Liquid Chromatography Analysis of Common Nutritional Components, in Feed and Food


Abstract
1. Introduction
2. Measurements of Commonly Consumed Food Commodities
2.1. Polyphenols
Method Application Experience
2.2. Capsaicinoids
2.2.1. Measurement of Capsaicin and Dehydrocapsaicin in Real Samples
2.2.2. Method Application Experience
2.3. Caffeine and Theobromine
2.3.1. Alkaloid Analysis and Reported Application to Real Samples
2.3.2. Alkaloid Bioavailability and Transference to Biological Samples
2.3.3. Method Application Experience
2.4. Cholesterol
Method Application Experience
3. Determinations Designed for Feed and Feed Ingredients
3.1. Mycotoxins
3.1.1. Recent Approaches for the Determination of Mycoxotins in Feeds
3.1.2. Agricultural by-Products as Feed Ingredients
3.2. Antibiotics
3.2.1. Recent Multiresidue and Multi-Class Analysis of Antibiotics in Feeds
3.2.2. Multiresidue Analysis of Antibiotics in Foods
3.2.3. Method Application Experience (Mycotoxins and Antibiotics)
3.3. Amino Acids
3.3.1. Fish Tissue
3.3.2. Filamentous Cyanobacteria, Spirulina sp.
3.3.3. Compound Feedstuff
3.3.4. Bacterial Cell Walls, Peptidoglycan, and Food-Extracted Peptides
3.3.5. Method Application Experience
3.4. Triphenylmethane Dyes
4. The Common Ground among Measurements Performed in Food and Feed Laboratories
4.1. Nitrates and Nitrites
4.1.1. Ion Exchange Chromatography
4.1.2. Ion Pairing and Reverse Phase Chromatography
4.1.3. Miscellaneous Methods for Nitrates and Nitrites
4.1.4. Method Application Experience
4.1.5. Legislation
4.2. Carotenoids
Method Application Experience
4.3. Carbohydrates and Sugars Soluble in Ethanol
4.3.1. Carbohydrate Measurement Using Amino-Based Columns
4.3.2. Carbohydrate Measurement Using Amide-Based Columns
4.3.3. Carbohydrate Measurement Using Ligand Exchange-Based Columns
4.3.4. Reverse Phase Columns and Sugar Derivatization Techniques
4.3.5. Aqueous Normal Phase Chromatography for Sugars
4.3.6. Complex Carbohydrates and Conjugates
4.3.7. Method Application Experience
4.4. Organic Acids
4.4.1. Reverse Phase Chromatography Analysis in Foods
4.4.2. Ion Exchange Chromatography Analysis in Foods
4.4.3. Ion Exclusion Chromatography Analysis in Foods
4.4.4. Silages
4.4.5. Method Application Experience
4.5. Vitamins
4.5.1. Fat-Soluble Vitamins
Sample Preparation
Chromatographic Analysis
4.5.2. Hydrosoluble Vitamins
4.5.3. Method Application Experience
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ercsey-Ravasz, M.; Toroczkai, Z.; Lakner, Z.; Baranyu, J. Complexity of the international agro-food trade network and its impact on food safety. PLoS ONE 2012, 7, e37810. [Google Scholar] [CrossRef]
- Fair, K.R.; Bauch, C.T.; Anand, M. Dynamics of the Global Wheat Trade Network and Resilience to Shocks. Sci. Rep. 2017, 7, 7177. [Google Scholar] [CrossRef] [PubMed]
- Keding, G.B.; Schneider, K.; Jordan, I. Production and processing of foods as core aspects of nutrition-sensitive agriculture and sustainable diets. Food Secur. 2013, 5, 825–846. [Google Scholar] [CrossRef]
- Canady, R.; Lane, R.; Paoli, G.; Wilson, M.; Bialk, H.; Hermansky, S.; Kobielush, B.; Li, J.-E.; Llewellyn, C.; Scimeca, J. Determining the applicability of threshold of toxicological concern approaches to substances found in foods. Crit. Rev. Food Sci. Nutr. 2013, 53, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Van der Fels-Klerx, H.J.; Adamse, P.; de Jong, J.; Hoogenboom, R.; de Nijs, M.; Bikker, P. A model for risk-based monitoring of contaminants in feed ingredients. Food Control 2017, 72, 211–218. [Google Scholar] [CrossRef]
- Allard, D.G. The ‘farm to plate’ approach to food safety—Everyone’s business. Can. J. Infect. Dis. 2002, 13, 185–190. [Google Scholar] [CrossRef]
- Kniel, K.E.; Kumar, D.; Thakur, S. Understanding the complexities of food safety using a “One Health” approach. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef]
- Krska, R.; de Nijs, M.; McNerney, O.; Pichler, M.; Gilbert, J.; Edwards, S.; Suman, M.; Magan, N.; Rossi, V.; van der Fels-Klerx, H.J.; et al. Safe food and feed through an integrated toolbox for mycotoxin management: The MyToolBox approach. World Mycotoxin J. 2016, 9, 487–495. [Google Scholar] [CrossRef]
- Wartella, W.A.; Lichtenstein, A.H.; Boon, C.S. Front-of-Package Nutrition Rating Systems and Symbols; Institute of Medicine of the National Academies Press: Washington, DC, USA, 2010; ISBN 978-0-309-15827-5. [Google Scholar]
- Association of American Feed Control Officials (AAFCO). Pet feed regulation. In AAFCO Official Publication; AAFCO: Atlanta, GA, USA, 2018. [Google Scholar]
- Yashin, Y.I.; Yashin, A.Y. Analysis of food products and beverages using high-performance liquid chromatography and ion chromatography with electrochemical detectors. J. Anal. Chem. 2004, 59, 1237–1243. [Google Scholar] [CrossRef]
- Nollet, L.M.L. Liquid chromatography in food analysis. In Encyclopedia of Analytical Chemistry; Meyers, R.A., McGorrin, R.J., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 2006. [Google Scholar] [CrossRef]
- Quitmann, H.; Fan, R.; Czermak, P. Acidic organic compounds in beverage, food, and feed production. In Biotechnology of Food and Feed Additives; Zorn, H., Czermak, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; ISBN 978-3-662-43760-5. [Google Scholar]
- Jiang, J.; Xiong, Y.L. Natural antioxidants as food and feed additives to promote health benefits and quality of meat products: A review. Meat Sci. 2016, 120, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Candan, T.; Bağdatl, A. Use of natural antioxidants in poultry meat. CBU J. Sci. 2017, 13, 279–291. [Google Scholar]
- Chanadang, S.; Koppel, K.; Aldrich, G. The impact of rendered protein meal oxidation level on shelf-life, sensory characteristics, and acceptability in extruded pet food. Animals 2016, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Sanches-Silva, A.; Costa, D.; Alburquerque, T.G.; Buonocore, G.G.; Ramos, F.; Castilho, M.C.; Machado, A.V.; Costa, H.S. Trends in the use of natural antioxidants in active food packaging: A review. Food Addit. Contam. Part A 2014, 31, 374–395. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.C.; Doyle, M.P. The challenges of eliminating or substituting antimicrobial preservatives in foods. Annu. Rev. Food Sci. Technol. 2017, 8, 371–390. [Google Scholar] [CrossRef]
- Gupta, C.; Prakash, D.; Gupta, S. A biotechnological approach to microbial based perfumes and flavours. J. Microbiol. Exp. 2015, 2. [Google Scholar] [CrossRef]
- Das, A.; Chakraborty, R. An introduction to sweeteners. In Sweeteners, Reference Series in Phytochemistry; Mérillon, J.-M., Ramawat, K.G., Eds.; Springer International Publishing: Basel, Switzerland, 2018; ISBN 978-3-319-27026-5. [Google Scholar]
- Mooradian, A.D.; Smith, M.; Tokuda, M. The role of artificial and natural sweeteners in reducing the consumption of table sugar: A narrative review. Clin. Nutr. ESPEN 2017, 18, 1–8. [Google Scholar] [CrossRef]
- Durán, S.; Dávila, L.A.; Contreras, M.C.E.; Rojas, D.; Costa, J. Noncaloric sweeteners in children: A controversial theme. BioMed Res. Int. 2018, 2018, 4806534. [Google Scholar]
- Subedi, B.; Kannan, K. Fate of Artificial Sweeteners in Wastewater Treatment Plants in New York State, USA. Environ. Sci. Technol. 2014, 48, 13668–13674. [Google Scholar] [CrossRef]
- Khan, S.A. Artificial sweeteners: Safe or unsafe? J. Pak. Med. Assoc. 2015, 25, 225–227. [Google Scholar]
- García-Almeida, J.M.; Conejo-Pareja, I.M.; Muñoz-Garach, A.; Gómez-Pérez, A.; García-Alemán, J. Sweeteners: Regulatory Aspects. In Sweeteners, Reference Series in Phytochemistry; Mérillon, J.-M., Ramawat, K.G., Eds.; Springer International Publishing: Basel, Switzerland, 2016; ISBN 978-3-319-27026-5. [Google Scholar]
- Shah, R.; de Jager, L.S. Recent analytical methods for the analysis of sweeteners in food: A regulatory perspective. Food Drug Adm. Papers 2017, 5, 13–31. [Google Scholar]
- Sharma, A.; Amarnath, S.; Thulasimani, M.; Ramaswamy, S. Artificial sweeteners as a sugar substitute: Are they really safe? Indian J. Pharmacol. 2016, 48, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Vanĕk, T.; Nepovím, A.; Valíček, P. Determination of stevioside in plant material and fruit teas. J. Food Compos. Anal. 2001, 14, 383–388. [Google Scholar] [CrossRef]
- Chaturvedula, V.S.P.; Zamora, J. Reversed-Phase HPLC analysis of steviol glycosides isolated from Stevia rebaudiana Bertoni. Food Nutr. Sci. 2014, 5, 1711–1716. [Google Scholar] [CrossRef]
- Wang, T.-H.; Avula, B.; Tang, W.; Wang, M.; Elsohly, M.A.; Khan, I.A. Ultra-HPLC method for quality and adulterant assessment of steviol glycosides sweeteners—Stevia rebaudiana and stevia products. Food Addit. Contam. Part A 2015, 32, 674–685. [Google Scholar]
- Martono, Y.; Riyanto, S.; Rohman, A.; Martono, S. Improvement method of fast and isocratic RP-HPLC analysis of major diterpene glycoside from Stevia rebaudiana leaves. AIP Conf. Proc. 2016, 1755, 080001–080008. [Google Scholar]
- Sádecka, J.; Polonský, J. Determination of inorganic ions in food and beverages by capillary electrophoresis. J. Chromatogr. 1999, 834, 401–417. [Google Scholar] [CrossRef]
- Rios, R.V.; Pessanha, M.D.F.; de Almeida, P.F.; Viana, C.L.; Lannes, S.C. Application of fats in some food products. Food Sci. Technol. 2014, 34, 3–15. [Google Scholar] [CrossRef]
- Li, H.; Zhou, X.; Gao, P.; Li, Q.; Li, H.; Huang, R.; Wu, M. Inhibition of lipid oxidation in foods and feeds and hydroxyl radical treated fish erythrocytes: A comparative study of Ginkgo biloba leaves extracts and synthetic antioxidants. Anim. Nutr. 2016, 2, 234–241. [Google Scholar] [CrossRef]
- Kerr, B.J.; Kellner, T.A.; Shurson, G.C. Characteristics of lipids and their feeding value in swine diets. J. Anim. Sci. Biotechnol. 2015, 6, 30. [Google Scholar] [CrossRef]
- Shurson, G.C.; Kerr, B.J.; Hanson, A.R. Evaluating the quality of feed fats and oils and their effects on pig growth performance. J. Anim. Sci. Biotechnol. 2015, 6, 10. [Google Scholar] [CrossRef]
- Pickova, J. Importance of knowledge on lipid composition of foods to support development towards consumption of higher levels of n-3 fatty acids via freshwater fish. Psychol. Res. 2009, 58, S39–S45. [Google Scholar]
- Benkerroum, N. Biogenic amines in dairy products: Origin, incidence, and control means. Compr. Rev. Food Sci. Food Saf. 2016, 15, 801–826. [Google Scholar] [CrossRef]
- Spano, G.; Russo, P.; Lonvaud-Funel, A.; Lucas, P.; Alexandre, H.; Grandvalet, C.; Coton, E.; Coton, M.; Barnavon, L.; Bach, B.; et al. Biogenic amines in fermented foods. Eur. J. Clin. Nutr. 2010, 64, S95–S100. [Google Scholar] [CrossRef] [PubMed]
- Landete, J.M.; Ferrer, S.; Polo, L.; Pardo, I. Biogenic amines in wines from three Spanish regions. J. Agric. Food Chem. 2005, 53, 1119–1124. [Google Scholar] [CrossRef]
- Biji, K.B.; Ravishankar, C.N.; Venkateswarlu, R.; Mohna, C.O.; Srinivasa, T.K. Biogenic amines in seafood: A review. J. Food Sci. Technol. 2016, 53, 2210–2218. [Google Scholar] [CrossRef] [PubMed]
- Karau, A.; Grayson, I. Amino acids in human and animal nutrition. Adv. Biochem. Eng. Biotechnol. 2014, 143, 189–228. [Google Scholar] [PubMed]
- Hardy, K.; Brand-Miller, J.; Brown, K.D.; Thomas, M.G.; Copeland, L. The importance of dietary carbohydrate in human evolution. Q. Rev. Biol. 2015, 90, 251–268. [Google Scholar] [CrossRef] [PubMed]
- Bach Knudsen, K.E.; Lærke, H.N.; Ingerslev, A.K.; Hedemann, M.S.; Nielsen, T.S.; Theil, P.K. Carbohydrates in pig nutrition—Recent advances. J. Anim. Sci. 2016, 94, 1–11. [Google Scholar] [CrossRef]
- McDowell, L.R. Vitamins in Animal and Human Nutrition; Iowa State University Press: Ames, IA, USA, 2000; pp. 15–217. ISBN 0-8138-2630-6. [Google Scholar]
- Koleva, I.I.; van Beek, T.A.; Soffers, A.E.M.F.; Dusemund, B.; Rietjens, I.M.C.M. Alkaloids in the human food chain—Natural occurrence and possible adverse effects. Mol. Nutr. Food Res. 2012, 56, 30–52. [Google Scholar] [CrossRef]
- Di Lorenzo, C.; Dos Santos, A.; Colombo, F.; Moro, E.; Dell’Agli, M.; Restani, P. Development and validation of HPLC method to measure active amines in plant food supplements containing Citrus aurantium L. Food Control 2014, 46, 136–142. [Google Scholar] [CrossRef]
- Endlová, L.; Laryšová, A.; Vrbovsky, V.; Navrátilová, Z. Analysis of alkaloids in poppy straw by high-performance liquid chromatography. IOSR J. Eng. 2015, 3, 1–7. [Google Scholar]
- Arai, K.; Terashima, H.; Aizaewa, S.; Taga, A.; Yamamoto, A.; Tsutsumiuchi, K.; Kodama, S. Simultaneous Determination of Trigonelline, Caffeine, Chlorogenic Acid and Their Related Compounds in Instant Coffee Samples by HPLC Using an Acidic Mobile Phase Containing Octanesulfonate. Anal. Sci. 2015, 31, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-B.; Qi, W.; Zhang, L.; Yuan, D. Qualitative and quantitative analyses of alkaloids in Uncaria species by UPLC-ESI-Q-TOF/MS. Chem. Pharm. Bull. 2014, 62, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Ramdani, D.; Chaudhry, A.S.; Seal, C.J. Alkaloid and polyphenol analysis by HPLC in green and black tea powders and their potential use as additives in ruminant diets. AIP Conf. Proc. 2018, 1927, 0300081–0300086. [Google Scholar]
- Yashin, A.; Yashin, Y.; Xia, X.; Nemzer, B. Chromatographic Methods for Coffee Analysis: A Review. Journal Food Res. 2017, 6, 60–82. [Google Scholar] [CrossRef]
- Pereira, C.A.M.; Rodrigues, T.R.; Yariwake, J.H. Quantification of Harman alkaloids in sour passion fruit pulp and seeds by a novel dual SBSE-LC/Flu (stir bar sorptive extraction-liquid chromatography with fluorescence detector) method. J. Braz. Chem. Soc. 2014, 25, 1472–1483. [Google Scholar] [CrossRef]
- Kowalczyk, E.; Patyra, E.; Grelik, A.; Kwiatek, K. Development and validation of an analytical method for determination of ergot alkaloids in animal feedingstuffs with high performance liquid chromatography-fluorescence detection. Pol. J. Vet. Sci. 2016, 19, 559–565. [Google Scholar] [CrossRef]
- Upadhyay, V.; Sharma, N.; Joshi, H.M.; Malik, A.; Mishra, M.; Singh, B.P.; Tripathi, S. Development and validation of rapid RP-HPLC method for estimation of piperine in Piper nigrum L. Int. J. Herb. Med. 2013, 1, 6–9. [Google Scholar]
- Granados-Chinchilla, F.; Rodriguez, C. Tetracyclines in food and feedingstuffs: From regulation to analytical methods, bacterial resistance, and environmental and health implications. J. Anal. Methods Chem. 2017, 2017, 1315497. [Google Scholar] [CrossRef]
- Alshannaq, A.; Yu, J.-H. Occurrence, toxicity, and analysis of major mycotoxins in food. Int. J. Environ. Res. Public Health 2017, 14, 632. [Google Scholar] [CrossRef]
- Granados-Chinchilla, F.; Molina, A.; Chavarría, G.; Alfaro-Cascante, M.; Bogantes-Ledezma, D.; Murillo-Williams, A. Aflatoxins occurrence through the food chain in Costa Rica: Applying the One Health approach to mycotoxin surveillance. Food Control 2017, 82, 217–226. [Google Scholar] [CrossRef]
- Oliveira, F.A.; Pereira, E.N.C.; Gobbi, J.M.; Soto-Blanco, B.; Melo, M.M. Multiresidue method for detection of pesticides in beef meat using liquid chromatography coupled to mass spectrometry detection (LC-MS) after QuEChERS extraction. Food Addit. Contam. Part A 2018, 35, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, K.; Schmitz, R.; von Soosten, D.; Frahm, J.; Kersten, S.; Meyer, U.; Breves, G.; Hackenberg, R.; Spitzke, M.; Dänicke, S. Effects of glyphosate residues and different concentrate feed proportions on performance, energy metabolism and health characteristics in lactating dairy cows. Arch. Anim. Nutr. 2017, 71, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Velkoska-Markovska, L.; Petanovska-Ilievska, B.; Markovski, A. Application of high performance liquid chromatography to the analysis of pesticide residues in apple juice. Contemp. Agric. 2018, 67, 93–102. [Google Scholar] [CrossRef]
- Recjczak, T.; Tuzimski, T. Application of high-performance liquid chromatography with diode array detector for simultaneous determination of 11 synthetic dyes in selected beverages and foodstuffs. Food Anal. Methods 2017, 10, 3572–3588. [Google Scholar] [CrossRef]
- Shehata, A.B.; Rizk, M.S.; Rend, E.A. Certification of caffeine reference material purity by ultraviolet/visible spectrophotometry and high-performance liquid chromatography with diode-array detection as two independent analytical methods. J. Food Drug Anal. 2016, 24, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Cancho Grande, B.; Falcón, M.S.G.; Comesaña, M.R.; Gándara, J.S. Determination of sulfamethazine and trimethoprim in liquid feed premixes by HPLC and diode array detection, with an analysis of the uncertainty of the analytical results. J. Agric. Food Chem. 2001, 49, 3145–3150. [Google Scholar] [CrossRef]
- Barbosa, J.; Moura, S.; Barbosa, R.; Ramos, F.; Silveira, M.I. Determination of nitrofurans in animal feeds by liquid chromatography-UV photodiode array detection and liquid chromatography-ionspray tandem mass spectrometry. Anal. Chim. Acta 2007, 586, 359–365. [Google Scholar] [CrossRef]
- Horigome, J.; Kozuma, M.; Shirasaki, T. Fluorescence pattern analysis to assist food safety––Food analysis technology driven by fluorescence fingerprints. Hitachi Rev. 2016, 65, 248–253. [Google Scholar]
- Borràs, S.; Companyó, R.; Guiteras, J. Analysis of sulfonamides in animal feeds by liquid chromatography with fluorescence detection. J. Agric. Food Chem. 2011, 59, 5240–5247. [Google Scholar] [CrossRef]
- Patyra, E.; Kwiatek, K. Determination of fluoroquinolones in animal feed by ion pair high-performance liquid chromatography with fluorescence detection. Anal. Lett. 2017, 50, 1711–1720. [Google Scholar] [CrossRef]
- Wacoo, A.P.; Wendiro, D.; Vuzi, P.C.; Hawumba, J.F. Methods for Detection of Aflatoxins in Agricultural Food Crops. J. Appl. Chem. 2014, 2014, 70629. [Google Scholar] [CrossRef]
- Shuib, N.S.; Makahleh, A.; Salhimi, S.M.; Saad, B. Determination of aflatoxin M1 in milk and dairy products using highperformance liquid chromatography-fluorescence with post column photochemical derivatization. J. Chromatogr. A 2017, 1510, 51–56. [Google Scholar] [CrossRef] [PubMed]
- González de la Huebra, M.J.; Vincent, U.; von Holst, C. Sample preparation strategy for the simultaneous determination of macrolide antibiotics in animal feedingstuffs by liquid chromatography with electrochemical detection (HPLC-ECD). J. Pharm. Biomed. Anal. 2007, 43, 1628–1637. [Google Scholar] [CrossRef] [PubMed]
- Gazdik, Z.; Ztka, O.; Petrlova, J.; Adam, V.; Zehnalek, J.; Horna, A.; Reznicek, V.; Beklova, M.; Kizek, R. Determination of vitamin C (ascorbic acid) using high performance liquid chromatography coupled with electrochemical detection. Sensors 2008, 8, 7097–7112. [Google Scholar] [CrossRef]
- Başaran, U.; Akkbik, M.; Mut, H.; Gülümser, E.; Doğrusöz, M.C.; Koçoğlu, S. High-Performance liquid chromatography with refractive index detection for the determination of inulin in chicory roots. Anal. Lett. 2018, 51, 83–95. [Google Scholar] [CrossRef]
- Kupina, S.; Roman, M. Determination of total carbohydrates in wine and wine-like beverages by HPLC with a refractive index detector: First action 2013.12. J. AOAC Int. 2014, 97, 498–505. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, Y. Determination of betaine, choline and trimethylamine in feed additive by ion-exchange liquid chromatography/non-suppressed conductivity detection. J. Chromatogr. A 2007, 1170, 114–117. [Google Scholar] [CrossRef]
- Wei, D.; Xang, X.; Wang, N.; Zhu, Y. A rapid ion chromatography column-switching method for online sample pretreatment and determination of L-carnitine, choline and mineral ions in milk and powdered infant formula. RSC Adv. 2017, 7, 5920–5927. [Google Scholar] [CrossRef]
- Troise, A.D.; Fiore, A.; Fogliano, V. Quantitation of acrylamide in foods by high-resolution mass spectrometry. J. Agric. Food Chem. 2014, 62, 74–79. [Google Scholar] [CrossRef]
- Patyra, E.; Nebot, C.; Gavilán, R.E.; Cepeda, A.; Kwiatek, K. Development and validation of an LC-MS/MS method for the quantification of tiamulin, trimethoprim, tylosin, sulfadiazine and sulfamethazine in medicated feed. Food Addit. Contam. Part A 2018, 35, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Lin, K.; Huang, X.; Chen, M. A simple and fast extraction method for the determination of multiclass antibiotics in eggs using LC-MS/MS. Journal of Agricultural and Food Chemistry. J. Agric. Food Chem. 2017, 65, 5064–5073. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Kersten, S.; Valenta, H.; Meyer, U.; Engelhardt, U.H.; Dänicke, S. Development of a multi-toxin method for investigating the carryover of zearalenone, deoxynivalenol and their metabolites into milk of dairy cows. Food Addit. Contam. Part A 2015, 32, 371–380. [Google Scholar]
- Botha, C.J.; Legg, M.J.; Truter, M.; Sulyok, M. Multitoxin analysis of Aspergillus clavatus-infected feed samples implicated in two outbreaks of neuromycotoxicosis in cattle in South Africa. Onderstepoort J. Vet. Res. 2014, 81, e1–e6. [Google Scholar] [CrossRef] [PubMed]
- Granados-Chinchilla, F.; Artavia, G. A straightforward LC approach using an amine column and single quad mass detector to determine choline chloride in feed additives and feeds. MethodsX 2017, 4, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Nassar, A.-E.F.; Bjorge, S.M. On-Line liquid chromatography-accurate radioisotope counting coupled with a radioactivity detector and mass spectrometer for metabolite identification in drug discovery and development. Anal. Chem. 2003, 75, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Bae, I.K.; Jeong, M.H.; Park, H.J.; Jung, J.S.; Kim, J.E. A new HPLC-ELSD method for simultaneous determination of N-acetylglucosamine and N-acetylgalactosamine in dairy foods. Int. J. Anal. Chem. 2015, 2015, 892486. [Google Scholar] [CrossRef]
