
Transcriptome–Metabolome Analysis Reveals That Crossbreeding Improves Meat Quality in Hu Sheep and Their F1-Generation Sheep

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. Muscle Staining
2.3. Transcriptome Sequencing and Bioinformatics Analysis
2.4. Metabolome Sequencing and Bioinformatics Analysis
2.5. Comprehensive Analysis of Transcriptome and Metabolome Data
2.6. Quantitative Real-Time PCR (qRT-PCR) Validation of DEGs
2.7. Statistical Analysis
3. Results
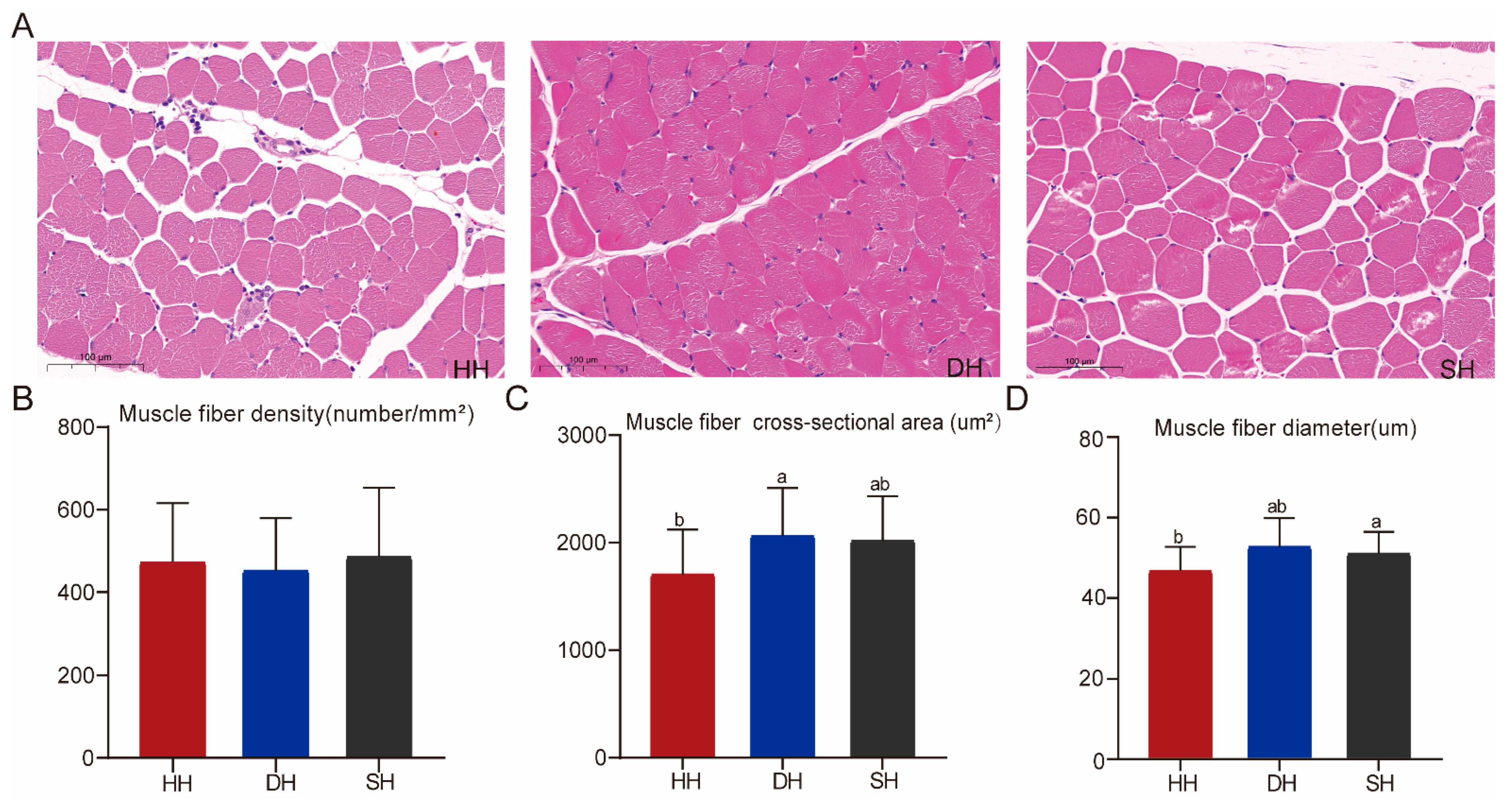
3.1. Determination of Muscle Fiber Characteristics of Hu Sheep and Their Crossbred F1-Generation Sheep
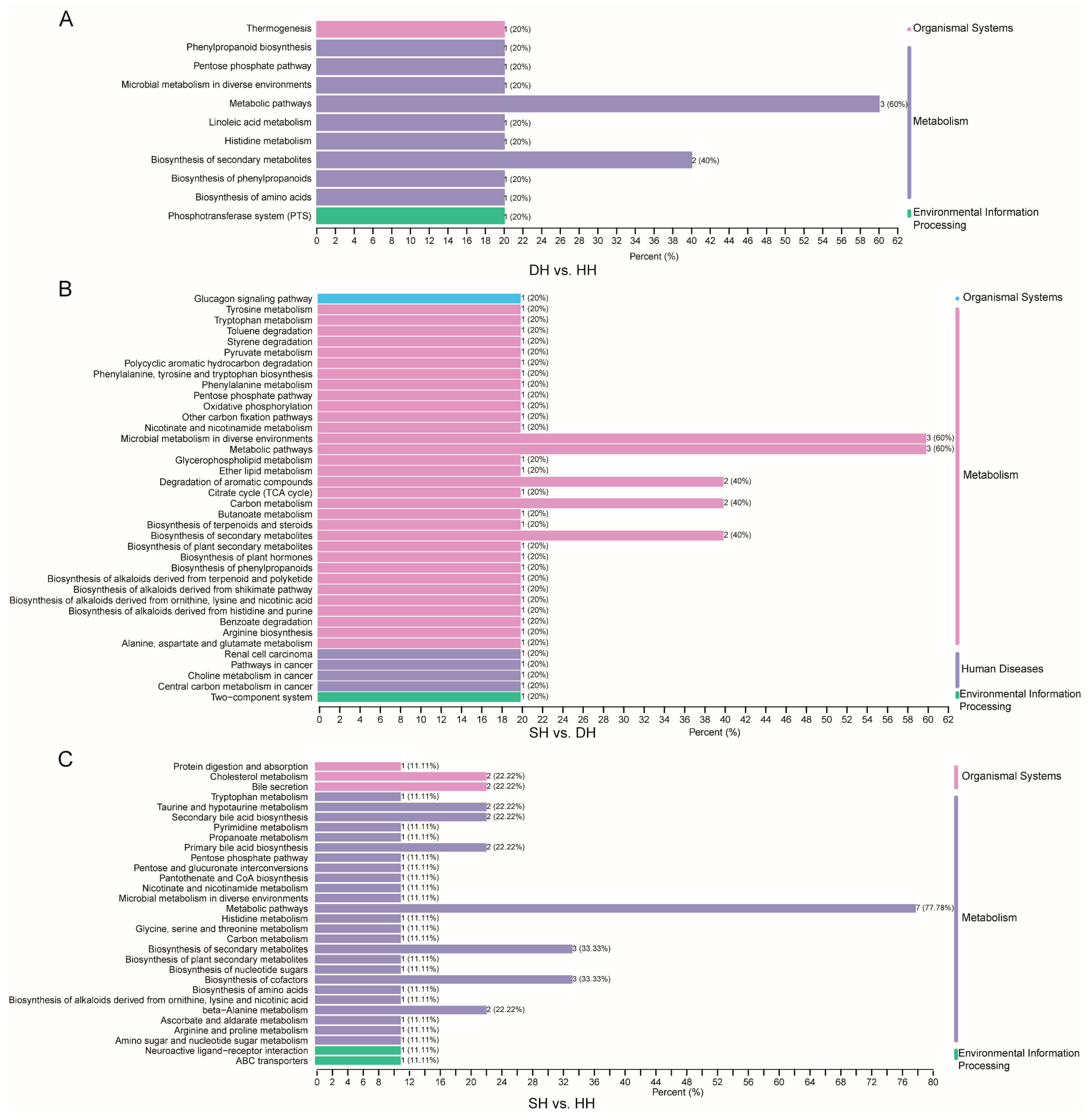
3.2. Transcriptome Profiling of Muscle
3.3. LC–MS Metabolomic Analysis in Muscle
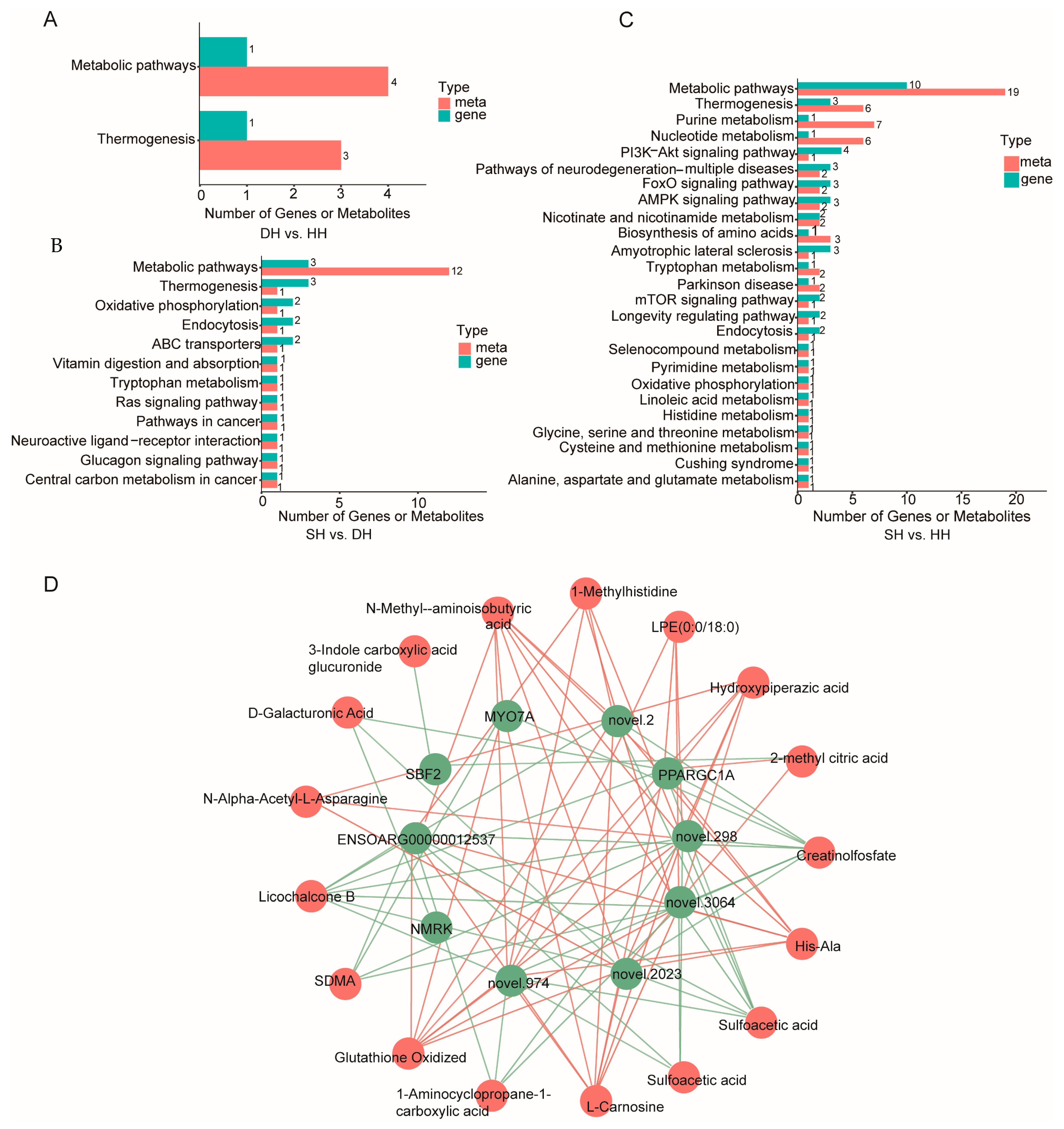
3.4. Transcriptome and Metabolome Integration Analysis
3.5. Detection of DEG Expression in Longissimus Dorsi by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kantono, K.; Hamid, N.; Ma, Q.; Chadha, D.; Oey, I. Consumers’ perception and purchase behaviour of meat in China. Meat Sci. 2021, 179, 108548. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, D.; Yin, L.; Li, Z.; Yu, C.; Du, H.; Jiang, X.; Yang, C.; Liu, Y. Integration analysis of metabolome and transcriptome profiles revealed the age-dependent dynamic change in chicken meat. Food Res. Int. 2022, 156, 111171. [Google Scholar] [CrossRef]
- Lu, Z.; Yue, Y.; Shi, H.; Zhang, J.; Liu, T.; Liu, J.; Yang, B. Effects of sheep sires on muscle fiber characteristics, fatty acid composition and volatile flavor compounds in F1 crossbred lambs. Foods 2022, 11, 4076. [Google Scholar] [CrossRef]
- Miao, X.; Luo, Q.; Zhao, H.; Qin, X. Comparison of alternative splicing (AS) events in adipose tissue of polled dorset versus small tail han sheep. Heliyon 2023, 9, e14938. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.; Campo, M.; Balado, J.; Sañudo, C. Effect of Texel crossbreeding on productive traits, carcass and meat quality of Segureña lambs. J. Sci. Food Agric. 2019, 99, 3335–3342. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Tang, Z.; Yang, F.; Shi, J.; Liu, T.; Cheng, S. Functional analysis of microorganisms and metabolites in the cecum of different sheep populations and their effects on production traits. Front. Microbiol. 2024, 15, 1437250. [Google Scholar] [CrossRef] [PubMed]
