Developing and Validating a Method for Separating Flavonoid Isomers in Common Buckwheat Sprouts Using HPLC-PDA

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. CSE Preparation
2.3. Flavonoids Analysis by Reversed-Phase HPLC
2.4. Optimizing the Chromatography Conditions
2.5. Method Validation
2.5.1. Linearity
2.5.2. Accuracy, Precision, and Recovery
2.5.3. Limits of Detection and Quantification
2.6. Statistical Analysis
3. Results and Discussion
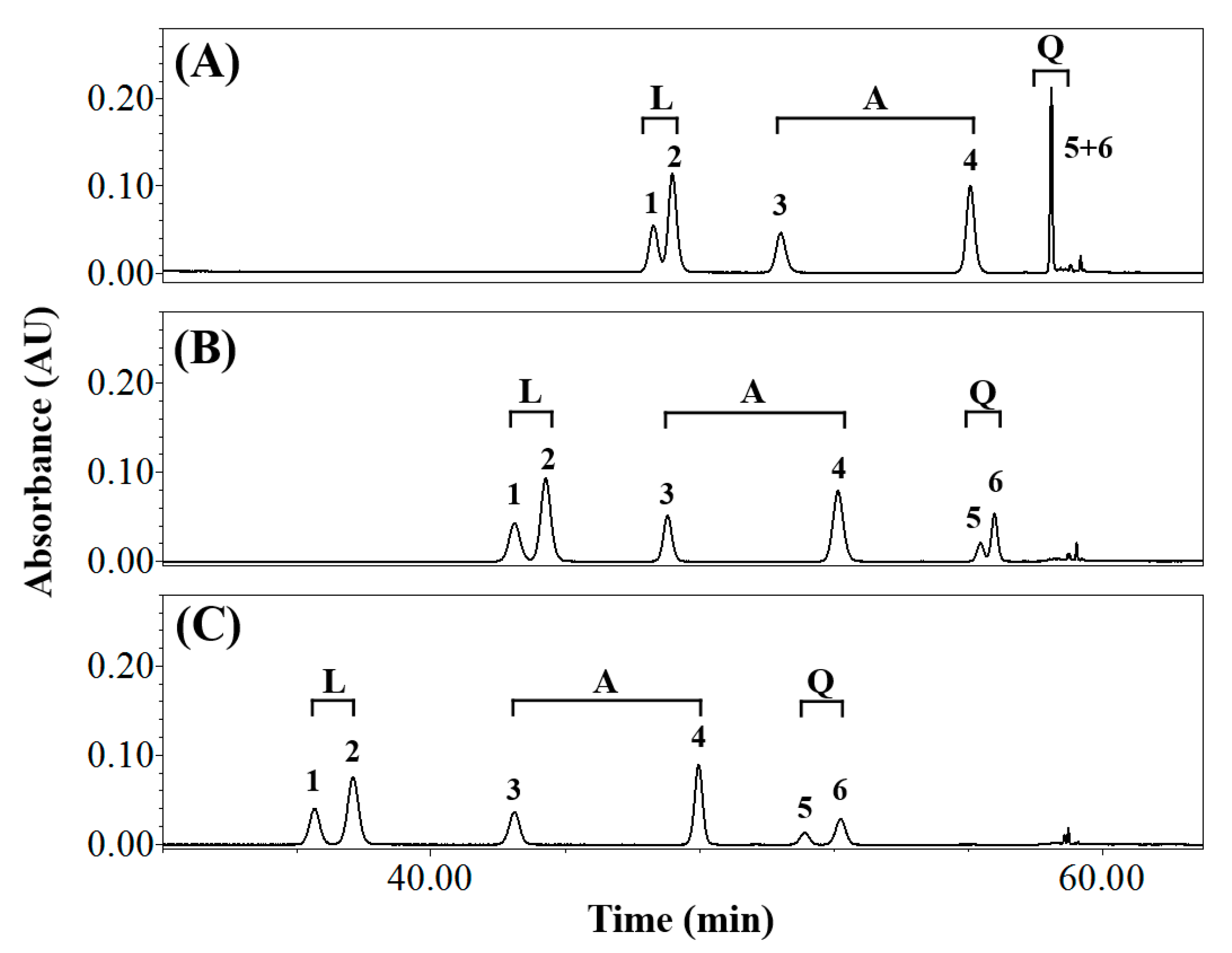
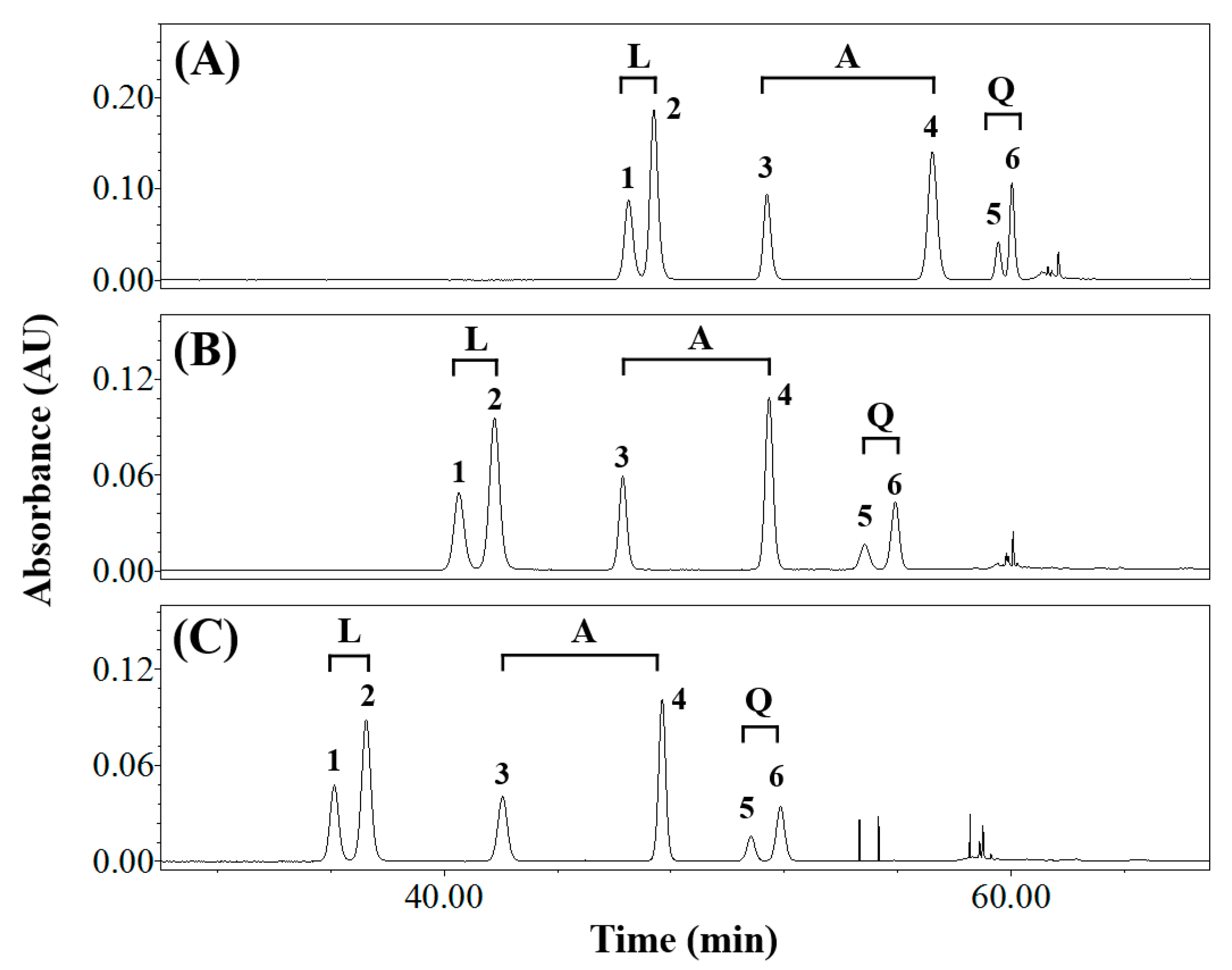
3.1. Effects of Elution Conditions on Isomer Separation
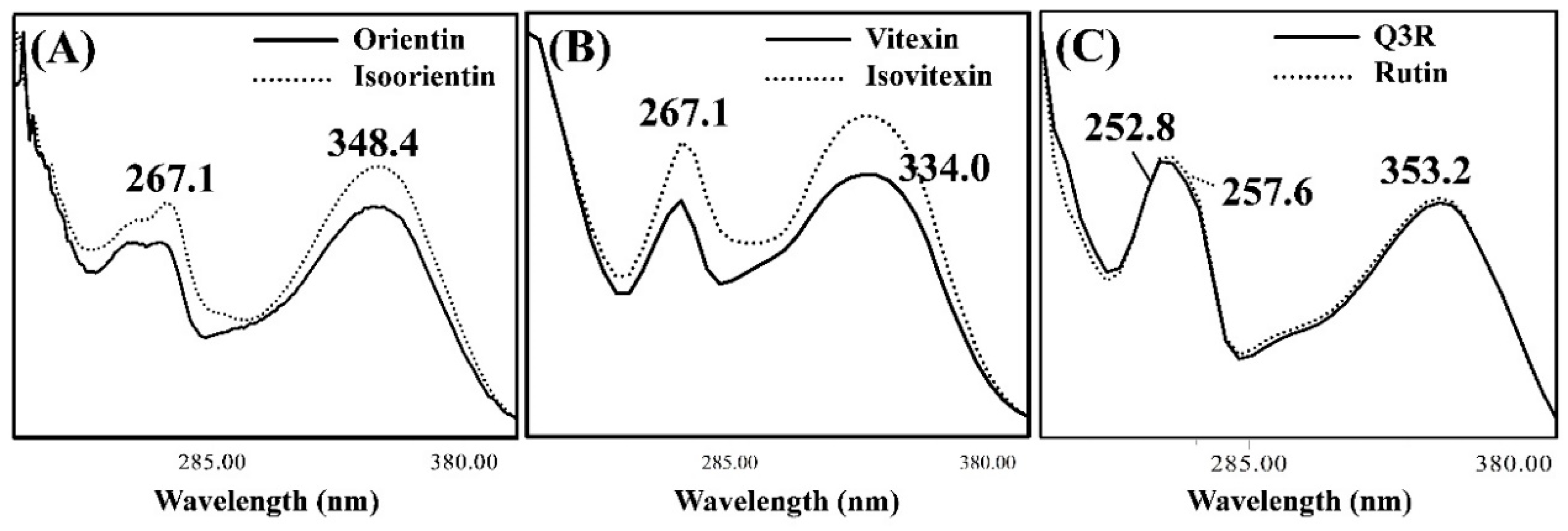
3.2. Isolating the Three Isomer Pairs from CS
3.3. Validating the Applied Method
3.3.1. Linearity and Range
3.3.2. Precision, Accuracy, and Recovery
3.3.3. Limits of Detection and Quantification
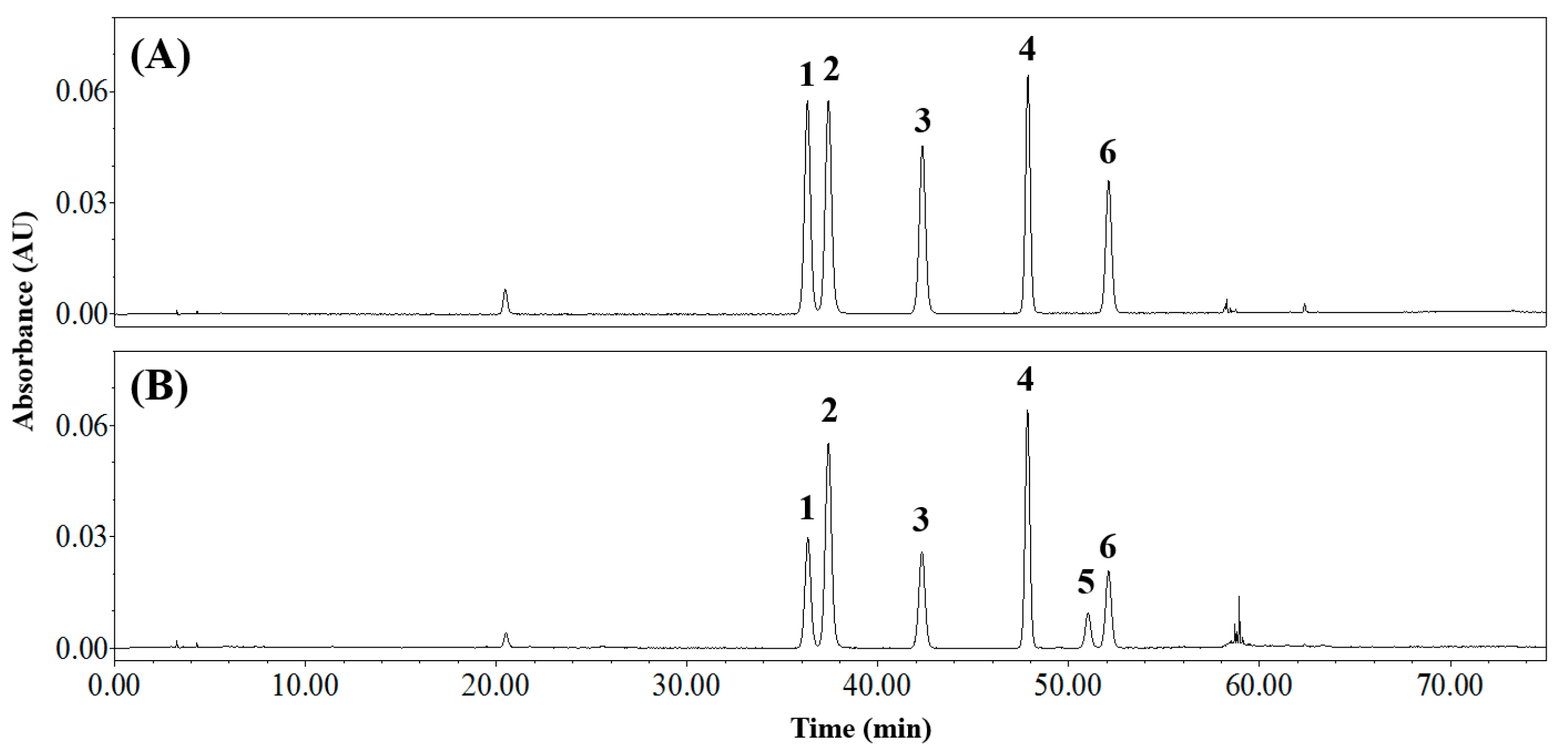
3.4. Applying the Analytical Method to CSE
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, Z.-L.; Zhou, M.-L.; Tang, Y.; Li, F.-L.; Tang, Y.-X.; Shao, J.-R.; Xue, W.-T.; Wu, Y.-M. Bioactive compounds in functional buckwheat food. Food Res. Int. 2012, 49, 389–395. [Google Scholar] [CrossRef]
- Giménez-Bastida, J.A.; Zieliński, H. Buckwheat as a functional food and its effects on health. J. Agric. Food Chem. 2015, 63, 7896–7913. [Google Scholar] [CrossRef] [PubMed]
