito, M.J. Physiologically based pharmacokinetic model for developmental exposures to TCDD in the rat. Toxicol. Sci. 2004, 80, 115–133. [Google Scholar] [CrossRef]
- Emond, C.; Birnbaum, L.S.; DeVito, M.J. Use of a physiologically based pharmacokinetic model for rats to study the influence of body fat mass and induction of CYP1A2 on the pharmacokinetics of TCDD. Environ. Health Perspect. 2006, 114, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Emond, C.; DeVito, M.; Warner, M.; Eskenazi, B.; Mocarelli, P.; Birnbaum, L.S. An assessment of dioxin exposure across gestation and lactation using a PBPK model and new data from Seveso. Environ. Int. 2016, 92–93, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Emond, C.; DeVito, M.J.; Diliberto, J.J.; Birnbaum, L.S. The Influence of Obesity on the Pharmacokinetics of Dioxin in Mice: An Assessment Using Classical and PBPK Modeling. Toxicol. Sci. 2018, 164, 218–228. [Google Scholar] [CrossRef]
- Emond, C.; Ruiz, P.; Mumtaz, M. Physiologically based pharmacokinetic toolkit to evaluate environmental exposures: Applications of the dioxin model to study real life exposures. Toxicol. Appl. Pharmacol. 2017, 315, 70–79. [Google Scholar] [CrossRef]
- Maruyama, W.; Yoshida, K.; Tanaka, T.; Nakanishi, J. Determination of tissue–blood partition coefficients for a physiological model for humans, and estimation of dioxin concentration in tissues. Chemosphere 2002, 46, 975–985. [Google Scholar] [CrossRef]
- USEPA. EPA’s Reanalysis of Key Issues Related to Dioxin Toxicity and Response to NAS Comments: In Support of Summary Information on the Integrated Risk Information System (IRIS); U.S. Environmental Protection Agency: Washington, DC, USA, 2012.
- Andersen, M.E.; Birnbaum, L.S.; Barton, H.A.; Eklund, C.R. Regional Hepatic CYP1A1 and CYP1A2 Induction with 2,3,7,8-Tetrachlorodibenzo-p-dioxin Evaluated with a Multicompartment Geometric Model of Hepatic Zonation. Toxicol. App. Pharmacol. 1997, 144, 145–155. [Google Scholar] [CrossRef]
- Leung, H.W.; Ku, R.H.; Paustenbach, D.J.; Andersen, M.E. A physiologically based pharmacokinetic model for 2,3,7,8-tetrachlorodibenzo-p-dioxin in C57BL/6J and DBA/2J mice. Toxicol. Lett. 1988, 42, 15–28. [Google Scholar] [CrossRef]
- Olson, J.R.; McGarrigle, B.P.; Gigliotti, P.J.; Kumar, S.; McReynolds, J.H. Hepatic Uptake and Metabolism of 2,3,7,8-Tetrachlorodibenzo-p-dioxin and 2,3,7,8-Tetrachlorodibenzofuran. Fundam. App. Toxicol. 1994, 22, 631–640. [Google Scholar] [CrossRef]
- Van den Berg, M.; Birnbaum, L.; Bosveld, A.T.; Brunström, B.; Cook, P.; Feeley, M.; Giesy, J.P.; Hanberg, A.; Hasegawa, R.; Kennedy, S.W.; et al. Toxic equivalency factors (TEFs) for PCBs, PCDDs, PCDFs for humans and wildlife. Environ. Health Perspect. 1998, 106, 775–792. [Google Scholar] [CrossRef]
- Haws, L.C.; Su, S.H.; Harris, M.; Devito, M.J.; Walker, N.J.; Farland, W.H.; Finley, B.; Birnbaum, L.S. Development of a refined database of mammalian relative potency estimates for dioxin-like compounds. Toxicol. Sci. 2006, 89, 4–30. [Google Scholar] [CrossRef]
- Geyer, H.J.; Schramm, K.W.; Feicht, E.A.; Behechti, A.; Steinberg, C.; Brüggemann, R.; Poiger, H.; Henkelmann, B.; Kettrup, A. Half-lives of tetra-, penta-, hexa-, hepta-, and octachlorodibenzo-p-dioxin in rats, monkeys, and humans—A critical review. Chemosphere 2002, 48, 631–644. [Google Scholar] [CrossRef]
