
Comparison of the Medical Uses and Cellular Effects of High and Low Linear Energy Transfer Radiation
 ,
, 
Abstract
1. Introduction
Characteristics of Ionizing Radiation
2. Medical Applications for High- and Low-LET Radiation
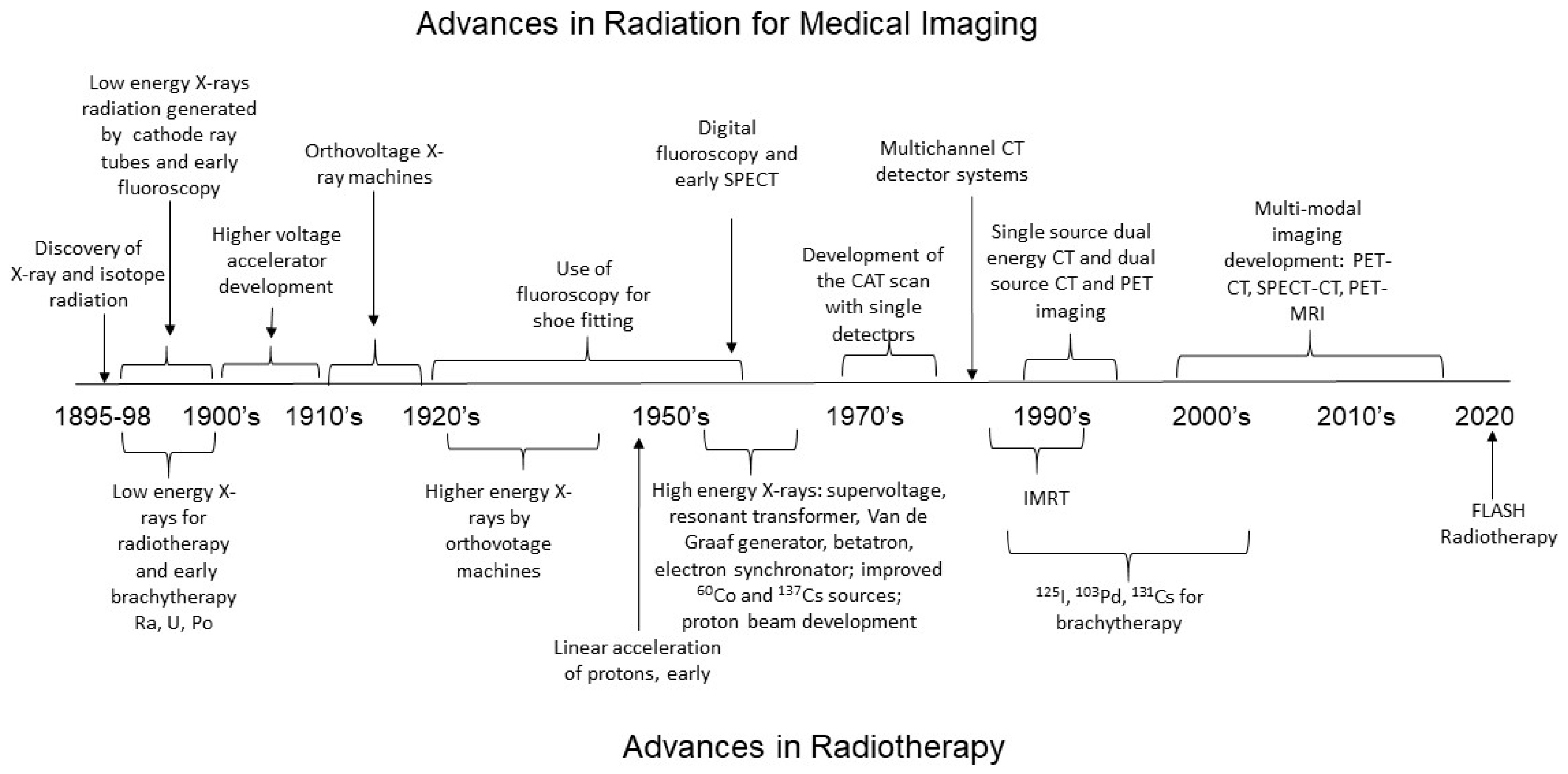
2.1. Medical Imaging and the Development of Low-LET Radiation Technology
2.1.1. Relationship between Radiation Energy and Medical Images
2.1.2. Radiation Requirements for Real-Time Imaging of Organ Function with Fluoroscopy and Positron Emission Tomography
2.1.3. Future Imaging Technique Development
2.2. Radiotherapy for Cancer Treatment
Development of Low-LET Photon Radiation for Cancer Treatment
2.3. High-LET Radiation for Cancer Treatment
2.3.1. Development of Brachytherapy Using Radioactive Isotopes
2.3.2. Hadron Therapy: High-LET Radiation Beams for Cancer Treatment
2.4. Techniques for Sparing Normal Tissues during X-ray Cancer Radiotherapy
3. Cellular Effects of High- and Low-LET Radiation
3.1. Cancer Cell Responses to High- and Low-LET Radiation
3.2. Normal, Non-Cancer Cell Responses to High- and Low-LET Radiation
4. Signal Transduction by High and Low LET
4.1. Pathways for DNA Repair
4.2. Regulation of the Cell Cycle: The Gateway for Cell Death or Accelerated Senescence
4.3. Regulation of the Protein Degradation, Endoplasmic Reticulum Stress, and the Unfolded Protein Response Pathway
5. Protein Expression and Gene Transcription by High- and Low-LET Radiation
5.1. Alterations in Protein Levels with Low- and HIGH-LET Radiation
5.2. Regulation of p53 and NF-κB Transcription Factors
5.3. Comparison of Low- and High-LET Radiation Using Genomic Analysis in Cancer Cells
5.4. Comparison of Low- and High-LET Radiation Using Genomic Analysis in Normal Cells
6. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pereira, G.C.; Traughber, M.; Muzic, R.F. The role of imaging in radiation therapy planning: Past, present, and future. BioMed Res. Int. 2014, 2014, 231090. [Google Scholar] [CrossRef] [PubMed]
- Howell, J.D. Early Clinical Use of the x-ray. Trans. Am. Clin. Clim. Assoc. 2016, 127, 341–349. [Google Scholar]
- Haas, L.F. Wilhelm Conrad Von Rȍntgen (1845–1923). J. Neurol. Neurosurg Psychiatry 2001, 70, 126. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bouten, R.M.; Young, E.F.; Selwyn, R.; Iacono, D.; Rittase, W.B.; Day, R.M. Effects of radiation on endothelial barrier and vascular integrity. In Tissue Barriers in Disease, Injury and Regeneration; Gorbunov, N.V., Ed.; Elsevier, Inc.: Amsterdam, The Netherlands, 2021; pp. 43–94. [Google Scholar]
- Hall, E.J.; Giacci, A.J. Radiobiology for the Radiologist, 8th ed.; Hall, E.J., Garcia, A.J., Eds.; Wolters Kluwer: Philadelphia, PA, USA, 2019. [Google Scholar]
- Abid, S.H.; Malhotra, V.; Perry, M.C. Radiation-induced and chemotherapy-induced pulmonary injury. Curr. Opin. Oncol. 2001, 13, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Radiation All around, Us. 26 April 2022. Available online: https://www.nrc.gov/about-nrc/radiation/around-us/doses-daily-lives.html (accessed on 20 May 2022).
- Preston, D.L.; Shimizu, Y.; Pierce, D.A.; Suyama, A.; Mabuchi, K. Studies of mortality of atomic bomb survivors. report 13: Solid cancer and noncancer disease mortality: 1950–1997. Radiat. Res. 2003, 160, 381–407. [Google Scholar] [CrossRef]
- Committee to Assess Health. Risks from Exposure to Low Levels of Ionizing Radiation, Health risks from exposure to low levels of ionizing radiation BEIR VII, Phase 2. In National Research Souncil of the National Academies; National Academies Press: Washington, DC, USA, 2006. [Google Scholar]
- Preston, D.L.; Ron, E.; Tokuoka, S.; Funamoto, S.; Nishi, N.; Soda, M.; Mabuchi, K.; Kodama, K. Solid cancer incidence in atomic bomb survivors: 1958–1998. Radiat. Res. 2007, 168, 1–64. [Google Scholar] [CrossRef]
- Cardis, E.; Vrijheid, M.; Blettner, M.; Gilbert, E.; Hakama, M.; Hill, C.; Howe, G.; Kaldor, J.; Muirhead, C.R.; Schubauer-Berigan, M.; et al. The 15-country collaborative study of cancer risk among radiation workers in the nuclear industry: Estimates of radiation-related cancer risks. Radiat. Res. 2007, 167, 396–416. [Google Scholar] [CrossRef]
- Matthews, E.P. Radiation physics, biology, and protection. Radiol. Technol. 2019, 90, 471–485. [Google Scholar]
- Curtis, R.A. Introduction to Ionizing Radiation. Ionizing Radiation-Hazard Recognition. 1999. Available online: https://www.osha.gov/ionizing-radiation/introduction/handout (accessed on 23 May 2022).
- Valentin, J. Relative biological effectiveness (RBE), quality factor (Q), and radiation weighting factor (wR). Ann. ICRP 2003, 33, 1–121. [Google Scholar] [CrossRef]
- Radiation Sources and Doses. 11 May 2022. Available online: https://www.epa.gov/radiation/radiation-sources-and-doses (accessed on 20 May 2022).