- Nam, T.G.; Kim, D.-O.; Eom, S.H. Effects of light sources on major flavonoids and antioxidant activity in common buckwheat sprouts. Food Sci. Biotechnol. 2018, 27, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-L.; Kim, S.-K.; Park, C.-H. Introduction and nutritional evaluation of buckwheat sprouts as a new vegetable. Food Res. Int. 2004, 37, 319–327. [Google Scholar] [CrossRef]
- Park, C.H.; Yeo, H.J.; Park, Y.E.; Chun, S.W.; Chung, Y.S.; Lee, S.Y.; Park, S.U. Influence of chitosan, salicylic acid and jasmonic acid on phenylpropanoid accumulation in germinated buckwheat (Fagopyrum esculentum Moench). Foods 2019, 8, 153. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-Y.; Peng, C.-C.; Yang, Y.-L.; Peng, R.Y. Optimization of bioactive compounds in buckwheat sprouts and their effect on blood cholesterol in hamsters. J. Agric. Food Chem. 2008, 56, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Zaidul, I.S.M.; Suzuki, T.; Mukasa, Y.; Hashimoto, N.; Takigawa, S.; Noda, T.; Matsuura-Endo, C.; Yamauchi, H. Comparison of phenolic compositions between common and tartary buckwheat (Fagopyrum) sprouts. Food Chem. 2008, 110, 814–820. [Google Scholar] [CrossRef]
- Nam, T.-G.; Lee, S.M.; Park, J.-H.; Kim, D.-O.; Baek, N.-I.; Eom, S.H. Flavonoid analysis of buckwheat sprouts. Food Chem. 2015, 170, 97–101. [Google Scholar] [CrossRef]