- Yan, W.; Wang, N.; Zhang, P.; Zhang, J.; Wu, S.; Zhu, Y. Simultaneous determination of sucralose and related compounds by high-performance liquid chromatography with evaporative light scattering detection. Food Chem. 2016, 204, 358–364. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, M.J.; Li, J.; Yao, S.C.; Xue, J.; Zou, W.B.; Hu, C.Q. determination of spectinomycin and related substances by HPLC coupled with evaporative light scattering detection. Acta Chromatogr. 2015, 207, 93–109. [Google Scholar] [CrossRef]
- Ligor, M.; Studzińska, S.; Horna, A.; Buszewski, B. Corona-charged aerosol detection: An analytical approach. Crit. Rev. Anal. Chem. 2013, 43, 64–78. [Google Scholar] [CrossRef]
- Vehovec, T.; Obreza, A. Review of operating principle and applications of the charged aerosol detector. J. Chromatogr. A 2010, 1217, 1549–1556. [Google Scholar] [CrossRef]
- Grembecka, M.; Lebiedzińska, A.; Szefer, P. Simultaneous separation and determination of erythritol, xylitol, sorbitol, mannitol, maltitol, fructose, glucose, sucrose and maltose in food products by high performance liquid chromatography coupled to charged aerosol detector. Microchem. J. 2014, 117, 77–82. [Google Scholar] [CrossRef]
- Szekeres, A.; Budai, A.; Bencsik, O.; Németh, L.; Bartók, T.; Szécsi, Á.; Mesterházy, Á.; Vágvölgyi, C. Fumonisin measurement from maize samples by high-performance liquid chromatography coupled with corona charged aerosol detector. J. Chromatogr. Sci. 2014, 52, 1181–1185. [Google Scholar] [CrossRef]
- Enri, F.; Steuer, W.; Bosshart, H. Automation and validation of HPLC-Systems. Chromatographia 1987, 24, 201–207. [Google Scholar]
- Oláh, E.; Tarnai, M.; Fekete, J. Possibility of large volume injection and band focusing in UHPLC. J. Chromatogr. Sci. 2013, 51, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Williamson, G. The role of polyphenols in modern nutrition. Nutr. Bull. 2017, 42, 226–235. [Google Scholar] [CrossRef]
- Pandey, K.B.; Rizvi, S.I. Plant polyphenols as dietary antioxidants in human health and disease. Oxidaive Med. Cell. Longev. 2009, 2, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, M.; Franke, K.; Fischer, K. Feeding on ripening and over-ripening fruit: Interactions between sugar, ethanol and polyphenol contents in a tropical butterfly. J. Exp. Biol. 2017, 220, 3127–3134. [Google Scholar] [CrossRef]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin Biosynthesis and Structure. Plant Physiol. 2010, 153, 895–905. [Google Scholar] [CrossRef]
- Bensalem, J.; Dal-Pen, A.; Gillard, E.; Fréderic, C.; Pallet, V. Protective effects of berry polyphenols against age-related cognitive impairment. Nutr. Aging 2015, 3, 89–106. [Google Scholar] [CrossRef]
- Kowalska, K.; Olejnik, A.; Szwajgier, D.; Olkowicz, M. Inhibitory activity of chokeberry, bilberry, raspberry and cranberry polyphenol-rich extract towards adipogenesis and oxidative stress in differentiated 3T3-L1 adipose cells. PLoS ONE 2017, 12, e0188583. [Google Scholar] [CrossRef] [PubMed]
- Khalifa, I.; Zhu, W.; Li, K.-K.; Li, C.-M. Polyphenols of mulberry fruits as multifaceted compounds: Compositions, metabolism, health benefits, and stability—A structural review. J. Funct. Foods 2018, 40, 28–43. [Google Scholar] [CrossRef]
- Pérez-Jiménez, J.; Neveu, V.; Vos, F.; Sclbert, A. Identification of the 100 richest dietary sources of polyphenols: An application of the Phenol-Explorer database. Eur. J. Clin. Nutr. 2010, 64, S112–S120. [Google Scholar] [CrossRef] [PubMed]
- Acosta, O.; Vaillant, F.; Pérez, A.M.; Dornier, M. Concentration of polyphenolic compounds in blackberry (Rubus adenotrichos Schltdl) juice by nanofiltration. J. Food Process Eng. 2017, 40, e12343. [Google Scholar] [CrossRef]
- Haminiuk, C.W.; Maciel, G.M.; Plata-Oviedo, M.S.V.; Peralta, R.M. Phenolic compounds in fruits—An overview. Int. J. Food Sci. Technol. 2012, 47, 2023–2044. [Google Scholar] [CrossRef]
- Karasawa, M.M.G.; Mohan, C. Fruits as Prospective Reserves of bioactive Compounds: A Review. Nat. Prod. Bioprospect. 2018, 8, 335–346. [Google Scholar] [CrossRef]
- Reynoso-Camacho, R.; Rufino, M.S.M.; Amaya-Cruz, D.M.; Pérez, A.M. Non-extractable polyphenols in tropical fruits: Occurrence and health-related properties. In Non-extractable Polyphenols and Carotenoids: Importance in Human Nutrition and Health; Saura-Calixto, F., Pérez-Jiménez, J., Eds.; Royal Society of Chemistry: Cambridge, UK, 2018; ISBN 978-1-78801-106-8. [Google Scholar]
- Abbas, M.; Saeed, F.; Anjum, F.M.; Afzaal, M.; Tufail, T.; Bashir, M.S.; Ishtiaq, A.; Hussain, S.; Suleria, H.A. Natural polyphenols: An overview. Int. J. Food Prop. 2016, 20, 1689–1699. [Google Scholar] [CrossRef]
- Kaşikci, M.B.; Bağdatlioğlu, N. High hydrostatic pressure treatment of fruit, fruit products and fruit juices: A review on phenolic compounds. J. Food Health Sci. 2016, 2, 27–39. [Google Scholar]
- Miletić, N.; Mitrović, O.; Popović, B.; Nedović, V.; Zlatković, B.; Kandić, M. Polyphenolic content and antioxidant capacity in fruits of plum (Prunus domestica L.) cultivars “Valjevka” and “Mildora” as influenced by air drying. J. Food Qual. 2013, 36, 229–237. [Google Scholar] [CrossRef]
- McSweeny, M.; Seetharaman, K. State of polyphenols in the drying process of fruits and vegetables. Crit. Rev. Food Sci. Nutr. 2015, 55, 660–669. [Google Scholar] [CrossRef]
- Koffi, E.; Sea, T.; Dodehe, Y.; Soro, S. Effect of solvent type on extraction of polyphenols from twenty three Ivorian plants. J. Anim. Plant Sci. 2010, 5, 550–558. [Google Scholar]
- Flores, G.; Dastmalchi, K.; Wu, S.-B.; Whalen, K.; Dabo, A.J.; Reynertson, K.A.; Foronjy, R.F.; D’Armiento, J.M.; Kennelly, E.J. Phenolic-rich extract from the Costa Rican Guava (Psidium friedrichsthalianum) pulp with antioxidant and anti-inflammatory activity. Potential for COPD therapy. Food Chem. 2013, 141, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Tresserra-Rimbau, E.; Arranz, S.; Vallverdu-Queralt, A. New Insights into the Benefits of Polyphenols in Chronic Diseases. Oxidative Med. Cell. Longev. 2017, 2017, 1432071. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Dwyer, J. Exploring Possible Health Effects of Polyphenols in Foods. Nutr. Today 2017, 52, 62–72. [Google Scholar] [CrossRef]
- Gordon, A.; Jungfer, E.; da Silva, B.A.; Maia, J.G.S.; Marx, F. Phenolic constituents and antioxidant capacity of four underutilized fruits from the Amazon region. J. Agric. Food Chem. 2011, 59, 7688–7699. [Google Scholar] [CrossRef] [PubMed]
- Assefa, A.D.; Jeong, Y.-J.; Kim, D.-J.; Jeon, Y.-A.; Ok, H.-C.; Baek, H.-J.; Sung, J.-S. Characterization, identification, and quantification of phenolic compounds using UPLC-Q-TOF-MS and evaluation of antioxidant activity of 73 Perilla frutescens accessions. Food Res. Int. 2018, 111, 153–167. [Google Scholar] [CrossRef]
- Anton, D.; Bender, I.; Kaart, T.; Roasto, M.; Heinonen, M. Changes in polyphenols contents and antioxidant capacities of organically and conventionally cultivated tomato (Solanum lycopersicum L.) fruits during ripening. Int. J. Anal. Chem. 2017, 2017, 2367453. [Google Scholar] [CrossRef]
- Radovanović, B.C.; Anđelković, A.S.M.; Radovanović, A.B.; Anđelković, M.Z. Antioxidant and antimicrobial activity of polyphenol extracts from wild berry fruits grown in southeast Serbia. Trop. J. Pharm. Res. 2013, 12, 813–819. [Google Scholar] [CrossRef]
- Veljković, J.N.; Pavlović, A.N.; Mitić, S.S.; Tošić, S.B.; Stojanović, G.S.; Kaličanin, B.M.; Stanović, D.M.; Stojković, M.B.; Mitić, M.N.; Brcanović, J.M. Evaluation of individual phenolic compounds and antioxidant properties of black, green, herbal and fruit tea infusions consumed in Serbia: Spectrophotometrical and electrochemical approaches. J. Food Nutr. Res. 2013, 52, 12–24. [Google Scholar]
- Miletić, N.; Popović, B.; Mitrović, O.; Kandić, M.; Leposavić, A. Phenolic compounds and antioxidant capacity of dried and candied fruits commonly consumed in Serbia. Czech J. Food Sci. 2014, 32, 360–368. [Google Scholar] [CrossRef]
- Rojas-Garbanzo, C.; Zimmermann, B.F.; Schulze-Kaysers, N.; Schieber, A. Characterization of phenolic and other polar compounds in peel and flesh of pink guava (Psidium guajava L. cv. ‘Criolla’) by ultra-high performance liquid chromatography with diode array and mass spectrometric detection. Food Res. Int. 2017, 100, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Azofeifa, G.; Quesada, S.; Pérez, A.M.; Vaillant, F.; Michel, A. Effect of an in vitro digestion on the antioxidant capacity of a microfiltrated blackberry juice (Rubus adenotrichos). Beverages 2018, 4, 30. [Google Scholar] [CrossRef]
- Georgé, S.; Brat, P.; Alter, P.; Amiot, M.J. Rapid determination of polyphenols and vitamin C in plant derived products. J. Agric. Food Chem. 2005, 53, 1370–1373. [Google Scholar] [CrossRef] [PubMed]
- Gouvêa, A.; de Araujo, M.C.; Schulz, D.F.; Pacheco, S.; Godoy, R.; Cabral, L. Anthocyanins standards (cyanidin-3-O-glucoside and cyanidin-3-O-rutinoside) isolation from freeze-dried açaí (Euterpe oleraceae Mart.) by HPLC. Ciênc. Tecnol. Aliment. 2012, 32, 43–46. [Google Scholar] [CrossRef]
- Teboukeu, G.B.; Djikeng, F.T.; Klang, M.J.; Ndomou, S.H.; Karuna, M.S.L.; Womeni, H.M. Polyphenol antioxidants from cocoa pods: Extraction optimization, effect of the optimized extract, and storage time on the stability on palm olein during thermoxidation. J. Food Process. Preserv. 2018, 42, e13592. [Google Scholar] [CrossRef]
- Ryu, W.-K.; Kim, H.-W.; Kim, G.-D.; Rhee, H.-I. Rapid determination of capsaicinoids by colorimetric method. J. Food Drug Anal. 2017, 25, 798–803. [Google Scholar] [CrossRef]
- Collins, M.D.; Wasmund, L.M.; Bosland, P.W. Improved method for quantifying capsaicinoids in Capsicum using high-performance liquid chromatography. Hortic. Sci. 1995, 30, 137–139. [Google Scholar]
- Guzmán, I.; Bosland, P.W. Sensory properties of chile pepper heat e and its importance to food quality and cultural preference. Appetite 2017, 117, 186–190. [Google Scholar] [CrossRef]
- Kobata, K.; Sugawara, M.; Mimura, M.; Yawaza, S.; Watanabe, T. Potent Production of Capsaicinoids and Capsinoids by Capsicum Peppers. J. Agric. Food Chem. 2013, 61, 11127–11132. [Google Scholar] [CrossRef]
- Garcés-Claver, A.; Gil-Ortega, R.; Álvarez-Fernández, A.; Arnedo-Andrés, M.S. Inheritance of capsaicin and dihydrocapsaicin, determined by HPLC-ESI/MS, in an intraspecific cross of Capsicum annuum L. J. Agric. Food Chem. 2007, 55, 6951–6957. [Google Scholar] [CrossRef]
- Goll, J.; Frey, A.; Minceva, M. Study of the separation limits of continuous solid support free liquid-liquid chromatography: Separation of capsaicin and dihydrocapsaicin by centrifugal partition chromatography. J. Chromatogr. A 2013, 1284, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Othman, Z.A.A.; Ahmed, Y.B.H.; Habila, M.A.; Ghafar, A.A. Determination of capsaicin and dihydrocapsaicin in Capsicum fruit samples using high performance liquid chromatography. Molecules 2011, 16, 8919–8929. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Yang, Q.; Matthäus, B.; Li, P.; Zhang, Q.; Zhang, L. Simultaneous determination of capsaicin and dihydrocapsaicin for vegetable oil adulteration by immunoaffinity chromatography cleanup coupled with LC-MS/MS. J. Chromatogr. B 2016, 1021, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Ma, D.; Cao, W.; Shi, H.; Jiang, Y. Fast simultaneous determination of capsaicin, dihydrocapsaicin and nonivamide for detecting adulteration in edible and crude vegetable oils by UPLC-MS/MS. Food Addit. Contam. Part A 2018, 35, 1447–1452. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Fiechter, G.; Fritz, E.-M.; Mayer, H.K. Quantitation of capsaicinoids in different chilies from Austria by a novel UPLC method. J. Food Compos. Anal. 2017, 60, 32–37. [Google Scholar] [CrossRef]
- Sganzerla, M.; Coutinho, J.P.; Tavares de Melo, A.M.; Godoy, E.T. Fast method of capsaicinoids analysis from Capsicum chinense fruits. Food Res. Int. 2014, 64, 718–725. [Google Scholar] [CrossRef]
- Dang, Y.M.; Hong, Y.S.; Lee, C.L.; Khan, N.; Park, S.; Jeong, S.-W.; Kim, K.S. Determination of Capsaicinoids in Red Pepper Products from South Korea by High-Performance Liquid Chromatography with Fluorescence Detection. Anal. Lett. 2018, 51, 1291–1303. [Google Scholar] [CrossRef]
- Lu, M.; Ho, C.-T.; Huang, Q. Extraction, bioavailability, and bioefficacy of capsaicinoids. J. Food Drug Anal. 2017, 25, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hu, J.; Sheng, L.; Li, Y. Simultaneous quantification of capsaicin and dihydrocapsaicin in rat plasma using HPLC coupled with tandem mass spectrometry. J. Chromatogr. B 2010, 878, 2292–2297. [Google Scholar] [CrossRef]
- Kuzma, M.; Fodor, K.; Maász, G.; Avar, P.; Mózsik, G.; Past, T.; Fischer, E.; Perjési, P. A validated HPLC-FLD method for analysis of intestinal absorption and metabolism of capsaicin and dihydrocapsaicin in the rat. J. Pharm. Biomed. Anal. 2015, 103, 59–66. [Google Scholar] [CrossRef]
- Baskaran, P.; Krishnan, V.; Ren, J.; Thyagarjan, B. Capsaicin induces browning of white adipose tissue and counters obesity by activating TRPV1 channel-dependent mechanisms. Br. J. Pharmacol. 2016, 173, 2369–2389. [Google Scholar] [CrossRef]
- Wolde, T. Effects of caffeine on health and nutrition: A Review. Food Sci. Qual. Manag. 2014, 30, 59–65. [Google Scholar]
- Martínez-Pinilla, E.; Oῆatibia-Asibia, A.; Franco, R. The relevance of theobromine for the beneficial effects of cocoa consumption. Front. Pharmacol. 2015, 6, 1–5. [Google Scholar] [CrossRef]
- Ahluwalia, N.; Herrick, K. Caffeine intake from food and beverage sources and trends among children and adolescents in the United States: Review of national quantitative studies from 1999 to 2011. Adv. Nutr. 2015, 6, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Verster, J.C.; Koenig, J. Caffeine intake and its sources: A review of national representative studies. Crit. Rev. Food Sci. Nutr. 2018, 58, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Simmons, R.; Mwongela, S.M. Two-stage course-embedded determination of caffeine and related compounds by HPLC in caffeine containing food, beverages and (or) related products. J. Lab. Chem. Educ. 2017, 5, 19–25. [Google Scholar]
- Lowry, J.A.; Pearce, R.E.; Gaedigk, A.; Venneman, M.; Talib, N.; Shaw, P.; Leeder, J.S.; Kearns, G.L. Lead and its effects on cytochromes P450. J. Drug Metab. Toxicol. 2012, S5, S004. [Google Scholar] [CrossRef]
- Grujić-Letić, N.; Rakić, B.; Šefer, E.; Milanović, M.; Nikšić, M.; Vujić, I.; Milić, N. Quantitative determination of caffeine in different matrices. Maced. Pharm. Bull. 2016, 62, 77–84. [Google Scholar]
- Gonçalves, E.S.; Rodrigues, S.V.; Vieira da Silva-Filho, E. The use of caffeine as a chemical marker of domestic wastewater contamination in surface waters: Seasonal and spatial variations in Teresópolis, Brazil. Rev. Ambient. Agua 2017, 12, 192–202. [Google Scholar] [CrossRef]
- Gliszczyńska-Świglo, A.; Rybicka, I. Simultaneous determination of caffeine and water-soluble vitamins in energy drinks by HPLC with photodiode array and fluorescence detection. Food Anal. Methods 2015, 8, 139–146. [Google Scholar] [CrossRef]
- Mazdeh, F.Z.; Moradi, Z.; Moghaddam, G.; Moradi-Khatoonabadi, Z.; Aftabdari, F.E.; Badaei, P.; Hajimahmoodi, M. Determination of synthetic food colors, caffeine, sodium benzoate and potassium sorbate in sports drinks. Trop. J. Pharm. Res. 2016, 15, 183–188. [Google Scholar] [CrossRef]
- Ortega, N.; Romero, M.-P.; Macià, A.; Reguant, J.; Anglès, N.; Morelló, J.-R.; Motilva, M.-J. Comparative study of UPLC-MS/MS and HPLC-MS/MS to determine procyanidins and alkaloids in cocoa samples. J. Food Compos. Anal. 2010, 23, 298–305. [Google Scholar] [CrossRef]
- Rodríguez-Carrasco, Y.; Gaspari, A.; Graziani, G.; Santini, A.; Ritieni, A. Fast analysis of polyphenols and alkaloids in cocoa-based products by ultra-high performance liquid chromatography and orbitral high resolution mass spectrometry (UHPLC-Q-Orbitrap-MS/MS). Food Res. Int. 2018, 111, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Oellig, C.; Schunck, J.; Schwack, W. Determination of caffeine, theobromine and theophylline in Mate beer and Mate soft drinks by high-performance thin-layer chromatography. J. Chromatogr. A 2018, 1533, 208–212. [Google Scholar] [CrossRef]
- Alvi, S.N.; Hammami, M.M. Validated HPLC method for determination of caffeine level in human plasma using synthetic plasma: Application to bioavailability studies. J. Chromatogr. Sci. 2011, 49, 292–296. [Google Scholar] [CrossRef]
- Begas, E.; Kouvaras, E.; Tsakalof, A.K.; Bounitsi, M.; Asprodini, E.K. Development and validation of a reverse-phase HPLC method for CYP1A2 phenotyping by use of a caffeine metabolite ratio in saliva. Biomed. Chromatogr. 2015, 29, 1657–1663. [Google Scholar] [CrossRef]
- Rodríguez, A.; Costa-Bauza, A.; Saenz-Torres, C.; Rodrigo, D.; Grases, F. HPLC method for urinary theobromine determination: Effect of consumption of cocoa products on theobromine urinary excretion in children. Clin. Biochem. 2015, 48, 1138–1143. [Google Scholar] [CrossRef]
- Shrestha, S.; Rijal, S.K.; Pokhrel, P.; Rai, K.P. A simple HPLC method for determination of caffeine content in tea and coffee. J. Food Sci. Technol. 2016, 9, 74–78. [Google Scholar]
- Fajara, B.E.P.; Susanti, H. HPLC determination of caffeine in coffee beverage. IOP Conf. Ser. Mater. Sci. Eng. 2017, 259, 01211. [Google Scholar] [CrossRef]
- Aşçi, B.; Zor, Ş.D.; Dönmez, Ö.A. Development and validation of HPLC method for the simultaneous determination of five food additives and caffeine in soft drinks. Int. J. Anal. Chem. 2016, 2016, 2879406. [Google Scholar] [CrossRef]
- Yamamoto, T.; Takahashi, H.; Suzuki, K.; Hirano, A.; Kamei, M.; Goto, T.; Takahashi, N.; Kawada, T. Theobromine enhances absorption of cacao polyphenol in rats. Biosci. Biotechnol. Biochem. 2014, 78, 2059–2063. [Google Scholar] [CrossRef] [PubMed]
- Romano, R.; Santini, A.; Le Grottaglie, L.; Manzo, N.; Visconti, A.; Ritieni, A. Identification markers based on fatty acid composition to differentiate between roasted Arabica and Canephora (Robusta) coffee varieties in mixtures. J. Food Compos. Anal. 2014, 35, 1–9. [Google Scholar] [CrossRef]
- López-Sánchez, R.C.; Lara-Díaz, V.J.; Aranda-Gutiérrez, A.; Martínez-Cardona, J.A.; Hernández, J.A. HPLC method for quantification of caffeine and its three major metabolites in human plasma using fetal bovine serum matrix to evaluate prenatal drug exposure. J. Anal. Methods Chem. 2018, 2018, 2085059. [Google Scholar] [CrossRef] [PubMed]
- Ptolemy, A.S.; Tzioumis, E.; Thomke, A.; Rifai, S.; Kellogg, M. Quantification of theobromine and caffeine in saliva, plasma and urine via liquid chromatography-tandem mass spectrometry: A single analytical protocol applicable to cocoa intervention studies. J. Chromatogr. A 2010, 878, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Ikeda, K.; Terada, H.; Mochizuki, M.; Sugiyama, H. HPLC determination of caffeine using a photodiode array detector and applying a derivative processing to chromatograms. Bull. Nippon Vet. Life Sci. Univ. 2014, 63, 48–57. [Google Scholar]
- Naviglio, D.; Gallo, M.; Le Grottaglie, L.; Scala, C.; Ferrara, L.; Santini, A. Determination of cholesterol in Italian chicken eggs. Food Chem. 2012, 132, 701–708. [Google Scholar] [CrossRef]
- Ribeiro, S.M.L.; Luz, S.S.; Aquino, R.S. The role of nutrition and physical activity in cholesterol and aging. Clin. Geriatr. Med. 2015, 31, 401–416. [Google Scholar] [CrossRef]
- Albuquerque, T.G.; Oliveira, M.B.P.P.; Sanches-Silva, A.; Costa, H.S. Cholesterol determination in foods: Comparison between high performance and ultra-high performance liquid chromatography. Food Chem. 2016, 193, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Gylling, H.; Simonen, P. Are plant sterols and plant stenols are a viable future treatment for dyslipidemia? Expert Rev. Cardiovasc. Ther. 2016, 4, 549–551. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, P.D.; Pollier, J.; Panda, S.; Szymanski, J.; Massalha, H.; Yona, M.; Unger, T.; Malitsky, S.; Arendt, P.; Powels, L.; et al. Plant cholesterol biosynthetic pathway overlaps with phytosterol metabolism. Nat. Plants 2016, 3, 16205. [Google Scholar] [CrossRef] [PubMed]
- Cruz, R.; Casal, S.; Mendes, E.; Costa, A.; Santos, C.; Morais, S. Validation of a single-extraction procedure for sequential analysis of vitamin E, cholesterol, fatty acids, and total fat in seafood. Food Anal. Methods 2012, 6, 1196–1204. [Google Scholar] [CrossRef]
- Saldanha, T.; Bragagnolo, N. Effect of grilling on cholesterol oxide formation and fatty acids alterations in fish. Ciênc. Tecnol. Aliment. 2010, 30, 385–390. [Google Scholar] [CrossRef]
- Bauer, L.C.; Santana, D.A.; Macedo, M.S.; Torres, A.G.; de Souza, N.E.; Simionato, J.I. Method validation for simultaneous determination of cholesterol and cholesterol oxides in milk by RP-HPLC-DAD. J. Braz. Chem. Soc. 2014, 25, 161–168. [Google Scholar] [CrossRef]
- Daneshfar, A.; Khezeli, T.; Lotfi, H.J. Determination of cholesterol in food samples using dispersive liquid-liquid micro extraction followed by HPLC-UV. J. Chromatogr. B 2009, 877, 456–460. [Google Scholar] [CrossRef]
- Georgiou, C.A.; Constantinou, M.S.; Kapnissi-Christodoulou, C.P. Sample preparation: A critical step in the analysis of cholesterol oxidation products. Food Chem. 2014, 145, 1918–1926. [Google Scholar] [CrossRef]
- Robinet, P.; Wang, Z.; Hazen, S.L.; Smith, J.D. A simple and sensitive enzymatic method for cholesterol quantification in macrophages and foam cells. J. Lipid Res. 2010, 51, 3364–3369. [Google Scholar] [CrossRef]
- Carvalho, P.O.; Campos, P.R.B.; Noffs, M.D.; Fegolente, P.B.L.; Fegolente, L.V. Enzymatic hydrolysis of salmon oil by native lipases: Optimization of process parameters. J. Braz. Chem. Soc. 2009, 20, 117–124. [Google Scholar] [CrossRef]
- Codex Alimentarius. Code of Practice for the Prevention and Reduction of Mycotoxin Contamination in Cereals (CAC/RCP 51-2003). 2003. Available online: http://www.fao.org/fao-who-codexalimentarius/codex-texts/codes-of-practice/es/ (accessed on 20 June 2018).