- Samara, E.; Abdoun, K.; Okab, A.; Al-Badwi, M.; El-Zarei, M.; Al-Seaf, A.M.; AI-Haidary, A. Assessment of heat tolerance and production performance of Aardi, Damascus, and their crossbred goats. Int. J. Biometeorol. 2016, 60, 1377–1387. [Google Scholar] [CrossRef]
- An, X.; Zhang, S.; Li, T.; Chen, N.; Wang, X.; Zhang, B.; Ma, Y. Transcriptomics analysis reveals the effect of Broussonetia papyrifera L. fermented feed on meat quality traits in fattening lamb. PeerJ 2021, 9, e11295. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Yang, X.; Zhang, Y.; Wang, S.; Jin, C.; Xia, W.; Chen, W.; Cai, B.; Zheng, C. PPM1J regulates meat quality feature and glycerophospholipids composition in broiler by modulating protein dephosphorylation. NPJ Sci. Food 2024, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yang, Y.; Wang, M.; Jiang, W.; Du, Y.; Hao, Z.; Lei, C.; Zhu, K.; Liu, B.; Niu, L.; et al. Effects of L-arginine on gut microbiota and muscle metabolism in fattening pigs based on omics analysis. Front. Microbiol. 2024, 15, 1490064. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, Q.; Wang, X.; Xu, X.; Wang, Y.; Wei, L.; Na, L.; Liu, H.; Hu, L.; Zhao, N.; et al. Targeted and untargeted metabolomics reveals meat quality in grazing yak during different phenology periods on the Qinghai-Tibetan Plateau. Food Chem. 2024, 447, 138855. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Cai, D.; Zhang, Z.; Cai, B.; Yang, X.; Kong, S.; Wu, R.; Lin, D.; Yuan, R.; Mo, Y.; et al. Metabolomic, lipidomic and transcriptomic reveal meat quality differences among hybrid, indigenous and commercial broiler. LWT-Food Sci. Technol. 2024, 209, 116765. [Google Scholar] [CrossRef]
- Zhao, L.; Li, F.; Zhang, X.; Zhang, D.; Li, X.; Zhang, Y.; Zhao, Y.; Song, Q.; Huang, K.; Xu, D.; et al. Integrative analysis of transcriptomics and proteomics of longissimus thoracis of the Hu sheep compared with the Dorper sheep. Meat Sci. 2022, 193, 108930. [Google Scholar] [CrossRef]