- Liu, C.-L.; Chen, Y.-S.; Yang, J.-H.; Chiang, B.-H. Antioxidant activity of tartary (Fagopyrum tataricum (L.) Gaertn.) and common (Fagopyrum esculentum Moench) buckwheat sprouts. J. Agric. Food Chem. 2008, 56, 173–178. [Google Scholar] [CrossRef]
- Ishii, S.; Katsumura, T.; Shiozuka, C.; Ooyauchi, K.; Kawasaki, K.; Takigawa, S.; Fukushima, T.; Tokuji, Y.; Kinoshita, M.; Ohnishi, M.; et al. Anti-inflammatory effect of buckwheat sprouts in lipopolysaccharide-activated human colon cancer cells and mice. Biosci. Biotechnol. Biochem. 2008, 72, 3148–3157. [Google Scholar] [CrossRef]
- Nam, T.G.; Lim, T.-G.; Lee, B.H.; Lim, S.; Kang, H.; Eom, S.H.; Yoo, M.; Jang, H.W.; Kim, D.-O. Comparison of anti-inflammatory effects of flavonoid-rich common and tartary buckwheat sprout extracts in lipopolysaccharide-stimulated RAW 264.7 and peritoneal macrophages. Oxid. Med. Cell. Longev. 2017, 2017, 9658030. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-K.; Park, S.-H.; Kim, S.-J. A time-course study of flavonoids in buckwheats (Fagopyrum species). Korean J. Agric. Sci. 2011, 38, 87–94. [Google Scholar] [CrossRef]
- Ozga, J.A.; Saeed, A.; Wismer, W.; Reinecke, D.M. Characterization of cyanidin-and quercetin-derived flavonoids and other phenolics in mature saskatoon fruits (Amelanchier alnifolia Nutt.). J. Agric. Food Chem. 2007, 55, 10414–10424. [Google Scholar] [CrossRef] [PubMed]
