
Fc-Binding Cyclopeptide Induces Allostery from Fc to Fab: Revealed Through in Silico Structural Analysis to Anti-Phenobarbital Antibody

 and
and 
Abstract
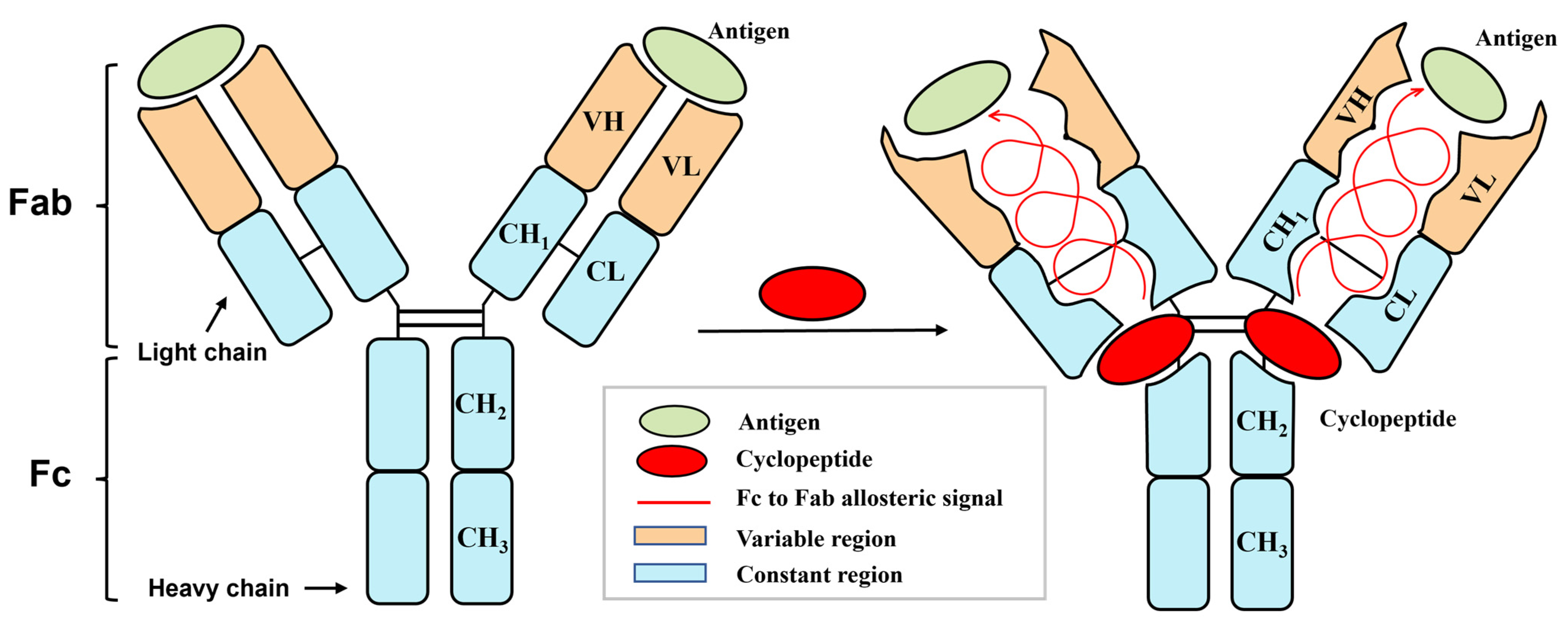
1. Introduction
2. Materials and Methods
2.1. Molecular Docking
2.2. System Construction and Molecular Dynamics Simulations
2.3. Analytical Metrics
2.3.1. Analysis of Antibody Backbone Structural Fluctuation (RMSD, RMSF, Rg)