- Sengbusch, E.; Pérez-Andújar, A.; DeLuca, P.M.; Mackie, T.R. Maximum proton kinetic energy and patient-generated neutron fluence considerations in proton beam arc delivery radiation therapy. Med. Phys. 2009, 36, 364–372. [Google Scholar] [CrossRef]
- Hall, N.; Rouge, B. The physics of proton therapy. Phys. Med. Biol. 2015, 60, R155. [Google Scholar] [CrossRef]
- Wilson, J.W.; Kim, M.-H.; De Angelis, G.; Cucinotta, F.A.; Yoshizawa, N.; Badavi, F.F. Implementation of Gy-Eq for Deterministic Effects Limitation in Shield Design. J. Radiat. Res. 2002, 43, S103–S106. [Google Scholar] [CrossRef]
- Mulford, D.A.; Scheinberg, D.A.; Jurcic, J.G. The promise of targeted {alpha}-particle therapy. J. Nucl. Med. 2005, 46, 199S–204S. [Google Scholar] [PubMed]
- Wang, B.; Yasuda, H. Relative biological effectiveness of high LET particles on the reproductive system and fetal development. Life 2020, 10, 298. [Google Scholar] [CrossRef] [PubMed]
- Zackrisson, B.; Johansson, B.; Ostbergh, P. Relative biological effectiveness of high-energy photons (up to 50 MV) and electrons (50 MeV). Radiat. Res. 1991, 128, 192. [Google Scholar] [CrossRef]
- Paganetti, H. Proton Relative Biological Effectiveness–Uncertainties and Opportunities. Int. J. Part. Ther. 2018, 5, 2–14. [Google Scholar] [CrossRef]
- Paganetti, H.; Giantsoudi, D. Relative biological effectiveness uncertainties and implications for beam arrangements and dose constraints in proton therapy. Semin. Radiat. Oncol. 2018, 28, 256–263. [Google Scholar] [CrossRef]
- Coia, L.R.; Moylan, D.J. (Eds.) Introduction to Clinical Radiation Oncology; Medical Physics Publishing: Madison, WI, USA, 1996; p. 24. [Google Scholar]
- Huh, H.D.; Kim, S. History of radiation therapy technology. Prog. Med. Phys. 2020, 31, 124–134. [Google Scholar] [CrossRef]
- McCollough, C.H.; Leng, S.; Yu, L.; Fletcher, J.G. Dual- and multi-energy CT: Principles, technical approaches, and clinical applications. Radiology 2015, 276, 637–653. [Google Scholar] [CrossRef]
- Grajo, J.R.; Patino, M.; Prochowski, A.; Sahani, D.V. Dual energy CT in practice: Basic principles and applications. Appl. Radiol. 2016, 45, 6–12. [Google Scholar] [CrossRef]
- Strickland, D.; Stranges, A.N. X-rays: Laying the foundation of modern radiology, 1896–1930. Med. Nei. Secoli. 1991, 3, 207–222. [Google Scholar]
- Kemerink, M.; Dierichs, T.J.; Dierichs, J.; Huynen, H.J.; Wildberger, J.E.; van Engelshoven, J.M.A.; Kemerink, G.J. Characteristics of a First-Generation X-ray System. Radiology 2011, 259, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.R. Orthovoltage therapy; is there still a need for it. Calif. Med. 1958, 89, 420–422. [Google Scholar] [PubMed]
- Hill, R.; Healy, B.; Holloway, L.; Kuncic, Z.; Thwaites, D.; Baldock, C. Advances in kilovoltage X-ray beam dosimetry. Phys. Med. Biol. 2014, 59, R183–R231. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.D.; Wu, X.; Liu, H. Image quality and dose efficiency of high energy phase sensitive X-ray imaging: Phantom studies. J. X-Ray Sci. Technol. 2014, 22, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Kotre, C.J.; Birch, I.P. Phase contrast enhancement of X-ray mammography: A design study. Phys. Med. Biol. 1999, 44, 2853–2866. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.C.; Nieves, L.M.; Betzer, O.; Sadan, T.; Noël, P.B.; Popovtzer, R.; Cormode, D.P. Nanoparticle contrast agents for X-ray imaging applications. WIREs Nanomed. Nanobiotechnol. 2020, 12, e1642. [Google Scholar] [CrossRef]
- Smith-Bindman, R.; Kwan, M.L.; Marlow, E.; Theis, M.K.; Bolch, W.; Cheng, S.Y.; Bowles, E.J.A.; Duncan, J.R.; Greenlee, R.T.; Kushi, L.H.; et al. Trends in use of medical imaging in US health care systems and in Ontario, Canada, 2000–2016. JAMA 2019, 322, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, K.B. Godfrey Newbold Hounsfield (1919-2004): The man who revolutionized neuroimaging. Ann. Indian Acad. Neurol. 2016, 19, 448–450. [Google Scholar] [CrossRef]
- Dilmanian, F.A.; Wu, X.Y.; Parsons, E.C.; Ren, B.; Kress, J.; Button, T.M.; Chapman, L.D.; Coderre, J.A.; Giron, F.; Greenberg, D.; et al. Single- and dual-energy CT with monochromatic synchrotron X-rays. Phys. Med. Biol. 1997, 42, 371–387. [Google Scholar] [CrossRef]
- Lusic, H.; Grinstaff, M. X-ray-Computed Tomography Contrast Agents. Chem. Rev. 2013, 113, 1641–1666. [Google Scholar] [CrossRef] [PubMed]
- Zopfs, D.; Graffe, J.; Reimer, R.P.; Schäfer, S.; Persigehl, T.; Maintz, D.; Borggrefe, J.; Haneder, S.; Lennartz, S.; Hokamp, N.G. Quantitative distribution of iodinated contrast media in body computed tomography: Data from a large reference cohort. Eur. Radiol. 2020, 31, 2340–2348. [Google Scholar] [CrossRef] [PubMed]
- Flohr, T.; Petersilka, M.; Henning, A.; Ulzheimer, S.; Ferda, J.; Schmidt, B. Photon-counting CT review. Phys. Medica 2020, 79, 126–136. [Google Scholar] [CrossRef]
- Henzler, T.; Fink, C.; Schoenberg, S.O.; Schoepf, U.J. Dual-Energy CT: Radiation dose aspects. Am. J. Roentgenol. 2012, 199, S16–S25. [Google Scholar] [CrossRef] [PubMed]
- Shalom, N.E.; Gong, G.X.; Auster, M. Fluoroscopy: An essential diagnostic modality in the age of high-resolution cross-sectional imaging. World J. Radiol. 2020, 12, 213–230. [Google Scholar] [CrossRef]
- Deng, C.; Cao, X.; Wu, D.; Ding, H.; You, R.; Chen, Q.; Chen, L.; Zhang, X.; Zhang, Q.; Wu, Y. Small lung lesions invisible under fluoroscopy are located accurately by three-dimensional localization technique on chest wall surface and performed bronchoscopy procedures to increase diagnostic yields. BMC Pulm. Med. 2016, 16, 166. [Google Scholar] [CrossRef]
- Hill, N.E.; Giampetro, D.M. Fluoroscopy Contrast Materials; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Li, X.; Hirsch, J.A.; Rehani, M.; Ganguli, S.; Yang, K.; Liu, B. Radiation effective dose above 100 msv from fluoroscopically guided intervention: Frequency and patient medical condition. Am. J. Roentgenol. 2020, 215, 433–440. [Google Scholar] [CrossRef]
- Choi, M.H.; Jung, S.E.; Oh, S.N.; Byun, J.Y. Educational effects of radiation reduction during fluoroscopic examination of the adult gastrointestinal tract. Acad. Radiol. 2018, 25, 202–208. [Google Scholar] [CrossRef]
- Nicholson, P.J.; Guest, W.C.; van Prooijen, M.; Farb, R.I. Digital subtraction myelography is associated with less radiation dose than CT-based techniques. Clin. Neuroradiol. 2020, 31, 627–631. [Google Scholar] [CrossRef]
- Smith-Bindman, R.; Lipson, J.; Marcus, R.; Kim, K.; Mahesh, M.; Gould, R.; de González, A.B.; Miglioretti, D.L. Radiation dose associated with common computed tomography examinations and the associated lifetime attributable risk of cancer. Arch. Intern. Med. 2009, 169, 2078–2086. [Google Scholar] [CrossRef]
- Mahesh, M. Computed Tomography Dose. Radiation Dose. 2021. Available online: https://www.radiologyinfo.org/en/info/safety-xray (accessed on 18 August 2022).