- Pawlowska, A.M.; Camangi, F.; Bader, A.; Braca, A. Flavonoids of Zizyphus jujuba L. and Zizyphus spina-christi (L.) Willd (Rhamnaceae) fruits. Food Chem. 2009, 112, 858–862. [Google Scholar] [CrossRef]
- Patel, O.; Muller, C.; Joubert, E.; Louw, J.; Rosenkranz, B.; Awortwe, C. Inhibitory interactions of Aspalathus linearis (rooibos) extracts and compounds, aspalathin and Z-2-(β-d-glucopyranosyloxy)-3-phenylpropenoic acid, on cytochromes metabolizing hypoglycemic and hypolipidemic drugs. Molecules 2016, 21, 1515. [Google Scholar] [CrossRef]
- Pereira, C.A.M.; Yariwake, J.H.; McCullagh, M. Distinction of the C-glycosylflavone isomer pairs orientin/isoorientin and vitexin/isovitexin using HPLC-MS exact mass measurement and in-source CID. Phytochem. Anal. 2005, 16, 295–301. [Google Scholar] [CrossRef]
- Willems, J.L.; Khamis, M.M.; Saeid, W.M.; Purves, R.W.; Katselis, G.; Low, N.H.; El-Aneed, A. Analysis of a series of chlorogenic acid isomers using differential ion mobility and tandem mass spectrometry. Anal. Chim. Acta 2016, 933, 164–174. [Google Scholar] [CrossRef]
- Geng, P.; Sun, J.; Zhang, M.; Li, X.; Harnly, J.M.; Chen, P. Comprehensive characterization of C-glycosyl flavones in wheat (Triticum aestivum L.) germ using UPLC-PDA-ESI/HRMSn and mass defect filtering. J. Mass Spectrom. 2016, 51, 914–930. [Google Scholar] [CrossRef]