- Song, G.; Solomon, A.; Zhu, T.; Li, Z.; Wang, S.; Song, B.; Dong, X.; Ren, Z. Spatial metabolomics, LC-MS and RNA-Seq reveal the effect of red and white muscle on rabbit meat flavor. Meat Sci. 2025, 219, 109671. [Google Scholar] [CrossRef]
- Zhang, R.; An, X.; Li, J.; Lu, Z.; Niu, C.; Xu, Z.; Zhang, J.; Geng, Z.; Yue, Y.; Yang, B. Comparative analysis of growth performance, meat productivity, and meat quality in Hu sheep and its hybrids. Acta Prataculturae Sin. 2024, 33, 186–197. [Google Scholar]
- Zhang, L.; Yue, Y.; An, X.; Li, J.; Yang, B.; Xu, Z.; Zhang, J.; Geng, Z.; Guo, Y.; Zhang, R. Comparative analysis of the contents of Amino Acids, Fatty Acids and Volatile Flavor Compounds in the muscles of Hu Sheep and their different hybrid combinations. Acta Vet. Zootech. Sin. 2024, 55, 4428–4442. [Google Scholar]
- Ding, W.; Lu, Y.; Xu, B.; Chen, P.; Li, A.; Jian, F.; Yu, G.; Huang, S. Meat of Sheep: Insights into Mutton Evaluation, Nutritive Value, Influential Factors, and Interventions. Agriculture 2024, 14, 1060. [Google Scholar] [CrossRef]
- Lee, S.; Joo, S.; Ryu, Y. Skeletal muscle fiber type and myofibrillar proteins in relation to meat quality. Meat Sci. 2010, 86, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Lang, Y.; Zhang, S.; Xie, P.; Yang, X.; Sun, B.; Yang, H. Muscle fiber characteristics and postmortem quality of longissimus thoracis, psoas major and semitendinosus from Chinese Simmental bulls. Food Sci. Nutr. 2020, 8, 6083–6094. [Google Scholar] [CrossRef] [PubMed]
- Listrat, A.; Lebret, B.; Louveau, I.; Astruc, T.; Bonnet, M.; Lefaucheur, L.; Picard, B.; Bugeon, J. How muscle structure and composition influence meat and flesh quality. Sci. World J. 2016, 2016, 3182746. [Google Scholar] [CrossRef]
- Du, M.; Yan, X.; Tong, J.F.; Zhao, J.; Zhu, M. Maternal obesity, inflammation, and fetal skeletal muscle development. Biol. Reprod. 2010, 82, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Li, F.; Guo, Q.; Zhou, Y.; Miao, Y.; Li, Y.; Wang, Z.; Jiang, R.; Dong, J.; Liu, X.; et al. Structural and functional plasticity of collagen fibrils. DNA Cell Biol. 2019, 38, 367–373. [Google Scholar] [CrossRef]