- Karakatsanis, N.A.; Fokou, E.; Tsoumpas, C. Dosage optimization in positron emission tomography: State-of-the-art methods and future prospects. Am. J. Nucl. Med. Mol. Imaging 2015, 5, 527–547. [Google Scholar] [PubMed]
- Taylor, K.; Lemon, J.A.; Boreham, D.R. Radiation-induced DNA damage and the relative biological effectiveness of 18F-FDG in wild-type mice. Mutagenesis 2014, 29, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Quinn, B.; Dauer, Z.; Pandit-Taskar, N.; Schoder, H.; Dauer, L.T. Radiation dosimetry of 18F-FDG PET/CT: Incorporating exam-specific parameters in dose estimates. BMC Med. Imaging 2016, 16, 41. [Google Scholar] [CrossRef]
- Seah, J.; Brady, Z.; Ewert, K.; Law, M. Artificial intelligence in medical imaging: Implications for patient radiation safety. Br. J. Radiol. 2021, 94, 20210406. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Ohno, Y.; Kauczor, H.U.; Hatabu, H. Radiation dose reduction in chest CT—Review of available options. Eur. J. Radiol. 2014, 83, 1953–1961. [Google Scholar] [CrossRef] [PubMed]
- Arndt, C.; Güttler, F.; Heinrich, A.; Bürckenmeyer, F.; Diamantis, I.; Teichgräber, U. Deep learning CT image reconstruction in clinical practice. RöFo-Fortschr. Auf Dem Geb. Röntgenstrahlen Bildgeb. Verfahr. 2020, 193, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Mould, R.F. Emil Herman Grubbe (1875–1960) with special reference to priorityfor X-ray cancer therapy. Nowotw. J. Oncol. 2018, 68, 286–289. [Google Scholar]
- Maturen, K.E.; Feng, M.U.; Wasnik, A.P.; Azar, S.F.; Appelman, H.D.; Francis, I.R.; Platt, J.F. Imaging effects of radiation therapy in the abdomen and pelvis: Evaluating “innocent bystander” tissues. RadioGraphics 2013, 33, 599–619. [Google Scholar] [CrossRef]
- Stea, B.; Hazard, L.J.; Gonzalez, V.; Hamilton, R. The role of radiation therapy in the control of locoregional and metastatic cancer. J. Surg. Oncol. 2011, 103, 627–638. [Google Scholar] [CrossRef]
- Lederman, M. The early history of radiotherapy: 1895–1939. Int. J. Radiat. Oncol. 1981, 7, 639–648. [Google Scholar] [CrossRef]
- Schaue, D.; McBride, W.H. Opportunities and challenges of radiotherapy for treating cancer. Nat. Rev. Clin. Oncol. 2015, 12, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.-W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Barnett, G.C.; West, C.; Dunning, A.M.; Elliott, R.M.; Coles, C.E.; Pharoah, P.D.P.; Burnet, N.G. Normal tissue reactions to radiotherapy: Towards tailoring treatment dose by genotype. Nat. Cancer 2009, 9, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Gianfaldoni, S.; Gianfaldoni, R.; Wollina, U.; Lotti, J.; Tchernev, G.; Lotti, T. An overview on radiotherapy: From its history to its current applications in dermatology. Open Access Maced. J. Med Sci. 2017, 5, 521–525. [Google Scholar] [CrossRef]
- Sarria, G.R.; Petrova, V.; Wenz, F.; Abo-Madyan, Y.; Sperk, E.; Giordano, F.A. Intraoperative radiotherapy with low energy X-rays for primary and recurrent soft-tissue sarcomas. Radiat. Oncol. 2020, 15, 110. [Google Scholar] [CrossRef]
- Berven, E. The Development and organization of therapeutic radiology in Sweden. Radiology 1962, 79, 829–841. [Google Scholar] [CrossRef]
- Podgorsak, E.B. Treatment machines for external beam radiotherapy. In Radiation Oncology Physics: A Handbook for Teachers and Students; Podgorsak, E.B., Ed.; International Atomic Energy Agency: Vienna, Austria, 2005; pp. 123–160. [Google Scholar]
- Adams, G.D. The Use of a 70-Mev Synchrotron in Cancer Therapy. Radiology 1964, 83, 785–796. [Google Scholar] [CrossRef]
- Fiorentino, E.; Letardi, T.; Marino, A. High-repetition-rate electron beam with resonant transformer. Il. Nuovo. Cim. 1982, 71, 205–217. [Google Scholar] [CrossRef]
- Huang, K.; Li, Y.F.; Li, D.Z.; Chen, L.M.; Tao, M.Z.; Ma, Y.; Zhao, J.R.; Li, M.H.; Chen, M.; Mirzaie, M.; et al. Resonantly enhanced betatron hard X-rays from ionization injected electrons in a laser plasma accelerator. Sci. Rep. 2016, 6, 27633. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, Y.; Ming, X.; Deng, J. Energy modulated photon radiotherapy: A Monte Carlo feasibility study. Bio. Med. Res. Int. 2016, 2016, 7319843. [Google Scholar] [CrossRef]
- Söderström, A.E.S.; Eklöf, A.; Brahme, A. Aspects on the optimal photon beam energy for radiation therapy. Acta Oncol. 1999, 38, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, G.H. Problems in clinical evaluation of radiotherapeutic methods. JAMA 1962, 179, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Mercado, R.; Sala, J.M. Comparison of conventional and supervoltage radiation in the management of cancer of the cervix. analysis of survival rates and complications. Radiology 1968, 90, 967–970. [Google Scholar] [CrossRef]
- Marion, J.B. The effects of nuclear radiations. In Energy in Perspective; Academic Press, Inc.: New York, NY, USA, 1974; pp. 140–169. [Google Scholar]
- Hobbs, R.F.; Howell, R.W.; Song, H.; Baechler, S.; Sgouros, G. Redefining Relative Biological Effectiveness in the Context of the EQDX Formalism: Implications for Alpha-Particle Emitter Therapy. Radiat. Res. 2014, 181, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Baiocco, G.; Barbieri, S.; Babini, G.; Morini, J.; Alloni, D.; Friedland, W.; Kundrát, P.; Schmitt, E.; Puchalska, M.; Sihver, L.; et al. The origin of neutron biological effectiveness as a function of energy. Sci. Rep. 2016, 6, 34033. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.; Underwood, T.S.A.; Carabe-Fernandez, A.; Timlin, C.; Dale, R.G. Fast neutron relative biological effects and implications for charged particle therapy. Br. J. Radiol. 2011, 84, S11–S18. [Google Scholar] [CrossRef]
- Halnan, K.E.; Hornsey, S. RBE values for neutron therapy: A simple method for converting acceptable photon dose to limiting neutron dose. Int. J. Radiat. Oncol. 1981, 7, 601–604. [Google Scholar] [CrossRef]
- Choi, J.; Kang, J.O. Basics of particle therapy II: Relative biological effectiveness. Radiat. Oncol. J. 2012, 30, 1–13. [Google Scholar] [CrossRef]
- Howard, M.E.; Beltran, C.; Anderson, S.; Tseung, W.C.; Sarkaria, J.N.; Herman, M.G. Investigating dependencies of relative biological effectiveness for proton therapy in cancer cells. Int. J. Part. Ther. 2017, 4, 12–22. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, Y.; Ahmad, M.; Ming, X.; Zhou, L.; Deng, J. Intermediate megavoltage photon beams for improved lung cancer treatments. PLoS ONE 2015, 10, e0145117. [Google Scholar] [CrossRef]
- Sun, M.; Ma, L. Treatment of exceptionally large prostate cancer patients with low-energy intensity-modulated photons. J. Appl. Clin. Med. Phys. 2006, 7, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, M.; Tripathi, D.; Kumar, L.; Chowdhary, R.L.; Kakria, A.; Kumar, P.; Mitra, S.; Gairola, M. Can a low-energy photon beam be suitable for the treatment of cervical malignancies? A dosimetric analysis. J. Curr. Oncol. 2020, 3, 55. [Google Scholar] [CrossRef]
- Kemikler, G. History of Brachytherapy. Turk. J. Oncol. 2019, 31, 1–10. [Google Scholar] [CrossRef]
- Eisenstein, M. The declining art of brachytherapy. Nature 2019, 574, S81. [Google Scholar] [CrossRef] [PubMed]
- Awan, S.B.; Hussain, M.; Dini, S.A.; Meigooni, A.S. Historical review of interstitial prostate brachytherapy. Int. J. Radiat. Res. 2008, 5, 153–168. [Google Scholar]
- Aronowitz, J.N. A century of brachytherapy (from the prostate’s perspective). In Brachytherapy; Devlin, P.M., Cormack, R.A., Steward, A.J., Eds.; Springer Puablishing: New York, NY, USA, 2015; pp. 1–34. [Google Scholar]
- Lee, C.D. Recent developments and best practice in brachytherapy treatment planning. Br. J. Radiol. 2014, 87, 20140146. [Google Scholar] [CrossRef]
- Fischer-Valuck, B.; Gay, H.A.; Patel, S.; Baumann, B.; Michalski, J.M. A brief review of low-dose rate (LDR) and high-dose rate (HDR) brachytherapy boost for high-risk prostate. Front. Oncol. 2019, 9, 1378. [Google Scholar] [CrossRef]