- Stalikas, C.D. Extraction, separation, and detection methods for phenolic acids and flavonoids. J. Sep. Sci. 2007, 30, 3268–3295. [Google Scholar] [CrossRef]
- Yáñez, J.A.; Andrews, P.K.; Davies, N.M. Methods of analysis and separation of chiral flavonoids. J. Chromatogr. B 2007, 848, 159–181. [Google Scholar] [CrossRef]
- Xu, F.; Liu, Y.; Zhang, Z.; Yang, C.; Tian, Y. Quasi-MSn identification of flavanone 7-glycoside isomers in Da Chengqi Tang by high performance liquid chromatography-tandem mass spectrometry. Chin. Med. 2009, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-H.; Ahn, J.-B.; Kim, H.-J.; Im, N.-K.; Kozukue, N.; Levin, C.E.; Friedman, M. Changes in free amino acid, protein, and flavonoid content in jujube (Ziziphus jujube) fruit during eight stages of growth and antioxidative and cancer cell inhibitory effects by extracts. J. Agric. Food Chem. 2012, 60, 10245–10255. [Google Scholar] [CrossRef] [PubMed]
- Ministry of Food and Drug Safety. Analytical Method Guideline about Validation of Drugs and etc.; Ministry of Food and Drug Safety: Cheongju, Korea, 2015; pp. 1–51. Available online: http://www.nifds.go.kr/brd/m_15/view.do?seq=10030&srchFr=&srchTo=&srchWord=&srchTp=&itm_seq_1=0&itm_seq_2=0&multi_itm_seq=0&company_cd=&company_nm=&page=45 (accessed on 7 May 2019).