- Roy, B.; Das, C.; Aalhus, J.; Bruce, H. Relationship between meat quality and intramuscular collagen characteristics of muscles from calf-fed, yearling-fed and mature crossbred beef cattle. Meat Sci. 2021, 173, 108375. [Google Scholar] [CrossRef]
- Lepetit, J. A theoretical approach of the relationships between collagen content, collagen cross-links and meat tenderness. Meat Sci. 2007, 76, 147–159. [Google Scholar] [CrossRef]
- Xu, M.; Chen, X.; Chen, D.; Yu, B.; Huang, Z. FoxO1: A novel insight into its molecular mechanisms in the regulation of skeletal muscle differentiation and fiber type specification. Oncotarget 2017, 8, 10662–10674. [Google Scholar] [CrossRef]
- Sandri, M.; Lin, J.; Handschin, C.; Yang, W.; Arany, Z.; Lecker, S.H.; Goldberg, A.; Spiegelman, B. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar] [CrossRef] [PubMed]
- Lefaucheur, L. A second look into fibre typing—Relation to meat quality. Meat Sci. 2010, 84, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Zhang, W.; Xie, Z.C.; Li, J.; Chen, H. CaMKII in cardiovascular diseases, Especially CaMKII-δ: Friends or enemies. Drug Des. Devel. Ther. 2024, 18, 3461–3476. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Zhou, Y.; Yang, J.; Li, J.; Peng, Y.; Zhang, X.; Miao, Y.; Jiang, W.; Bu, G.; Hou, L.; et al. Targeted overexpression of PPARγ in skeletal muscle by random insertion and CRISPR/Cas9 transgenic pig cloning enhances oxidative fiber formation and intramuscular fat deposition. FASEB J. 2021, 35, e21308. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Jian, T.; Li, J.; Lv, H.; Tong, B.; Li, J.; Meng, X.; Ren, B.; Chen, J. Chicoric acid ameliorates nonalcoholic fatty liver disease via the AMPK/Nrf2/NF-κB signaling pathway and restores gut microbiota in High-Fat-Diet-Fed mice. Oxid. Med. Cell. Longev. 2020, 2020, 9734560. [Google Scholar] [CrossRef]