- Njumbe Ediage, E.; Van Poucke, C.; De Saeger, S. A multi-analyte LC-MS/MS method for the analysis of 23 mycotoxins in different sorghum varieties: The forgotten sample matrix. Food Chem. 2015, 117, 397–404. [Google Scholar] [CrossRef]
- Binder, E. Managing the risk of mycotoxins in modern feed production. Anim. Feed Sci. Technol. 2007, 133, 149–166. [Google Scholar] [CrossRef]
- Dzuman, Z.; Zachariasova, M.; Lacina, O.; Veprokova, Z.; Slavokova, P.; Hajslova, J. A rugged high-throughput analytical approach for the determination and quantification of multiple mycotoxins in complex feed matrices. Talanta 2014, 121, 263–272. [Google Scholar] [CrossRef]
- Chavarría, G.; Molina, A.; Leiva, A.; Méndez, G.; Wong-González, E.; Cortés-Muñoz, M.; Rodríguez, C.; Granados-Chinchilla, F. Distribution, stability, and protein interactions of Aflatoxin M1 in fresh cheese. Food Control 2017, 73, 581–586. [Google Scholar] [CrossRef]
- Ostry, V.; Malir, F.; Toman, J.; Grosse, Y. Mycotoxins as human carcinogens, the IARC Monographs classification. Mycotoxins Res. 2016, 33, 65–73. [Google Scholar] [CrossRef]
- CAST. Mycotoxins: Risks in Plant, Animal, and Human Systems; Council for Agricultural Science and Technology: Ames, IA, USA, 2003; pp. 13–48. ISBN 1-887383-22-0. [Google Scholar]
- Molina Alvarado, A.; Zamora-Sanabria, R.; Granados-Chinchilla, F. A Focus on Aflatoxins in Feedstuffs: Levels of Contamination, Prevalence, Control Strategies, and Impacts on Animal Health. In Aflatoxin Control, Analysis, Detection and Health Risks; Lukman Bola Abdulra’uf, Ed.; IntechOpen: London, UK, 2017; pp. 115–152. [Google Scholar]
- Codex Alimentarius. Code of Practice for the Reduction of Aflatoxin B1 in Raw Materials and Supplemental Feedingstuffs for Milk Producing Animals (CAC/RCP 45-1997). 1997. Available online: http://www.fao.org/fao-who-codexalimentarius/codex-texts/codes-of-practice/es/ (accessed on 20 June 2018).
- Flores-Flores, M.; González-Peñas, E. Development and validation of a high performance liquid chromatographic-mass spectrometry method for the simultaneous quantification of 10 trichothecenes in ultra-high temperature processed cow milk. J. Chromatogr. A 2015, 1419, 37–44. [Google Scholar] [CrossRef]
- Njumbe Ediage, E.; Diana Di Mavungu, J.; Monbaliu, S.; Van Peteghem, C.; De Saeger, S. A Validated Multianalyte LC–MS/MS Method for Quantification of 25 Mycotoxins in Cassava Flour, Peanut Cake and Maize Samples. J. Agric. Food Chem. 2011, 59, 5173–5180. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, R.; Storm, I.; Rasmussen, P.; Smedsgaard, J.; Nielsen, K. Multi-mycotoxin analysis of maize silage by LC-MS/MS. Anal. Bioanal. Chem. 2010, 397, 765–776. [Google Scholar] [CrossRef]
- Chala, A.; Taye, W.; Ayalew, A.; Krska, R.; Sulyok, M.; Logrieco, A. Multimycotoxin analysis of sorghum (Sorghum bicolor L. Moench) and finger millet (Eleusine coracana L. Gaten) from Ethiopia. Food Control 2014, 45, 29–35. [Google Scholar] [CrossRef]
- Vishwanath, V.; Sulyok, M.; Labuda, R.; Bicker, W.; Krska, R. Simultaneous determination of 186 fungal and bacterial metabolites in indoor matrices by liquid chromatography/tandem mass spectrometry. Anal. Bioanal. Chem. 2009, 395, 1355–1372. [Google Scholar] [CrossRef]
- Alija, C.M.; Brar, S.K.; Verma, M.; Tyagi, R.D.; Godbout, S.; Valéro, J.R. Bio-processing of agro-byproducts to animal feed. Crit. Rev. Biotechnol. 2012, 32, 382–400. [Google Scholar]
- Mikušová, P.; Ritieni, A.; Santini, A.; Juhasová, G.; Šrobárová, A. Contamination by mould of grape berries in Slovakia. Food Addit. Contam. Part A 2010, 27, 738–747. [Google Scholar] [CrossRef]
- Santini, A.; Ferracane, R.; Meca, G.; Ritieni, A. Overview of analytical methods for beauvericin and fusaproliferin in food matrices. Anal. Bioanal. Chem. 2009, 395, 1253–1260. [Google Scholar] [CrossRef]
- Gil-Serna, J.; Vázquez, C.; González-Jaén, M.T.; Patiño, B. Wine contamination with ochratoxins: A Review. Beverages 2018, 4, 6. [Google Scholar] [CrossRef]
- Pizzutti, I.R.; de Kok, A.; Scholten, J.; Righi, L.W.; Cardoso, C.D.; Rohers, G.N.; da Silva, R.C. Development, optimization and validation of a multimethod for the determination of 36 mycotoxins in wines by liquid chromatography–tandem mass spectrometry. Talanta 2014, 129, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Nistor, E.; Dobre, A.; Dobre, A.; Bampidis, V.; Ciola, V. Grape pomace in sheep and dairy cows feeding. J. Hortic. For. Biotechnol. 2014, 18, 146–150. [Google Scholar]
- Guerra-Rivas, C.; Gallardo, B.; Mantecón, Á.R.; del Álamo-Sanza, M.; Manso, T. Evaluation of grape pomace from red winw by-products as feed for sheep. J. Sci. Food Agric. 2017, 97, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Kerasioti, E.; Terzopoulou, Z.; Komini, O.; Kafantaris, I.; Makri, S.; Stagos, D.; Gerasopoulos, K.; Anisimov, N.Y.; Tsatsakis, A.M.; Kouretas, D. Tissue specific effects of feeds supplemented with grape pomace or olive oil mill wastewater on detoxification enzymes in sheep. Toxicol. Rep. 2017, 4, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Avantaggiato, G.; Greco, D.; Damascelli, A.; Solfrizzo, M.; Visconti, A. Assessment of Multi-mycotoxin Adsorption Efficacy of Grape Pomace. J. Agric. Food Chem. 2014, 62, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Gambacorta, L.; Pinton, P.; Avantaggiato, G.; Oswald, I.P.; Solfrizzo, M. Grape Pomace, an Agricultural Byproduct Reducing Mycotoxin Absorption: In Vivo Assessment in Pig Using Urinary Biomarkers. J. Agric. Food Chem. 2016, 64, 6762–6771. [Google Scholar] [CrossRef]
- Anadón, A.; Martínez-Larrañaga, M.; Ares, I.; Martínez, M. Chapter 7—Regulatory Aspects for the Drugs and Chemicals Used in Food-Producing Animals in the European Union. In Veterinary Toxicology: Basic and Clinical Principles, 3rd ed.; Gupta, R.C., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 103–131. [Google Scholar]
- Decheng, S.; Peilong, W.; Yang, L.; Ruiguo, W.; Shulin, W.; Zhiming, X.; Su, Z. Simultaneous determination of antibiotics and amantadines in animal-derived feedstuffs by ultraperformance liquid chromatographic-tandem mass spectrometry. J. Chromatogr. B 2018, 1095, 183–190. [Google Scholar] [CrossRef]
- Molognoni, L.; Coelho de Souza, N.; Antunes de Sá Ploêncio, L.; Amadeu Micke, G.; Daguer, H. Simultaneous analysis of spectinomycin, halquizol, zilpaterol, and melamine in feedingstuffs by ion-pair liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2018, 1569, 110–117. [Google Scholar] [CrossRef]
- Cancho Grande, B.; García Falcón, M.S.; Simal Gándara, J. El uso de los antibióticos en la alimentación animal: Perspectiva actual. Cienc. Tecnol. Aliment. 2000, 3, 39–47. [Google Scholar]
- Rojek-Podgórska, B. EU Legislation in Progress: Review of Medicated feed Legislation. European Parliamentary Research Service (EPRS). 2016. Available online: http://www.europarl.europa.eu/RegData/etudes/BRIE/2016/583843/EPRS_BRI%282016%29583843_EN.pdf (accessed on 10 July 2018).
- Kang, H.; Lee, S.; Shin, D.; Jeong, J.; Hong, J.; Rhee, G. Occurrence of veterinary drug residues in farmed fishery products in South Korea. Food Control 2018, 85, 57–65. [Google Scholar] [CrossRef]
- Kumar Saxena, S.; Rangasamy, R.; Krishnan, A.; Singh, D.; Uke, S.; Kumar Malekadi, P.; Sengar, A.; Peer Mohamed, D. Simultaneous determination of multi-residue and multi-class antibiotics in aquaculture shrimps by UPLC-MS/MS. Food Chem. 2018, 260, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.; Ribeiro, C.; Barcellos Hoft, R.; Dalla Costa, T. A simple and high-throughput method for determination and confirmation of 14 coccidiostats in poultry muscle eggs using liquid chromatography-quadrupole linear ion trap–tandem mass spectrometry (HPLC-QqLIT-MS/MS): Validation according to European Union 2002/657/EC. Talanta 2017, 168, 43–51. [Google Scholar] [PubMed]
- World Health Organization. Global Action Plan on Antimicrobial Resistance. WHO Library Cataloguing 2015. Available online: http://www.wpro.who.int/entity/drug_resistance/resources/global_action_plan_eng.pdf (accessed on 18 July 2018).
- Camargo Valese, A.; Molognoni, L.; Coelho de Souza, N.; Antunes de Sá Ploêncio, L.; Oliveira Costa, A.; Barreto, F. Development, validation and different approaches for the measurement uncertainty of a multi-class veterinary drugs residues LC-MS method for feeds. J. Chromatogr. B 2017, 1053, 48–59. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Directive 2001/82/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to veterinary medicinal products. Off. J. Eur. Communities 2001, L 311, 1–66. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32001L0082&from=ES (accessed on 10 July 2018).
- European Commission. Regulation (EC) No 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorization and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency. Off. J. Eur. Communities 2004, L 136, 1–33. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32004R0726&from=ES (accessed on 10 July 2018).
- US Food and Drug Administration. Medicated Feeds. Available online: https://www.fda.gov/AnimalVeterinary/Products/AnimalFoodFeeds/MedicatedFeed/default.htm#license (accessed on 19 July 2018).
- Robert, C.; Basseur, P.-Y.; Dubois, M.; Delahaut, P.; Gillard, N. Development and validation of rapid multiresidue and multi-class analysis for antibiotics and anthelmintics in feed by ultra high performance liquid chromatography coupled to tandem mass spectrometry. Food Addit. Contam. Part A 2016, 33, 1312–1323. [Google Scholar] [CrossRef]
- Shendy, A.; Al-Ghobashy, M.; Gad Alla, S.; Lotfy, H. Development and validation of a modified QuEChERS protocol coupled to LC-MS/MS for simultaneous determination of multi-class antibiotic residues in honey. Food Chem. 2016, 190, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, C.; Guo, J.; Yuan, Y.; Wang, J.; Liu, L.; Yue, T. Development and application of method for the analysis of 9 mycotoxins in maize by HPLC-MS/MS. J. Food Sci. 2013, 78, 1752–1756. [Google Scholar] [CrossRef]
- Duelge, K.; Nishshanka, U.; De Alwis, H. An LC-MS/MS method for the determination of antibiotic residues in distillers grains. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2017, 1053, 81–86. [Google Scholar] [CrossRef]
- Mohanty, B.; Mahanty, A.; Ganguly, S.; Sankar, T.V.; Chakraborty, K.; Rangasamy, A.; Paul, B.; Sarma, D.; Mathew, S.; Kunnath Asha, K.; et al. Amino acid compositions of 27 food fishes and their importance in clinical nutrition. J. Amino Acids 2014, 2014, 269797. [Google Scholar] [CrossRef]
- 218Ribeiro Alvarenga, R.; Borges Rodrigues, P.; de Souza Cantarelli, V.; Gilberto Zangeronimo, M.; da Silva Júnior, J.; da Silva, L.; Moreira dos Santos, L.; Pereira, L. Energy values and chemical composition of spirulina (Spirulina platensis) evaluated with broilers. Rev. Bras. Zootec. 2011, 40, 992–996. [Google Scholar] [CrossRef]
- Campanella, L.; Russo, M.V.; Avino, P. Free and total amino acid composition in blue-green algae. Ann. Chim. 2002, 92, 343–352. [Google Scholar] [PubMed]
- Abdullah Al-Dhabi, N.; Valan Arasu, M. Quantification of Phytochemical from Commercial Spirulina Products and Their Antioxidant Activities. Evid. Based Complement. Alternat. Med. 2016, 2016, 7631864. [Google Scholar]
- Dziągwa-Becker, M.M.; Ramos, J.M.M.; Topolski, J.K.; Oleszek, W.A. Determination of free amino acids in plants by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). BioSci. Trends 2011, 5, 231–238. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, D.; Li, X.; Meng, L. Chromatographic method for determination of the free amino acid content of chamomile flowers. Pharmacogn. Mag. 2015, 11, 176–179. [Google Scholar] [PubMed]
- Salazar-Villanea, S.; Bruininx, E.M.A.M.; Gruppen, H.; Hendriks, W.H.; Carré, P.; Quinsac, A.; van der Poel, A.F.B. Physical and chemical changes of rapessed meal proteins during toasting and their effects on in vitro digestibility. J. Anim. Sci. Biotechnol. 2016, 7, 62. [Google Scholar] [CrossRef]
- Jajić, I.; Krstović, S.; Glamočić, D.; Jakšić, S.; Abramović, B. Validation of an HPLC method for the determination of amino acids in feed. J. Serbian Chem. Soc. 2013, 78, 839–850. [Google Scholar] [CrossRef]
- Szkudzińska, K.; Smutniak, H.; Rubaj, J.; Korol, W.; Bielecka, G. Method validation for determination of amino acids in feed by UPLC. Accredit. Qual. Assur. 2018, 22, 247–252. [Google Scholar] [CrossRef]
- Desmarais, S.M.; Cava, F.; de Pedro, M.A.; Casey Huang, K. Isolation and Preparation of Bacterial Cell Walls for Compositional Analysis by Ultra Performance Liquid Chromatography. J. Vis. Exp. 2014, 83, e51183. [Google Scholar] [CrossRef]
- Kühner, D.; Stahl, M.; Demircioglu, D.D.; Bertsche, U. From cells to muropeptide structures in 24 h: Peptidoglycan mapping by UPLC-MS. Sci. Rep. 2014, 4, 7494. [Google Scholar] [CrossRef]
- Marseglia, A.; Sforza, S.; Faccini, A.; Bencivenni, M.; Palla, G.; Caligian, A. Extraction, identification and semi-quantification of oligopeptides in cocoa beans. Food Res. Int. 2014, 63, 382–389. [Google Scholar] [CrossRef]
- Prados, I.M.; Marina, M.L.; García, M.C. Isolation and identification by high resolution liquid chromatography tandem mass spectrometry of novel peptides with multifunctional lipid lowering capacity. Food Res. Int. 2018, 111, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Holman, B.W.B.; Malau-Aduli, A.E.O. Spirulina as livestock supplement and animal feed. J. Anim. Physiol. Anim. Nutr. 2013, 97, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Nurcahya Dewi, E.; Amalia, U.; Mel, M. The effect of Different Treatments to the Amino Acid Contents of Micro Algae Spirulina sp. Aquatic Procedia 2016, 7, 59–65. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, K.; Li, P.; Zhou, J.; Chao, Y. Simultaneous determination of 20 underivatized amino acids by high performance liquid chromatography-evaporative light-scattering detection. Se Pu 2011, 29, 908–911. [Google Scholar] [PubMed]
- Prinsen, H.C.M.T.; Schiebergen–Bronkhorst, B.G.M.; Roeleveld, M.N.; Jans, J.J.M.; de Sain-van der Velden, M.G.M.; Visser, G.; van Hasselt, P.M.; Verhoeven-Duif, N.M. Rapid quantification of underivatized amino acids in plasma by hydrophilic interaction liquid chromatography (HILIC) coupled with tándem mass-spectrometry. J. Inherit. Metab. Dis. 2016, 39, 651–660. [Google Scholar] [CrossRef]
- Masuda, A.; Dohmae, N. Amino acid analysis of sub-picomolar amounts of proteins by pre-column fluorescence derivatization with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate. Biosc. Trends 2011, 5, 231–238. [Google Scholar] [CrossRef]
- Dhillon, M.K.; Kumar, S.; Gujar, G.T. A common HPLC-PDA method for amino acid analysis in insects and plants. Indian J. Exp. Biol. 2014, 52, 73–79. [Google Scholar]
- Artavia, G.; Rojas-Bogantes, L.; Granados-Chinchilla, F. Two alternative chromatography methods assisted by the sulfonic acid moiety for the determination of furosine in milk. MethodsX 2018, 5, 639–647. [Google Scholar] [CrossRef]
- Dolowy, M.; Pyka, A. Application of TLC, HPLC and GC methods to the study of amino acid and peptide enantiomers: A review. Biomed. Chromatogr. 2013. [Google Scholar] [CrossRef]
- Bartolomeo, M.; Maisano, F. Validation of a Reversed-Phase HPLC Method for Quantitative Amino Acid Analysis. J. Biomol. Tech. 2006, 17, 131–137. [Google Scholar]