- Xie, R.; Tu, M.; Wu, Y.; Adhikari, S. Improvement in HPLC separation of acetic acid and levulinic acid in the profiling of biomass hydrolysate. Bioresour. Technol. 2011, 102, 4938–4942. [Google Scholar] [CrossRef] [PubMed]
- Welch, C.J.; Brkovic, T.; Schafer, W.; Gong, X. Performance to burn? Re-evaluating the choice of acetonitrile as the platform solvent for analytical HPLC. Green Chem. 2009, 11, 1232–1238. [Google Scholar] [CrossRef]
- Giegold, S.; Teutenberg, T.; Tuerk, J.; Kiffmeyer, T.; Wenclawiak, B. Determination of sulfonamides and trimethoprim using high temperature HPLC with simultaneous temperature and solvent gradient. J. Sep. Sci. 2008, 31, 3497–3502. [Google Scholar] [CrossRef] [PubMed]
- Plumb, R.; Mazzeo, J.R.; Grumbach, E.S.; Rainville, P.; Jones, M.; Wheat, T.; Neue, U.D.; Smith, B.; Johnson, K.A. The application of small porous particles, high temperatures, and high pressures to generate very high resolution LC and LC/MS separations. J. Sep. Sci. 2007, 30, 1158–1166. [Google Scholar] [CrossRef]
- Praveena, R.; Sadasivam, K.; Deepha, V.; Sivakumar, R. Antioxidant potential of orientin: A combined experimental and DFT approach. J. Mol. Struct. 2014, 1061, 114–123. [Google Scholar] [CrossRef]
- Marston, A.; Hostettmann, K. Separation and quantification of flavonoids. In Flavonoids: Chemistry, Biochemistry, and Applications; Andersen, O.M., Markham, K.R., Eds.; CRC Press: Boca Raton, FL, USA, 2005; pp. 1–36. [Google Scholar]
- Naseem, B.; Sabri, A.; Hasan, A.; Shah, S.S. Interaction of flavonoids within organized molecular assemblies of anionic surfactant. Colloids Surf. B Biointerface 2004, 35, 7–13. [Google Scholar] [CrossRef]
- Anouar, E.H.; Gierschner, J.; Duroux, J.-L.; Trouillas, P. UV/Visible spectra of natural polyphenols: A time-dependent density functional theory study. Food Chem. 2012, 131, 79–89. [Google Scholar] [CrossRef]
- Jovanov, P.; Guzsvány, V.; Lazić, S.; Franko, M.; Sakač, M.; Šarić, L.; Kos, J. Development of HPLC-DAD method for determination of neonicotinoids in honey. J. Food Compos. Anal. 2015, 40, 106–113. [Google Scholar] [CrossRef]
- Lu, Q.-Y.; Lee, R.-P.; Huang, J.; Yang, J.; Henning, S.M.; Hong, X.; Heber, D.; Li, Z. Quantification of bioactive constituents and antioxidant activity of Chinese yellow wine. J. Food Compos. Anal. 2015, 44, 86–92. [Google Scholar] [CrossRef]
- Coelho, E.M.; da Silva Padilha, C.V.; Miskinis, G.A.; de Sá, A.G.B.; Pereira, G.E.; de Azevêdo, L.C.; Lima, M.D.S. Simultaneous analysis of sugars and organic acids in wine and grape juices by HPLC: Method validation and characterization of products from northeast Brazil. J. Food Compos. Anal. 2018, 66, 160–167. [Google Scholar] [CrossRef]
