
Long-Term Consumption of Food-Derived Chlorogenic Acid Protects Mice against Acetaminophen-Induced Hepatotoxicity via Promoting PINK1-Dependent Mitophagy and Inhibiting Apoptosis

Abstract
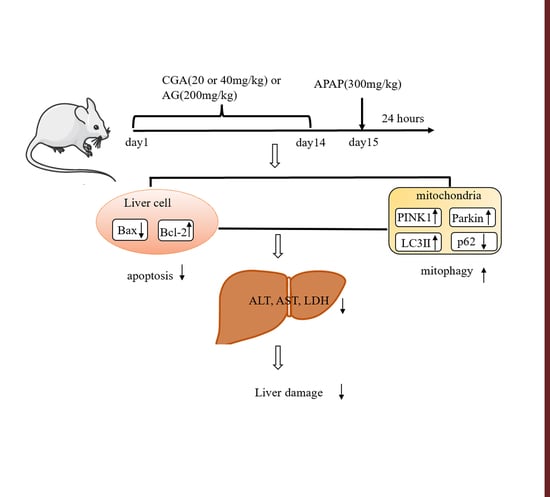
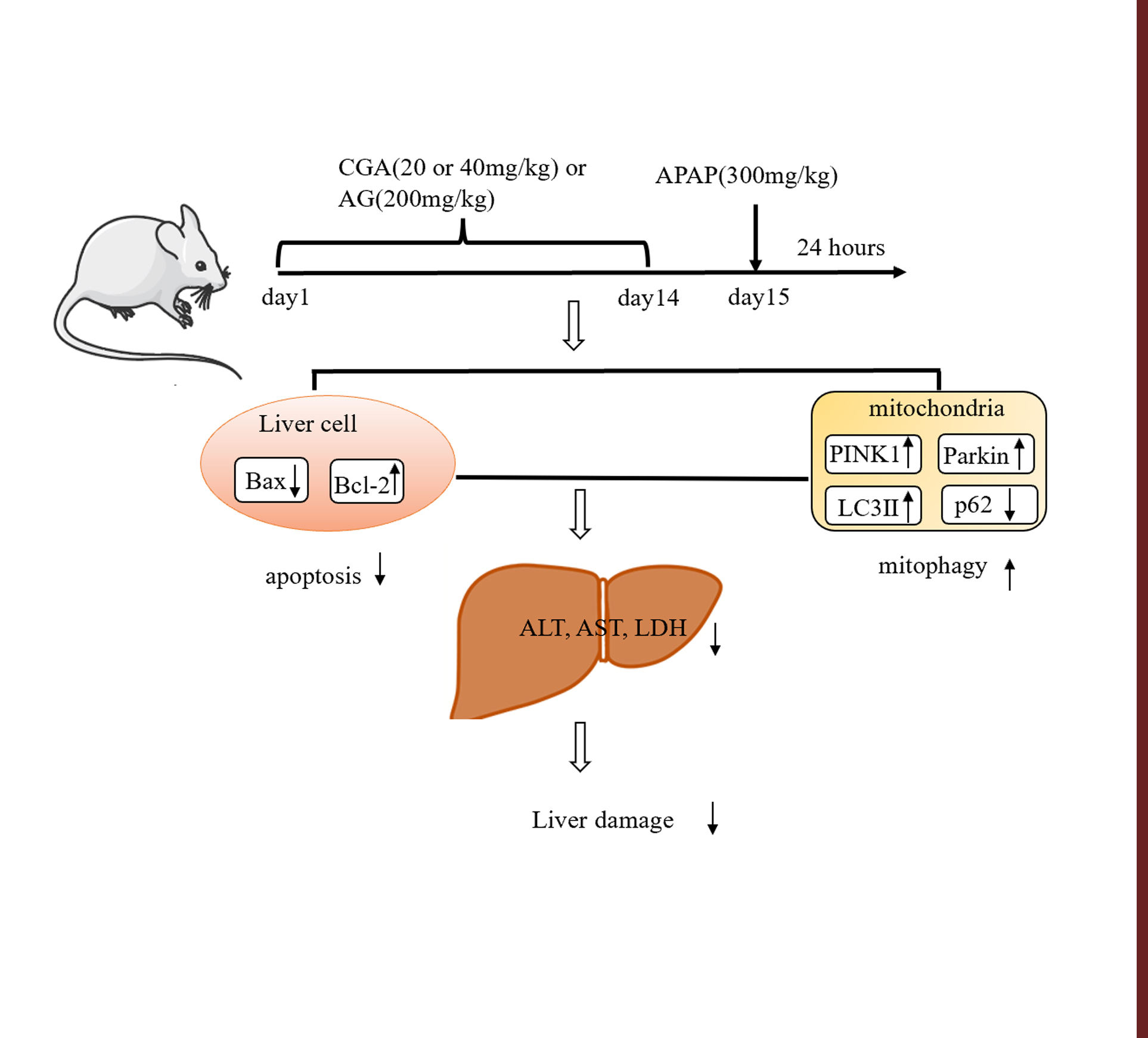{kind=link}
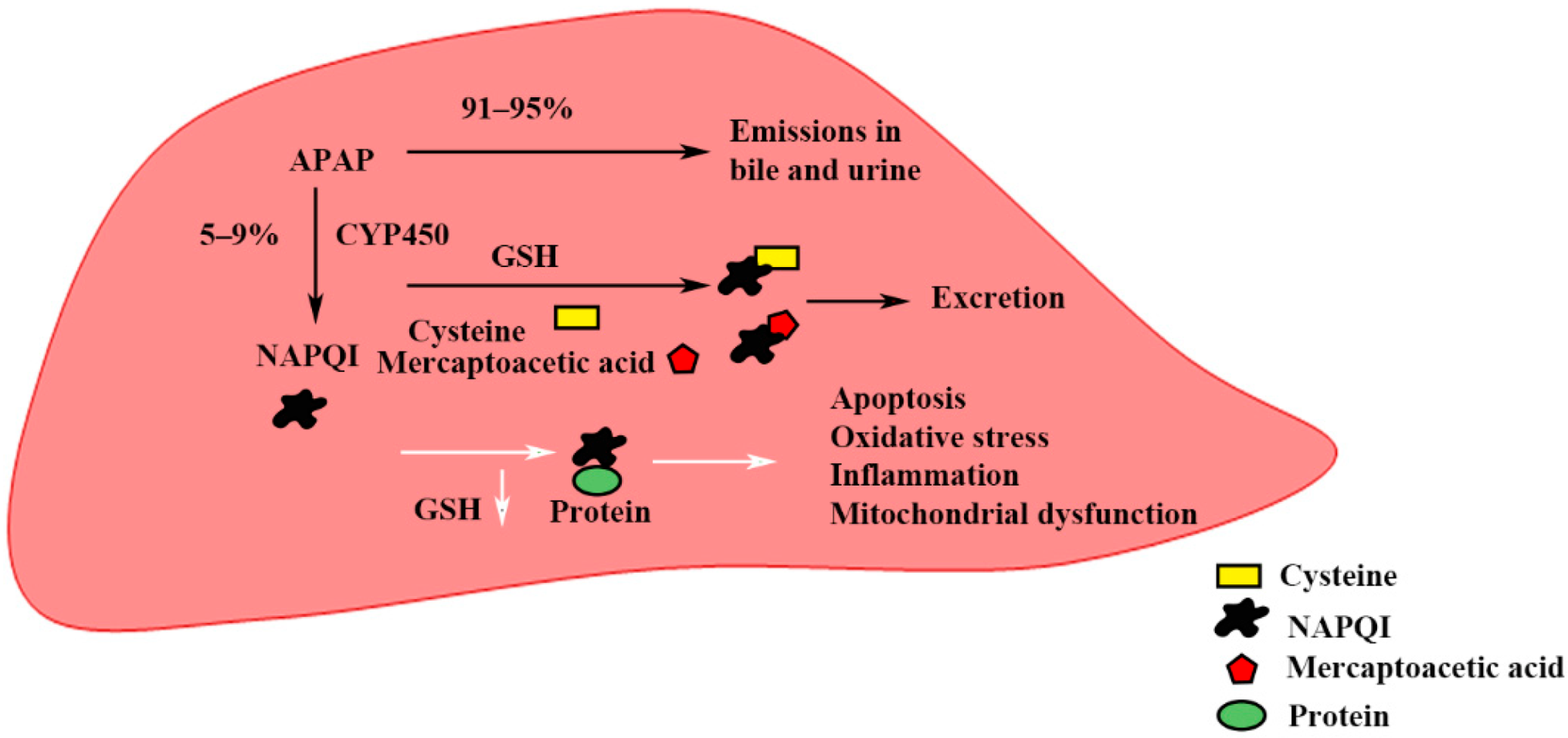{kind=link}
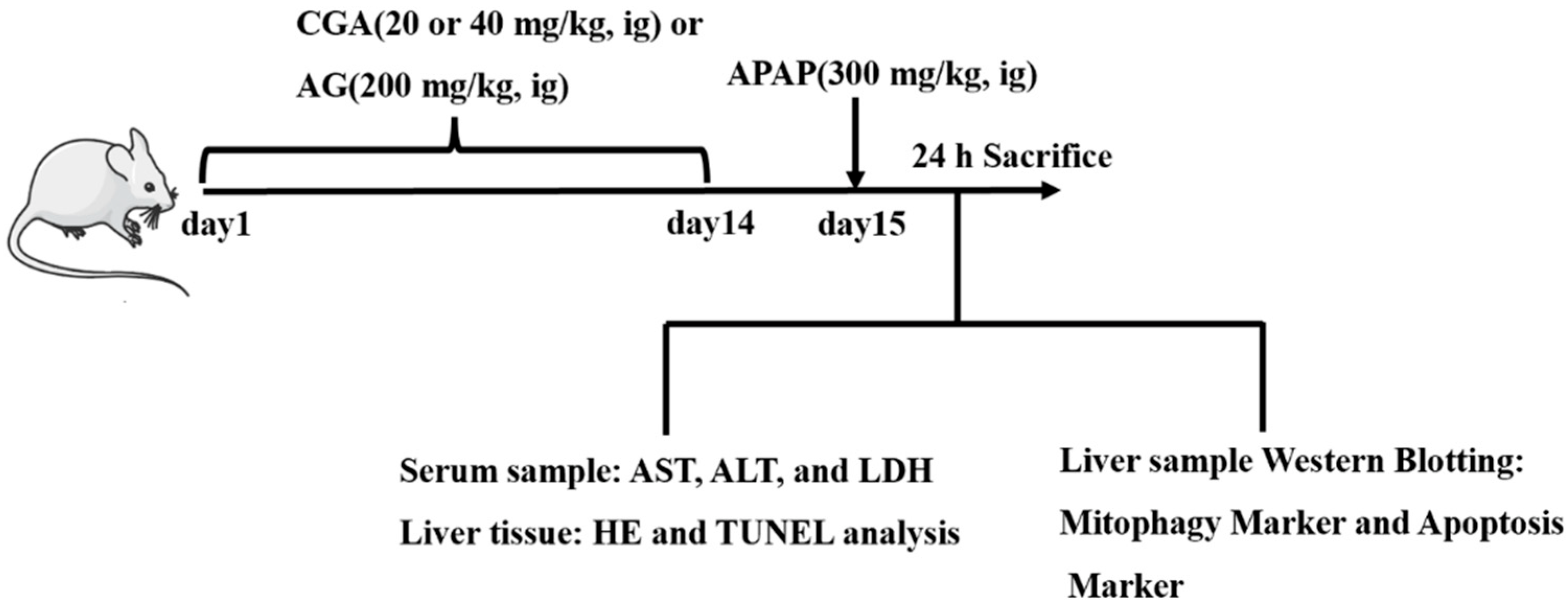{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Animals
2.3. Cell Culture
2.4. Biochemical Analysis for Serum
2.5. HE Staining and TUNEL Assay
2.6. PINK1 siRNA Transfection
2.7. RT-qPCR Analysis
2.8. Western Blotting Analysis
2.9. Immunofluorescence Analysis
2.10. Statistical Analysis
3. Results
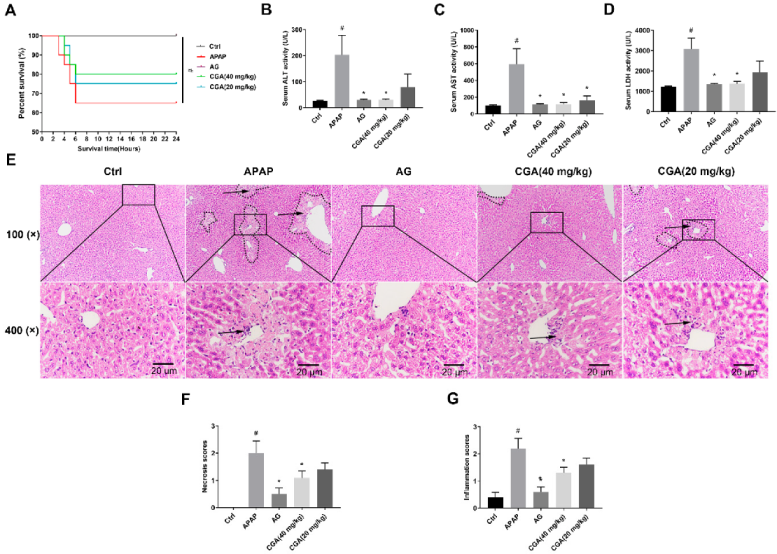
3.1. CGA Alleviated Hepatotoxicity in APAP-Induced Mice
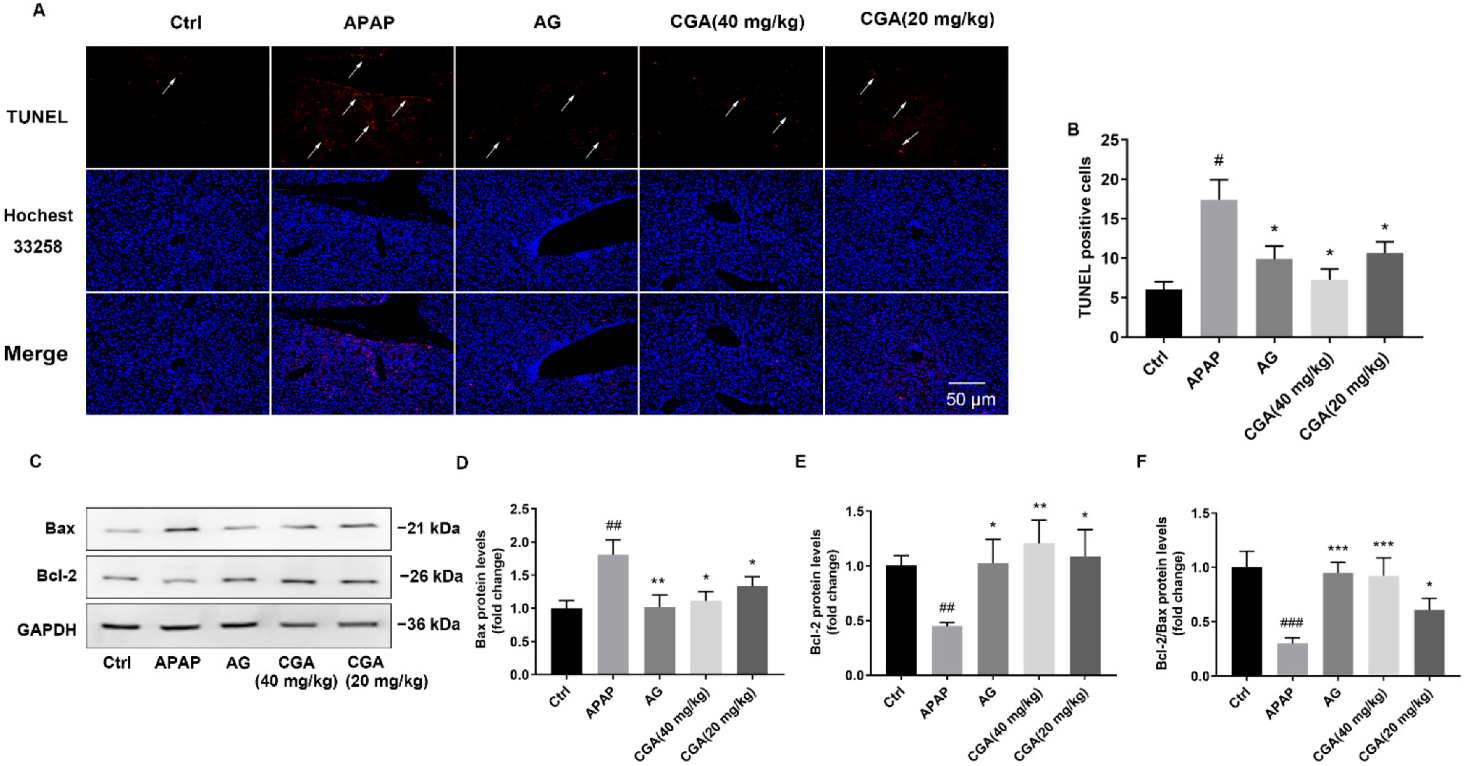
3.2. CGA Suppressed Liver Cell Apoptosis in APAP Hepatotoxicity Mice
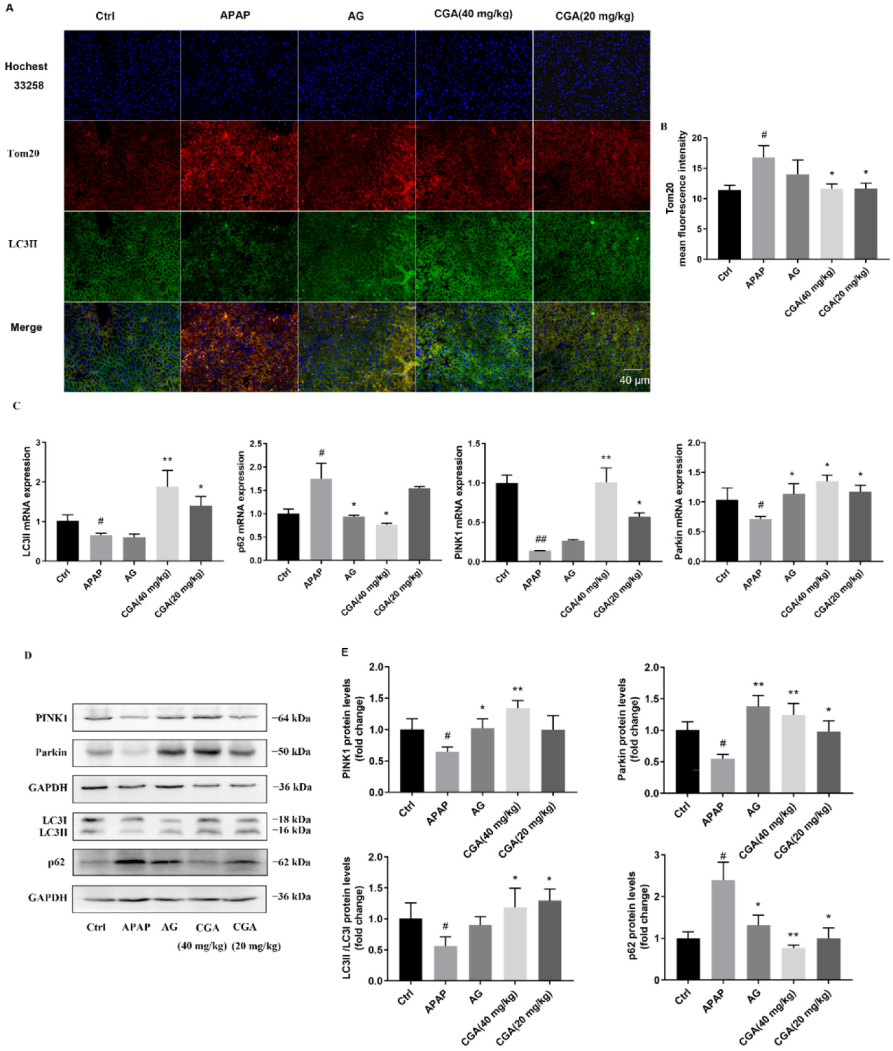
3.3. CGA Triggered PINK1-Dependent Mitophagy in APAP Hepatotoxicity Mice
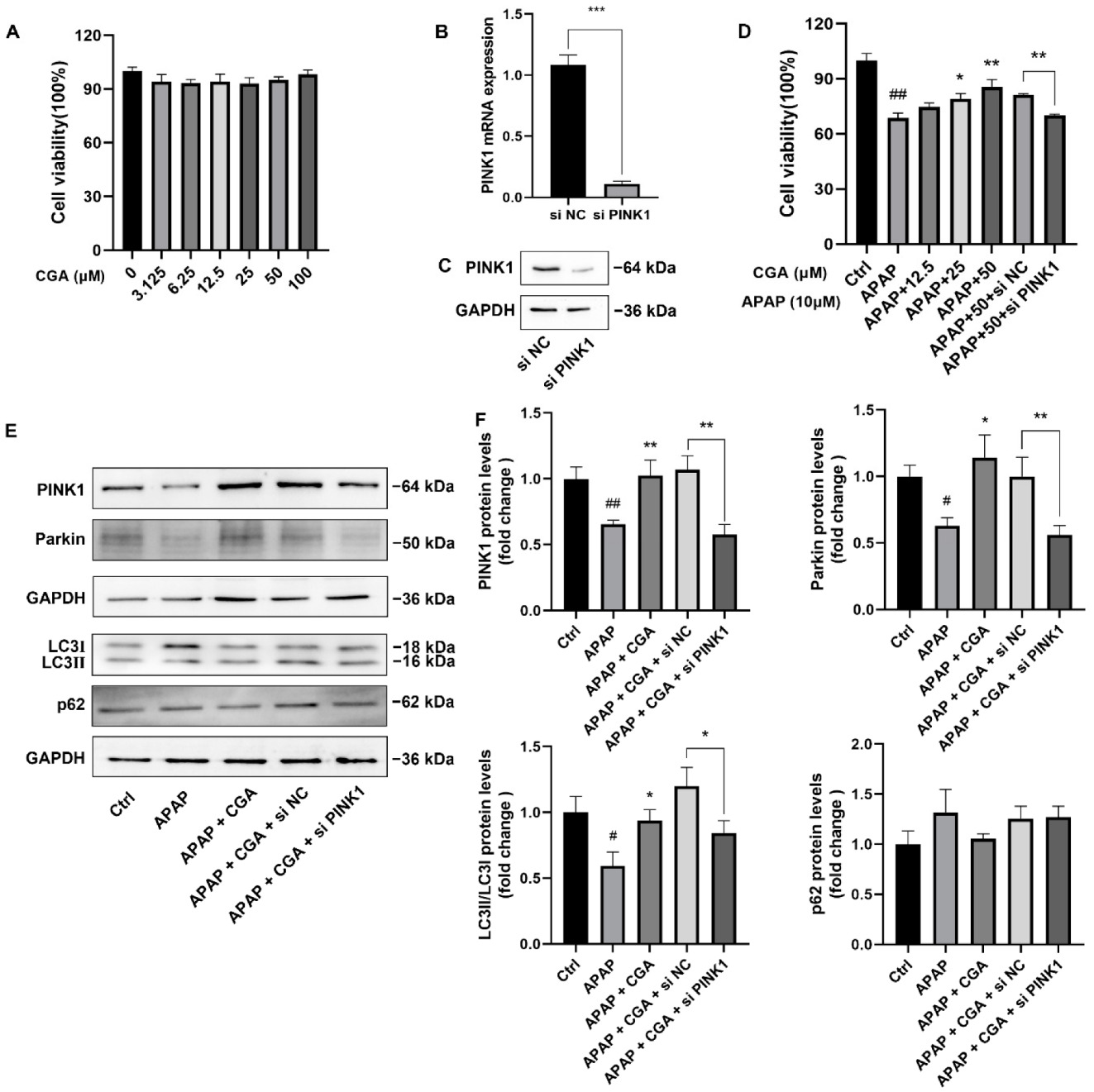
3.4. siPINK1 Reversed the Protective Effect of CGA
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ramachandran, A.; Jaeschke, H. Acetaminophen Toxicity: Novel Insights into Mechanisms and Future Perspectives. Gene Expr. 2018, 18, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Jung, Y.S.; Yun, J.; Han, S.B.; Roh, Y.S.; Song, M.J.; Hong, J.T. Peroxiredoxin 6 mediates acetaminophen-induced hepatocyte death through JNK activation. Redox Biol. 2020, 32, 101496. [Google Scholar] [CrossRef]
- Cai, Z.; Wang, B.; Zhou, Z.; Zhao, X.; Hu, L.; Ren, Q.; Deng, L.; Li, Z.; Wang, G. Discovery of a novel and orally active Farnesoid X receptor agonist for the protection of acetaminophen-induced hepatotoxicity. Chem. Biol. Drug Des. 2022, 99, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Jaeschke, H. Oxidant Stress and Acetaminophen Hepatotoxicity: Mechanism-Based Drug Development. Antioxid. Redox Signal. 2021, 35, 718–733. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Wang, J.; Xie, H.; Liu, Y.; Bai, Y.; Che, Q.; Cao, H.; Huang, G.; Guo, J.; Su, Z. Protective effect and mechanism of chitooligosaccharides on acetaminophen-induced liver injury. Food Funct. 2021, 12, 9979–9993. [Google Scholar] [CrossRef]
- Moyer, A.M.; Fridley, B.L.; Jenkins, G.D.; Batzler, A.J.; Pelleymounter, L.L.; Kalari, K.R.; Ji, Y.; Chai, Y.; Nordgren, K.K.; Weinshilboum, R.M. Acetaminophen-NAPQI hepatotoxicity: A cell line model system genome-wide association study. Toxicol. Sci. 2011, 120, 33–41. [Google Scholar] [CrossRef]