- van Ede, K.I.; van Duursen, M.B.; van den Berg, M. Evaluation of relative effect potencies (REPs) for dioxin-like compounds to derive systemic or human-specific TEFs to improve human risk assessment. Arch. Toxicol. 2016, 90, 1293–1305. [Google Scholar] [CrossRef]
- Bradfield, C.A.; Kende, A.S.; Poland, A. Kinetic and equilibrium studies of Ah receptor-ligand binding: Use of [125I]2-iodo-7,8-dibromodibenzo-p-dioxin. Mol. Pharmacol. 1988, 34, 229–237. [Google Scholar]
- Henry, E.C.; Gasiewicz, T.A. Transformation of the aryl hydrocarbon receptor to a DNA-binding form is accompanied by release of the 90 kDa heat-shock protein and increased affinity for 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. J. 1993, 294, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Bohonowych, J.E.; Denison, M.S. Persistent Binding of Ligands to the Aryl Hydrocarbon Receptor. Toxicol. Sci. 2007, 98, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Ramadoss, P.; Perdew, G.H. Use of 2-azido-3-[125I]iodo-7,8-dibromodibenzo-p-dioxin as a probe to determine the relative ligand affinity of human versus mouse aryl hydrocarbon receptor in cultured cells. Mol. Pharmacol. 2004, 66, 129–136. [Google Scholar] [CrossRef]
- Punt, A.; Ossendorp, B. Implementation and verification of PBPK modelling codes of TCDD in rats and humans into Berkeley Madonna. EFSA Support. Publ. 2018, 15, 1374E. [Google Scholar] [CrossRef]
- Michalek, J.E.; Ketchum, N.S.; Tripathi, R.C. Diabetes mellitus and 2,3,7,8-tetrachlorodibenzo-p-dioxin elimination in veterans of Operation Ranch Hand. J Toxicol Environ Health A 2003, 66, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Poiger, H.; Schlatter, C. Pharmacokinetics of 2,3,7,8-TCDD in man. Chemosphere 1986, 15, 1489–1494. [Google Scholar] [CrossRef]
- Geusau, A.; Schmaldienst, S.; Derfler, K.; Päpke, O.; Abraham, K. Severe 2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD) intoxication: Kinetics and trials to enhance elimination in two patients. Arch. Toxicol. 2002, 76, 316–325. [Google Scholar] [CrossRef]
- Baccarelli, A.; Giacomini, S.M.; Corbetta, C.; Landi, M.T.; Bonzini, M.; Consonni, D.; Grillo, P.; Patterson, D.G.; Pesatori, A.C.; Bertazzi, P.A. Neonatal thyroid function in Seveso 25 years after maternal exposure to dioxin. PLoS. Med. 2008, 5, e161. [Google Scholar] [CrossRef]
- Mocarelli, P.; Gerthoux, P.M.; Patterson, D.G., Jr.; Milani, S.; Limonta, G.; Bertona, M.; Signorini, S.; Tramacere, P.; Colombo, L.; Crespi, C.; et al. Dioxin exposure, from infancy through puberty, produces endocrine disruption and affects human semen quality. Environ. Health Perspect. 2008, 116, 70–77. [Google Scholar] [CrossRef]
- Connor, K.T.; Aylward, L.L. Human Response to Dioxin: Aryl Hydrocarbon Receptor (AhR) Molecular Structure, Function, and Dose-Response Data for Enzyme Induction Indicate an Impaired Human AhR. J. Toxicol. Environ. Health Part B 2006, 9, 147–171. [Google Scholar] [CrossRef]
- Haddad, S. Physiologically Based Pharmacokinetic Modeling of Chemical Mixtures. In Chemical Mixtures and Combined Chemical and Nonchemical Stressors: Exposure, Toxicity, Analysis, and Risk; Rider, C.V., Simmons, J.E., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 307–333. [Google Scholar]