- Jin, H.; Wang, H.; Wu, J.; Hu, M.; Zhou, X.; Yang, S.; Zhao, A.; He, K. Asparagine synthetase regulates the proliferation and differentiation of chicken skeletal muscle satellite cells. Anim. Biosci. 2024, 37, 1848–1862. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, Y.; Ohashi, K.; Otaka, N.; Kawanishi, H.; Takikawa, T.; Fang, l.; Takahara, K.; Tatsumi, M.; Ishihama, S.; Takefuji, M.; et al. Myonectin protects against skeletal muscle dysfunction in male mice through activation of AMPK/PGC1α pathway. Nat. Commun. 2023, 14, 4675. [Google Scholar] [CrossRef] [PubMed]
- Zylka, M.; Shearman, L.; Weaver, D.; Reppert, S. Three period homologs in mammals: Differential light responses in the suprachiasmatic circadian clock and oscillating transcripts outside of brain. Neuron 1998, 20, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Takumi, T.; Taguchi, K.; Miyake, S.; Sakakida, Y.; Takashima, N.; Matsubara, C.; Maebayashi, Y.; Okumura, K.; Takekida, S.; Yamamoto, S.; et al. A light-independent oscillatory gene mPer3 in mouse SCN and OVLT. EMBO J. 1998, 17, 4753–4759. [Google Scholar] [CrossRef]
- Li, Y.; Li, W.; Deng, J.; Yin, M. PER3 promoter hypermethylation correlates to the progression of pan-cancer. Clin. Epigenetics 2024, 16, 140. [Google Scholar] [CrossRef]
- Staszkiewicz, R.; Sobański, D.; Pulka, W.; Gładysz, D.; Gadzieliński, M.; Strojny, D.; Grabarek, B. Variances in the expression profile of circadian clock-related genes in astrocytic brain tumors. Cancers 2024, 16, 2335. [Google Scholar] [CrossRef]
- Xia, Q.; Li, Q.; Gan, S.; Guo, X.; Zhang, X.; Zhang, J.; Niu, M. Exploring the roles of fecundity-related long non-coding RNAs and mRNAs in the adrenal glands of Small-Tailed Han Sheep. BMC Genet. 2020, 21, 39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liang, S.; Nie, P.; Liao, Y.; Ai, Q.; Yan, X.; Liu, H.; Ji, H.; Zeng, Z. Efficacy of Kushen decoction on high-fat-diet-induced hyperlipidemia in rats. J. Tradit. Chin. Med. 2022, 42, 364–371. [Google Scholar]