- Chowdhury, A.; Nabila, J.; Adelusi Temitope, I.; Wang, S. Current etiological comprehension and therapeutic targets of acetaminophen-induced hepatotoxicity. Pharmacol. Res. 2020, 161, 105102. [Google Scholar] [CrossRef]
- Moles, A.; Torres, S.; Baulies, A.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial-Lysosomal Axis in Acetaminophen Hepatotoxicity. Front. Pharmacol. 2018, 9, 453. [Google Scholar] [CrossRef]
- Jaeschke, H. Acetaminophen hepatotoxicity and sterile inflammation: The mechanism of protection of Chlorogenic acid. Chem. Biol. Interact. 2016, 243, 148–149. [Google Scholar] [CrossRef][Green Version]
- Fisher, E.S.; Curry, S.C. Evaluation and treatment of acetaminophen toxicity. Adv. Pharmacol. 2019, 85, 263–272. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Arzuk, E.; Tokdemir, M.; Orhan, H. Mitochondrial versus microsomal bioactivation of paracetamol by human liver and kidney tissues. Toxicol. Lett. 2022, 363, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Sung, H.C.; Chuang, T.Y.; Lai, T.C.; Lee, T.L.; Lee, C.W.; Lee, I.T.; Chen, Y.L. Vitamin D3 decreases TNF-α-induced inflammation in lung epithelial cells through a reduction in mitochondrial fission and mitophagy. Cell Biol. Toxicol. 2022, 38, 427–450. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Yuan, Q.; Zhou, C.; Huang, W.; Yu, X. Mitochondrial stress response in drug-induced liver injury. Mol. Biol. Rep. 2021, 48, 6949–6958. [Google Scholar] [CrossRef]
- Ma, X.; McKeen, T.; Zhang, J.; Ding, W.X. Role and Mechanisms of Mitophagy in Liver Diseases. Cells 2020, 9, 837. [Google Scholar] [CrossRef]
- Jiang, Z.; Yang, X.; Han, Y.; Li, J.; Hu, C.; Liu, C.; Xiao, W. Sarmentosin promotes USP17 and regulates Nrf2-mediated mitophagy and cellular oxidative stress to alleviate APAP-induced acute liver failure. Phytomedicine 2022, 104, 154337. [Google Scholar] [CrossRef]
- Van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Wang, H.; Ni, H.M.; Chao, X.; Ma, X.; Rodriguez, Y.A.; Chavan, H.; Wang, S.; Krishnamurthy, P.; Dobrowsky, R.; Xu, D.X.; et al. Double deletion of PINK1 and Parkin impairs hepatic mitophagy and exacerbates acetaminophen-induced liver injury in mice. Redox Biol. 2019, 22, 101148. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Ding, W.X. Targeting Pink1-Parkin-mediated mitophagy for treating liver injury. Pharmacol. Res. 2015, 102, 264–269. [Google Scholar] [CrossRef]