- Holm, H.H. The history of interstitial brachytherapy of prostatic cancer. Semin. Surg. Oncol. 1997, 13, 431–437. [Google Scholar] [CrossRef]
- Park, D.S. Current status of brachytherapy for prostate cancer. Korean J. Urol. 2012, 53, 743–749. [Google Scholar] [CrossRef]
- Wilson, R.R. Radiological Use of Fast Protons. Radiology 1946, 47, 487–491. [Google Scholar] [CrossRef]
- Tian, X.; Liu, K.; Hou, Y.; Cheng, J.; Zhang, J. The evolution of proton beam therapy: Current and future status. Mol. Clin. Oncol. 2017, 8, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Tobias, C.A.; Lawrence, J.H.; Born, J.L.; McCombs, R.K.; Roberts, J.E.; Anger, H.O.; Low-Beer, B.V.A.; Huggins, C.B. Pituitary irradiation with high-energy proton beams: A preliminary report. Cancer Res. 1958, 18, 121–134. [Google Scholar]
- Palm, A.; Johansson, K.A. A review of the impact of photon and proton external beam radiotherapy treatment modalities on the dose distribution in field and out-of-field; implications for the long-term morbidity of cancer survivors. Acta Oncol. 2007, 46, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Jiang, L.; Cui, X.; Zhang, J.; Yu, J. Proton beam therapy for cancer in the era of precision medicine. J. Hematol. Oncol. 2018, 11, 136. [Google Scholar] [CrossRef]
- Michalski, J.M.; Pisansky, T.M.; Lawton, C.A.; Potters, L.; Kuban, D.A. Chapter 51-Prostate cancers. In Clinical Radiation Oncology, 3rd ed.; Gunderson, L.L., Tepper, J.E., Eds.; Elsevier, Inc.: Amsterdam, The Netherlands, 2012; pp. e51–e85. [Google Scholar]
- Levin, W.P.; Kooy, H.; Loeffler, J.S.; Delaney, T.F. Proton beam therapy. Br. J. Cancer 2005, 93, 849–854. [Google Scholar] [CrossRef]
- Lin, H.; Shi, C.; Huang, S.; Shen, J.; Kang, M.; Chen, Q.; Zhai, H.; McDonough, J.; Tochner, Z.; Deville, C.; et al. Applications of various range shifters for proton pencil beam scanning radiotherapy. Radiat. Oncol. 2021, 16, 146. [Google Scholar] [CrossRef]
- Mould, R. Priority for radium therapy of benign conditions and cancer. Curr. Oncol. 2007, 14, 118–122. [Google Scholar] [CrossRef]
- Thames, H.D., Jr. Early fractionation methods and the origin of NSD concept. Acta Oncol. 1988, 27, 89–103. [Google Scholar] [CrossRef]
- Pajonk, F.; Vlashi, E.; McBride, W.H. Radiation resistance of cancer stem cells: The 4 R’s of radiobiology revisited. Stem Cells 2010, 28, 639–648. [Google Scholar] [CrossRef]
- Ramroth, J.; Cutter, D.J.; Darby, S.C.; Higgins, G.S.; McGale, P.; Partridge, M.; Taylor, C.W. Dose and fractionation in radiation therapy of curative intent for non-small cell lung cancer: Meta-analysis of randomized trials. Int. J. Radiat. Oncol. 2016, 96, 736–747. [Google Scholar] [CrossRef]
- Demaria, S.; Guha, C.; Schoenfeld, J.; Morris, Z.; Monjazeb, A.; Sikora, A.; Crittenden, M.; Shiao, S.; Khleif, S.; Gupta, S.; et al. Radiation dose and fraction in immunotherapy: One-size regimen does not fit all settings, so how does one choose? J. Immunother. Cancer 2021, 9, e002038. [Google Scholar] [CrossRef]
- Yoon, S.M.; Chu, F.-I.; Ruan, D.; Steinberg, M.L.; Raldow, A.; Lee, P. Assessment of toxic effects associated with dose-fractionated radiotherapy among patients with cancer and comorbid collagen vascular disease. JAMA Netw. Open 2021, 4, e2034074. [Google Scholar] [CrossRef]
- Roach, M.C.; Bradley, J.D.; Robinson, C.G. Optimizing radiation dose and fractionation for the definitive treatment of locally advanced non-small cell lung cancer. J. Thorac. Dis. 2018, 10, S2465–S2473. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Gao, F.; Yang, Y.; Wu, D.; Zhang, Y.; Feng, G.; Dai, T.; Du, X. FLASH radiotherapy: History and future. Front. Oncol. 2021, 11, 644400. [Google Scholar] [CrossRef] [PubMed]
- Jones, B. The influence of hypoxia on LET and RBE relationships with implications for ultra-high dose rates and FLASH modelling. Phys. Med. Biol. 2022, 67, 25011. [Google Scholar] [CrossRef] [PubMed]
- Bourhis, J.; Sozzi, W.J.; Jorge, P.G.; Gaide, O.; Bailat, C.; Duclos, F.; Patin, D.; Ozsahin, M.; Bochud, F.; Germond, J.-F.; et al. Treatment of a first patient with FLASH-radiotherapy. Radiother. Oncol. 2019, 139, 18–22. [Google Scholar] [CrossRef]
- Reisz, J.A.; Bansal, N.; Qian, J.; Zhao, W.; Furdui, C.M. Effects of ionizing radiation on biological molecules—Mechanisms of damage and emerging methods of detection. Antioxid. Redox Signal. 2014, 21, 260–292. [Google Scholar] [CrossRef]
- Munro, T.R. The relative radiosensitivity of the nucleus and cytoplasm of chinese hamster fibroblasts. Radiat. Res. 1970, 42, 451. [Google Scholar] [CrossRef]
- Gulston, M. Clustered DNA damage induced by gamma radiation in human fibroblasts (HF19), hamster (V79-4) cells and plasmid DNA is revealed as Fpg and Nth sensitive sites. Nucleic Acids Res. 2002, 30, 3464–3472. [Google Scholar] [CrossRef]
- Bartesaghi, S.; Radi, R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. 2017, 14, 618–625. [Google Scholar] [CrossRef]
- Azzam, E.I.; Jay-Gerin, J.-P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Suit, H.D. Radiation Biology: The conceptual and practical impact on radiation therapy. Radiat. Res. 1983, 94, 10–40. [Google Scholar] [CrossRef] [PubMed]
- Pouget, J.-P.; Mather, S.J. General aspects of the cellular response to low- and high-LET radiation. Eur. J. Nucl. Med. 2001, 28, 541–561. [Google Scholar] [CrossRef] [PubMed]
- Withers, H.R. The 4Rs of radiotherapy. In Advances in Radiation Biology; Lett, J.T., Alder, H., Eds.; Academic Press: New York, NY, USA, 1975; pp. 241–249. [Google Scholar]
- Tamulevicius, P.; Wang, M.; Iliakis, G. Homology-directed repair is required for the development of radioresistance during S phase: Interplay between double-strand break repair and checkpoint response. Radiat. Res. 2007, 167, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Matsuya, Y.; McMahon, S.J.; Tsutsumi, K.; Sasaki, K.; Okuyama, G.; Yoshii, Y.; Mori, R.; Oikawa, J.; Prise, K.M.; Date, H. Investigation of dose-rate effects and cell-cycle distribution under protracted exposure to ionizing radiation for various dose-rates. Sci. Rep. 2018, 8, 8287. [Google Scholar] [CrossRef]
- Kim, C.; Kim, B. Anti-cancer natural products and their bioactive compounds inducing ER stress-mediated apoptosis: A review. Nutrients 2018, 10, 1021. [Google Scholar] [CrossRef]
- Sørensen, B.S.; Horsman, M.R. Tumor hypoxia: Impact on radiation therapy and molecular pathways. Front. Oncol. 2020, 10, 562. [Google Scholar] [CrossRef]
- Steel, G.G.; McMillan, T.J.; Peacock, J.H. The 5Rs of radiobiology. Int. J. Radiat. Biol. 1989, 56, 1045–1048. [Google Scholar] [CrossRef]
- Coco Martin, E.M.; Mooren, C.; Ottenheim, W.; Burrill, M.N.D.; Sprong, H.; Bartelink, A.B.J. Potential of radiation-induced chromosome aberrations to predict radiosensitivity in human tumour cells. Int. J. Radiat. Biol. 1999, 75, 1161–1168. [Google Scholar] [CrossRef]
- Hawkins, R.B. The relationship between the sensitivity of cells to high-energy photons and the rbe of particle radiation used in radiotherapy. Radiat. Res. 2009, 172, 761–776. [Google Scholar] [CrossRef]
- Roobol, S.J.; van den Bent, I.; Van Cappellen, W.A.; Abraham, T.E.; Paul, M.W.; Kanaar, R.; Houtsmuller, A.B.; Van Gent, D.C.; Essers, J. Comparison of high- and low-LET radiation-induced dna double-strand break processing in living cells. Int. J. Mol. Sci. 2020, 21, 6602. [Google Scholar] [CrossRef] [PubMed]
- Jezkova, L.; Zadneprianetc, M.; Kulikova, E.; Smirnova, E.; Bulanova, T.; Depes, D.; Falkova, I.; Boreyko, A.; Krasavin, E.; Davidkova, M.; et al. Particles with similar LET values generate DNA breaks of different complexity and reparability: A high-resolution microscopy analysis of γH2AX/53BP1 foci. Nanoscale 2017, 10, 1162–1179. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Kim, S.-H. Relative biological effectiveness of fast neutrons for apoptosis in mouse hair follicles. J. Veter.-Sci. 2007, 8, 335–340. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kysela, B.; Arrand, J.; Michael, B. Relative contributions of levels of initial damage and repair of double-strand breaks to the ionizing radiation-sensitive phenotype of the Chinese hamster cell mutant, XR-V15B. Part II. Neutrons. Int. J. Radiat. Biol. 1993, 64, 531–538. [Google Scholar] [CrossRef]