- Martín-Reyes, F.; Ho-Plagaro, A.; Rodríguez-Díaz, C.; Lopez-Gómez, C.; Garcia-Serrano, S.; Reyes, D.; Gonzalo, M.; Fernández-Garcia, J.; Montiel-Casado, C.; Fernández-Aguilar, J.; et al. Oleic acid regulates the circadian rhythm of adipose tissue in obesity. Pharmacol. Res. 2023, 187, 106579. [Google Scholar] [CrossRef]
- Bakhtiarizadeh, M.; Alamouti, A. RNA-Seq based genetic variant discovery provides new insights into controlling fat deposition in the tail of sheep. Sci. Rep. 2020, 10, 13525. [Google Scholar] [CrossRef]
- Albarran, L.; Lopez, J.; Jardin, I.; Sanchez-Collado, J.; Berna-Erro, A.; Smani, T.; Camello, P.; Salido, G.; Rosado, J. EFHB is a novel cytosolic Ca2+ sensor that modulates STIM1-SARAF interaction. Cell. Physiol. Biochem. 2018, 51, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Kang, X.; Liu, Y.; Liu, X.; Chan, S.; Wang, Y.; Li, Z.; Ling, Y.; Feng, D.; Li, M.; et al. Integrated analysis of the whole transcriptome of skeletal muscle reveals the ceRNA regulatory network related to the formation of muscle fibers in Tan sheep. Front. Genet. 2022, 13, 991606. [Google Scholar] [CrossRef] [PubMed]
- Halling, J.; Pilegaard, H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl. Physiol. Nutr. Metab. 2020, 45, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Salazar, G.; Cullen, A.; Huang, J.; Zhao, Y.; Serino, A.; Hilenski, L.; Patrushev, N.; Forouzandeh, F.; Hwang, H. SQSTM1/p62 and PPARGC1A/PGC-1α at the interface of autophagy and vascular senescence. Autophagy 2020, 16, 1092–1110. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Cai, B.; Kong, S.; Zhou, Z.; Zhang, J.; Zhang, X.; Nie, Q. PPARGC1A is a moderator of skeletal muscle development regulated by miR-193b-3p. Int. J. Mol. Sci. 2022, 23, 9575. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lim, K.; Lee, E.; Lee, K.; Kim, T.; Ryu, Y.; Hong, K. Polymorphisms of the 5′ regulatory region of the porcine PPARGC1A gene and the effects on muscle fiber characteristics and meat quality. Mol. Biol. Rep. 2012, 39, 3933–3942. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, G.; Cinar, M.; Ponsuksili, S.; Wimmers, K.; Tesfaye, D.; Looft, C.; Jüngst, H.; Tholen, E.; Phatsara, C.; Schellander, K.; et al. Association of PPARGC1A and CAPNS1 gene polymorphisms and expression with meat quality traits in pigs. Meat Sci. 2011, 89, 478–485. [Google Scholar] [CrossRef]