- Sun, J.; Yu, F.; Wang, T.; Bian, J.; Liu, Z.; Zou, H. The role of DRP1-PINK1-Parkin-mediated mitophagy in early cadmium-induced liver damage. Toxicology 2022, 466, 153082. [Google Scholar] [CrossRef]
- Shi, Q.; Zhao, G.; Wei, S.; Guo, C.; Wu, X.; Zhao, R.C.; Di, G. Pterostilbene alleviates liver ischemia/reperfusion injury via PINK1-mediated mitophagy. J. Pharmacol. Sci. 2022, 148, 19–30. [Google Scholar] [CrossRef]
- Ntamo, Y.; Ziqubu, K.; Chellan, N.; Nkambule, B.B.; Nyambuya, T.M.; Mazibuko-Mbeje, S.E.; Gabuza, K.B.; Marcheggiani, F.; Tiano, L.; Dludla, P.V. Drug-Induced Liver Injury: Clinical Evidence of N-Acetyl Cysteine Protective Effects. Oxid. Med. Cell Longev. 2021, 2021, 3320325. [Google Scholar] [CrossRef]
- Ershad, M.; Naji, A.; Vearrier, D. N Acetylcysteine. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar] [PubMed]
- Gao, Z.; Yi, W.; Tang, J.; Sun, Y.; Huang, J.; Lan, T.; Dai, X.; Xu, S.; Jin, Z.G.; Wu, X. Urolithin A protects against acetaminophen-induced liver injury in mice via sustained activation of Nrf2. Int. J. Biol. Sci. 2022, 18, 2146–2162. [Google Scholar] [CrossRef]
- Akakpo, J.Y.; Ramachandran, A.; Curry, S.C.; Rumack, B.H.; Jaeschke, H. Comparing N-acetylcysteine and 4-methylpyrazole as antidotes for acetaminophen overdose. Arch. Toxicol. 2022, 96, 453–465. [Google Scholar] [CrossRef]
- Morita, A.; Omoya, Y.; Ito, R.; Ishibashi, Y.; Hiramoto, K.; Ohnishi, S.; Yoshikawa, N.; Kawanishi, S. Glycyrrhizin and its derivatives promote hepatic differentiation via sweet receptor, Wnt, and Notch signaling. Biochem. Biophys. Rep. 2021, 28, 101181. [Google Scholar] [CrossRef]
- Yu, J.; Jiang, Y.S.; Jiang, Y.; Peng, Y.F.; Sun, Z.; Dai, X.N.; Cao, Q.T.; Sun, Y.M.; Han, J.C.; Gao, Y.J. Targeted metabolomic study indicating glycyrrhizin’s protection against acetaminophen-induced liver damage through reversing fatty acid metabolism. Phytother. Res. 2014, 28, 933–936. [Google Scholar] [CrossRef]
- Yan, T.; Wang, H.; Zhao, M.; Yagai, T.; Chai, Y.; Krausz, K.W.; Xie, C.; Cheng, X.; Zhang, J.; Che, Y.; et al. Glycyrrhizin Protects against Acetaminophen-Induced Acute Liver Injury via Alleviating Tumor Necrosis Factor α-Mediated Apoptosis. Drug Metab. Dispos. 2016, 44, 720–731. [Google Scholar] [CrossRef]
- Yang, H.; Jiang, T.; Li, P.; Mao, Q. The protection of glycyrrhetinic acid (GA) towards acetaminophen (APAP)-induced toxicity partially through fatty acids metabolic pathway. Afr. Health Sci. 2015, 15, 1023–1027. [Google Scholar] [CrossRef]