- Newman, C.; Prise, K.M.; Folkard, M.; Michael, B.D.H. DNA double-strand break distributions in X-ray and alpha-particle irradiated V79 cells: Evidence for non-random breakage. Int. J. Radiat. Biol. 1997, 71, 347–363. [Google Scholar] [CrossRef]
- Lorat, Y.; Brunner, C.U.; Schanz, S.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Nanoscale analysis of clustered DNA damage after high-LET irradiation by quantitative electron microscopy–The heavy burden to repair. DNA Repair 2015, 28, 93–106. [Google Scholar] [CrossRef]
- Barnard, S.; Bouffler, S.; Rothkamm, K. The shape of the radiation dose response for DNA double-strand break induction and repair. Genome Integr. 2013, 4, 1. [Google Scholar] [CrossRef]
- Dugan, L.C.; Bedford, J.S. Are chromosomal instabilities induced by exposure of cultured normal human cells to low- or high-LET radiation? Radiat. Res. 2003, 159, 301–311. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Chang, D.S.; Lasley, F.D.; Das, I.J.; Mendonca, M.S.; Dynlacht, J.R. Radiation survival models, SLD, PLD, and dose rate. In Basic Radiotherapy Physics and Biology; Springer Publishing: New York, NY, USA, 2021; pp. 243–253. [Google Scholar]
- Kim, B.M.; Hong, Y.; Lee, S.; Liu, P.; Lim, J.H.; Lee, Y.H.; Lee, T.H.; Chang, K.T.; Hong, Y. Therapeutic Implications for Overcoming Radiation Resistance in Cancer Therapy. Int. J. Mol. Sci. 2015, 16, 26880–26913. [Google Scholar] [CrossRef]
- Panganiban, R.-A.M.; Snow, A.L.; Day, R.M. Mechanisms of radiation toxicity in transformed and non-transformed cells. Int. J. Mol. Sci. 2013, 14, 15931–15958. [Google Scholar] [CrossRef] [PubMed]
- Bylicky, M.A.; Mueller, G.P.; Day, R.M. Radiation resistance of normal human astrocytes: The role of non-homologous end joining DNA repair activity. J. Radiat. Res. 2019, 60, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef] [PubMed]
- Sertorio, M.; Nowrouzi, A.; Akbarpour, M.; Chetal, K.; Salomonis, N.; Brons, S.; Mascia, A.; Ionascu, D.; McCauley, S.; Kupneski, T.; et al. Differential transcriptome response to proton versus X-ray radiation reveals novel candidate targets for combinatorial PT therapy in lymphoma. Radiother. Oncol. 2021, 155, 293–303. [Google Scholar] [CrossRef]
- Amundson, S.S.A. Gene Expression Studies for the Development of Particle Therapy. Int. J. Part. Ther. 2018, 5, 49–59. [Google Scholar] [CrossRef]
- Capasso, S.; Alessio, N.; Squillaro, T.; Di Bernardo, G.; Melone, M.A.; Cipollaro, M.; Peluso, G.; Galderisi, U. Changes in autophagy, proteasome activity and metabolism to determine a specific signature for acute and chronic senescent mesenchymal stromal cells. Oncotarget 2015, 6, 39457–39468. [Google Scholar] [CrossRef]
- Mladenova, V.; Mladenov, E.; Stuschke, M.; Iliakis, G. DNA damage clustering after ionizing radiation and consequences in the processing of chromatin breaks. Molecules 2022, 27, 1540. [Google Scholar] [CrossRef]
- Penninckx, S.; Pariset, E.; Cekanaviciute, E.; Costes, S.V. Quantification of radiation-induced DNA double strand break repair foci to evaluate and predict biological responses to ionizing radiation. NAR Cancer 2021, 3, zcab046. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X.; Wang, P.; Yu, X.; Essers, J.; Chen, D.; Kanaar, R.; Takeda, S.; Wang, Y. Characteristics of DNA-binding proteins determine the biological sensitivity to high-linear energy transfer radiation. Nucleic Acids Res. 2010, 38, 3245–3251. [Google Scholar] [CrossRef]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ Is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef]
- Ackerson, S.M.; Romney, C.; Schuck, P.L.; Stewart, J.A. To join or not to join: Decision points along the pathway to double-strand break repair vs. chromosome end protection. Front. Cell Dev. Biol. 2021, 9, 708763. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, I.; van Gent, D.C. Pathway choice in DNA double strand break repair: Observations of a balancing act. Genome Integr. 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.; Clemente-Blanco, A. Cell cycle and dna repair regulation in the damage response: Protein phosphatases take over the reins. Int. J. Mol. Sci. 2020, 21, 446. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.A.; Harper, J.V.; Cucinotta, F.A.; O’Neill, P. Participation of DNA-PKcs in DSB Repair after Exposure to High- and Low-LET Radiation. Radiat. Res. 2010, 174, 195–205. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Löbrich, M. Contribution of DNA repair and cell cycle checkpoint arrest to the maintenance of genomic stability. DNA Repair 2006, 5, 1192–1198. [Google Scholar] [CrossRef]
- Gerelchuluun, A.; Manabe, E.; Ishikawa, T.; Sun, L.; Itoh, K.; Sakae, T.; Suzuki, K.; Hirayama, R.; Asaithamby, A.; Chen, D.J.; et al. The major DNA repair pathway after both proton and carbon-ion radiation is NHEJ, but the HR pathway is more relevant in carbon ions. Radiat. Res. 2015, 183, 345–356. [Google Scholar] [CrossRef]
- Brahme, A. A DNA Repair-Based Model of Cell Survival with Important Clinical Consequences. Radiat. Res. 2020, 194, 202–235. [Google Scholar] [CrossRef]
- Antonelli, F.; Campa, A.; Esposito, G.; Giardullo, P.; Belli, M.; Dini, V.; Meschini, S.; Simone, G.; Sorrentino, E.; Gerardi, S.; et al. Induction and repair of DNA DSB as revealed by h2ax phosphorylation foci in human fibroblasts exposed to low- and high-LET radiation: Relationship with early and delayed reproductive cell death. Radiat. Res. 2015, 183, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Macaeva, E.; Tabury, K.; Michaux, A.; Janssen, A.; Averbeck, N.; Moreels, M.; De Vos, W.H.; Baatout, S.; Quintens, R. High-LET carbon and iron ions elicit a prolonged and amplified p53 signaling and inflammatory response compared to low-LET X-Rays in human peripheral blood mononuclear cells. Front. Oncol. 2021, 11, 768493. [Google Scholar] [CrossRef]
- Allen, C.; Hirakawa, H.; Nakajima, N.I.; Moore, S.; Nie, J.; Sharma, N.; Sugiura, M.; Hoki, Y.; Araki, R.; Abe, M.; et al. Low- and high-LET ionizing radiation induces delayed homologous recombination that persists for two weeks before resolving. Radiat. Res. 2017, 188, 82–93. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Zhang, P.; Wang, Y. The Ku-dependent non-homologous end-joining but not other repair pathway is inhibited by high linear energy transfer ionizing radiation. DNA Repair 2008, 7, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-H.; Lin, Y.-T.; Laiakis, E.; Goudarzi, M.; Weber, W.; Fornace, A. Serum Metabolomic Alterations Associated with Cesium-137 Internal Emitter Delivered in Various Dose Rates. Metabolites 2020, 10, 270. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-H.; Wang, Y.-W.; Chen, R.; Zhou, B.; Ashwell, J.D.; Fornace, A.J. Ionizing radiation impairs T Cell activation by affecting metabolic reprogramming. Int. J. Biol. Sci. 2015, 11, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Day, R.M.; Snow, A.L.; Panganiban, R.A.M. Radiation-induced accelerated senescence: A fate worse than death? Cell Cycle 2014, 13, 2011–2012. [Google Scholar] [CrossRef]
- Panganiban, R.A.M.; Mungunsukh, O.; Day, R.M. X-irradiation induces ER stress, apoptosis, and senescence in pulmonary artery endothelial cells. Int. J. Radiat. Biol. 2012, 89, 656–667. [Google Scholar] [CrossRef]
- Wang, J.; Shen, T.; Zhu, W.; Dou, L.; Gu, H.; Zhang, L.; Yang, Z.; Chen, H.; Zhou, Q.; Sánchez, E.R.; et al. Protein phosphatase 5 and the tumor suppressor p53 down-regulate each other’s activities in mice. J. Biol. Chem. 2018, 293, 18218–18229. [Google Scholar] [CrossRef]
- Li, H.-H.; Cai, X.; Shouse, G.; Piluso, L.G.; Liu, X. A specific PP2A regulatory subunit, B56γ, mediates DNA damage-induced dephosphorylation of p53 at Thr55. EMBO J. 2007, 26, 402–411. [Google Scholar] [CrossRef]