- Zhou, X.; Zhang, J.; Pan, Z.; Li, D. Review of Methods for the Detection and Determination of Malachite Green and Leuco-Malachite Green in Aquaculture. Crit. Rev. Anal. Chem. 2018, 14, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Adel, M.; Dadar, M.; Oliveri Conti, G. Antibiotics and malachite green in farmed rainbow trout (Oncorhynchus mykiss) from Iranian markets: A risk assessment. Int. J. Food Prop. 2017, 20, 402–408. [Google Scholar] [CrossRef]
- Sudova, E.; Machova, J.; Svobodova, Z.; Vesely, T. Negative effects of malachite green and possibilities of its replacement in the treatment of fish eggs and fish: A review. Vet. Med. 2007, 52, 527–539. [Google Scholar] [CrossRef]
- Hidayah, N.; Abu Bakar, F.; Mahyudin, N.A.; Faridah, S.; Nur-Azura, M.S.; Zaman, M.Z. Detection of malachite green and leuco-malachite green in fishery industry. Int. Food Res. J. 2013, 20, 1511–1519. [Google Scholar]
- Xie, J.; Peng, T.; Chen, D.; Zhang, Q.; Wang, G.; Wang, X.; Guo, Q.; Jiang, F.; Chen, D.; Deng, J. Determination of malachite green, crystal violet and their leuco-metabolites in fish by HPLC-VIS detection after immunoaffinity column clean-up. J. Chromatogr. B 2013, 913–914, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Miao, S. HPLC Determination and MS Confirmation of Malachite Green, Gentian Violet, and Their Leuco Metabolite Residues in Channel Catfish Muscle. J. Agric. Food Chem. 2010, 58, 7109–7114. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liao, K.; Huang, X.; Yuan, D. Simultaneous determination of malachite green, crystal violet and their leuco-metabolites in aquaculture water samples using monolithic fiber-based solid-phase microextraction coupled with high performance liquid chromatography. Anal. Methods 2015, 7, 8138. [Google Scholar] [CrossRef]
- Bae Lee, J.; Yun Kim, H.; Mi Jang, Y.; Young Song, J.; Min Woo, S.; Sun Park, M.; Sook Lee, H.; Kyu Lee, S.; Kim, M. Determination of malachite green and crystal violet in processed fish products. Food Addit. Contam. Part A 2010, 27, 953–961. [Google Scholar] [CrossRef]
- Chengyun, Z.; Jie, W.; Xuefang, D.; Zhimou, G.; Mingyang, L.; Xinmiao, L. Fast analysis of malachite green, leucomalachite green, crystal violet and leucocrystal violet in fish tissue based on a modified QuEChERS procedure. Chin. J. Chromatogr. 2014, 4, 419–425. [Google Scholar]
- Turnipseed, S.B.; Andersen, W.C.; Roybal, J.E. Determination and Confirmation of Leucomalachite Green in Salmon using No-Discharge Atmospheric Pressure Chemical Ionization LC-MS. J. AOAC Int. 2005, 88, 1312–1317. [Google Scholar]
- Abro, K.; Mahesar, S.A.; Iqbal, S.; Perveen, S. Quantification of malachite green in fish feed utilizing liquid chromatography-tandem mass spectrometry with a monolithic column. Food Addit. Contam. Part A 2014, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- EFSA. Malachite green in food, EFSA Panel on Contaminants in the Food Chain (CONTAM). EFSA J. 2016, 14, 4530. [Google Scholar] [CrossRef]
- Furusawa, N. An isocratic Toxic Chemical-Free Mobile Phase HPLC-PDA analysis of Malachite Green and Leuco-Malachite Green. Chromatography 2014, 1, 75–81. [Google Scholar] [CrossRef]
- Bilandžić, N.; Varenina, I.; Solomun Kolanović, B.; Oraić, D.; Zrnčić, S. Malachite green residues in farmed fish in Croatia. Food Control 2012, 26, 393–396. [Google Scholar] [CrossRef]
- Barani, A.; Tajik, H. Malachite green residue in farmed fish in north-west part of Iran. Int. J. Food Prop. 2017, 20, S580–S585. [Google Scholar] [CrossRef]
- Bedale, W.; Sindelar, J.J.; Milkowski, A.L. Dietary nitrate and nitrite: Benefits, risks, and evolving perceptions. Meat Sci. 2016, 120, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Wakida, F.T.; Lerner, D.N. Non-agricultural sources of groundwater nitrate: Review and case study. Water Res. 2005, 39, 3–16. [Google Scholar] [CrossRef]
- Deutsche Forschungsgemeinschaft. Nitrate and Nitrite in Diet: An Approach to Assess Benefit and Risk for Human Health; Institut für Lebensmitteltoxikol: Hannover, Germany, 2014; p. 42. [Google Scholar]
- Iammarino, M.; Di Taranto, A.; Cristino, M. Monitoring of nitrites and nitrates levels in leafy vegetables (spinach and lettuce): A contribution to risk assessment. J. Sci. Food Agric. 2014, 94, 773–778. [Google Scholar] [CrossRef]
- Lundberg, J.O.; Gladwin, M.T.; Ahluwalia, A.; Benjamin, N.; Bryan, N.S.; Butler, A.; Cabrales, P.; Fago, A.; Feelisch, M.; Ford, P.C.; et al. Nitrate and nitrite in biology, nutrition and therapeutics. Nat. Chem. Biol. 2009, 5, 865–869. [Google Scholar] [CrossRef]
- Shiva, S. Nitrite: A physiological store of nitric oxide and modulator of mitochondrial function. Redox Biol. 2013, 1, 40–44. [Google Scholar] [CrossRef]
- Espejo-Herrera, N.; Gràcia-Lavedan, E.; Boldo, E.; Aragonés, N.; Pérez-Gómez, B.; Pollán, M.; Molina, A.J.; Fernández, T.; Martín, V.; La Vecchia, C.; et al. Colorectal cancer risk and nitrate exposure through drinking water and diet. Int. J. Cancer 2016, 139, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.H. Too Much of Good Thing? Nitrate from Nitrogen Fertilizers and Cancer. Rev. Environ. Health 2009, 24, 357–363. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Regulation (EC) No 1881/2006 of 19 December 2006 setting levels for certain contaminants in foodstuff. Off. J. Eur. Communities 2006, 016.011, 1–35. [Google Scholar]
- Brkić, D.; Bošnir, J.; Bevardi, M.; Gross Bošković, A.; Miloš, S.; Lasić, D.; Krivohlavek, A.; Racz, A.; Mojsović-Ćuić, A.; Uršulin Trstenjak, N. Nitrate in leafy green vegetables and estimated intake. Afr. J. Tradit. Complement. Altern. Med. 2017, 14, 31–41. [Google Scholar] [PubMed]
- Pardo, O.; Yusà, V.; Villalba, P.; Perez, J.A. Monitoring programme on nitrate in vegetables and vegetable-based baby foods marketed in the Region of Valencia: Levels and estimated daily intake. Food Addit. Contam. 2010, 27, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Quijano, L.; Yusà, V.; Font, G.; McAllister, C.; Torres, C.; Pardo, O. Risk assessment and monitoring programme of nitrates through vegetables in the Region of Valencia (Spain). Food Chem. Toxicol. 2017, 100, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.; Arcot, J.; Lee, N.A. Nitrate and nitrite quantification from cured meat and vegetables and their estimated dietary intake in Australians. Food Chem. 2009, 115, 334–339. [Google Scholar] [CrossRef]
- Dumitru Croitoru, M. Nitrite and nitrate can be accurately measured in samples of vegetal and animal origin using an HPLC-UV/VIS technique. J. Chromatogr. B 2012, 911, 154–161. [Google Scholar] [CrossRef]
- Tsikas, D. Analysis of nitrite and nitrate I biological fluids by assays based on the Griess reaction: Appraisal of the Griess reaction in the L-arginine/nitric oxide area of research. J. Chromatogr. B 2007, 851, 51–70. [Google Scholar] [CrossRef]
- Dumitru Croitoru, M.; Fülöp, I.; Miklos, A.; Hosszú, B.; Tátar, V.; Muntean, D. Presence of nitrate and nitrite in vegetables grown for self-consumption. Farmacia 2015, 63, 530–533. [Google Scholar]
- Yagoub Abdulkair, B.; Elzupir, A.O.; Alamer, A.S. An Ultrasound Assessed Extraction Combined with Ion-Pair HPLC Method and Risk Assessment of Nitrite and Nitrate in Cured Meat. J. Anal. Methods Chem. 2018, 2018, 1907151. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Chung, J.; Hwang, D. A High Performance Liquid Chromatography Method for Determining Nitrate and Nitrite Levels in Vegetables. J. Food Drug Anal. 2003, 11, 233–238. [Google Scholar]
- Nemade, K.; Fegade, U.; Ingle, S.; Attarde, S. High Performance Liquid Chromatography Method for Determination of Nitrite and Nitrate in Vegetable and Water samples. Int. J. Adv. Sci. Tech. Res. 2014, 4, 238–250. [Google Scholar]
- Scheeren, M.B.; Arul, J.; Gariépy, C. Comparison of different method for nitrite and nitrate determination in meat products. In Proceedings of the 59th International Congress of Meat Science and Technology, Izmir, Turkey, 18–23 August 2013. [Google Scholar]
- Dos Santos Baião, D.; Conte-Junior, C.; Flosi Paschoalin, V.; Silveira Alvares, T. Quantitative and Comparative Contents of Nitrate and Nitrite in Beta vulgaris L. by Reversed. Phase High-Performance Liquid Chromatography-Fluorescence. Food Anal. Methods 2016, 9, 1002–1008. [Google Scholar] [CrossRef]
- Casanova, J.A.; Gross, L.K.; McMullen, S.E.; Schenck, F.J. Use of Greiss reagent containing Vanadium(III) fro post-column derivatization and simultaneous determination of nitrite and nitrate in baby food. J. AOAC Int. 2006, 89, 447–451. [Google Scholar] [PubMed]
- Marcus, Y. Thermodynamics of Solvation of Ions. J. Chem. Soc. Faraday Trans. 1991, 87, 2995–2999. [Google Scholar] [CrossRef]
- Oruc, H.H.; Akkoc, A.; Uzunoglu, I.; Kennerman, E. Nitrate Poisoning in Horses Associated with Ingestion of Forage and Alfalfa. J. Equine Vet. Sci. 2010, 30, 159–162. [Google Scholar] [CrossRef]
- Merino, L.; Örnemark, U.; Toldrá, F. Chapter Three—Analysis of Nitrite and Nitrate in Foods: Overview of Chemical, Regulatory and Analytical Aspects. Adv. Food Nutr. Res. 2017, 81, 65–107. [Google Scholar] [PubMed]
- Wang, Q.; Yu, L.; Liu, Y.; Lin, L.; Lu, R.; Zhu, J.; He, L.; Lu, Z. Methods for the detection and determination of nitrite and nitrate: A review. Talanta 2017, 165, 709–720. [Google Scholar] [CrossRef]
- O’Neil, C.A.; Schwartz, S.J. Chromatographic analysis of cis/trans carotenoid isomers. J. Chromatogr. 1992, 624, 235–252. [Google Scholar] [CrossRef]
- Wilberg, V.C.; Rodriguez-Amaya, D.B. HPLC Quantitation of Major Carotenoids of Fresh and Processed Guava, Mango and Papaya. LWT-Food Sci. Technol. 1995, 28, 474–480. [Google Scholar] [CrossRef]
- Zanatta, C.F.; Mercadante, A.Z. Carotenoid composition from the Brazilian tropical fruit camu–camu (Myrciaria dubia). Food Chem. 2007, 101, 1526–1532. [Google Scholar] [CrossRef]
- Chen, J.P.; Tai, C.Y.; Chen, B.H. Improved liquid chromatographic method for determination of carotenoids in Taiwanese mango (Mangifera indica L.). J. Chromatogr. A 2004, 29, 261–268. [Google Scholar] [CrossRef]
- Inbaraj, B.S.; Chien, J.T.; Chen, B.H. Improved High Performance Liquid Chromatographic Method for Determination of Carotenoids in the Microalga Chlorella pyrenoidosa. J. Chromatogr. A 1102, 1102, 193–199. [Google Scholar] [CrossRef] [PubMed]
- McGraw, K.J.; Toomey, M.B. Carotenoid accumulation in the tissues of zebra finches: Predictors of integumentary pigmentation and implications for carotenoid allocation strategies. Physiol. Biochem. Zool. 2010, 83, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Aluç, Y.; Kankılıç, G.B.; Tüzün, I. Determination of carotenoids in two algae species from the saline water of Kapulukaya reservoir by HPLC. J. Liquid Chromatogr. Relat. Technol. 2018, 41, 1–8. [Google Scholar] [CrossRef]
- Shih-Chuan, L.; Jau-Tien, L.; Deng-Jye, Y. Determination of cis- and trans- α- and β-carotenoids in Taiwanese sweet potatoes (Ipomoea batatas (L.) Lam.) harvested at various times. Food Chem. 2009, 116, 605–610. [Google Scholar]
- Van Jaarsveld, P.J.; Marais, D.W.; Harmse, E.; Nestel, P.; Rodriguez-Amaya, D.B. Retention of β-carotene in boiled, mashed orange-fleshed sweet potato. J. Food Compos. Anal. 2006, 19, 321–329. [Google Scholar] [CrossRef]
- Huck, C.; Popp, M.; Scherz, H.; Bonn, G.K. Development and Evaluation of a New Method for the Determination of the Carotenoid Content in Selected Vegetables by HPLC and HPLC–MS–MS. J. Chromatogr. Sci. 2000, 38, 441–449. [Google Scholar] [CrossRef]
- Lessin, W.J.; Catigani, G.L.; Schwartz, S.J. Quantification of cis-trans Isomers of Provitamin A Carotenoids in Fresh and Processed Fruits and Vegetables. J. Agric. Food Chem. 1997, 45, 3728–3732. [Google Scholar] [CrossRef]
- Gayosso-García, L.E.; Yahia, E.M.; González-Aguila, G.A. Identification and quantification of phenols, carotenoids, and vitamin C from papaya (Carica papaya L., cv. Maradol) fruit determined by HPLC-DAD-MS/MS-ESI. Food Res. Int. 2011, 44, 1284–1291. [Google Scholar] [CrossRef]
- Schweiggert RFSteingass, C.B.; Esquivel, P.; Carle, R. Chemical and Morphological Characterization of Costa Rican Papaya (Carica papaya L.) Hybrids and Lines with Particular Focus on Their Genuine Carotenoid Profiles. J. Agric. Food Chem. 2012, 60, 2577–2585. [Google Scholar]
- Chacón-Ordóñez, T.; Schweiggert, R.M.; Bosy-Westphal, A.; Jiménez, V.M.; Carle, R.; Esquivel, P. Carotenoids and carotenoid esters of orange- and yellow-fleshed mamey sapote (Pouteria sapota (Jacq.) H.E. Moore & Stearn) fruit and their postprandial absorption in humans. Food Chem. 2017, 221, 673–682. [Google Scholar] [PubMed]
- Rojas-Garbanzo, C.; Gleichenhagen, M.; Heller, A.; Esquivel, P.; Schulze-Kaysers, N.; Schieber, A. Carotenoid profile, antioxidant capacity, and chromoplasts of pink guava (Psidium guajava L. Cv. ‘Criolla’) during fruit ripening. J. Agric. Food Chem. 2017, 65, 3737–3747. [Google Scholar] [CrossRef] [PubMed]
- Irias-Mata, A.; Jiménez, V.M.; Steingass, C.B.; Schweiggert, R.M.; Carle, R.; Esquivel, P. Carotenoids and xanthophyll esters of yellow and red nance fruits (Byrsonima crassifolia (L.) Kunth) from Costa Rica. Food Res. Int. 2018, 111, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Marutti, L.R.B.; Mercadante, A.Z. Carotenoid esters analysis and occurrence: What do we know so far? Arch. Biochem. Biophys. 2018, 648, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Hempel, J.; Schweiggert, R.M.; Ni, Y.; Carle, R. Carotenoids and carotenoid esters of red and yellow Physalis (Physalis alkekengi L. and P. pubescens L.) fruits and calyces. J. Agric. Food Chem. 2017, 65, 6140–6151. [Google Scholar] [CrossRef] [PubMed]
- De Rosso, V.; Mercadante, A. Identification and Quantification of Carotenoids, by HPLC-PDA-MS/MS, from Amazonian Fruits. J. Agric. Food Chem. 2007, 55, 5062–5072. [Google Scholar] [CrossRef]
- Ligor, M.; Kováčová, J.; Gadzała-Kopciuch, R.; Studzińska, S.; Bocian, S.; Lehotay, J.; Buszewski, B. Study of RP HPLC Retention Behaviours in Analysis of Carotenoids. Chromatographia 2014, 77, 1047–1057. [Google Scholar] [CrossRef]
- Schex, R.; Lieb, V.; Jiménez, V.M.; Esquivel, P.; Schweiggert, R.; Carle, R.; Steingrass, C.B. HPLC-DAD-APCI/ESI-MSn analysis of carotenoids and α-tocopherol in Costa Rican Acrocomia aculeata fruits of varying maturity stages. Food Res. Int. 2018, 105, 645–653. [Google Scholar] [CrossRef]
- Schweiggert, R.M.; Vargas, E.; Conrad, J.; Hempel, J.; Gras, C.C.; Ziegler, J.U.; Mayer, A.; Jiménez, V.; Esquivel, P.; Carle, R. Carotenoids, carotenoid esters, and anthocyanins of yellow-, orange-, and red-peeled cashew apples (Anacardium occidentale L.). Food Chem. 2016, 200, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Chacón-Ordoñez, T.; Esquivel, P.; Jiménez, V.M.; Carle, R.; Schweiggert, R.M. Deposition Form and Bioaccessibility of Keto-Carotenoids from Mamey Sapote (Pouteria sapota), Red Bell Pepper (Capsicum annuum), and Sockeye Salmon (Oncorhynchus nerka) Filet. J. Agric. Food Chem. 2016, 64, 1989–1998. [Google Scholar] [CrossRef] [PubMed]
- Dolan, J.W. How Much Can I Inject? Part II: Injecting in Solvents Other than Mobile Phase. LCGC N. Am. 2014, 32, 854–859. [Google Scholar]