- Yu, Y.; Wu, Y.; Yan, H.Z.; Xia, Z.R.; Wen, W.; Liu, D.Y.; Wan, L.H. Rosmarinic acid ameliorates acetaminophen-induced acute liver injury in mice via RACK1/TNF-α mediated antioxidant effect. Pharm. Biol. 2021, 59, 1286–1293. [Google Scholar] [CrossRef]
- Abdel, D.M.; Abushouk, A.I.; Reggi, R.; Yarla, N.S.; Palmery, M.; Peluso, I. Association of antioxidant nutraceuticals and acetaminophen (paracetamol): Friend or foe? J. Food Drug Anal. 2018, 26, S78–S87. [Google Scholar] [CrossRef] [PubMed]
- Eugenio, P.D.; Montes de Oca-Solano, H.A.; Pedraza-Chaverri, J. Role of food-derived antioxidant agents against acetaminophen-induced hepatotoxicity. Pharm. Biol. 2016, 54, 2340–2352. [Google Scholar] [CrossRef]
- Wu, J.T. Bioactivity and application of chlorogenic acid. Mod. Agric. Sci. Technol. 2009, 19, 349–350. [Google Scholar]
- Yan, Y.; Zhou, X.; Guo, K.; Zhou, F.; Yang, H. Chlorogenic Acid Protects Against Indomethacin-Induced Inflammation and Mucosa Damage by Decreasing Bacteroides-Derived LPS. Front. Immunol. 2020, 11, 1125. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Dilnashin, H.; Singh, R.; Singh, S.P. Neuroprotective Effect of Chlorogenic Acid on Mitochondrial Dysfunction-Mediated Apoptotic Death of DA Neurons in a Parkinsonian Mouse Model. Oxid. Med. Cell. Longev. 2020, 2020, 6571484. [Google Scholar] [CrossRef]
- Owumi, S.E.; Olusola, J.K.; Arunsi, U.O.; Oyelere, A.K. Chlorogenic acid abates oxido-inflammatory and apoptotic responses in the liver and kidney of Tamoxifen-treated rats. Toxicol. Res. 2021, 10, 345–353. [Google Scholar] [CrossRef]
- Hu, F.; Guo, Q.; Wei, M.; Huang, Z.; Shi, L.; Sheng, Y.; Ji, L. Chlorogenic acid alleviates acetaminophen-induced liver injury in mice via regulating Nrf2-mediated HSP60-initiated liver inflammation. Eur. J. Pharmacol. 2020, 883, 173286. [Google Scholar] [CrossRef]
- Jaeschke, H.; Duan, L.; Akakpo, J.Y.; Farhood, A.; Ramachandran, A. The role of apoptosis in acetaminophen hepatotoxicity. Food Chem. Toxicol. 2018, 118, 709–718. [Google Scholar] [CrossRef]
- Ji, L.; Jiang, P.; Lu, B.; Sheng, Y.; Wang, X.; Wang, Z. Chlorogenic acid, a dietary polyphenol, protects acetaminophen-induced liver injury and its mechanism. J. Nutr. Biochem. 2013, 24, 1911–1919. [Google Scholar] [CrossRef]
- Nguyen, N.U.; Stamper, B.D. Polyphenols reported to shift APAP-induced changes in MAPK signaling and toxicity outcomes. Chem. Biol. Interact. 2017, 277, 129–136. [Google Scholar] [CrossRef]