- Kong, L.; Yue, Y.; Li, J.; Yang, B.; Chen, B.; Liu, J.; Lu, Z. Transcriptomics and metabolomics reveal improved performance of Hu sheep on hybridization with Southdown sheep. Food Res. Int. 2023, 173, 113240. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, C.; Kong, Y.; Li, F.; Yue, X. Effects of intramuscular fat on meat quality and its regulation mechanism in Tan sheep. Front. Nutr. 2022, 9, 908355. [Google Scholar] [CrossRef]
- Guo, H.; Khan, R.; Abbas Raza, S.; Suhail, S.; Khan, H.; Khan, S.; EI-Aziz, A.; Zan, L. RNA-Seq reveals function of Bta-miR-149-5p in the regulation of bovine adipocyte differentiation. Animals 2021, 11, 1207. [Google Scholar] [CrossRef]
- Khan, M.I.; Jo, C.; Tariq, M.R. Meat flavor precursors and factors influencing flavor precursors—A systematic review. Meat Sci. 2015, 110, 278–284. [Google Scholar] [CrossRef]
- Antonelo, D.; Cônsolo, N.; Gómez, J.; Beline, M.; Goulart, R.; Corte, R.; Calnago, L.; Schilling, M.; Gerrard, D.; Silva, S. Metabolite profile and consumer sensory acceptability of meat from lean Nellore and Angus × Nellore crossbreed cattle fed soybean oil. Food Res. Int. 2020, 132, 109056. [Google Scholar] [CrossRef] [PubMed]
- D’Astous-Pagé, J.; Gariépy, C.; Blouin, R.; Cliché, V.; Sullivan, B.; Fortin, F.; Palin, M.-F. Carnosine content in the porcine longissimus thoracis muscle and its association with meat quality attributes and carnosine-related gene expression. Meat Sci. 2017, 124, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, L.; Li, S.; Ning, C.; Zhou, C. L-Arginine/L-lysine improves emulsion stability of chicken sausage by increasing electrostatic repulsion of emulsion droplet and decreasing the interfacial tension of soybean oil-water. Food Hydrocoll. 2019, 89, 492–502. [Google Scholar] [CrossRef]