- Pereira da Costa, M.; Conte-Junior, C.A. Chromatographic methods for the determination of carbohydrates and organic acids in foods of animal origin. Compr. Rev. Food Sci. Food Saf. 2015, 14, 586–600. [Google Scholar] [CrossRef]
- De Goeij, S. Quantitative Analysis Methods for Sugars. Master’s Thesis, Universiteit van Amsterdam, Amsterdam, The Netherlands, August 2013. [Google Scholar]
- Agius, C.; von Tucher, S.; Poppenberger, B.; Rozhon, W. Quantification of sugars and organic acids in tomato fruits. MethodsX 2018, 5, 2537–2550. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liang, L.; Zhu, M. Determination of Sugars in Molasses by HPLC Following Solid-Phase Extraction. Int. J. Food Prop. 2015, 18, 547–557. [Google Scholar] [CrossRef]
- Koh, D.-W.; Park, J.-W.; Lim, J.-H.; Yea, M.-J.; Bang, D.-Y. A rapid method for simultaneous quantification of 13 sugars and sugar alcohols in food products by UPLC-ELSD. Food Chem. 2018, 240, 694–700. [Google Scholar] [CrossRef]
- Zielinkski, A.A.F.; Braga, C.M.; Demiate, I.M.; Beltrame, F.L.; Nogueira, A.; Wosiaki, G. Development and optimization of a HPLC-RI method for the determination of major sugars in apple juice and evaluation of the effect of the ripening stage. Food Sci. Technol. 2014, 34, 38–43. [Google Scholar] [CrossRef]
- Duarte-Delgado, D.; Narváez-Cuenca, C.E.; Restrepo-Sánchez, L.P.; Kushalappa, A.; Mosquera-Vásquez, T. Development and validation of a liquid chromatographic method to quantify sucrose, glucose, and fructose in tubers of Solanum tuberosum Group Phureja. J. Chromatogr. B 2015, 975, 18–23. [Google Scholar] [CrossRef]
- Shindo, T.; Sadamasu, Y.; Suzuki, K.; Tanaka, Y.; Togawa, A.; Uematsu, Y. Method of quantitative analysis by HPLC and confirmation by LC-MS of sugar alcohols in foods. Shokuhin Eiseigaku Zasshi 2013, 54, 358–363. [Google Scholar] [CrossRef]
- Canesin, R.C.F.S.; Isique, W.D.; Buzetti, S.; Aparecida de Souza, J. Derivation method for determining sorbitol in fruit trees. Am. J. Plant Sci. 2014, 5, 3457–3463. [Google Scholar] [CrossRef]
- Hung, W.-T.; Chen, Y.-T.; Wang, S.-H.; Liu, Y.-C.; Yang, W.-B. A new method for aldo-sugar analysis in beverages and dietary foods. Funct. Food Health Dis. 2016, 6, 234–245. [Google Scholar]
- Valliydan, B.; Shi, H.; Nguyen, H.T. A simple analytical method for high-throughput screening of major sugars from soybean by normal-phase hplc with evaporative light scattering detection. Chromatogr. Res. Int. 2015, 2015, 757649. [Google Scholar] [CrossRef]
- Wieß, K.; Alt, M. Determination of single sugars, including inulin, in plants and feed materials by high-performance liquid chromatography and refraction index detection. Fermentation 2017, 3, 36. [Google Scholar]
- Verspreet, J.; Pollet, A.; Cuyvers, S.; Vergauwen, R.; Van den Ende, W.; Delcour, J.A.; Courtin, C.M. A simple and accurate method for determining wheat grain fructan content and average degree of polymerization. J. Agric. Food Chem. 2012, 60, 2102–2107. [Google Scholar] [CrossRef] [PubMed]
- Correia, D.M.; Dias, L.G.; Veloso, A.C.A.; Dias, T.; Rocha, I.; Rodrigues, L.R.; Peres, A.M. Dietary sugars analysis: Quantification of fructooligosaccharides during fermentation by HPLC-RI method. Front. Nutr. 2014, 1, 11. [Google Scholar] [CrossRef]
- Salazar Murillo, M.M.; Granados-Chinchilla, F. Total starch in animal feeds and silages based on the chromatographic determination of glucose. MethodsX 2018, 5, 83–89. [Google Scholar] [CrossRef]
- Bai, W.; Fang, X.; Zhao, W.; Huang, S.; Zhang, H.; Qian, M. Determination of oligosaccharides and monosaccharides in Hakka rice wine by precolumn derivation high-performance liquid chromatography. J. Food Drug Anal. 2015, 23, 645–651. [Google Scholar] [CrossRef]
- Madhuri, K.V.; Prabhakar, K.V. Recent trends in the characterization of microbial exopolysaccharides. Orient. J. Chem. 2014, 30, 895–904. [Google Scholar] [CrossRef]
- Corradini, C.; Cavazza, A.; Bignardi, C. High-performance anion-exchange chromatography coupled with pulsed electrochemical detection as a powerful tool to evaluate carbohydrates of food interest: Principles and applications. Int. J. Carbohydr. Chem. 2012, 2012, 487564. [Google Scholar] [CrossRef]
- Dvořáčková, E.; Šnóblová, M.; Hrdlička, P. Carbohydrate analysis: From sample preparation to HPLC on different stationary phases coupled with evaporative light-scattering detection. J. Sep. Sci. 2014, 37, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Rippe, J.M.; Angelopoulos, T.J. Added sugars and risk factors for obesity, diabetes and heart disease. Int. J. Obes. 2016, 40, S22–S27. [Google Scholar] [CrossRef] [PubMed]
- Louie, J.C.Y.; Moshtaghian, H.; Boylan, S.; Flood, V.M.; Rangan, A.M.; Barclay, A.W.; Brand-Miller, J.C.; Gill, T.P. A systematic methodology to estimate added sugar content of foods. Eur. J. Clin. Nutr. 2015, 69, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Uçar, G.; Balaban, M. Hydrolysis of polysaccharides with 77% sulfuric acid for quantitative saccharification. Turk. J. Agric. For. 2003, 27, 361–365. [Google Scholar]
- Yan, X. High performance liquid chromatography for carbohydrate analysis. In High-Performance Liquid Chromatography (HPLC): Principles, Practices and Procedures; Zhuo, Y., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2014; ISBN 978-1-62948-854-7. [Google Scholar]
- Thøgersen, R.; Castro-Mejía, J.L.; Sundekilde, U.K.; Hansen, L.H.; Hansen, A.K.; Nielsen, D.S.; Bertram, H.C. Ingestion of an inulin-enriched pork sausage product positively modulates the gut microbiome and metabolome of healthy rats. Mol. Nutr. Food Res. 2018, 13, e1800608. [Google Scholar] [CrossRef]
- Szpylka, J.; Thiex, N.; Acevedo, B.; Albizu, A.; Angrish, P.; Austin, S.; Bach Knudsen, K.E.; Barber, C.A.; Berg, D.; Bhandari, S.D.; et al. Standard Method Performance Requirements (SMPRs®) 2018.002: Fructans in Animal Food (Animal Feed, Pet Food, and Ingredients). J. AOAC Int. 2018, 101, 1283–1284. [Google Scholar] [CrossRef] [PubMed]
- Longland, A.C.; Byrd, B.M. Pasture nonstructural carbohydrates and equine laminitis. J. Nut. 2006, 136, 2099S–2102S. [Google Scholar] [CrossRef]
- Stöber, P.; Bénet, S.; Hischenhuber, C. Simplified Enzymatic High-Performance Anion Exchange Chromatographic Determination of Total Fructans in Food and Pet Foods-Limitations and Measurement Uncertainty. J. Agric. Food Chem. 2004, 52, 2137–2146. [Google Scholar] [CrossRef]
- Rühmann, B.; Schmid, J.; Sieber, V. High throughput exopolysaccharide screening platform: From strain cultivation to monosaccharide composition and carbohydrate fingerprinting in one day. Carbohydr. Polym. 2015, 122, 212–220. [Google Scholar] [CrossRef]
- Affonso, R.C.L.; Voytena, A.P.L.; Fanan, S.; Pitz, H.; Coelho, D.S.; Hortmann, A.L.; Pereira, A.; Uarrota, V.G.; Hillmann, M.C.; Varela, L.A.C.; et al. Phytochemical composition, antioxidant activity, and the effect of the aqueous extract of coffee (Coffea arabica L.) bean residual press cake on the skin wound healing. Oxidative Med. Cell. Longev. 2016, 2016, 1923754. [Google Scholar] [CrossRef]
- Shao, J.; Cao, W.; Qu, H.; Pan, J.; Gong, X. A novel quality by design approach for developing an HPLC method to analyze herbal extracts: A case study of sugar content analysis. PLoS ONE 2018, 13, e019515. [Google Scholar] [CrossRef] [PubMed]
- Swetwiwathana, A.; Visessanguan, W. Potential of bacteriocin-producing lactic acid bacteria for safety improvements of traditional Thai fermented meat and human health. Meat Sci. 2015, 109, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Gurtler, J.B.; Mai, T.L. Traditional preservatives: Organic acids. Encycl. Food Microbiol. 2014, 3, 119–130. [Google Scholar]
- Anyasi, T.A.; Jideani, A.I.O.; Edokpayi, J.N.; Anokwuru, C.P. Application of Organic Acids in Food Preservation. In Organic Acids, Characteristics, Properties and Synthesis; Vargas, C., Ed.; Nova Sciences Publishers, Inc.: New York, NY, USA, 2016; pp. 1–47. ISBN 9781634859523. [Google Scholar]
- Neffe-Skocińska, K.; Sionek, B.; Ścibisz, I.; Kotoźyn-Krajewska, D. Acid contents and the effect of fermentation condition of Kombucha tea beverages on physicochemical, microbiological and sensory properties. CyTA-J. Food 2017, 15. [Google Scholar] [CrossRef]
- Nour, V.; Trandafir, I.; Ionica, M.E. HPLC Organic Acid Analysis in Different Citrus Juices under Reversed Phase Conditions. Not. Bot. Hort. Agrobot. Cluj 2010, 38, 44–48. [Google Scholar]
- Lobo Roriz, C.; Barros, L.; Carvalho, A.M.; Ferreira, I.C.F.R. HPLC-Profiles of Tocopherols, Sugars, and Organic Acids in Three Medicinal Plants Consumed as Infusions. Int. J. Food Sci. 2014, 2014, 241481. [Google Scholar] [CrossRef]
- Scherer, R.; Rybka, A.C.P.; Ballus, C.A.; Dillenburg Meinhart, A.; Texeira Filho, J.; Texeira Godoy, H. Validation of HPLC method for simultaneous determination of main organic acids in fruits and juices. Food Chem. 2012, 135, 150–154. [Google Scholar] [CrossRef]
- Llano, T.; Quijorna, N.; Andrés, A.; Coz, A. Sugar, acid and furfural quantification in a sulphite pulp mill: Feedstock, product and hydrolysate analysis by HPLC/RID. Biotechnol. Rep. 2017, 15, 75–83. [Google Scholar] [CrossRef]
- Jain, I.; Kumar, V.; Satyanarayana, T. Xylooligosaccharides: An economical prebiotic from agroresidues and their health benefits. Indian J. Exp. Biol. 2015, 53, 131–142. [Google Scholar]
- Saleh Zaky, A.; Pensupa, N.; Andrade-Eiroa, Á.; Tucker, G.A.; Du, C. A new HPLC method for simultaneously measuring chloride, sugars, organic acids and alcohols in food samples. J. Food Compos. Anal. 2017, 56, 25–33. [Google Scholar] [CrossRef]
- Ergönül, P.G.; Nergiz, C. Determination of Organic Acids in Olive Fruit by HPLC. Czech J. Food Sci. 2010, 28, 202–205. [Google Scholar] [CrossRef]
- Tašev, K.; Stefova, M.; Ivanova-Petropulos, V. HPLC method validation and application for organic acid analysis in wine after solid-phase extraction. Maced. J. Chem. Chem. Eng. 2016, 35. [Google Scholar] [CrossRef]
- Mihaljević Žulj, M.; Puhelek, I.; Jagatić Korenika, A.M.; Maslov Bandić, L.; Pavlešić, T.; Jeromel, A. Organic Acid Composition in Croatian Predicate Wines. Agric. Conspec. Sci. 2015, 80, 113–117. [Google Scholar]
- Sánchez-Machado, D.I.; López-Cervantes, J.; Martínez-Cruz, O. Quantification of Organic Acids in Fermented Shrimp Waste by HPLC. Food Technol. Biotechnol. 2008, 46, 456–460. [Google Scholar]
- Ke, W.C.; Ding, W.R.; Xu, D.M.; Ding, L.M.; Zhang, P.; Li, F.D.; Guo, X.S. Effects of addition of malic or citric acids on fermentation quality and chemical characteristics of alfalfa silage. J. Dairy Sci. 2017, 100, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fasciano, J.M.; Mansour, F.R.; Danielson, N.D. Ion-Exclusion High-Performance Liquid Chromatography of Aliphatic Organic Acids Using a Surfactant-Modified C18 Column. J. Chromatogr. Sci. 2016, 54, 958–970. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Cheng, W.; Zhao, Z.; Cao, J. HPLC method for the simultaneous quantification of the major organic acids in Angeleno plum fruit. IOP Conf. Ser. Mater. Sci. Eng. 2014, 62, 012035. [Google Scholar] [CrossRef]
- Coblentz, W.K.; Akins, M.S. Silage review: Recent advances and future technologies for baled silages. J. Dairy Sci. 2018, 101, 4075–4092. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Yu, P.; Ali, M.; Cone, J.W.; Hendriks, W.H. Nutritive value of maize silage in relation to dairy cow performance and milk quality. J. Sci. Food Agric. 2015, 95, 238–258. [Google Scholar] [CrossRef]
- Silva, V.P.; Pereira, O.G.; Leandro, E.S.; Da Silva, K.G.; Ribeiro, K.G.; Mantovani, H.C.; Santos, S.A. Effects of lactic acid bacteria with bacteriocinogenic potential on the fermentation profile and chemical composition of alfalfa silage in tropical conditions. J. Dairy Sci. 2016, 99, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, M.J.; Kim, H.J.; Cho, S.K. Development of HPLC-UV method for detection and quantification of eight organic acids in animal feed. J. Chromatogr. Sep. Tech. 2017, 8, 385. [Google Scholar] [CrossRef]
- Grune, T.; Lietz, G.; Palou, A.; Ross, A.C.; Stahl, W.; Tang, G.; Thurnham, D.; Yin, S.A.; Biesalski, H.K. β-Carotene Is an Important Vitamin A Source for Humans. J. Nutr. 2010, 140, 2268S–2285S. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C. What is vitamin A and why do we need it? Community Eye Health 2013, 26, 65. [Google Scholar] [PubMed]
- Ding, Z.; Saldeen, T.G.P.; Mathur, P.; Mehta, J.L. Mixed tocopherols are better than alpha-tocopherol as antioxidant as good as statins. Curr. Res. Cardiol. 2016, 3, 128–129. [Google Scholar] [CrossRef]
- Alshahrani, F.; Aljohani, N. Vitamin D: Deficiency, Sufficiency and Toxicity. Nutrients 2013, 5, 3605–3616. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Boyer, F.; Fernandez-Romero, J.M.; Luque de Castro, M.D.; Quesada, J.M. Determination of vitamins D2, D3, K1 and K3 and some hydroxy metabolites of vitamin D3 in plasma using a continuous clean-up–preconcentration procedure coupled on-line with liquid chromatography–UV detection. Analyst 1999, 124, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Schwalfenberg, G.K. Vitamins K1 and K2: The emerging group of vitamins required for human health. J. Nutr. Metab. 2017, 345, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Harshman, S.G.; Finnan, E.G.; Barger, K.J.; Bailey, R.L.; Haytowitz, D.B.; Gilhooly, C.H.; Booth, S.L. Vegetables and Mixed Dishes Are Top Contributors to Phylloquinone Intake in US Adults: Data from the 2011–2012 NHANES. J. Nutr. 2017, 96, 149–154. [Google Scholar] [CrossRef]
- Beulens, J.; Booth, S.; van den Heuvel, E.; Stoecklin, E.; Baka, A.; Vermeer, C. The role of menaquinones (vitamin K2) in human health. Br. J. Nutr. 2013, 110, 1357–1368. [Google Scholar] [CrossRef]
- Chavez-Servín, J.; Castellote, A.; Lopez-Sabater, M.C. Simultaneous analysis of Vitamins A and E in infant milk-based formulae by normal-phase high-performance liquid chromatography-diode array detection using a short narrow-bore column. J. Chromatogr. A 2003, 1122, 138–143. [Google Scholar] [CrossRef]
- Płonka, J.; Toczek, A.; Tomczyk, V. Multivitamin Analysis of Fruits, Fruit–Vegetable Juices, and Diet Supplements. Food Anal. Methods 2012, 5, 1167–1176. [Google Scholar] [CrossRef]
- Sami, R.; Li, Y.; Qi, B.; Wang, S.; Zhang, Q.; Han, F.; Ma, Y.; Jing, J.; Jiang, L. HPLC Analysis of Water-Soluble Vitamins (B2, B3, B6, B12, and C) and Fat-Soluble Vitamins (E, K, D, A, and β-Carotene) of Okra (Abelmoschus esculentus). J. Chem. 2014, 2014, 831357. [Google Scholar] [CrossRef]
- Xue, X.; You, J.; He, P. Simultaneous Determination of Five Fat-Soluble Vitamins in Feed by High-Performance Liquid Chromatography Following Solid-Phase Extraction. J. Chromatogr. Sci. 2012, 46, 345–350. [Google Scholar] [CrossRef]
- Qian, H.; Sheng, M. Simultaneous determination of fat-soluble vitamins A, D and E and pro-vitamin D2 in animal feeds by one-step extraction and high-performance liquid chromatography analysis. J. Chromatogr. A 1998, 825, 127–133. [Google Scholar] [CrossRef]
- Lee, H.; Kwak, B.; Ahn, J.; Jeong, S.; Shim, S.; Kim, K.; Yoon, T.; Leem, D.; Jeong, J. Simultaneous Determination of Vitamin A and E in Infant Formula by HPLC with Photodiode Array Detection. Korean J. Food Sci. Anim. Resour. 2011, 31, 191–199. [Google Scholar] [CrossRef]