- Haddad, S.; Krishnan, K. Physiological modeling of toxicokinetic interactions: Implications for mixture risk assessment. Environ. Health Perspect. 1998, 106 (Suppl. 6), 1377–1384. [Google Scholar] [CrossRef]
- Russel, F.G.; Wouterse, A.C.; van Ginneken, C.A. Physiologically based pharmacokinetic model for the renal clearance of phenolsulfonphthalein and the interaction with probenecid and salicyluric acid in the dog. J. Pharm. Biopharm. 1987, 15, 349–368. [Google Scholar] [CrossRef]
- Isaacs, K.K.; Evans, M.V.; Harris, T.R. Visualization-based analysis for a mixed-inhibition binary PBPK model: Determination of inhibition mechanism. J. Pharm. Pharmacodyn. 2004, 31, 215–242. [Google Scholar] [CrossRef]
- Niu, Z.; Zang, X.; Zhang, Y. Using physiologically based pharmacokinetic models to estimate the health risk of mixtures of trihalomethanes from reclaimed water. J. Hazard Mater. 2015, 285, 190–198. [Google Scholar] [CrossRef]
- Ali, N.; Tardif, R. Toxicokinetic Modeling of the Combined Exposure to Toluene and n-Hexane in Rats and Humans. J. Occupational Health 1999, 41, 95–103. [Google Scholar] [CrossRef]
- Haddad, S.; Tardif, R.; Charest-Tardif, G.; Krishnan, K. Physiological modeling of the toxicokinetic interactions in a quaternary mixture of aromatic hydrocarbons. Toxicol. Appl. Pharmacol. 1999, 161, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Haddad, S.; Charest-Tardif, G.; Tardif, R.; Krishnan, K. Validation of a physiological modeling framework for simulating the toxicokinetics of chemicals in mixtures. Toxicol. Appl. Pharmacol. 2000, 167, 199–209. [Google Scholar] [CrossRef]
- Ruiz, P.; Emond, C.; McLanahan, E.D.; Joshi-Barr, S.; Mumtaz, M. Exploring Mechanistic Toxicity of Mixtures Using PBPK Modeling and Computational Systems Biology. Toxicol. Sci. 2019, 174, 38–50. [Google Scholar] [CrossRef]
- Tardif, R.; Charest-Tardif, G.; Brodeur, J.; Krishnan, K. Physiologically based pharmacokinetic modeling of a ternary mixture of alkyl benzenes in rats and humans. Toxicol. Appl. Pharmacol. 1997, 144, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Sasso, A.F.; Isukapalli, S.S.; Georgopoulos, P.G. A generalized physiologically-based toxicokinetic modeling system for chemical mixtures containing metals. Theor. Biol. Med. Model. 2010, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Wall, R.J.; Fernandes, A.; Rose, M.; Bell, D.R.; Mellor, I.R. Characterisation of chlorinated, brominated and mixed halogenated dioxins, furans and biphenyls as potent and as partial agonists of the Aryl hydrocarbon receptor. Environ. Int. 2015, 76, 49–56. [Google Scholar] [CrossRef]
- Sridhar, J.; Foroozesh, M.; Stevens, C.L. QSAR models of cytochrome P450 enzyme 1A2 inhibitors using CoMFA, CoMSIA and HQSAR. SAR QSAR Environ. Res. 2011, 22, 681–697. [Google Scholar] [CrossRef]
- Farag, E.S.M.; Ayman, O.S.E.-K.; Khaled, B. Targeting the Aryl Hydrocarbon Receptor (AhR): A Review of the In-Silico Screening Approaches to Identify AhR Modulators. In High-Throughput Screening for Drug Discovery; Shailendra, K.S., Ed.; IntechOpen: Rijeka, Croatia, 2021; p. Ch. 5. [Google Scholar]