- Pang, C.; Sheng, Y.C.; Jiang, P.; Wei, H.; Ji, L.L. Chlorogenic acid prevents acetaminophen-induced liver injury: The involvement of CYP450 metabolic enzymes and some antioxidant signals. J. Zhejiang Univ. Sci. B 2015, 16, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Zheng, Z.; Shi, L.; Jin, Y.; Ji, L. Natural Polyphenol Chlorogenic Acid Protects Against Acetaminophen-Induced Hepatotoxicity by Activating ERK/Nrf2 Antioxidative Pathway. Toxicol Sci. 2018, 162, 99–112. [Google Scholar] [CrossRef]
- Zheng, Z.; Sheng, Y.; Lu, B.; Ji, L. The therapeutic detoxification of chlorogenic acid against acetaminophen-induced liver injury by ameliorating hepatic inflammation. Chem. Biol. Interact. 2015, 238, 93–101. [Google Scholar] [CrossRef]
- Akakpo, J.Y.; Jaeschke, M.W.; Ramachandran, A.; Curry, S.C.; Rumack, B.H.; Jaeschke, H. Delayed administration of N-acetylcysteine blunts recovery after an acetaminophen overdose unlike 4-methylpyrazole. Arch. Toxicol. 2021, 95, 3377–3391. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Sanchez-Guerrero, G.; Jaeschke, H.; Ramachandran, A. Activation of the adenosine A2B receptor even beyond the therapeutic window of N-acetylcysteine accelerates liver recovery after an acetaminophen overdose. Food Chem. Toxicol. 2022, 163, 112911. [Google Scholar] [CrossRef] [PubMed]
- Korver, S.; Bowen, J.; Pearson, K.; Gonzalez, R.J.; French, N.; Park, K.; Jenkins, R.; Goldring, C. The application of cytokeratin-18 as a biomarker for drug-induced liver injury. Arch. Toxicol. 2021, 95, 3435–3448. [Google Scholar] [CrossRef]
- Ge, M.; Yao, W.; Yuan, D.; Zhou, S.; Chen, X.; Zhang, Y.; Li, H.; Xia, Z.; Hei, Z. Brg1-mediated Nrf2/HO-1 pathway activation alleviates hepatic ischemia-reperfusion injury. Cell Death Dis. 2017, 8, e2841. [Google Scholar] [CrossRef]
- Ren, X.; Chen, L.; Xie, J.; Zhang, Z.; Dong, G.; Liang, J.; Liu, L.; Zhou, H.; Luo, P. Resveratrol Ameliorates Mitochondrial Elongation via Drp1/Parkin/PINK1 Signaling in Senescent-Like Cardiomyocytes. Oxid. Med. Cell. Longev. 2017, 2017, 4175353. [Google Scholar] [CrossRef]
- Li, Y.Q.; Zhang, F.; Yu, L.P.; Mu, J.K.; Yang, Y.Q.; Yu, J.; Yang, X.X. Targeting PINK1 Using Natural Products for the Treatment of Human Diseases. Biomed. Res. Int. 2021, 2021, 4045819. [Google Scholar] [CrossRef]