- Kastan, M.B.; Zhan, Q.; El-Deiry, W.S.; Carrier, F.; Jacks, T.; Walsh, W.V.; Plunkett, B.S.; Vogelstein, B.; Fornace, A.J. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 1992, 71, 587–597. [Google Scholar] [CrossRef]
- El-Deiry, W.S. p21(WAF1) Mediates cell-cycle inhibition, relevant to cancer suppression and therapy. Cancer Res. 2016, 76, 5189–5191. [Google Scholar] [CrossRef]
- Panganiban, R.A.M.; Day, R.M. Inhibition of IGF-1R prevents ionizing radiation-induced primary endothelial cell senescence. PLoS ONE 2013, 8, e78589. [Google Scholar] [CrossRef]
- Yim, J.-H.; Yun, J.M.; Kim, J.Y.; Lee, I.K.; Nam, S.Y.; Kim, C.S. Phosphoprotein profiles of candidate markers for early cellular responses to low-dose γ-radiation in normal human fibroblast cells. J. Radiat. Res. 2017, 58, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.E.; Ekedahl, J.; Joseph, B.; Castro, J.; Harms-Ringdahl, M.; Zhivotovsky, B.; Lewensohn, R. High-LET radiation induces apoptosis in lymphoblastoid cell lines derived from ataxia-telangiectasia patients. Int. J. Radiat. Biol. 2001, 77, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Yu, D.; Wang, Z.; Li, S. Relationship between p53 status and the bioeffect of ionizing radiation (Review). Oncol. Lett. 2021, 22, 661. [Google Scholar] [CrossRef] [PubMed]
- Niemantsverdriet, M.; van Goethem, M.-J.; Bron, R.; Hogewerf, W.; Brandenburg, S.; Langendijk, J.A.; van Luijk, P.; Coppes, R.P. High and low LET radiation differentially induce normal tissue damage signals. Int. J. Radiat. Oncol. 2012, 83, 1291–1297. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mori, E.; Takahashi, A.; Yamakawa, N.; Kirita, T.; Ohnishi, T. High LET heavy ion radiation induces p53-independent apoptosis. J. Radiat. Res. 2009, 50, 37–42. [Google Scholar] [CrossRef]
- Yamakawa, N.; Takahashi, A.; Mori, E.; Imai, Y.; Furusawa, Y.; Ohnishi, K.; Kirita, T.; Ohnishi, T. High LET radiation enhances apoptosis in mutated p53 cancer cells through Caspase-9 activation. Cancer Sci. 2008, 99, 1455–1460. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Matsumoto, H.; Yuki, K.; Yasumoto, J.-I.; Kajiwara, A.; Aoki, M.; Furusawa, Y.; Ohnishi, K.; Ohnishi, T. High-LET radiation enhanced apoptosis but not necrosis regardless of p53 status. Int. J. Radiat. Oncol. Biol. Phys. 2004, 60, 591–597. [Google Scholar] [CrossRef]
- Sia, J.; Szmyd, R.; Hau, E.; Gee, H.E. Molecular mechanisms of radiation-induced cancer cell death: A primer. Front. Cell Dev. Biol. 2020, 8, 41. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Cristofalo, V.J.; Lorenzini, A.; Allen, R.; Torres, C.; Tresini, M. Replicative senescence: A critical review. Mech. Ageing Dev. 2004, 125, 827–848. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of cellular senescence: Cell cycle arrest and senescence associated secretory phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Cao, K.; Xia, Y.; Li, Y.; Hou, Y.; Wang, L.; Li, L.; Chang, L.; Li, W. Cellular senescence in ionizing radiation (Review). Oncol. Rep. 2019, 42, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Lafargue, A.; Degorre, C.; Corre, I.; Alves-Guerra, M.-C.; Gaugler, M.-H.; Vallette, F.; Pecqueur, C.; Paris, F. Ionizing radiation induces long-term senescence in endothelial cells through mitochondrial respiratory complex II dysfunction and superoxide generation. Free Radic. Biol. Med. 2017, 108, 750–759. [Google Scholar] [CrossRef]
- Wyld, L.; Bellantuono, I.; Tchkonia, T.; Morgan, J.; Turner, O.; Foss, F.; George, J.; Danson, S.; Kirkland, J.L. Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers 2020, 12, 2134. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Liu, X.; Zhao, T.; Li, F.; Wang, Q.; Zhang, P.; Hirayama, R.; Chen, W.; Jin, X.; Zheng, X.; et al. Comparable radiation sensitivity in p53 wild-type and p53 deficient tumor cells associated with different cell death modalities. Cell Death Discov. 2021, 7, 184. [Google Scholar] [CrossRef]
- Zhang, X.; Ye, C.; Sun, F.; Wei, W.; Hu, B.; Wang, J. Both complexity and location of DNA damage contribute to cellular senescence induced by ionizing radiation. PLoS ONE 2016, 11, e0155725. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Datta, K.; Fornace, A.J.; Suman, S. Total body proton and heavy-ion irradiation causes cellular senescence and promotes pro-osteoclastogenic activity in mouse bone marrow. Heliyon 2021, 8, e08691. [Google Scholar] [CrossRef]
- Werner, E.; Wang, H.; Doetsch, P.W. Role of pro-inflammatory cytokines in radiation-induced genomic instability in human bronchial epithelial cells. Radiat. Res. 2015, 184, 621–629. [Google Scholar] [CrossRef]
- Yasui, L.; Owens, K. Necrosis is not induced by gadolinium neutron capture in glioblastoma multiforme cells. Int. J. Radiat. Biol. 2012, 88, 980–990. [Google Scholar] [CrossRef]
- Cornelissen, M.; Thierens, H.; De Ridder, L. Interphase death in human peripheral blood lymphocytes after moderate and high doses of low and high LET radiation: An electron microscopic approach. Anticancer. Res. 2002, 22, 241–245. [Google Scholar]
- Takahashi, A.; Yano, T.; Matsumoto, H.; Wang, X.; Ohnishi, K.; Tamamoto, T.; Tsuji, K.; Yukawa, O.; Ohnishi, T. Effects of accelerated carbon-ions on growth inhibition of transplantable human esophageal cancer in nude mice. Cancer Lett. 1998, 122, 181–186. [Google Scholar] [CrossRef]
- Strojan, P.; Hutcheson, K.A.; Eisbruch, A.; Beitler, J.J.; Langendijk, J.A.; Lee, A.W.; Corry, J.; Mendenhall, W.M.; Smee, R.; Rinaldo, A.; et al. Treatment of late sequelae after radiotherapy for head and neck cancer. Cancer Treat. Rev. 2017, 59, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Nonoguchi, N.; Kawabata, S.; Miyatake, S.-I.; Kuroiwa, T. Delayed brain radiation necrosis: Pathological review and new molecular targets for treatment. Med. Mol. Morphol. 2015, 48, 183–190. [Google Scholar] [CrossRef]
- El-Rabbany, M.; Duchnay, M.; Raziee, H.R.; Zych, M.; Tenenbaum, H.; Shah, P.S.; Azarpazhooh, A. Interventions for preventing osteoradionecrosis of the jaws in adults receiving head and neck radiotherapy. Cochrane Database Syst. Rev. 2019, 2019, CD011559. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-C.; Barshishat-Kupper, M.; McCart, E.A.; Mueller, G.P.; Day, R.M. Bone Marrow Protein Oxidation in Response to Ionizing Radiation in C57BL/6J Mice. Proteomes 2014, 2, 291–302. [Google Scholar] [CrossRef]
- Barshishat-Kupper, M.; McCart, E.; Freedy, J.G.; Tipton, A.J.; Nagy, V.; Kim, S.-Y.; Landauer, M.R.; Mueller, G.P.; Day, R.M. Protein oxidation in the lungs of C57BL/6J mice following X-irradiation. Proteomes 2015, 3, 249–265. [Google Scholar] [CrossRef]
- Barshishat-Kupper, M.; Tipton, A.J.; McCart, E.; McCue, J.; Mueller, G.P.; Day, R.M. Effect of ionizing radiation on liver protein oxidation and metabolic function in C57BL/6J mice. Int. J. Radiat. Biol. 2014, 90, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Gonon, G.; Groetz, J.-E.; de Toledo, S.M.; Howell, R.W.; Fromm, M.; Azzam, E.I. Nontargeted stressful effects in normal human fibroblast cultures exposed to low fluences of high charge, high energy (HZE) particles: Kinetics of biologic responses and significance of secondary radiations. Radiat. Res. 2013, 179, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.A.; Riggs, P.K.; Yang, T.C.; Pedemonte, C.H.; Clarke, M.S.F.; Feeback, D.L.; Au, W.W. Ionizing radiation-induced bioeffects in space and strategies to reduce cellular injury and carcinogenesis. Aviat. Space, Environ. Med. 2007, 78, A67–A78. [Google Scholar]
- Suman, S.; Rodriguez, O.C.; Winters, T.A.; Fornace, A.J., Jr.; Albanese, C.; Datta, K. Therapeutic and space radiation exposure of mouse brain causes impaired DNA repair response and premature senescence by chronic oxidant production. Aging 2013, 5, 607–622. [Google Scholar] [CrossRef]