- Calabrò, M.L.; Galtieri, V.; Cutroneo, P.; Tommasini, S.; Ficarra, P.; Ficarra, R. Study of the extraction procedure by experimental design and validation of a LC method for determination of flavonoids in Citrus bergamia juice. J. Pharm. Biomed. Anal. 2004, 35, 349–363. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition of Mobile Phase (Solvent A/Solvent B) | Resolution (Rs) | Symmetry Factor | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Luteolin Derivatives 1 | Apigenin Derivatives 2 | Quercetin Derivatives 3 | Orientin | Isoorientin | Vitexin | Isovitexin | Q3R 4 | Rutin | ||
| Water/Methanol | 1.23 ± 0.00 c,5 | 3.66 ± 0.03 c | 0.00 ± 0.00 c | n.d. 6 | n.d. | n.d. | n.d. | n.d. | n.d. | |
| Water/Acetonitrile | 2.53 ± 0.03 a | 10.17 ± 0.06 b | 2.09 ± 0.04 a | 1.17 ± 0.04 | 1.18 ± 0.05 | 0.96 ± 0.01 | 0.99 ± 0.01 | 1.12 ± 0.05 | 1.05 ± 0.02 | |
| Acidic water 7/Methanol | 1.19 ± 0.00 c | 3.71 ± 0.01 c | 0.00 ± 0.00 c | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | |
| Acidic water/Acetonitrile | 1.87 ± 0.00 b | 10.30 ± 0.03 a | 1.93 ± 0.02 b | 1.03 ± 0.02 | 1.03 ± 0.01 | 0.99 ± 0.02 | 0.99 ± 0.01 | 1.02 ± 0.02 | 1.00 ± 0.02 | |
| Flavonoid | Range (μg mL−1) | Calibration Curve | Correlation Coefficient (R) |
|---|---|---|---|
| Orientin | 6.25–200.00 | y = 13074.0x + 3111.6 | 0.9999 |
| Isoorientin | 6.25–200.00 | y = 13895.0x + 1876.0 | 0.9999 |
| Vitexin | 6.25–200.00 | y = 10604.0x + 4006.4 | 0.9999 |
| Isovitexin | 6.25–200.00 | y = 11450.0x + 6941.8 | 0.9999 |
| Rutin | 6.25–200.00 | y = 7751.5x + 4289.6 | 0.9999 |
| Flavonoid | tR 1 (min) | RSD 2 (%) of tR | LOD 3 (μg mL−1) | LOQ 4 (μg mL−1) | Spiking Level (μg mL−1) | Accuracy (%) | Precision (RSD; %) | ||
|---|---|---|---|---|---|---|---|---|---|
| Recovery (n = 3) | Confidence Interval (95%) | Intra-Day Repeatability (n = 3) | Inter-Day Reproducibility (n = 3 × 3 Days) | ||||||
| Orientin | 36.4 | 0.13 | 0.32 | 0.96 | 25 | 97.10 | 96.91–103.59 | 3.74 | 4.04 |
| 50 | 102.96 | 3.78 | 3.13 | ||||||
| 100 | 100.69 | 2.80 | 4.30 | ||||||
| Isoorientin | 37.5 | 0.14 | 0.09 | 0.26 | 25 | 97.05 | 96.87–103.54 | 3.33 | 3.56 |
| 50 | 102.88 | 4.18 | 2.85 | ||||||
| 100 | 100.69 | 2.67 | 4.35 | ||||||
| Vitexin | 42.4 | 0.12 | 0.26 | 0.77 | 25 | 96.67 | 96.48–103.83 | 3.84 | 4.55 |
| 50 | 103.09 | 3.69 | 3.46 | ||||||
| 100 | 100.70 | 2.72 | 4.22 | ||||||