- Ghallab, A.; Hassan, R.; Hofmann, U.; Friebel, A.; Hobloss, Z.; Brackhagen, L.; Begher-Tibbe, B.; Myllys, M.; Reinders, J.; Overbeck, N.; et al. Interruption of bile acid uptake by hepatocytes after acetaminophen overdose ameliorates hepatotoxicity. J. Hepatol. 2022, 77, 71–83. [Google Scholar] [CrossRef]
- Qu, L.; Fu, R.; Ma, X.; Fan, D. Hepatoprotective effects of ginsenoside Rk3 in acetaminophen-induced liver injury in mice by activation of autophagy. Food Funct. 2021, 12, 9128–9140. [Google Scholar] [CrossRef]
- Yao, Y.; Li, R.; Liu, D.; Long, L.; He, N. Rosmarinic acid alleviates acetaminophen-induced hepatotoxicity by targeting Nrf2 and NEK7-NLRP3 signaling pathway. Ecotoxicol. Environ. Saf. 2022, 241, 113773. [Google Scholar] [CrossRef]
- Gungor, H.; Ekici, M.; Ates, M.B. Lipid-lowering, anti-inflammatory, and hepatoprotective effects of isorhamnetin on acetaminophen-induced hepatotoxicity in mice. Drug Chem. Toxicol. 2022, 2, 1–9. [Google Scholar] [CrossRef]
- Shen, P.; Han, L.; Chen, G.; Cheng, Z.; Liu, Q. Emodin Attenuates Acetaminophen-Induced Hepatotoxicity via the cGAS-STING Pathway. Inflammation 2022, 45, 74–87. [Google Scholar] [CrossRef]
- Yao, L.; Zhang, J.; Jin, J.; Li, H.; Li, L.; Han, X.; Raza, H.K.; Li, X.; Mao, Y. An analysis of the efficacy and safety of compound glycyrrhizin injections in the treatment of drug-induced liver injury using a nationwide database. Int. J. Clin. Pharm. 2022, 44, 731–740. [Google Scholar] [CrossRef]
- Liang, S.B.; Hou, W.B.; Zheng, R.X.; Liang, C.H.; Yan, L.J.; Wang, H.N.; Cao, H.J.; Han, M.; Robinson, N.; Liu, J.P. Compound glycyrrhizin injection for improving liver function in children with acute ic-teric hepatitis: A systematic review and meta-analysis. Integr. Med. Res. 2022, 11, 100772. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, L.; Chen, R.; Zhao, Y.; Ren, D.; Yang, X. Chlorogenic acid inhibits trimethylamine-N-oxide formation and remodels intestinal microbiota to alleviate liver dysfunction in high L-carnitine feeding mice. Food Funct. 2021, 12, 10500–10511. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, J.; Zhang, J.; Deng, Y.; Xiong, G.; Fu, J.; Wei, L.; Lu, H. Chlorogenic acid alleviates thioacetamide-induced toxicity and promotes liver development in zebrafish (Danio rerio) through the Wnt signaling pathway. Aquat. Toxicol. 2022, 242, 106039. [Google Scholar] [CrossRef]
- Shi, H.; Dong, L.; Bai, Y.; Zhao, J.; Zhang, Y.; Zhang, L. Chlorogenic acid against carbon tetrachloride-induced liver fibrosis in rats. Eur. J. Pharmacol. 2009, 623, 119–124. [Google Scholar] [CrossRef]