- Sharma, R.K. Phosphorylation and characterization of bovine heart calmodulin-dependent phosphodiesterase. Biochemistry 1991, 30, 5963–5968. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, X.; Jamali, M.; Fang, R.; Peng, Z. Influence of L-histidine and L-lysine on the phosphorylation of myofibrillar and sarcoplasmic proteins from chicken breast in response to salting. LWT-Food Sci. Technol. 2020, 133, 110125. [Google Scholar]
- Abudupataer, M.; Zou, W.; Zhang, W.; Ding, S.; Zhou, Z.; Chen, J.; Hui, L.; Zhang, Z.; Wang, C.; Ge, J.; et al. Histamine deficiency delays ischaemic skeletal muscle regeneration via inducing aberrant inflammatory responses and repressing myoblast proliferation. J. Cell. Mol. Med. 2019, 23, 8392–8409. [Google Scholar] [CrossRef] [PubMed]
- Basaranoglu, M.; Basaranoglu, G.; Bugianesi, E. Carbohydrate intake and nonalcoholic fatty liver disease: Fructose as a weapon of mass destruction. Hepatobiliary Surg. Nutr. 2015, 4, 109–116. [Google Scholar] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; An, X.; Xu, Z.; Niu, C.; Geng, Z.; Zhang, J.; Shi, H.; Chen, Z.; Zhang, R.; Yue, Y. Transcriptome–Metabolome Analysis Reveals That Crossbreeding Improves Meat Quality in Hu Sheep and Their F1-Generation Sheep. Foods 2025, 14, 1384. https://doi.org/10.3390/foods14081384
Zhang L, An X, Xu Z, Niu C, Geng Z, Zhang J, Shi H, Chen Z, Zhang R, Yue Y. Transcriptome–Metabolome Analysis Reveals That Crossbreeding Improves Meat Quality in Hu Sheep and Their F1-Generation Sheep. Foods. 2025; 14(8):1384. https://doi.org/10.3390/foods14081384
Chicago/Turabian StyleZhang, Liwa, Xuejiao An, Zhenfei Xu, Chune Niu, Zhiguang Geng, Jinxia Zhang, Haina Shi, Zhenghan Chen, Rui Zhang, and Yaojing Yue. 2025. "Transcriptome–Metabolome Analysis Reveals That Crossbreeding Improves Meat Quality in Hu Sheep and Their F1-Generation Sheep" Foods 14, no. 8: 1384. https://doi.org/10.3390/foods14081384
APA StyleZhang, L., An, X., Xu, Z., Niu, C., Geng, Z., Zhang, J., Shi, H., Chen, Z., Zhang, R., & Yue, Y. (2025). Transcriptome–Metabolome Analysis Reveals That Crossbreeding Improves Meat Quality in Hu Sheep and Their F1-Generation Sheep. Foods, 14(8), 1384. https://doi.org/10.3390/foods14081384