- Yoon, M.; Campbell, J.L.; Andersen, M.E.; Clewell, H.J. Quantitative in vitro to in vivo extrapolation of cell-based toxicity assay results. Crit. Rev. Toxicol. 2012, 42, 633–652. [Google Scholar] [CrossRef]
- Moreau, M.; Mallick, P.; Smeltz, M.; Haider, S.; Nicolas, C.I.; Pendse, S.N.; Leonard, J.A.; Linakis, M.W.; McMullen, P.D.; Clewell, R.A.; et al. Considerations for Improving Metabolism Predictions for In Vitro to In Vivo Extrapolation. Front. Toxicol. 2022, 4, 894569. [Google Scholar] [CrossRef]
- Koonrungsesomboon, N.; Khatsri, R.; Wongchompoo, P.; Teekachunhatean, S. The impact of genetic polymorphisms on CYP1A2 activity in humans: A systematic review and meta-analysis. Pharm. J. 2018, 18, 760–768. [Google Scholar] [CrossRef]
- Rowlands, J.C.; Staskal, D.F.; Gollapudi, B.; Budinsky, R. The human AHR: Identification of single nucleotide polymorphisms from six ethnic populations. Pharm. Genom. 2010, 20, 283–290. [Google Scholar] [CrossRef]
- Bernillon, P.; Bois, F.Y. Statistical Issues in Toxicokinetic Modeling: A Bayesian Perspective. Environ. Health Perspect. 2000, 108, 883–893. [Google Scholar] [CrossRef]
- van Ede, K.I.; Andersson, P.L.; Gaisch, K.P.J.; van den Berg, M.; van Duursen, M.B.M. Comparison of intake and systemic relative effect potencies of dioxin-like compounds in female rats after a single oral dose. Arch.Toxicol. 2014, 88, 637–646. [Google Scholar] [CrossRef]
- van Ede, K.I.; Andersson, P.L.; Gaisch, K.P.; van den Berg, M.; van Duursen, M.B. Comparison of intake and systemic relative effect potencies of dioxin-like compounds in female mice after a single oral dose. Environ. Health Perspect. 2013, 121, 847–853. [Google Scholar] [CrossRef][Green Version]
- DeVito, M.J.; Ménache, M.G.; Diliberto, J.J.; Ross, D.G.; Birnbaum, L.S. Dose-response relationships for induction of CYP1A1 and CYP1A2 enzyme activity in liver, lung, and skin in female mice following subchronic exposure to polychlorinated biphenyls. Toxicol. Appl. Pharmacol. 2000, 167, 157–172. [Google Scholar] [CrossRef]
- DeVito, M.J.; Diliberto, J.J.; Ross, D.G.; Menache, M.G.; Birnbaum, L.S. Dose-response relationships for polyhalogenated dioxins and dibenzofurans following subchronic treatment in mice. I. CYP1A1 and CYP1A2 enzyme activity in liver, lung, and skin. Toxicol. Appl. Pharmacol. 1997, 147, 267–280. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, R.; Zacharewski, T.R.; Conolly, R.B.; Zhang, Q. A Physiologically Based Pharmacokinetic (PBPK) Modeling Framework for Mixtures of Dioxin-like Compounds. Toxics 2022, 10, 700. https://doi.org/10.3390/toxics10110700
Liu R, Zacharewski TR, Conolly RB, Zhang Q. A Physiologically Based Pharmacokinetic (PBPK) Modeling Framework for Mixtures of Dioxin-like Compounds. Toxics. 2022; 10(11):700. https://doi.org/10.3390/toxics10110700
Chicago/Turabian StyleLiu, Rongrui, Tim R. Zacharewski, Rory B. Conolly, and Qiang Zhang. 2022. "A Physiologically Based Pharmacokinetic (PBPK) Modeling Framework for Mixtures of Dioxin-like Compounds" Toxics 10, no. 11: 700. https://doi.org/10.3390/toxics10110700
APA StyleLiu, R., Zacharewski, T. R., Conolly, R. B., & Zhang, Q. (2022). A Physiologically Based Pharmacokinetic (PBPK) Modeling Framework for Mixtures of Dioxin-like Compounds. Toxics, 10(11), 700. https://doi.org/10.3390/toxics10110700