- Awwad, S.; Issa, R.; Alnsour, L.; Albals, D.; Al-Momani, I. Quantification of Caffeine and Chlorogenic Acid in Green and Roasted Coffee Samples Using HPLC-DAD and Evaluation of the Effect of Degree of Roasting on Their Levels. Molecules 2021, 26, 7502. [Google Scholar] [CrossRef]
- Rashid, R.; Mohd Wani, S.; Manzoor, S.; Masoodi, F.A.; Masarat, D.M. Green extraction of bioactive compounds from apple pomace by ultrasound assisted natural deep eutectic solvent extraction: Optimisation, comparison and bioactivity. Food Chem. 2023, 398, 133871. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Meng, H.; Liu, T.; Feng, Y.; Qi, Y.; Zhang, D.; Wang, H. Blueberry Phenolics Reduce Gastrointestinal Infection of Patients with Cerebral Venous Thrombosis by Improving Depressant-Induced Autoimmune Disorder via miR-155-Mediated Brain-Derived Neurotrophic Factor. Front Pharmacol. 2017, 8, 853. [Google Scholar] [CrossRef] [PubMed]
- Nam, E.J.; Hayashida, K.; Aquino, R.S.; Couchman, J.R.; Kozar, R.A.; Liu, J.; Park, P.W. Syndecan-1 limits the progression of liver injury and promotes liver repair in acetaminophen-induced liver injury in mice. Hepatology 2017, 66, 1601–1615. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Li, Y.; Cao, H.; Guo, H.; Alahmadi, T.A.; Alharbi, S.A.; Yu, J. Hepatoprotective potential of kirenol on ethanol-induced liver toxicity in albino rats and acetaminophen-induced oxidative stress-mediated apoptosis in hepatic HepG2 cells. J. Biochem. Mol. Toxicol. 2021, 35, e22786. [Google Scholar] [CrossRef] [PubMed]
- Oz, H.S.; Chen, T.S. Green-tea polyphenols downregulate cyclooxygenase and Bcl-2 activity in acetaminophen-induced hepatotoxicity. Dig. Dis. Sci. 2008, 53, 2980–2988. [Google Scholar] [CrossRef]
- Wang, K. Autophagy and apoptosis in liver injury. Cell Cycle 2015, 14, 1631–1642. [Google Scholar] [CrossRef]
- Stavropoulos, A.; Divolis, G.; Manioudaki, M.; Gavriil, A.; Kloukina, I.; Perrea, D.N.; Sountoulidis, A.; Ford, E.; Doulou, A.; Apostolidou, A.; et al. Coordinated activation of TGF-β and BMP pathways promotes autophagy and limits liver injury after acetaminophen intoxication. Sci. Signal. 2022, 15, eabn4395. [Google Scholar] [CrossRef]
- Qian, H.; Bai, Q.; Yang, X.; Akakpo, J.Y.; Ji, L.; Yang, L.; Rülicke, T.; Zatloukal, K.; Jaeschke, H.; Ni, H.M.; et al. Dual roles of p62/SQSTM1 in the injury and recovery phases of acetaminophen-induced liver injury in mice. Acta Pharm. Sin. B 2021, 11, 3791–3805. [Google Scholar] [CrossRef]