- Lim, H.; Woo, S.; Sig Kim, H.; Jong, S.; Lee, J. Comparison of extraction methods for determining tocopherols in soybeans. Eur. J. Lipid Sci. Technol. 2007, 109, 1124–1127. [Google Scholar] [CrossRef]
- Odes, S.; Hisil, Y. Analysis of vitamin A in eggs by high pressure liquid chromatography. Die Nahr. 1991, 35, 391–394. [Google Scholar]
- Reynolds, S.; Judd, H. Rapid procedure for the determination of vitamins A and D in fortified skimmed milk powder using high-performance liquid chromatography. Analyst 1989, 109, 489–492. [Google Scholar] [CrossRef]
- Turner, C.; Persson, M.; Mathiasson, L.; Adlercreutz, P.; King, J.W. Lipase-catalyzed reactions in organic and supercritical solvents: Application to fat-soluble vitamin determination in milk powder and infant formula. Enzym. Microb. Technol. 2001, 29, 111–121. [Google Scholar] [CrossRef]
- Jedlička, A.; Klimeš, J. Determination of Water- and Fat-Soluble Vitamins in Different Matrices Using High-Performance Liquid Chromatography. Chem. Papers 2005, 59, 202–222. [Google Scholar] [CrossRef]
- Rupérez, F.J.; Martín, D.; Herrera, E.; Barbas, C. Chromatographic analysis of alpha-tocopherol and related compounds in various matrices. J. Chromatogr. A 2001, 935, 45–69. [Google Scholar] [CrossRef]
- Stefanov, I.; Vlaeminck, B.; Fievez, V. A novel procedure for routine milk fat extraction based on dichloromethane. J. Food Compos. Anal. 2010, 23, 852–855. [Google Scholar] [CrossRef]
- Hung, G.W.C. Determination of Vitamins D2 and D3 in Feedingstuffs by High Performance Liquid Chromatography. J. Liquid Chromatogr. 1988, 11, 953–969. [Google Scholar] [CrossRef]
- Lee, S.; Morrisa, Y.; Grossa, L.; Portera, F.; Ortiz-Colona, F.; Farrowa, J.; Waqara, A.; Phifera, E.; Kerdahia, K. Development and Single-Lab Validation of an UHPLC-APCI-MS/MS Method for Vitamin K1 in Infant Formulas and Other Nutritional Formulas. J. Regul. Sci. 2015, 2, 27–35. [Google Scholar]
- Wagner, D.; Rousseau, D.; Sidhom, G.; Pouliot MAudet, T.; Vieth, R. Vitamin D3 Fortification, Quantification, and Long-Term Stability in Cheddar and Low-Fat Cheeses. J. Agric. Food Chem. 2008, 56, 7964–7969. [Google Scholar] [CrossRef]
- Kienen, V.; Costa, W.; Visentainer, J.; Souza, N.; Oliveira, C. Development of a green chromatographic method for determination of fat-soluble vitamins in food and pharmaceutical supplement. Talanta 2008, 75, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Gomis, D.B.; Fernández, M.P.; Gutiérrez, M.D. Simultaneous determination of fat-soluble vitamins and provitamins in milk by microcolumn liquid chromatography. J. Chromatogr. A 2000, 891, 109–114. [Google Scholar] [CrossRef]
- Koivu, T.; Piironen, V.; Henttonen, S.; Mattila, P. Determination of Phylloquinone in Vegetables, Fruits, and Berries by High-Performance Liquid Chromatography with Electrochemical Detection. J. Chromatogr. A 1997, 45, 4644–4649. [Google Scholar] [CrossRef]
- Shammugasamy, B.; Ramakrishnan, Y.; Ghazali, H.; Muhammad, K. Tocopherol and tocotrienol contents of different varieties of rice in Malaysia. J. Sci. Food Agric. 2015, 95, 672–678. [Google Scholar] [CrossRef]
- Kim, H.O. Development of an ion-pairing reagent and HPLC-UV method for the detection and quantification of six water-soluble vitamins in animal feed. Int. J. Anal. Chem. 2016, 2016, 8357358. [Google Scholar] [CrossRef]
- Midttun, Ø.; McCann, A.; Aarseth, O.; Kokeide, M.; Kvalheim, G.; Meyer, K.; Ueland, P.M. Combined measurement of 6 fat-soluble vitamins and 26 water-soluble functional vitamin markers and amino acids in 50 μL of serum or plasma by high-throughput mass spectrometry. Anal. Chem. 2016, 88, 10427–10436. [Google Scholar] [CrossRef] [PubMed]
- Kakitani, A.; Inoue, T.; Matsumoto, K.; Watanabe, J.; Nagatomi, Y.; Mochizuki, N. Simultaneous determination of water-soluble vitamins in beverages and dietary supplements by LC-MS/MS. Food Addit. Contam. Part A 2014, 31, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, W.-E.; Yan, J.-Q.; Liu, M.; Zhou, Y.; Shen, X.; Ma, Y.-L.; Feng, X.-S.; Yang, J.; Li, G.-H. A Review of the Extraction and Determination Methods of Thirteen Essential Vitamins to the Human Body: An Update from 2010. Molecules 2018, 23, 1484. [Google Scholar] [CrossRef]
- Abano, E.E.; Dadzie, R.G. Simultaneous detection of water-soluble vitamins using the High Performance Liquid Chromatography (HPLC)—A review. Croat. J. Food Sci. Technol. 2014, 6, 116–123. [Google Scholar] [CrossRef]















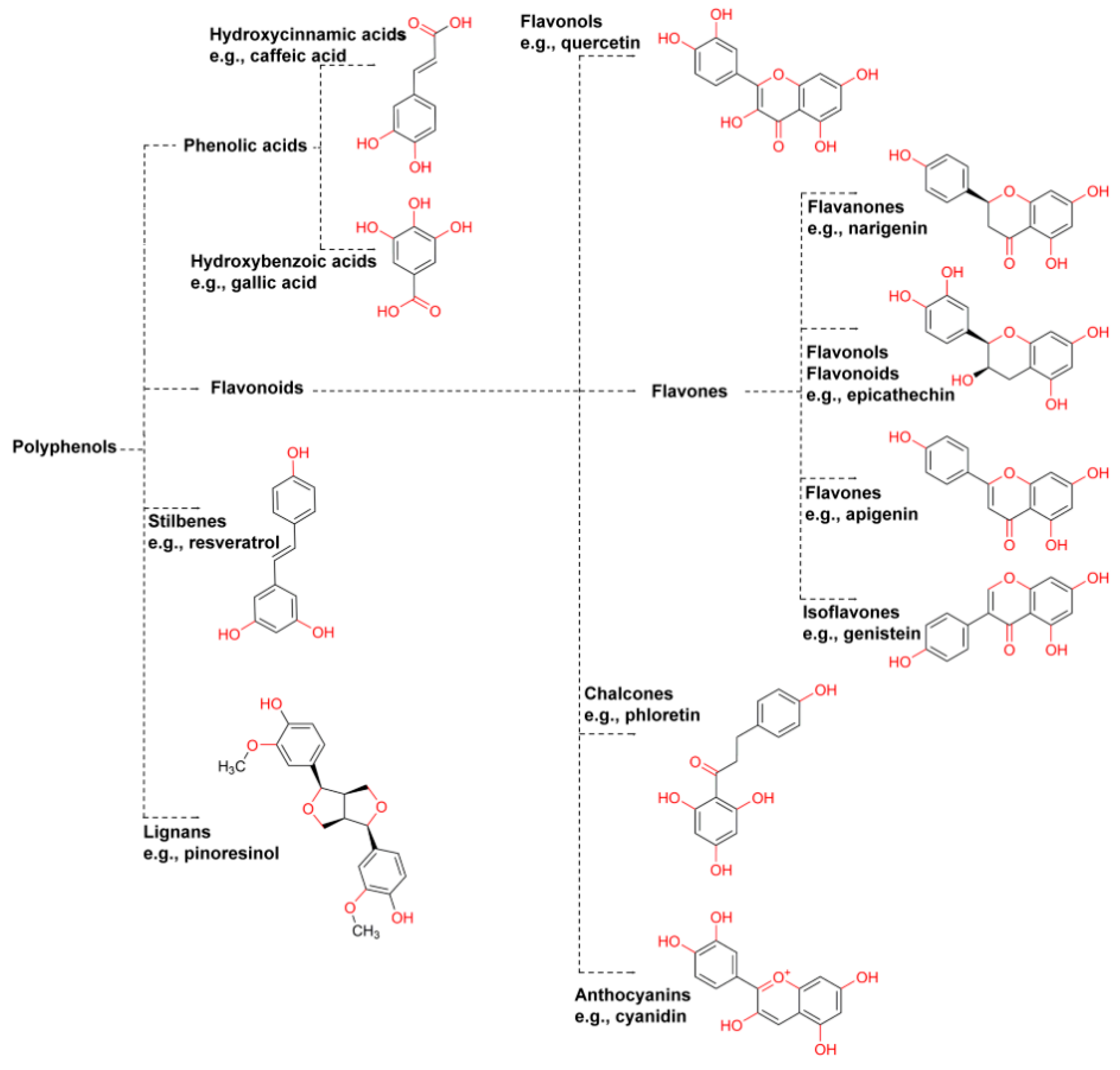{kind=link}
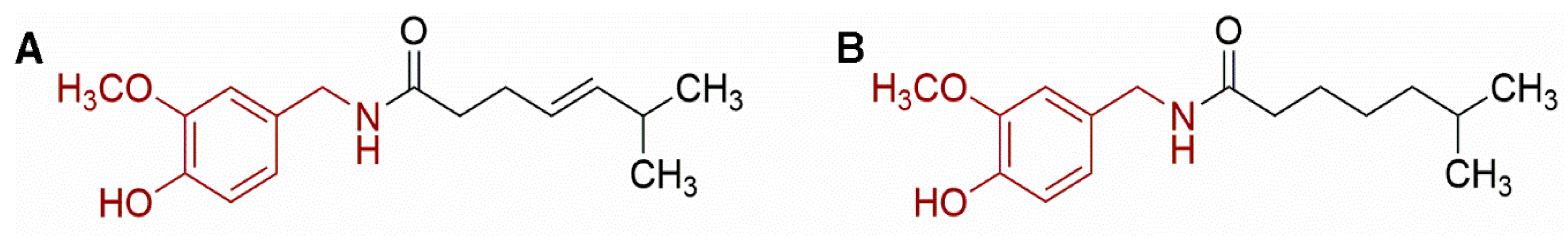{kind=link}
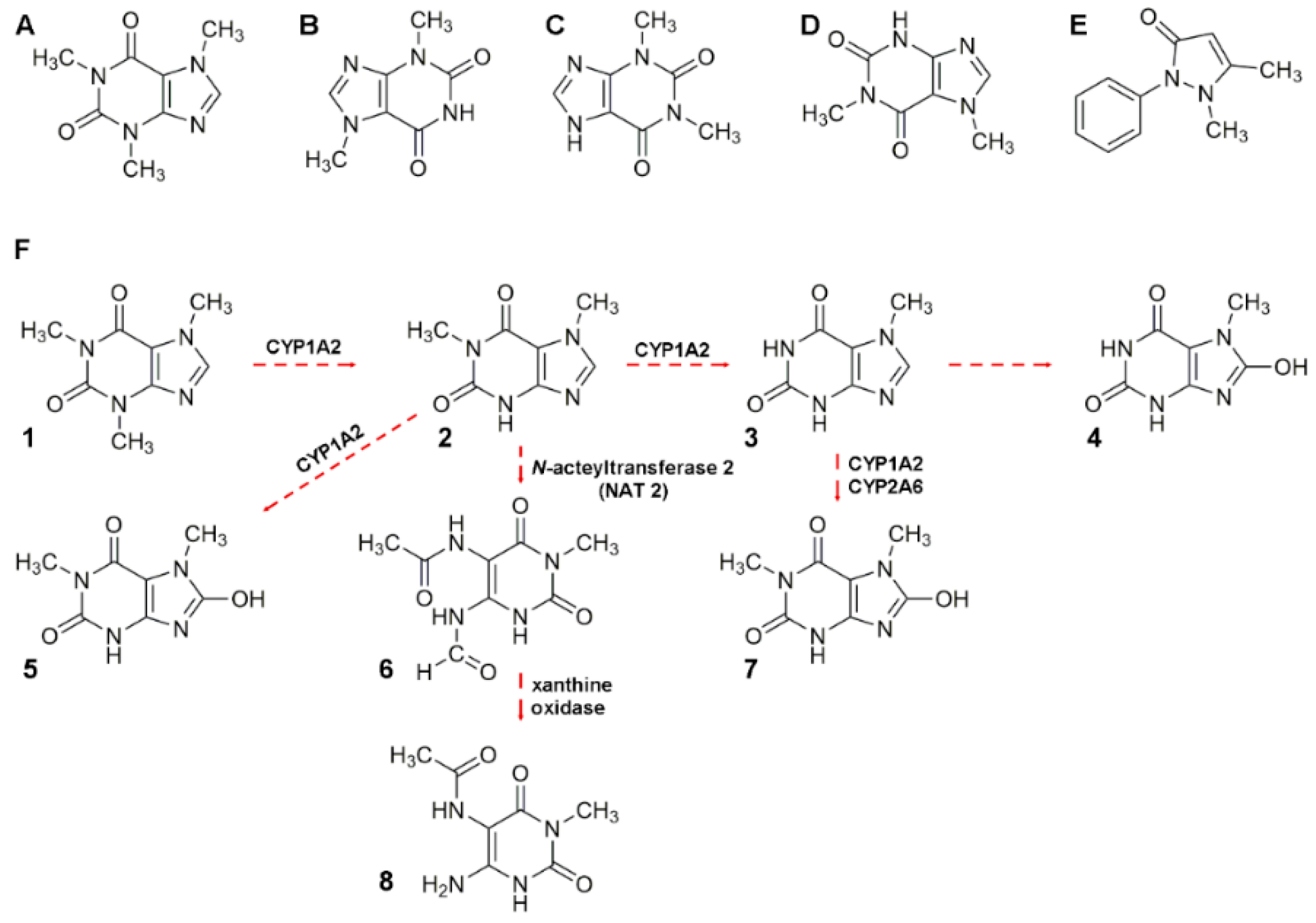{kind=link}
{kind=link}
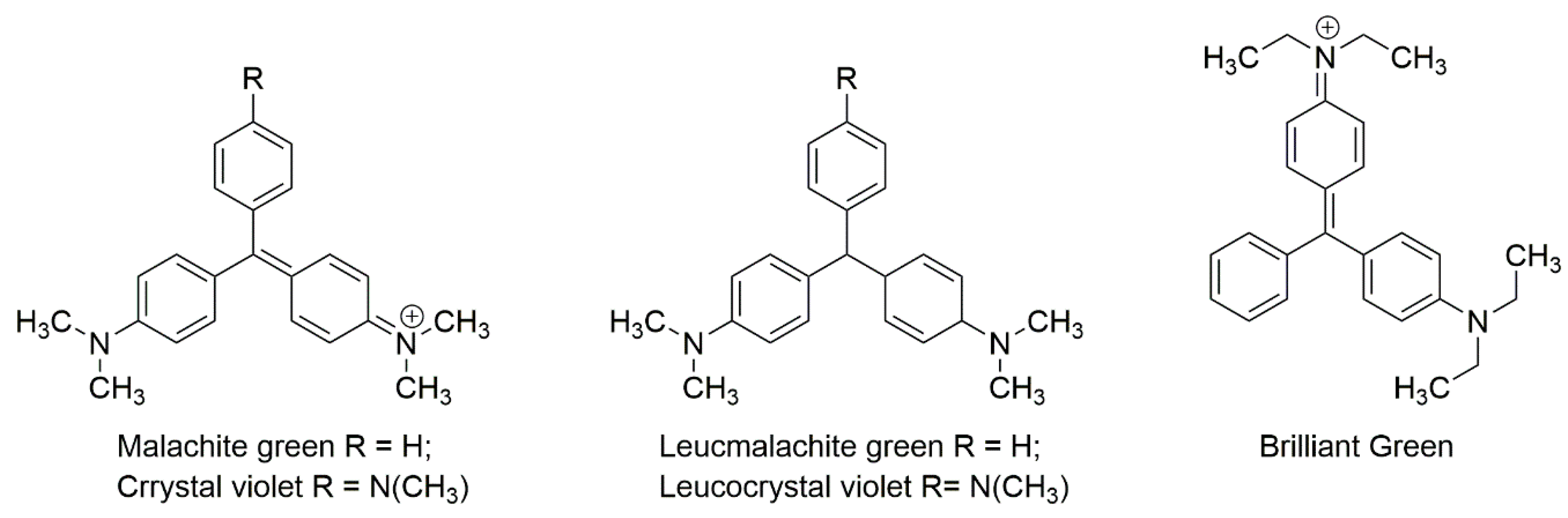{kind=link}
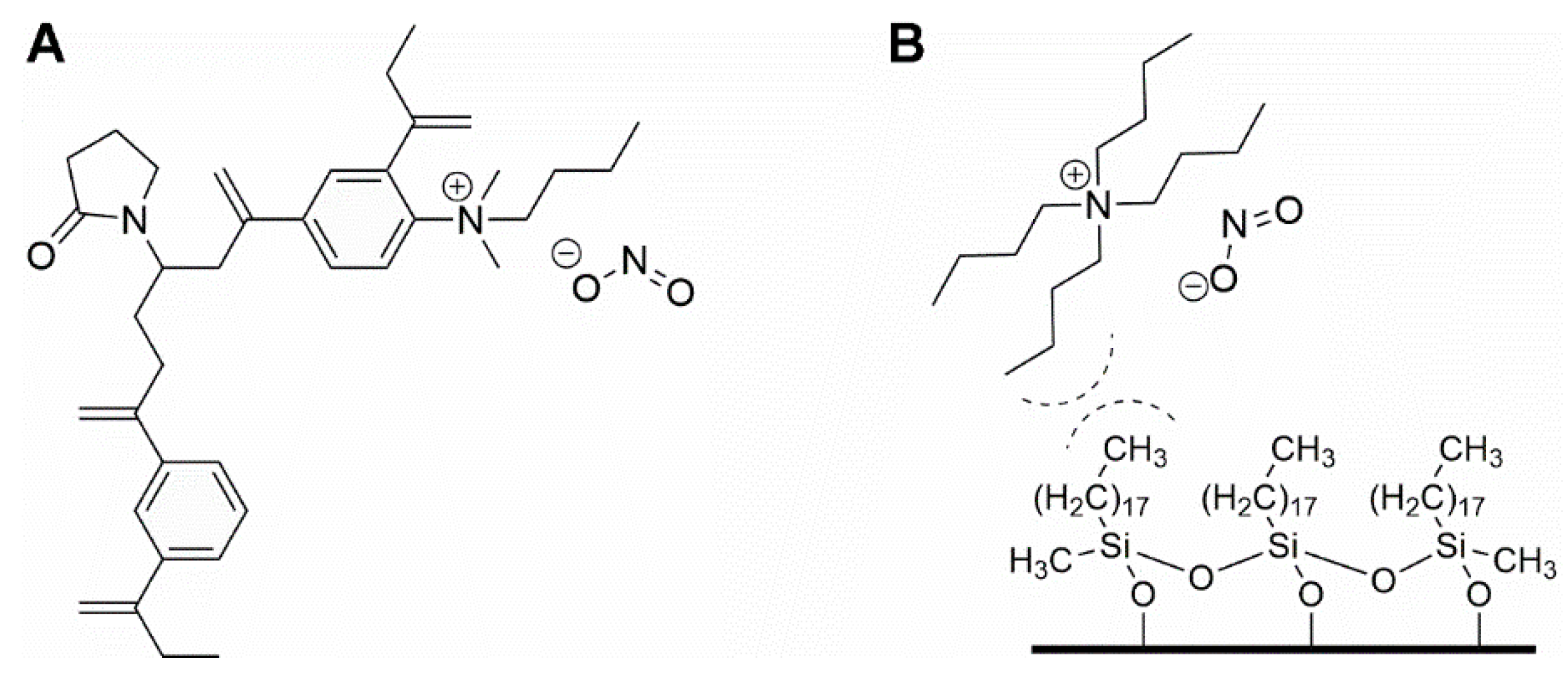{kind=link}
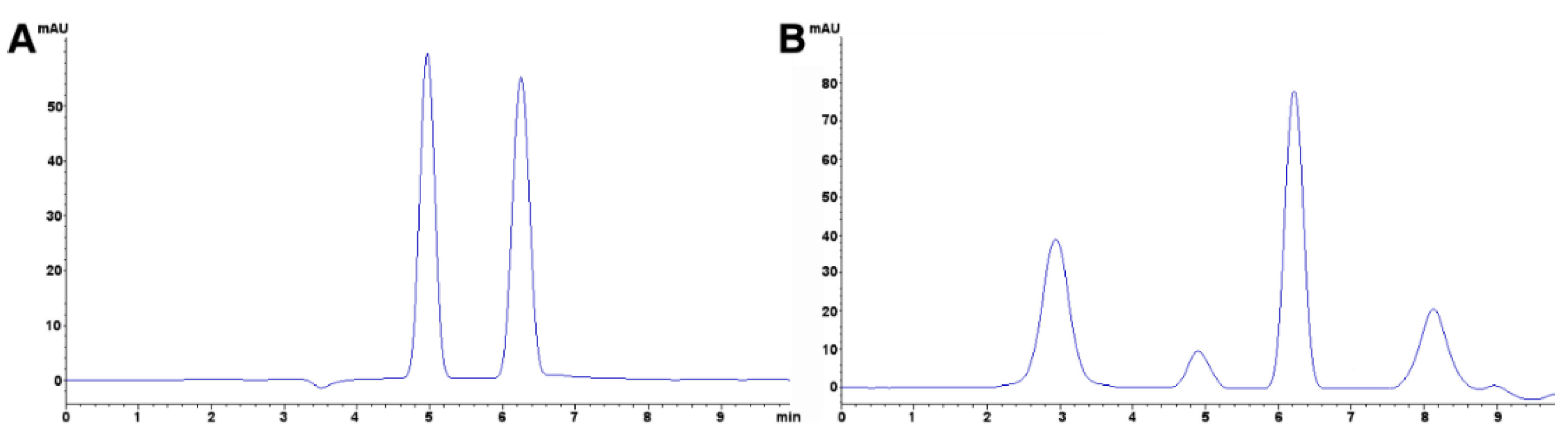{kind=link}
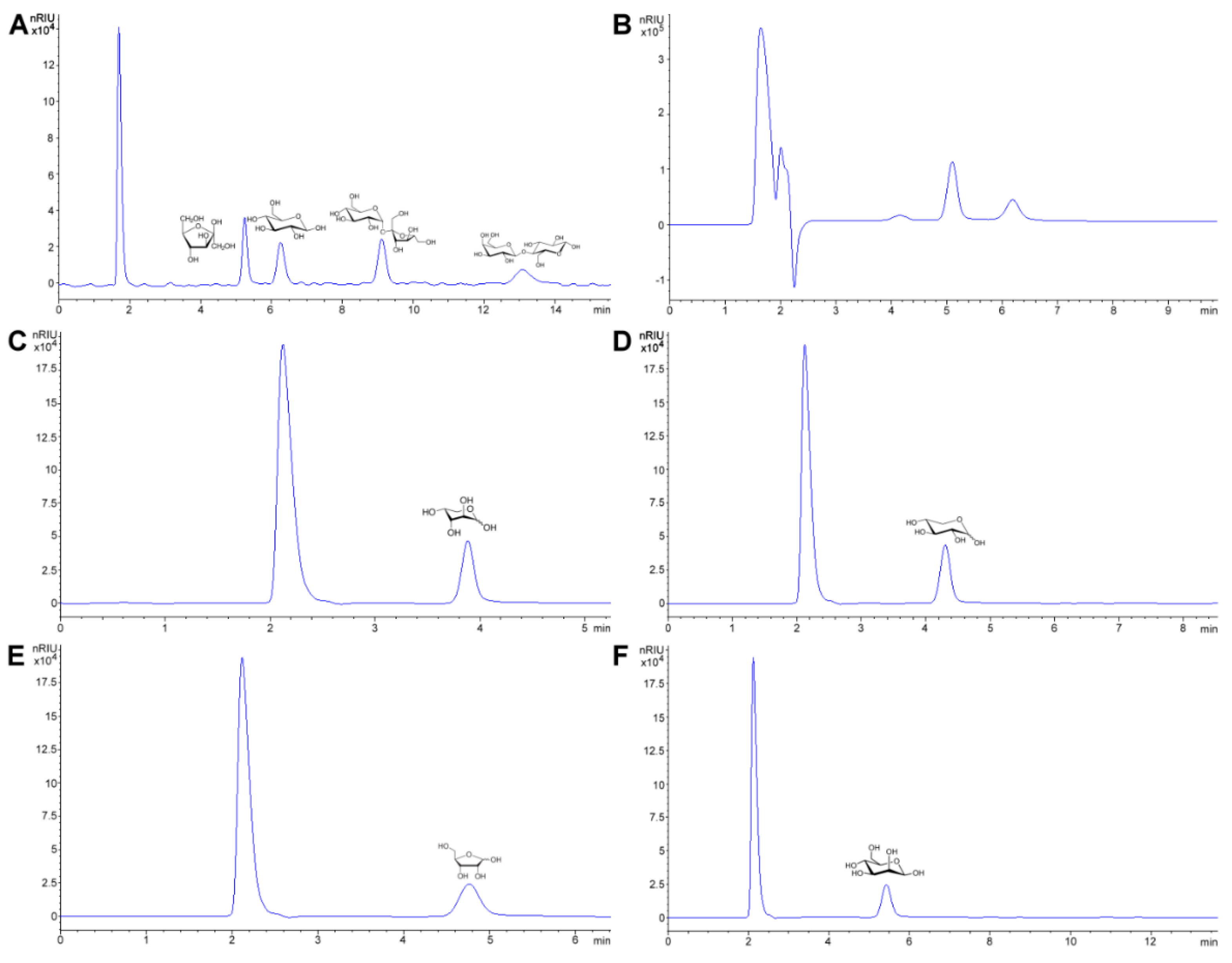{kind=link}
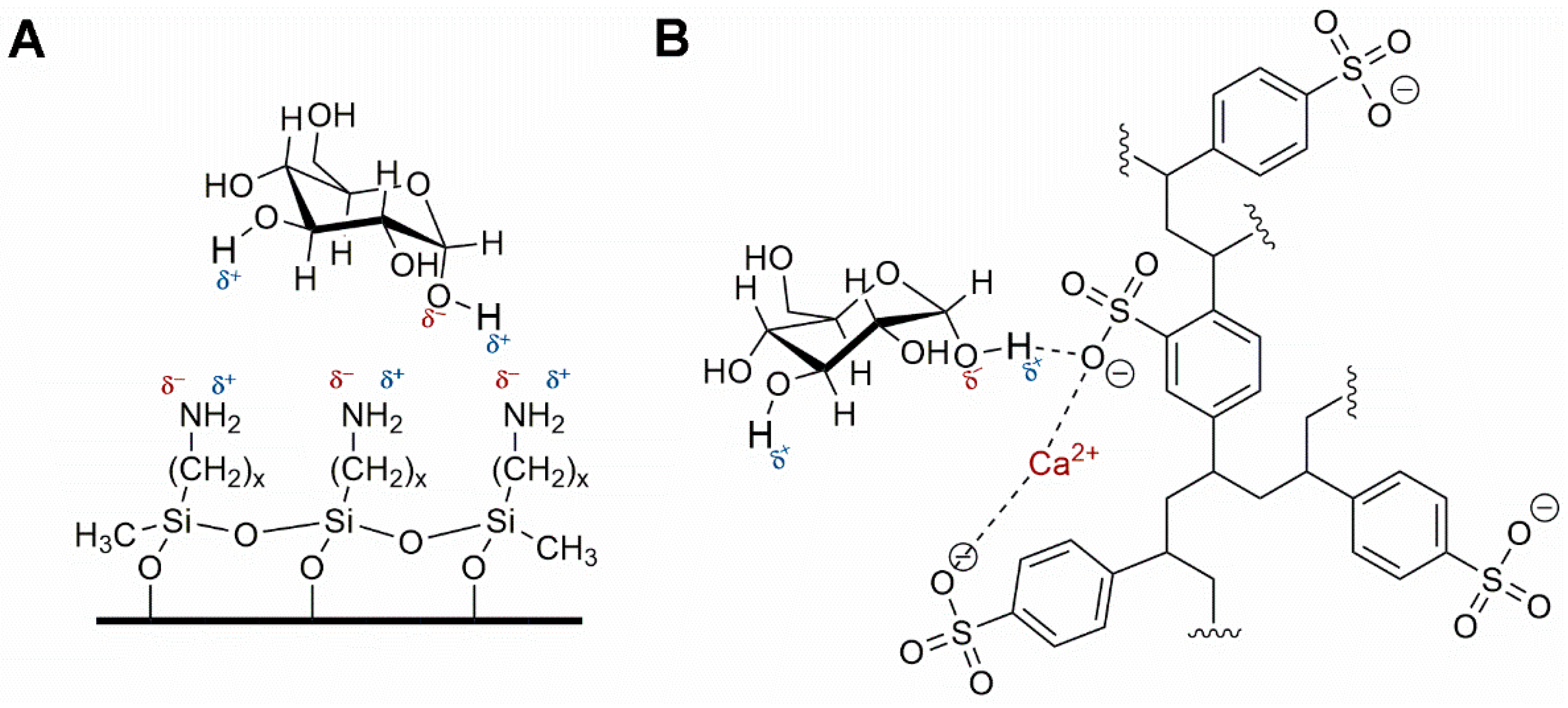{kind=link}
{kind=link}
{kind=link}
{kind=link}
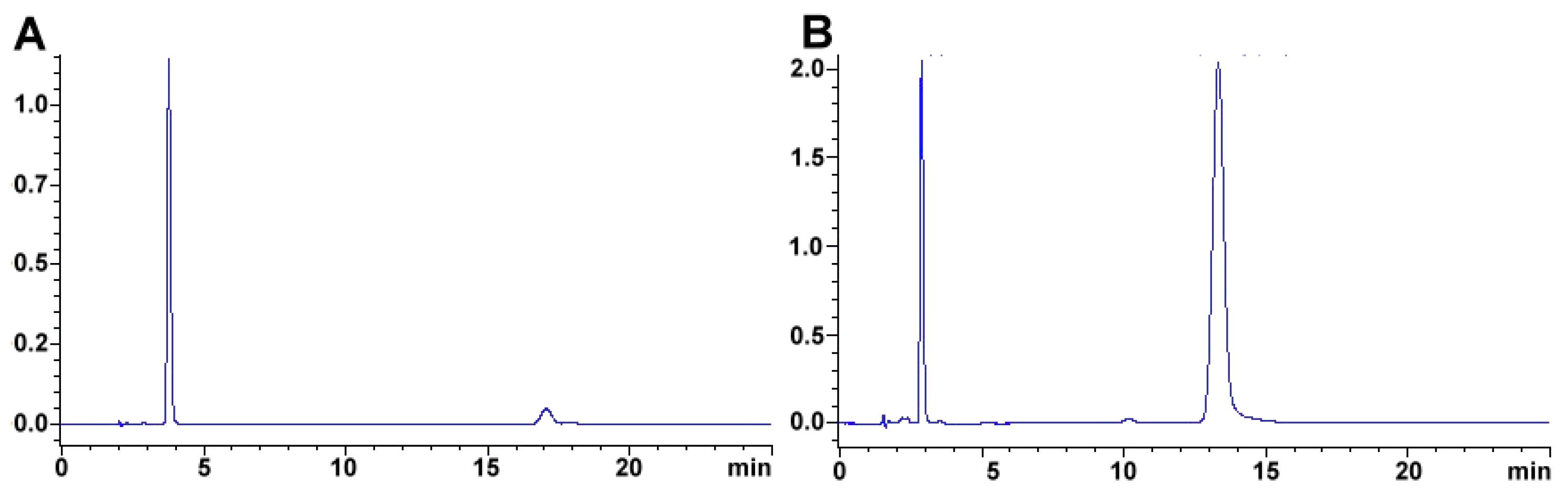{kind=link}
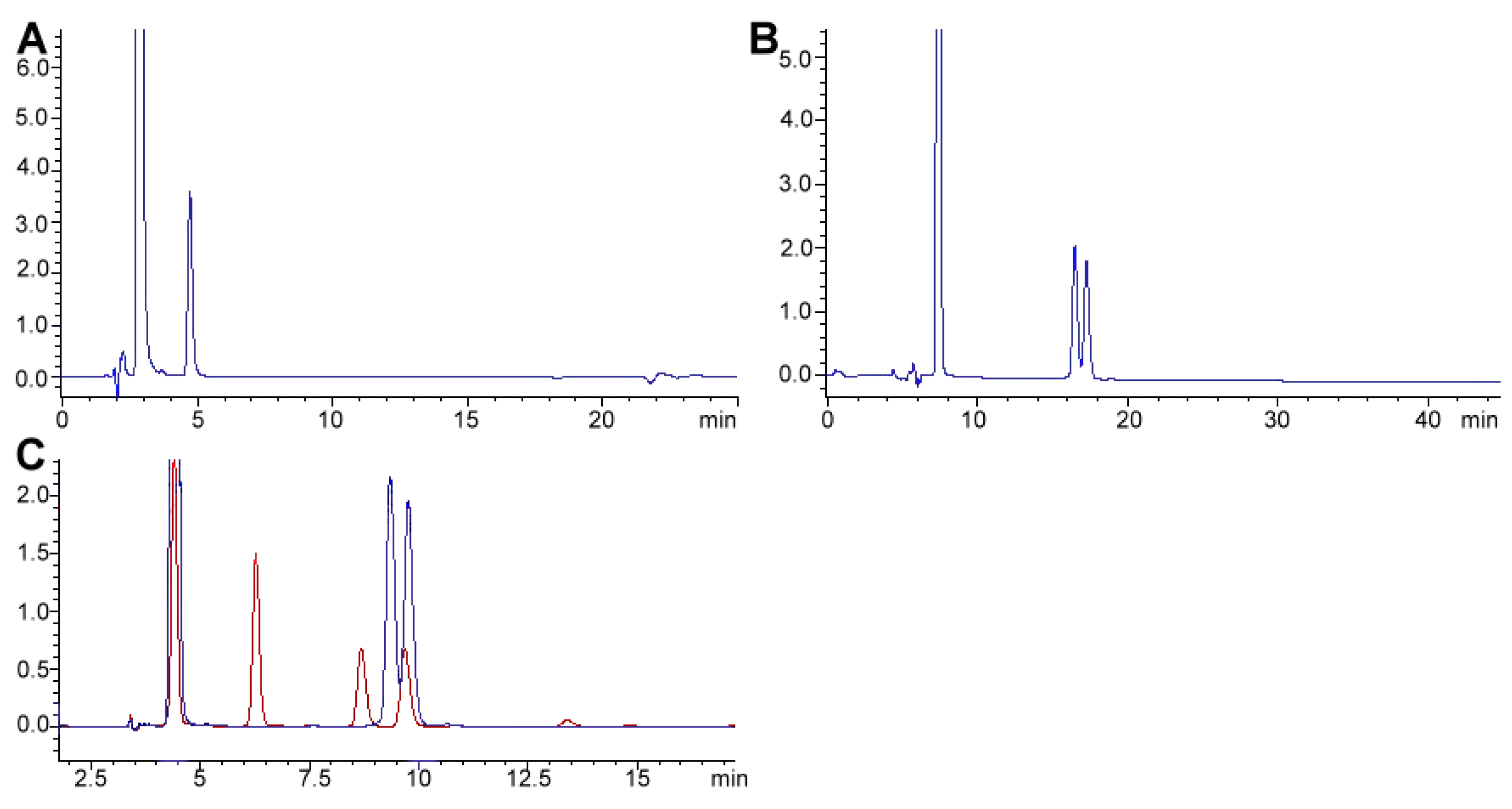{kind=link}
| Additives | |
| Analyte Category Examples | Relevance in Feed and Food Quality |
| Acidulants | Used in beverage, food, and feed production, are part of the primary metabolism, are often produced by fermentation. Acidic additives serve as buffers to regulate acidity, antioxidants, preservatives, flavor enhancers, and sequestrants. Related to beneficial effects on animal health and growth performance as feed additives. |
| Acetic acid, lactic acid, and citric acid [13]. | |
| Antioxidants | Lipid and protein oxidation can impact meat quality, nutrition, safety, and organoleptic properties. Antioxidants are added during animal production and meat processing to enhance the nutritional and health benefits of meat and minimize the formation of carcinogens for the chemical safety of cooked and processed meats [14,15]. They can also be used to extend food [16] and feed [17] shelf life. |
| Gallic, rosemarinic, canosic, and caffeic acids, glabrene, procyanidins, quercetin, catechin α-, β-, γ-, and δ-tocopherols, Eugenol, Carnosine, Tyr-Phe-Glu, and Tyr-Ser-Thr-Ala. | |
| Preservatives | Usually, act as bacteriostatic and bactericidal agents to prevent microbial spoilage, antimicrobials not only extend shelf life, but they also enhance the product’s safety [18]. |
| Acetates, bacteriocins, benzoates (p-hydroxybenzoic acid), borates, carbonates, lactates, nitrates/nitrites, parabens, propionates, sorbates, and sulfites. | |
| Flavors and fragrances | Widely used in food, beverage, feed, cosmetic, detergent, chemical and pharmaceutical formulations [19]. |
| Alcohols, methyl ketones, 2,3-butanedione, lactone, butanoic acid, esters, isovaleric acid, pyrazines, geosmin, vanillin, benzaldehyde, terpenes. | |
| Sweeteners | Non-nutritive sweeteners have become an essential part of daily life and are in increasing demand as it is used in a wide variety of dietary and medicinal products [20]. They play a role in the reduction of table sugar [21]. In the case of artificial sweeteners, their use is controversial as they have associated with health risks [20,22] and water pollution [23]; currently, the use of natural sweeteners is supported as an alternative [24]. Sweetened products must be subject to verification to ensure the presence of the sweetener. Furthermore, sweeteners are regulated food additives [25] unless recognized as safe [26,27]. |
| Approved as food additives: saccharin, aspartame, acesulfame potassium, sucralose, neotame, advantame. Generally regarded as safe (GRAS): Steviosides [28,29,30,31]. | |
| Natural Components | |
| Analyte Category Examples | Relevance in Feed and Food Quality |
| Inorganic ions | Essential in both raw and processed products, related to food nutritional quality, preservation, technological processing, and safety [32]. |
| Sulfites, sulfates, phosphate, polyphosphate, nitrate and nitrite, N-nitroso compounds, cyanide, bromide, bromate, chloride, chlorite, fluoride, iodide. | |
| Lipids and fatty acids | Major constituents of foods and feeds, of dietary importance as a significant source of energy. Provide essential fat-soluble nutrients. Are prone to peroxidation. Part of biological membranes. |
| C1:0 (formic/methanoic), C2:0 (acetic/ethanoic), C3:0 (propionic/propanoic), C4:0 (butyric/butanoic), C6:0 (caproic/hexanoic), C8:0 (caprylic/octanoic), C10:0 (capric/decanoic), C12:0 (lauric/dodecanoic), C12:0 (myristic/tetradecanoic), C16:0 (palmitic/hexadecanoic), 9c-C16:1 (palmitoleic/(9Z)-hexa-dec-9-enoic), stearic, oleic, ricinoleic, vaccenic, linoleic, α-linoleic, γ-linoleic, arachidic, eicosapentaenoic, behenic, erucic, docosahexaenoic, lignoceric, cholesterol [33,34,35,36,37]. | |
| Biogenic amines | Nitrogen-based toxic compounds, mainly formed through decarboxylation of amino acids. Relevant for quality and safety of diverse foods such as dairy products [38], fermented goods [39] including wines [40], fishery commodities [41]. |
| Putrescine, histamine, cadaverine. | |
| Amino acids | Part of a protein-containing diet, and as supplemented individual products. Amino acids are used in medical (parenteral) nutrition and dietary supplements [42]. |
| The main fermentative amino acids for animal nutrition are L-lysine, L-threonine, and L-tryptophan. DL-Methionine. | |
| Carbohydrates | The most abundant feed energy in diets for some species of animals [43,44]. |
| Glucose is the primary energy source for fetal growth, and lactose is crucial for the development of human and animal infants alike. | |
| Vitamins | Complex unrelated compounds present in minute amounts in natural foodstuffs. Essential to normal metabolism; their deficiency causes disease. |
| Fat-soluble: retinol (retinol (vitamin A) and retinyl acetate, and palmitate), tocopherols (α- (vitamin E), β-, γ-, and δ- and their acetates), ergocalciferol (vitamin D2), cholecalciferol (vitamin D3), phylloquinone (vitamin K1), menaquinone (vitamin K2), 7-dehydrocholesterol, β-carotene. Hydrosoluble or B complex vitamins: thiamine (B1), Riboflavin (B2), flavin mononucleotide or riboflavin-5′-phosphate, niacin/nicotinamide riboside/niacinamide (B3), pantothenic acid (B5), pyridoxine/pyridoxamine/pyridoxal (B6), biotin (B7), folates (B9), cobalamines (B12) [45]. | |
| Alkaloids | Alkaloids are natural compounds with a characteristic cyclic structure and a nitrogen atom [46]. Alkaloid-containing plants are an essential part of the regular diet, present as natural constituents of several food products [46]. The most common use for alkaloid-containing plants is as stimulants increased concentrations of these compounds can be attained within the food chain as a result of food processing, as food contaminants or as food flavorings [46]. |
| Octopamine, synephrine, tyramine, N-methyl-tyramine, hordenine in bitter orange products [47], morphine, codeine, thebaine, papaverine, and noscapine in poppy straw [48], caffeine and trigonelline in coffee [49], indole and oxindole alkaloids in Uncaria sp. [50], theobromine and caffeine in tea [51] and coffee [52], Harman alkaloids (harmane and harmine) in passion fruit [53], ergot alkaloids in animal feed (ergometrine, ergotamine, ergocornine, ergocryptine, ergocristine [54], piperine [55]. | |
| Residues and Contaminants | |
| Analyte Category Examples | Relevance in Feed and Food Quality |
| Chemotherapeutics and antiparasitic drugs | Antibiotics are extensively utilized in productive animals with therapeutic, prophylactic, metaphylactic, growth promoting, and food effectiveness enhancing ends. These practices that have been reflected in veterinary residues in products for human consumption (meat, eggs, and milk) and is also related to directly with allergies and antimicrobial resistance. |
| Tetracyclines [56]. | |
| Mycotoxins | Mycotoxins are practically ubiquitous contaminants, classified as teratogenic, carcinogenic and immunosuppressive, and that affects a great variety of grains, fruits and seeds, as well as eggs, dairy products, compounds feeds, and other feed ingredients [57]. |
| Aflatoxins [58]. | |
| Pesticides | Used for crop protection and to treat infestations in livestock. Their poor use results in contamination of the environment and the food itself, impacting human health. Residues usually found in vegetables, fruits, honey, fish, eggs, milk, and meat, serving as potential sources of contamination to consumers [59,60,61]. |
| Atrazine, glyphosate, aminomethylphosphonic acid, phenoxy herbicides. | |
| Type of Liquid Chromatography | Micro | Semi-Micro | Conventional | Semi-Preparative | Preparative | Process |
|---|---|---|---|---|---|---|
| Column internal diameter, mm | 0.3 < x ≤ 1.0 | 1.0 < x ≤ 3.0 | 4.0 < x ≤ 8.0 | 8.0 < x ≤ 20.0 | 20.0 < x ≤ 50.0 | x > 50.0 |
| Eluent flow rate, mL min−1 | 0.001 < x ≤ 0.1 | 0.1 < x ≤ 0.4 | 0.4 < x ≤ 2.0 | 2.0 < x ≤ 10.0 | 10.0 < x ≤ 150.0 | x > 150.0 |
| Detector Type Range of Applications, Attributes, and Minimal Detectable Quantity Limitations | Applications in Feed and Food Quality |
|---|---|
| Non-Destructive Detectors | |
| Photodiode-Array (PDA)/Variable wavelength (VW)/UV-vis | Sulfonated azo dyes in beverages, hard candy and fish roe samples [62], purity of caffeine reference material [63], sulfamethazine and trimethoprim in liquid feed premixes [64], nitrofurans animal feed [65]. |
| Selective; universal at low wavelengths, 3D spectra comparison can determine peak purity, can detect nanograms. Chromophore must be presesent, solvents transparent to the wavelength used must be provided. | |
| Fluorescence (FL) | |
| Very selective and specific; monitors two wavelengths simultaneously, 3D fluorescent spectra, fluorescent fingerprinting/fluorescence pattern analysis [66] gradients do not affect baseline significantly, can detect low picograms. Fluorophore must be present, derivative formation and quenching are often needed. | Sulfonamides [67] and fluoroquinolones [68] in animal feed, aflatoxins in agricultural food crops [69] and milk [70]. |
| Electrochemical (EC) | Macrolide antibiotics in animal feeds [71], vitamin C in oranges and apples [72]. |
| Very selective; oxidation or reduction must be possible, can detect from femtograms to nanograms. Conductive mobile phase, susceptible to background noise and electrode degradation. | |
| Refractive index (RI) | Inulin in chicory roots [73], total carbohydrates in wine and wine-like beverages [74]. |
| Universal (All compounds affect refractive properties) and versatile; solvent compatible, relatively simple, can detect micrograms. Gradient incompatible, high S/N ratio when the pump is mixing two or more solvents, susceptible to temperature and flow variation. | |
| Conductivity | Choline, and trimethylamine in feed additives [75], L-carnitine, choline, and metal ions in infant formula [76]. |
| Selective; an ionic form of compound necessary can detect low pictograms. Suppression of mobile phase background conductivity, special equipment required. | |
| Destructive detectors | |
| Mass spectrometry | Analysis of acrylamide in food [77], tiamulin, trimethoprim, tylosin, sulfadiazine, and sulfamethazine residues in medicated feed [78], multiclass antibiotics in eggs [79], zearalenone and deoxynivalenol metabolites in milk [80], cattle feed analysis of Aspergillus clavatus mycotoxins [81], choline chloride in feed and feed premixes [82]. |