- Castro, J.P.; Jung, T.; Grune, T.; Siems, W. 4-Hydroxynonenal (HNE) modified proteins in metabolic diseases. Free Radic. Biol. Med. 2017, 111, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Vladykovskaya, E.; Sithu, S.D.; Haberzettl, P.; Wickramasinghe, N.S.; Merchant, M.L.; Hill, B.; McCracken, J.; Agarwal, A.; Dougherty, S.; Gordon, S.A.; et al. Lipid Peroxidation Product 4-Hydroxy-trans-2-nonenal Causes Endothelial Activation by Inducing Endoplasmic Reticulum Stress. J. Biol. Chem. 2012, 287, 11398–11409. [Google Scholar] [CrossRef] [PubMed]
- Botzen, D.; Grune, T. Degradation of HNE-modified proteins–possible role of ubiquitin. Redox Rep. 2007, 12, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Höhn, A.; König, J.; Grune, T. Protein oxidation in aging and the removal of oxidized proteins. J. Proteom. 2013, 92, 132–159. [Google Scholar] [CrossRef]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin–proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Zois, C.E.; Koukourakis, M.I. Radiation-induced autophagy in normal and cancer cells: Towards novel cytoprotection and radio-sensitization policies? Autophagy 2009, 5, 442–450. [Google Scholar] [CrossRef]
- Moretti, L.; Cha, Y.I.; Niermann, K.J.; Lu, B. Switch Between Apoptosis and Autophagy: Radiation-Induced Endoplasmic Reticulum Stress? Cell Cycle 2007, 6, 793–798. [Google Scholar] [CrossRef]
- Chaurasia, M.; Bhatt, A.N.; Das, A.; Dwarakanath, B.S.; Sharma, K. Radiation-induced autophagy: Mechanisms and consequences. Free Radic. Res. 2016, 50, 273–290. [Google Scholar] [CrossRef]
- Pervan, M.; Iwamoto, K.S.; McBride, W.H. Proteasome structures affected by ionizing radiation. Mol. Cancer Res. 2005, 3, 381–390. [Google Scholar] [CrossRef]
- Pajonk, F.; McBride, W.H. Ionizing radiation affects 26s proteasome function and associated molecular responses, even at low doses. Radiother. Oncol. 2001, 59, 203–212. [Google Scholar] [CrossRef]
- Wang, Y.; Guan, H.; Xie, D.-F.; Xie, Y.; Liu, X.-D.; Wang, Q.; Sui, L.; Song, M.; Zhang, H.; Zhou, J.; et al. Proteomic Analysis Implicates Dominant Alterations of RNA Metabolism and the proteasome pathway in the cellular response to carbon-ion irradiation. PLoS ONE 2016, 11, e0163896. [Google Scholar] [CrossRef]
- Broustas, C.G.; Harken, A.D.; Garty, G.; Amundson, S.A. Identification of differentially expressed genes and pathways in mice exposed to mixed field neutron/photon radiation. BMC Genom. 2018, 19, 504. [Google Scholar] [CrossRef] [PubMed]
- Stankova, K.; Ivanova, K.; Nikolov, V.; Aneva, N.; Georgieva, R.; Boteva, R. Proteasome inhibition protects human peripheral blood mononuclear cells from radiation-induced oxidative stress. Int. J. Radiat. Biol. 2013, 89, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Edmonds, M.J.; Parsons, J.L. Regulation of base excision repair proteins by ubiquitylation. Exp. Cell Res. 2014, 329, 132–138. [Google Scholar] [CrossRef]
- Parsons, J.L.; Preston, B.D.; O’Connor, T.R.; Dianov, G.L. DNA polymerase δ-dependent repair of DNA single strand breaks containing 3′-end proximal lesions. Nucleic Acids Res. 2007, 35, 1054–1063. [Google Scholar] [CrossRef]
- Markkanen, E.; van Loon, B.; Ferrari, E.; Parsons, J.L.; Dianov, G.L.; Hübscher, U. Regulation of oxidative DNA damage repair by DNA polymerase λ and MutYH by cross-talk of phosphorylation and ubiquitination. Proc. Natl. Acad. Sci. USA 2011, 109, 437–442. [Google Scholar] [CrossRef]
- Dianov, G.L.; Meisenberg, C.; Parsons, J. Regulation of DNA repair by ubiquitylation. Biochemistry 2011, 76, 69–79. [Google Scholar] [CrossRef]
- Carter, R.J.; Parsons, J.L. Base Excision Repair, a Pathway Regulated by Posttranslational Modifications. Mol. Cell. Biol. 2016, 36, 1426–1437. [Google Scholar] [CrossRef]
- Carter, R.J.; Nickson, C.M.; Thompson, J.M.; Kacperek, A.; Hill, M.A.; Parsons, J.L. Complex DNA damage induced by high linear energy transfer alpha-particles and protons triggers a specific cellular dna damage response. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 776–784. [Google Scholar] [CrossRef]
- Nickson, C.M.; Fabbrizi, M.R.; Carter, R.J.; Hughes, J.R.; Kacperek, A.; Hill, M.A.; Parsons, J.L. USP9X Is Required to Maintain Cell Survival in Response to High-LET Radiation. Front. Oncol. 2021, 11, 671431. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, B.; Zhang, H.; Katsube, T.; Xie, Y.; Gan, L. Apoptosis induction by iron radiation via inhibition of autophagy in Trp53+/− mouse testes: Is chronic restraint-induced stress a modifying factor? Int. J. Biol. Sci. 2018, 14, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Y.; Lee, Y.-J.; Sai, S.; Ohno, T.; Kong, C.-B.; Lim, S.H.; Kim, E.H. The unfolded protein response: Neutron-induced therapy autophagy as a promising treatment option for osteosarcoma. Int. J. Mol. Sci. 2020, 21, 3766. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, T.-K.; Zeng, C.-H.; Yang, J.; Wang, Y.; Lu, J.; Zhu, G.-Y.; Guo, J.-H. Inhibition of endoplasmic reticulum stress-mediated autophagy enhances the anticancer effect of iodine-125 seed radiation on esophageal squamous cell carcinoma. Radiat. Res. 2020, 194, 236–245. [Google Scholar] [CrossRef]
- Persson, H.L. Iron-dependent lysosomal destabilization initiates silica-induced apoptosis in murine macrophages. Toxicol. Lett. 2005, 159, 124–133. [Google Scholar] [CrossRef]
- Persson, H.L.; Kurz, T.; Eaton, J.W.; Brunk, U.T. Radiation-induced cell death: Importance of lysosomal destabilization. Biochem. J. 2005, 389, 877–884. [Google Scholar] [CrossRef]
- Fushimi, K.; Uzawa, K.; Ishigami, T.; Yamamoto, N.; Kawata, T.; Shibahara, T.; Ito, H.; Mizoe, J.-E.; Tsujii, H.; Tanzawa, H. Susceptible genes and molecular pathways related to heavy ion irradiation in oral squamous cell carcinoma cells. Radiother. Oncol. 2008, 89, 237–244. [Google Scholar] [CrossRef]
- Higo, M.; Uzawa, K.; Kouzu, Y.; Bukawa, H.; Nimura, Y.; Seki, N.; Tanzawa, H. Identification of candidate radioresistant genes in human squamous cell carcinoma cells through gene expression analysis using DNA microarrays. Oncol. Rep. 2005, 14, 1293–1298. [Google Scholar] [CrossRef]
- Ishigami, T.; Uzawa, K.; Higo, M.; Nomura, H.; Saito, K.; Kato, Y.; Nakashima, D.; Shiiba, M.; Bukawa, H.; Yokoe, H.; et al. Genes and molecular pathways related to radioresistance of oral squamous cell carcinoma cells. Int. J. Cancer 2007, 120, 2262–2270. [Google Scholar] [CrossRef]
- Singh, V.; Gupta, D.; Arora, R. NF-kB as a key player in regulation of cellular radiation responses and identification of radiation countermeasures. Discoveries 2015, 3, e35. [Google Scholar] [CrossRef]
- Chishti, A.A.; Baumstark-Khan, C.; Koch, K.; Kolanus, W.; Feles, S.; Konda, B.; Azhar, A.; Spitta, L.F.; Henschenmacher, B.; Diegeler, S.; et al. Linear energy transfer modulates radiation-induced NF-kappa B activation and expression of its downstream target genes. Radiat. Res. 2018, 189, 354–370. [Google Scholar] [CrossRef]
- Di Maggio, F.M.; Minafra, L.; Forte, G.I.; Cammarata, F.P.; Lio, D.; Messa, C.; Gilardi, M.C.; Bravatà, V. Portrait of inflammatory response to ionizing radiation treatment. J. Inflamm. 2015, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, M.; Panyutin, I.G.; Neumann, R. Genome-wide gene expression changes in normal human fibroblasts in response to low-LET gamma-radiation and high-LET-like 125IUdR exposures. Radiat. Prot. Dosim. 2006, 122, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Kurpinski, K.; Jang, D.-J.; Bhattacharya, S.; Rydberg, B.; Chu, J.; So, J.; Wyrobek, A.; Li, S.; Wang, D. Differential effects of X-Rays and high-energy 56Fe ions on human mesenchymal stem cells. Int. J. Radiat. Oncol. 2009, 73, 869–877. [Google Scholar] [CrossRef]
- Ding, L.-H.; Park, S.; Peyton, M.; Girard, L.; Xie, Y.; Minna, J.D.; Story, M.D. Distinct transcriptome profiles identified in normal human bronchial epithelial cells after exposure to γ-rays and different elemental particles of high Z and energy. BMC Genom. 2013, 14, 372. [Google Scholar] [CrossRef] [PubMed]