- Song, S.; Tan, J.; Miao, Y.; Li, M.; Zhang, Q. Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J. Cell. Physiol. 2017, 232, 2977–2984. [Google Scholar] [CrossRef]
- Chao, X.; Wang, H.; Jaeschke, H.; Ding, W.X. Role and mechanisms of autophagy in acetaminophen-induced liver injury. Liver Int. 2018, 38, 1363–1374. [Google Scholar] [CrossRef]
- Kang, L.; Liu, S.; Li, J.; Tian, Y.; Xue, Y.; Liu, X. Parkin and Nrf2 prevent oxidative stress-induced apoptosis in intervertebral endplate chondrocytes via inducing mitophagy and anti-oxidant defenses. Life Sci. 2020, 243, 117244. [Google Scholar] [CrossRef]
- Wang, Q.; Jia, F.; Guo, C.; Wang, Y.; Zhang, X.; Cui, Y.; Song, M.; Cao, Z.; Li, Y. PINK1/Parkin-mediated mitophagy as a protective mechanism against AFB1-induced liver injury in mice. Food Chem. Toxicol. 2022, 164, 113043. [Google Scholar] [CrossRef]
- Zeb, A.; Choubey, V.; Gupta, R.; Kuum, M.; Safiulina, D.; Vaarmann, A.; Gogichaishvili, N.; Liiv, M.; Ilves, I.; Tämm, K.; et al. A novel role of KEAP1/PGAM5 complex: ROS sensor for inducing mitophagy. Redox Biol. 2021, 48, 102186. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, B.; Li, J.; Gong, D.; Dai, Y.; Wang, P.; Wan, L.; Xu, S. Long-Term Consumption of Food-Derived Chlorogenic Acid Protects Mice against Acetaminophen-Induced Hepatotoxicity via Promoting PINK1-Dependent Mitophagy and Inhibiting Apoptosis. Toxics 2022, 10, 665. https://doi.org/10.3390/toxics10110665
Hu B, Li J, Gong D, Dai Y, Wang P, Wan L, Xu S. Long-Term Consumption of Food-Derived Chlorogenic Acid Protects Mice against Acetaminophen-Induced Hepatotoxicity via Promoting PINK1-Dependent Mitophagy and Inhibiting Apoptosis. Toxics. 2022; 10(11):665. https://doi.org/10.3390/toxics10110665
Chicago/Turabian StyleHu, Bangyan, Jin Li, Daoyin Gong, Yuan Dai, Ping Wang, Lihong Wan, and Shijun Xu. 2022. "Long-Term Consumption of Food-Derived Chlorogenic Acid Protects Mice against Acetaminophen-Induced Hepatotoxicity via Promoting PINK1-Dependent Mitophagy and Inhibiting Apoptosis" Toxics 10, no. 11: 665. https://doi.org/10.3390/toxics10110665
APA StyleHu, B., Li, J., Gong, D., Dai, Y., Wang, P., Wan, L., & Xu, S. (2022). Long-Term Consumption of Food-Derived Chlorogenic Acid Protects Mice against Acetaminophen-Induced Hepatotoxicity via Promoting PINK1-Dependent Mitophagy and Inhibiting Apoptosis. Toxics, 10(11), 665. https://doi.org/10.3390/toxics10110665