| Selective and specific; based on a specified mass/charge ratio, ion fractionation, can detect low nanograms. Expensive, expert users are needed for equipment and data manipulation. | |
| Radioactivity | Drug metabolite identification [83]. |
| Selective; Distribution and mass balance wide response range can detect pictograms. Large-volume flow cells can produce peak broadening and decreased the resolution. | |
| Evaporative light scattering (ELS) | N-acetylglucosamine and N-acetylgalactosamine in dairy foods [84], sucralose and related compounds [85], spectinomycin and associated substances [86]. |
| Universal; Nonvolatile analyte nebulization, can detect in the range of nanograms. Volatile buffers required, poor reproducibility and limited dynamic range. | |
| Corona-Charged aerosol [87] | |
| Universal; can detect non-ultraviolet and weakly ultraviolet active compounds [88], ionized particles measured by an electrometer, can detect low nanograms. Volatile buffers required. | Erythritol, xylitol, sorbitol, mannitol, maltitol, fructose, glucose, sucrose, and maltose in food products [89], fumonisins in maize [90]. |
| Matrix | Analytes Identified | Extraction Method | Measurement Method, Chromatographic Column | Reference |
|---|---|---|---|---|
| Berries | Anthocyanins (68.6%), hydroxycinnamic acids (23.9%), flavonols (4.4%) | H2O, membrane ultrafiltration | Preparative LC: 250 × 20 mm Eurospher 100–5 C18 Identification: was HPLC/DAD/ESI±-MSn, 150 × 2.1 mm, 5 μm. λ 280, 325, 360 and 520 nm | [98] |
| Costa Rican guava | Ellagic acid, myricetin, quercitrin, and quercetin | MeOH/H2O (70 mL/100 mL). Freeze-dryed pulp, mechanical dispersion | LC-TOF-ESI± (m/z range 100–1000), Synergi Hydro RP 80A 250 × 4.6 mm, 4 μm. | [110] |
| Brazilian guava, jambolan, nance, and lúcuma | Hydrolyzable and condensed tannins, flavonols, and flavanols | Acetone/H2O/HCOOH (70:29:1). Freeze-dryed pulp, accelerated solvent extraction | HPLC-DAD-ESI−-MSn, Aqua RP18 150 × 2.0 mm, 3 μm. | [113] |
| Perilla frutescents (L.) Britton | Rosmarinic acid (12.7–85.3%), scutellarein-7-O-glucuronide (6.5–45.1%), caffeic acid, apigenin-7-O-diglucuronide, and apigenin-7-O-glucuronide | Ethanol (EtOH)/H2O (75 mL/100 mL). Accelerate solvent extraction (N2 1200 psi 70 °C) | UPLC-PDA-ESI−-TOF-MS, Kintex XB C18 column 150 × 2.1 mm, 1.7 μm | [114] |
| Solanum lycopersicum L. | e.g., caffeic acid hexosides, homovanillic acid hexoside, and dicaffeoylquinic acid (increasing trend) | Methanol (MeOH)/H2O (80 mL/100 mL) | HPLC-DAD-ESI−-MS/MS, Zorbax 300SB-C18 column (2.1 × 150 mm; 5 μm) | [115] |
| Rubus fruticosus L., Prunus spinos L. and Cornus mas L. | Gallic acid (138.0–443.5 mg kg−1 fresh weight), rutin (13.9–22.8 mg kg−1 fresh weight) | HCOOH/MeOH/H2O (0.1/70/29.9) | LC-FLD λex 280, 320, 322 nm λem 360 nm. Eclipse XDB C18 150 × 4.6 mm | [116] |
| Green, herbal and fruit teas | Gallic acid, caffeic acid (+)-catechin, (–)-epicatechin, (–)-epigallocatechin, procyanidin B1, and procyanidin B2 contribute to 43.6–99.9% | 95 °C for 10 min | LC-PDA/FLD scan 260–400 nm absorbance matching Zorbax Eclipse XDB-C18, 150 × 4.6 mm, 5 μm | [117] |
| Dried and candied fruit | Vanillic, ellagic, gallic, p-coumaric, chlorogenic, caffeic, ferulic, rosmarinic acids, and myricetin, quercetin, kaempferol, delphinidin, cyanidin, and pelargonidin | MeOH/H2O (62.5 mL/100 mL). Sonication | HPLC-DAD at 260, 280, 329, and 520 nm. Zorbax Eclipse Plus C18 column 150 × 4.6 mm, 3.5 µm | [118] |
| Pink guava | Ellagitannins, flavones, flavonols, flavanols, proanthocyanidins, dihydrochalcones, and anthocyanidins, and non-flavonoids such as phenolic acid derivatives, stilbenes, acetophenones, and benzophenones | Freeze dried pulp, MeOH/H2O (90:10), sonication | UHPLC-DAD-ESI+-MS/MS, BHE Shield RP18 150 × 2.1 mm, 1.7 μm. | [119] |
| Blackberry juice | Microfiltrate (tubular ceramic membrane) | HPLC-DAD-ESI+-IT-MS/MS Lichrosrb ODS-2 250 × 4.6 mm, 5 µm | [120] |
| Matrix | Extraction Method | Measurement Method, Chromatographic Column | Sensitivity, mg L−1 or mg kg−1 Fruit Dry Weight | Reference |
|---|---|---|---|---|
| Peppers Capsicum annuum L. | ACN, mechanical shaking | RP-LC/MS-TOF/ESI−, pseudo-molecular ions [M-H]− 304.2 and 306.2 m/z. IS 4,5-dimethoxybenzyl)-4-methyloctamide, 250 × 4.6, 5 µm | 0.06 | [128] |
| Natural capsaicinoid mixture (capsaicin/dihydrocapsaicin 67:33) | C7H16/EtAOc/MeOH/H2O (1:1:1:1) | 1. Sequential centrifugal partition chromatography. 2. Nucleosil 100-5 C18 column (125 × 3 mm, 5 µm, UV 280 nm | Preparative chemistry | [129] |
| Hot chilies, green peppers, red peppers, and yellow peppers | EtOH | HPLC-UV using a wavelength of 222 nm and a Betasil C18 150 × 4.6 mm, 3 μm column | 0.10 | [130] |
| Vegetable and waste oils | Immunoaffinity column, SPE loading solvent, 5 mL MeOH/H2O (5:95), washing solvent PBS, MeOH for elution | LC-ESI+-MS/MS, Hypersil Gold, 100 × 2.1 mm, 3.0 µm | 0.03 | [131] |
| Edible and crude vegetable oils | SPE C18, MeOH | IS capsaicin-d3, and dihydrocapsaicin-d3. RP-UPLC-ESI-MS/MS, ZORBAX Eclipse Plus C18 50 × 2.1 mm, 1.8 µm) | 0.5 | [132] |
| Austrian chili peppers | ACN/H2O (35:65) | UV and FLD λex 280 and λem 310 nm, UPLCTM BEH C18 50 × 2.1 mm, 1.7 μm | 0.136 | [133] |
| Brazilian Capsicum chinense Jacq. | MeOH sonication | UHPLC–DAD–APCI-MS/MS, Hypersil Gold C18 100 × 3 mm, 1.9 μm | 0.0027 | [134] |
| South Korean red peppers | MeOH/H2O (95:5), 80 °C 2 h | FLD λex 280 and λem 325 nm, Zorbax Eclipse XDB-C18 75 × 3 mm, 3.5 µm) | 0.06 | [135] |
| Matrix | Extraction Method | Measurement Method, Chromatographic Column | Sensitivity, mg L−1 or mg kg−1 | Reference |
|---|---|---|---|---|
| Food Samples | ||||
| Energy drinks | Sonication for degassing | DAD 270 nm (caffeine) Nova-Pak C18 150 × 3.9 mm, 5 μm, mobile phase: MeOH, NaH2PO4/hexanesulfonic acid (C6H13SO3H) | 0.023 | [148] |
| Energy drinks | Sonication for degassing. “Dilute and shoot” | 25 mmol L−1 NaAOc/HAOC buffer, pH 6.0, an inertsil OctaDecylSilane-3V 250 × 4.6 mm, 5 μm, UV 230 nm | 0.19 | [149] |
| Cocoa | Defat with C6H14, Acetone/H2O/HAOc (70/29.5/0.5) | 1. HPLC 250 × 4.60 mm, 5 μm 2. UPLC Acquity HSS T3 100 mm × 2.1, 1.8 μm | 0.001 for both LCs | [150] |
| Cocoa-based products | Defat by mechanical dispersion with C6H14, MeOH/H2O (80:20) | UHPLC-Q-Orbitrap-MS/MS polyphenols (n = 35, ESI−) and alkaloids (n = 2, ESI+) Kinetex biphenyl 100 × 2.1 mm, 1.7 µm | Theobromine 0.03, caffeine 0.04 | [151] |
| Mate beer and mate soft drinks | Sonication for degassing, ACN. | HP-TLC LiChrospher silica gel plates, fluorescence indicator and mobile phase acetone/toluene/chloroform (4:3:3) UV 274 nm | 0.4 | [152] |
| Biological Samples | ||||
| Human and synthetic plasma | Ultracentrifugation, 12,000 rpm | Waters Atlantis C18 150 × 4.6 mm, 5 µm. Mobile phase: 15 mmol L−1 PBS (pH 3.5)/ACN (83:17). PDA 274 nm, IS: antipyrine | 0.02 | [153] |
| Human saliva | Chloroform/isopropanol (85:15) | Mobile phase: H2O/HAOc/MeOH/ACN (79:1:20:2), Kromasil 100 C18 250 × 4.6 mm, 5 μm, 30 °C, UV 273 nm | 0.032 | [154] |
| Human and neonate plasma | SPE polymeric 96-well plates Strata-X™. Elution: MeOH/H2O/HAOc (70:29:1) | 10 mmol L−1 PBS (pH 6.8)/ACN (gradient mode). Zorbax® SB-Aq narrow bore RR 100 × 2.1 mm, 3.5 μm), 40 °C, UV 273 nm | 0.1 | [155] |
| Matrix | Extraction Method | Measurement Method, Chromatographic Column | Sensitivity, mg L−1 or mg kg−1 | Reference |
|---|---|---|---|---|
| Egg-, dairy-and meat-based products | ACN/2-propanol | 1. SupelcosilTM LC-18-DB 150 × 4.6 mm, 3 μm | 3 | [166] |
| 2. Acquity UPLC® BEH C18 50 × 2.1 mm, 1.7 µm, UV 210 nm | 0.7 | |||
| Seafood | 1. In situ: KOH 2 mol L−1/MeOH, 80 °C, N2, C6H14 | Vitamin E: FLD λex 290 λem 330). Cholesterol: UV 210 nm, Supelcosil™ LCSI 75 × 3.0 mm, 3 μm, mobile phase: n-hexane and 1,4-dioxane (97.5:2.5) IS: tocol | Vitamin E: 0.05 Cholesterol: 10 | [169] |
| 2. Modified Folch: MeOH/CH2Cl2 (1:2), saponification | ||||
| 3. Smedes: 2-propanol/cyclohexane (1:1.25), saponification | ||||
| Seafood | KOH 50 g/100 g/EtOH, 25 °C, 22 h, in the dark, C6H14 | 1. Nova Pack CN HP 300 × 3.9 mm, 4 μm, n-hexane/2-propanol (97:3), UV 210 nm (cholesterol oxides), RID (cholesterol and epoxides) | 0.01 | [170] |
| 2. Confirmation: HPLC-APCI-MS QTRAP® | ||||
| Dairy product | KOH 50 g/100 g/EtOH, 25 °C, 22 h, in the dark, C6H14 | Restek C18 150 × 6 mm, 5μm, mobile phase: ACN/2-propanol (95:5), UV 202 nm 25-hydroxy and cholesterol, 227 nm 7-ketocholesterol | 11.10 | [171] |
| Egg and dairy product and vegetable oil | 1. Egg yolk and milk: pretreatment with ACN | CLC-ODS-C8 150 × 6 mm, 5μm. Mobile phase: ACN/EtOH (50:50), HPLC-UV 210 nm, | 0.01 | [172] |
| 2. Liquid–liquid dispersion (DLLME) EtOH (800 µL)/CCl4 (35 µL). |
| Matrix | Number of Analytes/Execution Time (min) | Extraction Method | Measurement Method, Chromatographic Column | Reference |
|---|---|---|---|---|
| Cassava meal, peanut cakes, cornmeal, and different sorghum varieties | 25/28 | MeOH/CH3CO2CH2CH3/H2O (70:20:10), cleanup was performed using amino SPE cartridges | LC: Symmetry RP-18 150 × 2.1 mm, 5 µm, Identification: MS/MS/ESI+ | [177,186] |
| Cereals, compound feed and silages | 56/50 | Modified QuEChERS method | LC: Acquity UP3 HSS T3 100 × 2.1 mm, 1.8 µm, Identification: MS/MS/ESI± | [179] |
| Bovine milk | 10/30 | Acid acidified ACN and sodium acetate was used to separate the aqueous from the hydrophilic phase from milk | LC: Ascentis Express C18, 150 × 2.1 mm, 2.7 µm, Identification: MS/MS/ESI+ | [185] |
| Silage | 27/44 | Modified QuEChERS method | LC: Gemini® C6-Phenyl 100 × 2.0 mm, 3 μm, Identification: MS/MS/ESI± | [187] |
| Millet and Sorghum | 84 and 62 respectively/Not Indicated | ACN/H2O/CH3COOH (79:20:1) mixture | LC: Gemini® C18, 150 × 4.6 mm, 5 μm, Identification: MS/MS/ESI± | [188,189] |
| Matrix | Number of Analytes/Execution Time | Extraction Method | Measurement Method, Chromatographic Column | Reference |
|---|---|---|---|---|
| Recent Multiresidue and Multi-Class Analysis of Antibiotics in Feeds | ||||
| Rendering products | 40/Not Indicated | During extraction, fat was removed and clean up performed using an SPE PRiME HLB cartridge, eluate evaporated to dryness and reconstituted with ACN and formic acid | BEH C18 column Identification: HPLC-MS/MS/ESI+ | [201] |
| Compound feed | 3/Not Indicated | Formic acid/H2O (80:10) | Hypersil Gold HILIC (150 × 3.0 mm, 5 µm) and C18 (2.1 × 50 mm, 3.5 µm). Identification: HPLC-MS/MS/ESI+ | [202] |
| Pig, poultry, and cattle feed | 62/13 | ACN/H2O (90:10) acidified with CH3COOH. | C18 Vensusil XBP (50 × 2.1 mm, 3.0 μm, 100 Å). Identification: HPLC-MS/MS/ESI+ | [209] |
| Feed | 10/Not Indicated | Acidic extraction with hydrochloric acid (0.5 mol L−1 aqueous solution), and purified by SPE cartridge | Acquity UPLC HSS T3 (150 × 2.1 mm, 1.7 μm). Identification: HPLC-MS/MS/ESI± | [213] |
| Multiresidue Analysis of Antibiotics in Foods | ||||
| Fish muscle | 41/20 | Extraction with ammonium formate and ACN/H2O (80:20) | X-SELECT C18 (150 × 2.1 mm, 3.5 μm) Identification: HPLC-MS/MS/ESI± | [205] |
| Shrimp | 24/8 | Extraction with formic acid in water and ACN | XBridge BEH C18 (100 × 2.1 mm, 2.5 μm). Identification HPLC-MS/MS/ESI+ | [206] |
| Poultry muscle tissue and eggs | 14/14 | ACN extraction Centrifugation at 0 °C 45 min | Poroshell 120 ECC18 (50 × 3.0 mm, 2.7 μm) Identification: HPLC-MS/MS/ESI± (quadrupole linear ion trap) | [207] |
| Honey | 6/Not Indicated | Modified QuEChERS method Extraction was performed using ACN and MgSO4 and NaCl | ZORBAX Eclipse XDB C-18 (150 × 4.6 mm, 5 µm). Identification: HPLC-MS/MS/ESI+ | [214] |
| Matrix | Hydrolysis | Derivatization | Measurement Method, Chromatographic Column | Reference | |
|---|---|---|---|---|---|
| Applications in Feed and Related Matrices | |||||
| Spirulina sp. | Various physical methods | 2-mercaptoethanol | Licrospher 100 RP 18 125 × 4 mm, FLD λex 360 λem 460 nm | [218] | |
| Spirulina sp. | 1. Total AA: HClO4 8 mol L−1, 150 °C for 2 h, 140 °C for 4 h, 120 °C for 8 h, and 110 °C for 22 h. 2. Free AA: CCl3COOH, sodium deoxycholate | Triethylendiamine (TEA), phenylisothiocianate (separation of protonated species) | Supelcosil LC18-DB 250 × 4.6 mm, 5 µm. Gradient 0.7 mol L−1 acetate buffer pH 6.4/TEA, H2O and ACN/H2O (80:20). UV λ 254 nm | [219] | |
| Spirulina sp. | Pyrogallol, HCl 8.3 mol L−1 70–80 °C 2 h, IS triundecanoin | o-phtaldialdehyde (OPA) | Zorbax AAA at 40 °C 40 mmol L−1 NaH2PO4 pH 7.8, ACN/MeOH/H2O (45:45:10), 2.0 mL min−1, UV λ 338 nm | [220] | |
| Plants | Soncation, EZ:faastTM Free Amino Acid Kit | propyl chloroformate | UHPLC EZ:faastTM 4u AAA-MS, 250 × 2.0 mm, 3 µm. IS: homoarginine, methionine-d3, and homophenylalanine | [221] | |
| Chamomile flowers | Free amino acids: Sonication | AccQ Fluor, 55 °C | Shimpack column (250 × 4.6 mm, 5 μm) FLD λex 250 λem 395 nm | [222] | |
| Rapeseed meal | HCl 6 mol L−1, 110 °C 23 h | Ninhydrin | Ion exchange chromatography, Vis 570 nm (Pro 440 nm), IS: Norleucine | [223] | |
| Feed ingredients | HCl 6 mol L−1 0.1 g/100 mL phenol, 150 °C 6 h, Reacti-ThermTM | Borate buffer pH 10, OPA (primary-) and FMOC (secondary amines) | Zorbax Eclipse-AAA 40 °C, λex 262 λem 338 nm | [224] | |
| Feed | 6 mol L−1 HCl 110 °C 16–23 h, peformic acid, HBr, | AQC, borate buffer | AccQ-Tag Ultra C-18 100 × 2.1 mm, 1.7 µm). UPLC PDA 260 nm, IS: DL-2-aminobutyric acid | [225] | |
| Fish tissue | 6 mol L−1 HCl 110 °C closed vessel 24 h | AccQ-Fluor Reagent (AQC in 0.2 mol L−1 borate buffer pH 8.8) | FLD λex 250 λem 395 nm. RP C18 | [217] | |
| Selected Applications | |||||
| Matrix | Hydrolysis and treatment | Measurement method, chromatographic column | Reference | ||
| Lipoprotein | Sodium dodecyl sulfate (SDS), enzymatic digestion (e.g., pronase E, muramidase) | UPLC BEH C18 50 × 2.1 mm, 1.7 µm, 130 Å, UV 202–208 nm. Phosphate buffer 50 mmol L−1 pH 4.35/sodium azide and Phosphate buffer 75 mmol L−1 pH 4.95/MeOH (85:15) | [226] | ||
| Peptidoglycan | SDS, sonication, DNAse, RNAse, and trypsin. HCl for teichoic acids. Hydrolases (mutanolysin) | CF3COOH/MeOH, UPLC-TOF/MS-ESI+ CSH C18 100 × 2.1 mm, 1.7 µm, UV 210 nm | [227] | ||
| Cocoa beans | Fermentation, HCl 0.1 mol L−1, mechanical dispersion, ethyl ether | UPLC-ESI+-MS Acquity UPLC BEH C18 150 × 2.1 mm, 1.7 µm and LC/ESI+-MS/MS Aeris Peptide XB-C18 150 × 2.1 mm, 3µm | [228] | ||
| Olive seeds | n-hexane defat, Tris/HCl pH 7.5, SDS, dithiothreitol, high-intensity focus ultrasound, acetone precipitation, alcalase hydrolysis | RP-HPLC Jupiter Proteo 250 × 10 mm, 4 µm FLD λex 280 λem = 360 nm RP-HPLC-ESI+-QTOF-MS, Ascentis Express Peptide ES-C18 100 × 2.1 mm, 2.7 μm | [229] | ||
| Matrix | Extraction Method | Measurement Method, Chromatographic Column | Reference |
|---|---|---|---|
| Fresh fish muscles | Extraction with 0.1 mol L−1 NH4O2C2H3 buffer, pH 4.5, HAH solution 0.25 g mL−1, 1 mol L−1 p-TSA solution and can | LC: Cloversil-C18 250 × 4.6 mm, 5 µm. Identification: MS/MS/ESI+ | [243] |
| Channel Catfish muscle | Extraction with McIlvaine buffer, TSA, and TMPD. Oasis MCX SPE columns | LC: Prodigy ODS-3 C18 150 × 4.6 mm, 3 μm. Identification: MS/MS/ESI+ | [244] |