- Hellweg, C.E.; Spitta, L.F.; Koch, K.; Chishti, A.A.; Henschenmacher, B.; Diegeler, S.; Konda, B.; Feles, S.; Schmitz, C.; Berger, T.; et al. The role of the nuclear factor κb pathway in the cellular response to low and high linear energy transfer radiation. Int. J. Mol. Sci. 2018, 19, 2220. [Google Scholar] [CrossRef]
- Natarajan, M.; Aravindan, N.; Meltz, M.; Herman, T. Post-translational modification of I-kappa B alpha activates NF-κB in human monocytes exposed to 56Fe ions. Radiat. Environ. Biophys. 2002, 41, 139–144. [Google Scholar] [CrossRef]
- Hildebrandt, G.; Seed, M.P.; Freemantle, C.N.; Alam, C.A.S.; Colville-Nash, P.R.; Trott, K.R. Mechanisms of the anti-inflammatory activity of low-dose radiation therapy. Int. J. Radiat. Biol. 1998, 74, 367–378. [Google Scholar] [CrossRef]
- Kumari, A.; Simon, S.S.; Moody, T.D.; Garnett-Benson, C. Immunomodulatory effects of radiation: What is next for cancer therapy? Futur. Oncol. 2016, 12, 239–256. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, J.; Chen, Y.; Jia, Q.; Chu, Q. The roles of CC chemokines in response to radiation. Radiat. Oncol. 2022, 17, 63. [Google Scholar] [CrossRef]
- Higo, M.; Uzawa, K.; Kawata, T.; Kato, Y.; Kouzu, Y.; Yamamoto, N.; Shibahara, T.; Mizoe, J.-E.; Ito, H.; Tsujii, H.; et al. Enhancement of SPHK1 in vitro by carbon ion irradiation in oral squamous cell carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2006, 65, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, X.; Li, Y.; Zhang, H.; Xie, Y.; Zhang, X.; Ren, H.; Wang, Y.; Liao, S.; He, M.; et al. Early Effects of Low Dose12C6+Ion or X-ray Irradiation on Peripheral Blood Lymphocytes of Patients with Alimentary Tract Cancer. Dose-Response 2010, 9, 356–368. [Google Scholar] [CrossRef]
- McKelvey, K.J.; Hudson, A.L.; Back, M.; Eade, T.; Diakos, C.I. Radiation, inflammation and the immune response in cancer. Mamm. Genome 2018, 29, 843–865. [Google Scholar] [CrossRef] [PubMed]
- Crapo, J. Oxidative stress as an initiator of cytokine release and cell damage. Eur. Respir. J. 2003, 22, 4s–6s. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Nakayama, H.; Yoshida, R.; Hirosue, A.; Nagata, M.; Tanaka, T.; Kawahara, K.; Sakata, J.; Arita, H.; Nakashima, H.; et al. IL-6 controls resistance to radiation by suppressing oxidative stress via the Nrf2-antioxidant pathway in oral squamous cell carcinoma. Br. J. Cancer 2016, 115, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Unverricht-Yeboah, M.; Giesen, U.; Kriehuber, R. Comparative gene expression analysis after exposure to 123I-iododeoxyuridine, γ- and α-radiation—Potential biomarkers for the discrimination of radiation qualities. J. Radiat. Res. 2018, 59, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Sakakura, C.; Miyagawa, K.; Kuriu, Y.; Kin, S.; Nakase, Y.; Hagiwara, A.; Mitsufuji, S.; Okazaki, Y.; Hayashizaki, Y.; et al. Differential gene expression profiles of radioresistant oesophageal cancer cell lines established by continuous fractionated irradiation. Br. J. Cancer 2004, 91, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Bouten, R.M.; Dalgard, C.L.; Soltis, A.R.; Slaven, J.E.; Day, R.M. Transcriptomic profiling and pathway analysis of cultured human lung microvascular endothelial cells following ionizing radiation exposure. Sci. Rep. 2021, 11, 24214. [Google Scholar] [CrossRef]
- Wu, C.-T.; Chen, M.-F.; Chen, W.-C.; Hsieh, C.-C. The role of IL-6 in the radiation response of prostate cancer. Radiat. Oncol. 2013, 8, 159. [Google Scholar] [CrossRef]
- Hellweg, C.E. The Nuclear Factor κB pathway: A link to the immune system in the radiation response. Cancer Lett. 2015, 368, 275–289. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Radiation | RBE | Energy Range | References |
|---|---|---|---|
| Alpha particles | 4–20 | 3.2–9 MeV | [19,74,75] |
| Beta particles | 1–3.5 | 0.019–1.7 MeV | [20] |
| Slow neutrons | ~2.5–20 | ~10–100 KeV | [18,76,77,78] |
| Fast neutrons | ~5–20 | 0.1–3 MeV | [18,76,77,78] |
| Protons | ~0.89–3.1 * | 50–1000 MeV | [22,23,79,80] |
| Gamma rays | ~1 | 1.2–6 MeV | [19,20] |
| X-rays | ~1–1.1 | 200–50 MeV | [19,20,21] |
| Isotope | Radiation Type | Half-Life |
|---|---|---|
| 103Pd | 21 KeV gamma * | 17 days |
| 125I | 27–35 KeV gamma * | 60.25 days |
| 131Cs | 29.5–33.5 KeV gamma * | 9.7 days |
| 192Ir | 206–485 KeV gamma * | 74.17 days |
| 198Au | 314 KeV beta, 412 KeV gamma * | 2.7 days |
| 226Ra | 47–2450 KeV gamma | 1600 years |
| “R” | Definition |
|---|---|
| Repair | Sublethal DNA damage repair |
| Redistribution/reassortment | Redistribution of tumor cells into phases of the cell cycle |
| Repopulation | Tumor cell proliferation, symmetrical or asymmetrical division |
| Reoxygenation | Normalization of the hypoxic tumor microenvironment |
| Radiosensitivity | Susceptibility to radiation-induced cell death due to chromosome number alterations or mutations |
| Repair Pathway | Initiating Proteins | DNA Repair | References |
|---|---|---|---|
| HRR | MRN/CtIP | RPA/BRCA2/RAD51 | [133,144,146,147] |
| NHEJ | Ku70/80 | DNA PKcs/XRCC4/Artemis/Pol µ or Pol γ | [133,144,147] |
| Alt-EJ | MRN/CtIP | DNA ligase III/PARP1/pol θ | [133,147,148] |
| Cell Type | Radiation | Time | Genes | References |
|---|---|---|---|---|
| Oral squamous cell carcinoma (High v Low LET) * | X-ray (2, 4, 6 Gy) LET ~ 1 KeV/µm 12C (290 MeV/n) (1, 4, 7 Gy) LET = 75 KeV/µm 22Ne (400 MeV/n) (1, 4, 7 Gy) LET = 75 KeV/µm | 4 h | ↑TGFBR2, ↑SMURF2, ↓BMP7, ↑CCND1, ↑F2F3, ↑SPHK1 | [222,236] |
| Lymphoma | X-ray (6 MeV, 5 Gy) LET ~ 1 KeV/µm | 24 h | ↑CCL5, ↑ CCL17, ↑CCL22, ↑GNG8, ↑HMOX1, ↑IL32 | [139] |
| Lymphoma | Proton (129.3–148.2 MeV/n (5 Gy) LET = 3.5 KeV/µm | 24 h | ↑CCL5, ↑CCL17, ↑CCL22, ↑GNG8, ↑HMOX1, ↑IL32, ↑LRK2, ↑TNF | [139] |
| Cell Type | Radiation | Time | Genes | Reference |
|---|---|---|---|---|
| Peripheral blood mononuclear cells 1 | Gamma (250 keV, 1 Gy) LET ~ 1 KeV/µm | 8 h | ↑PCNA, ↑GADD45A, ↑ASTN2, ↑FDXR, ↑RPS27L, ↑VWCE, ↑PTPN14, ↑EDA2R, ↑CDKN1A, ↑IKBIP, ↑ANKRA2 | [154] |
| 12C (114.6–158.4 MeV/n, 0.25, 1 Gy) (1, 4, 7 Gy) LET = 60–80 KeV/µm 56Fe (1 GeV/n, 0.25, 1 Gy) LET = 155 KeV/µm | 8 h | ↑PCNA, ↑GADD45A, ↑ASTN2, ↑FDXR, ↑RPS27L, ↑VWCE, ↑PTPN14, ↑EDA2R, ↑CD80, ↑BCL2L1 | [154] | |
| Human bronchial epithelial cells 1 | Gamma (662 KeV 1,3 Gy) LET = 0.2 KeV/µm | 1, 4, 12, 24 h | ↑CDKN1A, ↑CCNA1, ↑ BTG2, ↑TRIM22, ↑ INPP5D, ↑GLUL, ↑THBS1, ↓SH3GL3 | [230] |
| 56Fe (1 GeV/n, 0.5, 1 Gy) LET = 150 KeV/µm 28SI (1 GeV/n, 0.5, 1 Gy) LET = 44 KeV/µm | 1, 4, 12, 24 h | ↑CDKN1A, ↑CCNA1, ↑ BTG2, ↑TRIM22, ↑INPP5D, ↑GLUL, ↓APH1B, ↑BLNK, ↑PLD1, ↑PLD3 | [230] | |
| HEK 2 | X-rays (4, 8 Gy, 200 keV) LET ~ 1 KeV/µm | 6 h | ↑TNF, ↑CXCL1, ↑CXCL2, ↑CXCL8, ↑CXCL10, ↑CCL2, ↑CD83, ↑NFKB2, ↑VCAM1, ↑NFKBIA, ↑BIRK3, ↓MAP2K6 | [226] |
| 22Ne ions (4 Gy, 80 MeV/n) LET = 92 KeV/µm | 6 h | ↑TNF, ↑CXCL1, ↑CXCL8, ↑CXCL10, ↑CCL2, ↑CD83, ↑NFKB2, ↑NFKBIA, ↑VCAM1 | [226] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russ, E.; Davis, C.M.; Slaven, J.E.; Bradfield, D.T.; Selwyn, R.G.; Day, R.M. Comparison of the Medical Uses and Cellular Effects of High and Low Linear Energy Transfer Radiation. Toxics 2022, 10, 628. https://doi.org/10.3390/toxics10100628
Russ E, Davis CM, Slaven JE, Bradfield DT, Selwyn RG, Day RM. Comparison of the Medical Uses and Cellular Effects of High and Low Linear Energy Transfer Radiation. Toxics. 2022; 10(10):628. https://doi.org/10.3390/toxics10100628
Chicago/Turabian StyleRuss, Eric, Catherine M. Davis, John E. Slaven, Dmitry T. Bradfield, Reed G. Selwyn, and Regina M. Day. 2022. "Comparison of the Medical Uses and Cellular Effects of High and Low Linear Energy Transfer Radiation" Toxics 10, no. 10: 628. https://doi.org/10.3390/toxics10100628
APA StyleRuss, E., Davis, C. M., Slaven, J. E., Bradfield, D. T., Selwyn, R. G., & Day, R. M. (2022). Comparison of the Medical Uses and Cellular Effects of High and Low Linear Energy Transfer Radiation. Toxics, 10(10), 628. https://doi.org/10.3390/toxics10100628