| Isovitexin | 47.9 | 0.07 | 0.29 | 0.88 | 25 | 97.12 | 96.84–104.20 | 3.61 | 4.13 |
| 50 | 103.60 | 3.76 | 3.44 | ||||||
| 100 | 100.83 | 2.69 | 4.24 | ||||||
| Rutin | 52.1 | 0.10 | 0.42 | 1.28 | 25 | 97.25 | 96.97–103.90 | 5.21 | 5.40 |
| 50 | 103.35 | 4.25 | 3.57 | ||||||
| 100 | 100.71 | 2.96 | 4.03 | ||||||
| Orientin | Isoorientin | Vitexin | Isovitexin | Q3R 1 | Rutin | ||
|---|---|---|---|---|---|---|---|
| tR 2 (min) | 36.4 | 37.5 | 42.4 | 47.9 | 51.1 | 52.1 | |
| Content (mg g−1 DW 4) | No. 1 | 6.63 ± 0.17 d,3 | 12.63 ± 0.21 a | 5.90 ± 0.11 e | 10.48 ± 0.18 b | 3.50 ± 0.06 f | 9.98 ± 0.21 c |
| No. 2 | 6.99 ± 0.04 d | 13.37 ± 0.20 a | 6.69 ± 0.32 d | 11.62 ± 0.25 b | 4.12 ± 0.07 e | 10.53 ± 0.23 c | |
| No. 3 | 7.02 ± 0.08 d | 13.48 ± 0.29 a | 6.89 ± 0.23 d | 12.14 ± 0.29 b | 3.69 ± 0.04 e | 10.74 ± 0.44 c | |
| No. 4 | 7.64 ± 0.11 e | 14.11 ± 0.27 b | 9.50 ± 0.06 c | 16.59 ± 0.22 a | 3.42 ± 0.07 f | 8.60 ± 0.08 d | |
| No. 5 | 8.16 ± 0.44 c | 15.24 ± 0.71 a | 7.93 ± 0.24 c | 13.83 ± 0.68 a | 4.56 ± 0.64 d | 12.18 ± 0.53 b | |
| No. 6 | 8.90 ± 0.22 c | 16.67 ± 0.33 a | 9.84 ± 0.25 b | 17.09 ± 0.50 a | 3.65 ± 0.03 d | 9.22 ± 0.11 bc | |
| No. 7 | 7.33 ± 0.20 c | 14.00 ± 0.62 a | 7.29 ± 0.26 c | 12.92 ± 0.44 a | 4.45 ± 0.25 d | 11.74 ± 0.44 b | |
| No. 8 | 6.99 ± 0.77 b | 13.27 ± 1.28 a | 7.26 ± 0.72 b | 13.34 ± 1.02 a | 3.90 ± 0.46 c | 10.70 ± 1.05 b | |
| No. 9 | 8.48 ± 0.26 d | 15.83 ± 0.46 b | 9.65 ± 0.32 c | 16.80 ± 0.50 a | 4.21 ± 0.13 e | 9.84 ± 0.28 c |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, D.; Jung, Y.S.; Kim, M.-S.; Oh, S.E.; Nam, T.G.; Kim, D.-O. Developing and Validating a Method for Separating Flavonoid Isomers in Common Buckwheat Sprouts Using HPLC-PDA. Foods 2019, 8, 549. https://doi.org/10.3390/foods8110549
Jang D, Jung YS, Kim M-S, Oh SE, Nam TG, Kim D-O. Developing and Validating a Method for Separating Flavonoid Isomers in Common Buckwheat Sprouts Using HPLC-PDA. Foods. 2019; 8(11):549. https://doi.org/10.3390/foods8110549
Chicago/Turabian StyleJang, Davin, Young Sung Jung, Mi-Seon Kim, Seung Eel Oh, Tae Gyu Nam, and Dae-Ok Kim. 2019. "Developing and Validating a Method for Separating Flavonoid Isomers in Common Buckwheat Sprouts Using HPLC-PDA" Foods 8, no. 11: 549. https://doi.org/10.3390/foods8110549
APA StyleJang, D., Jung, Y. S., Kim, M.-S., Oh, S. E., Nam, T. G., & Kim, D.-O. (2019). Developing and Validating a Method for Separating Flavonoid Isomers in Common Buckwheat Sprouts Using HPLC-PDA. Foods, 8(11), 549. https://doi.org/10.3390/foods8110549