| Aquaculture water | Not indicated | LC: Phenomenex C18 140, 250 × 4.6 mm, 5 μm. Identification: UV 558 nm (malachite green and crystal violet), FLD λex 265, λem 360 nm (leuco forms) | [245] |
| Processed fish products | Extraction with ammonium acetate buffer, HAH solution, p-TsOH solution, and can | LC: 250 × 4.6 mm, 5 μm Capcell PAK C18 Identification: MS/MS/ESI+ | [246] |
| Fish tissue | Modified QuEChERS Extraction: NH4O2CH and can | LC: XCharge C18 column | [247] |
| Salmon | Extraction C2H3O2− buffer, p-TSA solution, hydroxylamine and can | YMC phenyl 3-4-5 50 × 4.0 mm, 3 µm Identification: LC-MS/ESI+/APCI | [248] |
| Fish feed | Extraction with ACN/CH3OH/CH3COOH | Chromolith® Performance RP-18e (100 × 4.6 mm) Identification: MS/MS/ESI+ | [249] |
| Matrix | Mobile Phase Composition | Measurement Method, Chromatographic Column | Reference | |
|---|---|---|---|---|
| Ion Exchange Chromatography | ||||
| Leafy greens | 10 g L−1 of KH2PO4, pH 3.0 | Waters IC-PAK HC anion exchanger (150 × 4.6 mm), UV λ 214 nm | [263] | |
| Baby foods | Phosphate 5 mmol L−1 (pH 6.5) | Waters IC-PAK HC anion exchanger (150 × 4.6 mm), UV λ 214 nm | [264] | |
| Vegetables | Phosphate 5 mmol L−1 (pH 6.5) | Waters IC-PAK HC anion exchanger 150 × 4.6 mm, 10 µm, UV λ 214 nm | [265] | |
| Reverse Phase Chromatography | ||||
| Matrix | Ion pair reagent | Mobile phase composition | Measurement method, chromatographic column | Reference |
| Cured meat and vegetables | Tetrabutyl ammonium (TBA) | MeOH:H2O (75:25) | Phenomenex C18 110 Å Gemini 250 × 4.6 mm, 5 µm. PDA λ 214 nm | [266] |
| Vegetables | TBA, Greiss reagent | Gradient MeOH/ACN/H2O | X Bridge C18, 50 × 2.1 mm, 2.5 µm. UV-Vis λ 222 (nitrate) and 520 nm (nitrite) | [267] |
| Cured meats | 3 mmol L−1 TBA | ACN/2 mmol L−1 HPO42− pH 4 | RP-thermophenyl hexyl, 150 × 4.6 mm, 3 μm, UV λ 205 nm | [270] |
| Vegetables | 0.1 mol L−1 octyl ammonium salt | OA buffer pH 7.0/MeOH (70:30) | Phenomenex Luna C18 250 × 4.6 mm, 5 μm, UV λ 213 nm | [271] |
| Dried vegetables and water | Triethylamine (TEA) | C6H13SO3H, H2PO4−, TEA pH 3.0/MeOH (80:20) | C13 250 × 4.6 mm, 5 μm, UV λ 222 nm. | [272] |
| Ham | n-octylamine/TBA | 0.01 mol L−1 n-octylamine/5 mmol L−1 TBA pH 6.5 | AcclaimTM Polar Advantage and C18 Thermo Scientific™, HyPURITY™, 250 × 4.6 mm, 5 μm | [273] |
| Matrix | Extraction Method | Measurement Method, Chromatographic Column | Reference |
|---|---|---|---|
| Camu–camu (Myrciaria dubia (Kunth) Macvaugh) | Extracted from the crushed peel with acetone transferred to petroleum ether/diethyl ether and saponified with 10% KOH methanolic | HPLC-PDA Quantitative: C18 Nova-Pak ODS 300 × 3.9 mm, 4 µm set at 29 °C, mobile phase ACN/H2O/ethyl acetate For Qualitative: C30 YMC Carotenoid 250 × 4.6 mm, 3 µm at 33 °C. Mobile phase MeOH/MTBE (methyl tert-butyl ether) | [282] |
| Algae species, Chlorella vulgaris, and Scenedesmus regularis | Extraction with n-hexane–EtOH–acetone–toluene (10:6:7:7) 1 h, Saponification: 40 g/100 mL methanolic KOH at 25 °C in the dark for 16 h | PDA, YMC Carotenoid (250 × 4.6 mm, 5 µm, MeOH/ACN/H2O (84:14:2) and CH2Cl2 gradient UV λ 450 nm | [284] |
| Tissues of a species of colored bird (Taeniopygia guttata) | Plasma and liver extract: n-hexane:MTBE (1:1) Adipose tissue, retina, beak, legs: Saponification 0.02 mol L−1 methanolic KOH for 6 h, organic solvent extraction | PDA, YMC Carotenoid 250 × 4.6 mm, 5 µm, MeOH:ACN:CH2Cl2 linear gradient) | [285] |
| Taiwanese sweet potatoes (Ipomoea batatas (L.) Lam.) | Extraction with hexane/acetone/EtOH (2/1/1) containing MgCO3 and BHT (butylated hydroxytoluene) by 0.5 h, Saponification with 40 g/100 mL methanolic KOH for 3 h under nitrogen gas at 25 °C | PDA, YMC Carotenoid 250 × 4.6 mm, 5 µm, at 25 °C, MeOH–ACN–H2O (84:14:2) and CH2Cl2, UV λ 450 nm | [287] |
| Mashed orange-fleshed sweet potato | Extraction with acetone, THF, and THF:MeOH (1:1) | PDA Phenomenex LUNA C18 ODS, 250 × 4.6 mm, 5 µm, ACN:THF:MeOH: 1 g/100 mL NH4C2H3O2 (68:22:7:3) at room temperature and 450 nm | [288] |
| Selected vegetables | Extraction with THF and MeOH (1:1), petroleum ether containing 0.1 g/100 mL BHT and 50 mL 10 g/100 mL NaCl | HPLC–APCI±–MS, Phenomenex Luna Si C18 column (250 × 2 mm, 5 μm), ACN 0.1 g/100 mL/MeOH (0.05 mol L−1 NH4C2H3O2, 0.05 mL/100 mL TEA)/CHCl3 (0.1 g/100 mL BHT)/n-heptane (0.1 g/100 mL BHT), ambient temperature | [289] |
| Fresh and Processed Fruits and Vegetables | Extraction under subdued yellow light. 50:50 acetone/hexane Saponification: saturated methanolic KOH SPE Alumina N Sep Pak | UV, YMC Carotenoid 250 × 4.6 mm, 5 µm, mobile phase 89:11 MeOH/MTBE | [290] |
| Papaya (Carica papaya L., cv. Maradol) | Freeze-dried papaya homogenized in hexane: CH2Cl2 (1:1). Organic phase was separated and saponified with methanolic KOH 40 g/100 mL (1:1) for 1 h at 50 °C | HPLC-DAD-MS/MS-ESI+, C30 YMC Carotenoid 250 × 4.6 mm, 3 µm, at 15 °C. The mobile phase MeOH and MTBE | [291] |
| Papaya (Carica papaya L.) | MeOH ethyl acetate, and light petroleum (bp 40−60 °C) containing 0.1 g/L of both BHT and BHA (butylated hydroxyanisole) | DAD, YMC Carotenoid 250 × 3.0 mm, 3 µm at 25 °C. Mobile phase MeOH/MTBE/H2O (91:5:4) and MeOH/MTBE/H2O (6:90:4) | [292] |
| Yellow and red nance fruits (Byrsonima crassifolia (L.) Kunth) | Sample with CaCO3, NaCl solution (30 g/100 mL, were extracted MeOH/ethyl acetate/light petroleum 1:1:1 Saponification: methanolic KOH 30 g/100 mL stirring for 23 h | HPLC-PDA-MS/ESI+, C30 YMC Carotenoid 250 × 3.0 mm, 3 µm, MeOH/H2O/aqueous NH4C2H3O2 1 mol L−1 (90:8:2) and MTBE/methanol/aqueous NH4C2H3O2 1 mol L−1 (78:20:2), gradient, at 40 °C. UV λ 450 nm | [295] |
| Red and Yellow Physalis (Physalis alkekengi L. and P. pubescens L.) Fruits and Calyces | Extraction: CaCO3, MeOH/ethyl acetate/petroleum ether (1:1:1), containing 0.1 g L−1 BHA and BHT., sonication | HPLC-PDA-MS/ESI±, C30 YMC Carotenoid (250 × 4.6 mm, 3 µm, mobile phase MeOH/MTBE/H2O (80:18:2) and MeOH/MTBE/H2O (8:90:2), both 0.4 g L−1 NH4C2H3O2.Gradient. | [297] |
| Matrix | Sample Pretreatment, Extraction | Mobile Phase Composition | Measurement Method, Chromatographic Column | Reference |
|---|---|---|---|---|
| Amine-Based Columns | ||||
| Molasses | 1. SPE Sep-Pak C18 2. Microporous resin discoloration (Seplite LX), 30 °C | ACN:H2O (75:25) | RID, IS: maltose 1. Zorbax Carbohydrate 2. UltimateTM XB-NH2; both 250 × 4.6 mm, 5 µm | [307] |
| Tomato | Filtration, ACN/H2O (45:55), SPE Chromabond NH2 | ACN:H2O (80:20) | Nucleodur 100-5 NH2 125 × 4 mm, RID, IS: lactose | [306] |
| Amide-Based Columns | ||||
| Confectionery, chocolate products, snacks | Defat (when applicable), H2O 80 °C (+EtOH for chocolate products) | gradient ACN/H2O + 0.05 mL/100 mL ethanol- and triethyl- amine | UPLC-ELSD, Acquity BEH Amide (50, 100, 150) × 2.1, 1.7 µm, 85 °C | [308] |
| Apple Juice | Filtration | H2O | RID, Sugar PakTM 300 × 6.5 mm, 10 µm, 80 °C | [309] |
| Ligand-Based Columns | ||||
| Tubers | 1. H2O 92 °C 2. Reflux MeOH/H2O (50:50) 3. Activated Charcoal/MeOH 4. 2x·MeOH/H20 50:50 SPE Bond-Elut C18 | 10 mmol L−1 H2SO4 | RID, UHPLC Aminex HPX 87H 300 × 7.8 mm, 9 µm, 18 °C | [310] |
| Foods | Liquids: H2O/EtOH (50:50) Solids: H2O 65 °C, sonication, +EtOH Fat-/Protein-rich: H2O/EtOH (20:80), sonication | 1. H2O 2. ACN:H2O (9:1) | 1. HPLC-RID Ultron PS-80P 300 × 6.5 mm, 10 µm, 50 °C 2. LC-MS-ESI± Unison UK-Amino 150 × 2.0 mm, 3 µm | [311] |
| Derivatization-Based Approaches | ||||
| Fruit tree buds | MeOH extraction, benzyl alcohol/NaOH 8 mol L−1, SPE C18 | Gradient ACN/H2O | PDA λ 228 and 248 nm, Shim-pack C18 250 × 4.6 mm, 5 µm | [312] |
| Foods | 2,3-naphtalenediamine, iodine, HAOc | NH4O2CH3 50 mmol L−1 pH 5.0 in ACN/MeOH (70:30) | RID (sucrose/fructose)/UV λ 310, FLD λex 320 λem 360 nm, C18 250 × 4.6 mm | [313] |
| Normal Phase | ||||
| Glycine max (L.) Merr | H2O 55 °C, ACN | H2O/ACN + Acetone (75:25) | ELSD, PrevailTM Carbohydrate ES 250 × 4.6 mm, 5 µm | [314] |
| Complex Carbohydrates (e.g., Inulin and Fructans) | ||||
| Plants and feed materials | H2O | H2O or H2SO4 0.01 mol L−1 | 1. Knauer Eurokat Pb 2. Nucleosil CHO 620 3. Nucleosil CHO 682 (Pb) 4. Biorad Aminex HPX-87C. All columns 300 × 7.8 mm, RID | [315] |
| Wheat | HCl 60 mmol L−1 70 °C, quenching Na2CO3 | 90 mmol L−1 NaOH | Carbopac-PA-100 250 × 4.0 mm, PAD | [316] |
| Fungus sucrose fermentation | Filtration | ACN/0.04 g/100 mL NH4OH (70:30) | RID, Knauer Eurospher 100-5 NH2 Vertex 25 × 4.6 mm | [317] |
| Starch from feeds | heat stable amylase and amyloglucosidase | ACN/H2O (80:20) | RID, Zorbax Carbohydrate 150 × 4.6 mm, 5 µm | [318] |
| Wine | Diluted 1:9 with EtOH 70 mL/100 mL and 1-phenyl-3-methyl-5-pyrazolone derivatization | ACN/0.1 mol L−1 g/100 mL NH4C2H3O2 (70:30) | UV λ 245 nm, Eclipse XDB-C18 250 × 4.6 mm, 5 µm | [319] |
| Bacterial Exopolysaccharide | Microplate polysaccharide hydrolysis 4 mol L−1 CF3COOH, 90 min at 121 °C. Derivatization 1-phenyl-3-methyl-5-pyrazolone | 5 mmol L−1 NH4C2H3O2 pH 5.6/ACN gradient | Gravity C18, 100 × 2 mm, 1.8 µm, HPLC-UV-Ion trap/ESI+-MS | [320] |
| Matrix | Sample Pretreatment, Extraction | Mobile Phase Composition | Measurement Method, Chromatographic Column | Reference |
|---|---|---|---|---|
| Reverse Phase-Based Columns | ||||
| Kombucha | H2O 70–80 °C, filtration | 20 mmol H2PO4− pH 2.4/MeOH (97:3) | Luna C18 250 × 4.6 mm, 5 µm 30 °C UV λ 210 nm | [337] |
| Fresh fruits | Juice extraction, depulping, centrifugation | 50 mmol L−1 H2PO4− pH 2.8 | Hypersil Gold aQ 250 × 4.6 mm, 5 µm 10 °C UV λ 214 nm (254 ascorbic acid) | [338] |
| Medicinal plants infusions | (HPO3)n extraction | 3.6 mmol L−1 H2SO4 | Sphere-Clone C18 250 × 4.6 mm, 5 µm, 35 °C UV λ 215 nm (254 ascorbic acid) | [339] |
| Fruit juices | Filtration | 0.01 mol L−1 KH2PO4 pH 2.6 | RP-C18 150 × 4.6 mm, 3 µm, UV λ 210 nm | [340] |
| Wine | SPE C18 Elution: mobile phase (for acid protonation) | 0.005 mol L−1 H3PO4 pH 2.1, 1 mL/100 mL CAN | Lichrosorb RP-C18 150 × 4.0 mm, 5 µm, UV λ 210 nm | [345] |
| Ion Exchange-Based Columns | ||||
| Sulfite pulp mill | Filtration | 1. Ultrapure water 79 °C 2. Ultrapure water 68 °C 3. H2SO4 0.005 mol L−1 30 °C 4. H2SO4 0.005 mol L−1 60 °C | RID for all cases, 1. Aminex HPX-87P Pb2+ 300 × 7.8 mm, 9 µm 2. Transgenomic® CHO-782 Pb2+ 300 × 7.8 mm, 7 µm 3. Bio-rad Aminex HPX-87H 300 × 7.8 mm, 9 µm 4. Shodex SH-1011 H+ 300 × 8.0 mm, 6 µm | [341] |
| Food samples | Liquid samples: filtration Solid samples: H2O 85 °C | 0.005 mol L−1 H2SO4 35 °C | Hi-Plex H 300 × 8.0 mm, 6 µm, RID | [343] |
| Olive fruits | Maceration in H2O MeOH (75:25) | 0.1 g/100 mL H3PO4 | Shodex RSpak KC-118 300 × 8.0 mm, UV λ 214 nm | [344] |
| Wines | Filtration, SPE strong anion exchange | 0.065 mL/100 mL H3PO4 | Aminex HPX-87H 300 × 7.8 mm, 9 µm, 65 °C, UV λ 210 nm | [346] |
| Fermented shrimp waste | Centrifugation, sonication, filtration | (HPO3)n pH 2.1 | SS Exil ODS 250 × 4.0 mm, 5 µm, UV λ 210 nm | [347] |
| Silage | H2O 100 °C | 6 mmol L−1 HClO4 | Shodex KC 811 300 × 8.0 mm, 7 µm 50 °C, UV λ 210 | [348] |
| Ion Exclusion-Based Analysis | ||||
| Drinks | Filtration, heat-aided degassing | Precondition: 10 mmol L−1 SDS 3 h 0.3 mL min−1 Elution: 1.84 mmol L−1 H2SO4 pH 2.43 | Kinetex XB-C18 150 × 4.6 mm, 2.6 µm, UPLC-UV λ 210 nm | [349] |
| Matrix | Vitamin | Extraction Method | Measurement Method, Chromatographic Column | Reference |
|---|---|---|---|---|
| Infant and nutritional formulas | K1 | Lipase at 37 °C 2 h in PBS Extraction with hexane and concentrate with N2, reconstituted with MeOH | LC-MS/MS-ESI+ C18 50 × 2.1 mm, 2.6 µm 40 °C. Mobile phase: H2O/ACN 50:50 with 0.1 mL/100 mL HCOOH and ACN/MeOH 75:25, with 2.5 mmol L−1 NH4CO2H, gradient system | [377] |
| Additives, premixes, complete feed | A, E, K3, D3 | Extraction with 0.2 g/100 mL NH3 and EtOH, sonication 40–50 °C for 20–30 min in the dark, SPE (OASIS HLB) | HPLC-PDA C18 150 × 4.6 mm, 5 μm. Mobile phase MeOH/H2O (98:2), UV λ 230 nm | [366] |
| Okra (Abelmoschus esculentus) | B2, B3, B6, B12, C, E, K, D, A, β-Carotene | Vitamin B Analyses. 0.1 mol L−1 H2SO4 incubation 30 min at 121 °C, pH 4.5, 2.5 mol L−1 NaC2H3O2, Takadiastase enzyme Ascorbic Acid Analyses Extraction 0.3 mol L−1 metaphosphoric acid and 1.4 mol L−1 HAOc Saponification KOH 50 g/10 g in EtOH in reflux at 50 °C for 40 min. Extraction with ether | Vitamin B Analyses. Agilent ZORBAX Eclipse Plus C18 250 × 4.6 mm, 5 µm. Mobile phase MeOH/0.023 mol L−1 H3PO4 pH 3.54 (33:67) UV λ 270 nm at room temperature Ascorbic Acid Analyses PDA, 0.1 mol L−1 KC2H3O2 pH 4.9 and ACN/H2O (33:67). UV λ 254 nm at room temperature β-carotene: Agilent TC-C18 250 × 4.6 mm, 5 µm, ACN/MeOH/ethyl acetate (88:10:2) at 453 nm. Fat-soluble vitamins Eclipse XDB-C18 150 × 4.6 mm, 5 μm, MeOH at: 325, 265, 290, 244 nm | [365] |
| Fruits, juices, and supplements | A, D3, E, B1, B2, B3, B5, B6, B9, B12 C | Extraction with Carrez I and Carrez II solutions Cleaned by SPE (RP18 Bakerbond) | PDA, TSKGel ODS-100V, 150 × 4.6 mm, 5 μm at 30 °C. Gradient 0.01 mL/100 mL TFA in H2O and MeOH at 320, 275, 253, 290, 258, 218, 289, 360, and 262 nm | [364] |
| Cheeses | D3 | Saponification with KOH (60% g/100 mL in H2O). Extraction: MeOH/CHCl3 | DAD, at 228 and 266. C18 200 × 4.6 mm, 5 µm ambient temperature Mobile phase: MeOH/ACN/H2O (49.5:49.5:1) | [378] |
| Dairy and soybean oil | K1, D3, E, A | Saponification: KOH/EtOH + ascorbic acid. Mechanical shaking. Extraction: hexane, evaporated at 40 °C and re-dissolved in MeOH | PDA, C18 250 mm × 4.6 mm, 5 μm at 30 °C, 3.0 g/100 mL SDS and 0.02 mol L−1 PBS at pH 7, with 15.0 mL/100 mL butyl alcohol (organic solvent modifier). 230 and 300 nm (K1, D3 and E vitamins), 280 nm (A, E, D3, and K1) | [379] |
| Milk sample | K1, D3, D2, E, A | Extraction: hexane, concentrate with N2 and reconstituted with MeOH | HPLC-UV-Vis 325, 264, and 280 nm (A, D, K and E), C18 DBS 150 × 3 mm, 3 μm at room temperature; MeOH/H2O (99:1) and MeOH/THF (70:30) | [380] |
| Vegetables, fruits, and berries | K1 | Extraction with 2-propanol/hexane and CHCl3/MeOH | HPLC-EC Vydac 201 TP54 150 × 4.6 mm, 5 μm. Mobile phase: MeOH/0.05 mol L−1 NaC2H3O2, pH 3 (96:4) | [381] |
| Rice | E | Saponification KOH (600 g L−1) in EtOH with pyrogallol for 45 min at 70 °C, mechanical shaking. Microextraction: CCl4/ACN (1:10) | FLD, COSMOSIL π-NAP 250 × 4.6 mm, 5 μm at 25 °C. Mobile phase: MeOH/H2O/ACN (80:13:7). Vitamin E isomers λex 290 and λem 330 nm | [382] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortés-Herrera, C.; Artavia, G.; Leiva, A.; Granados-Chinchilla, F. Liquid Chromatography Analysis of Common Nutritional Components, in Feed and Food. Foods 2019, 8, 1. https://doi.org/10.3390/foods8010001
Cortés-Herrera C, Artavia G, Leiva A, Granados-Chinchilla F. Liquid Chromatography Analysis of Common Nutritional Components, in Feed and Food. Foods. 2019; 8(1):1. https://doi.org/10.3390/foods8010001
Chicago/Turabian StyleCortés-Herrera, Carolina, Graciela Artavia, Astrid Leiva, and Fabio Granados-Chinchilla. 2019. "Liquid Chromatography Analysis of Common Nutritional Components, in Feed and Food" Foods 8, no. 1: 1. https://doi.org/10.3390/foods8010001
APA StyleCortés-Herrera, C., Artavia, G., Leiva, A., & Granados-Chinchilla, F. (2019). Liquid Chromatography Analysis of Common Nutritional Components, in Feed and Food. Foods, 8(1), 1. https://doi.org/10.3390/foods8010001