2.3.2. Analysis of Antibody Dynamic Conformational Ensembles (PCA and FEL)
2.3.3. Analysis of Antibody Residue Correlation (DCCM and RCM)
2.3.4. Analysis of the Allosteric Transduction Pathway Within Antibody
2.3.5. Analysis of Non-Covalent Interactions Within Antibody
3. Results and Discussion
3.1. Identification of Antibody Allosteric Site
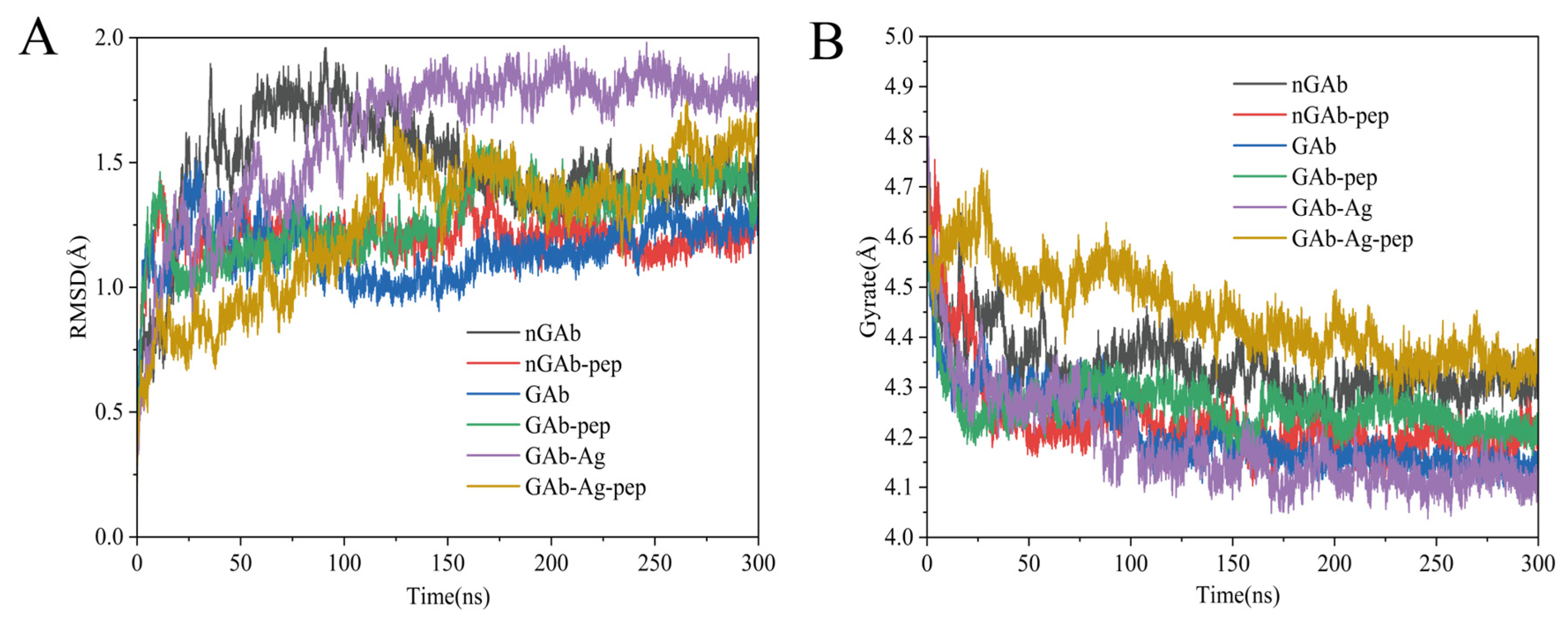
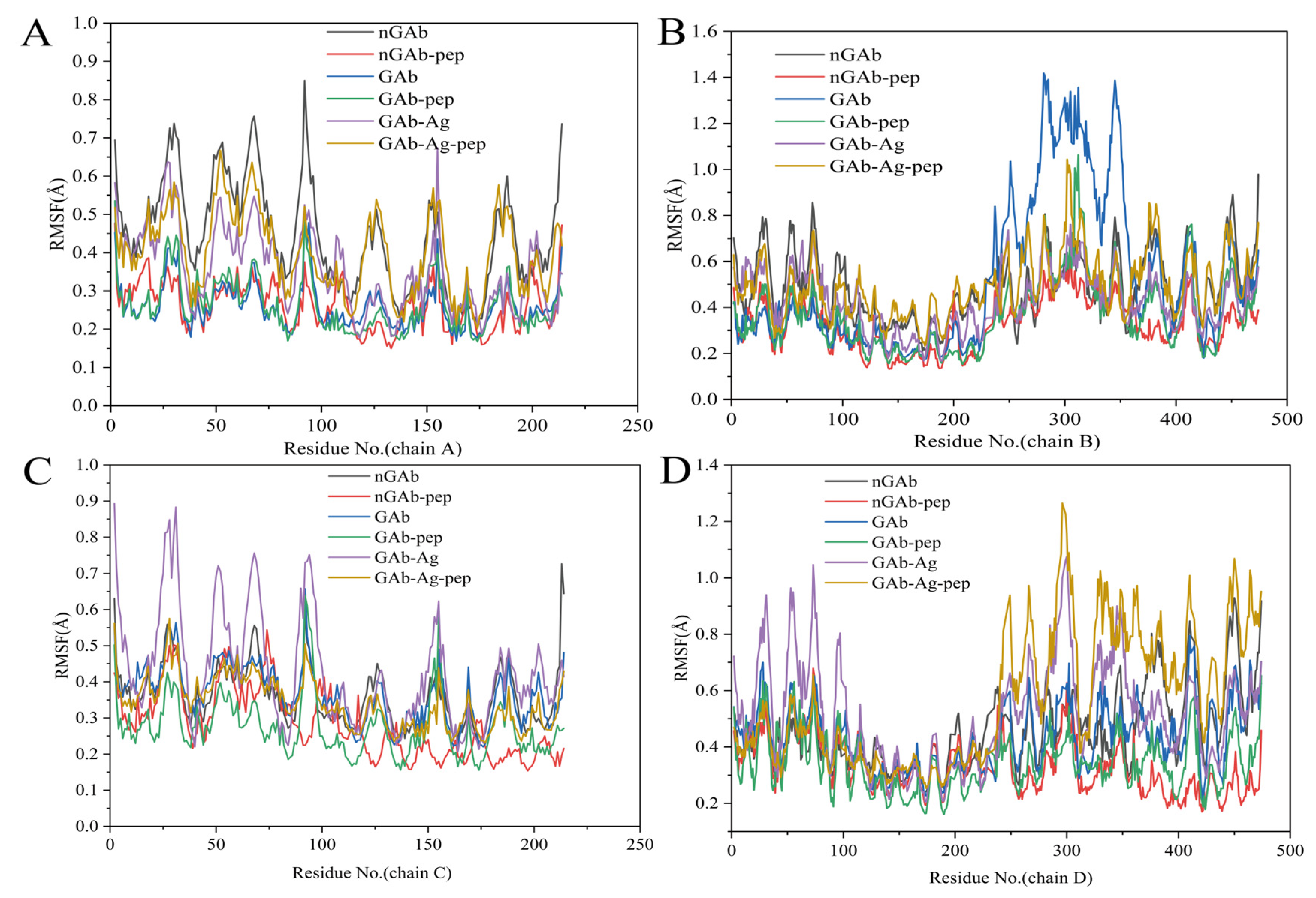
3.2. Influence of Cyclopeptide on Antibody Backbone Structure
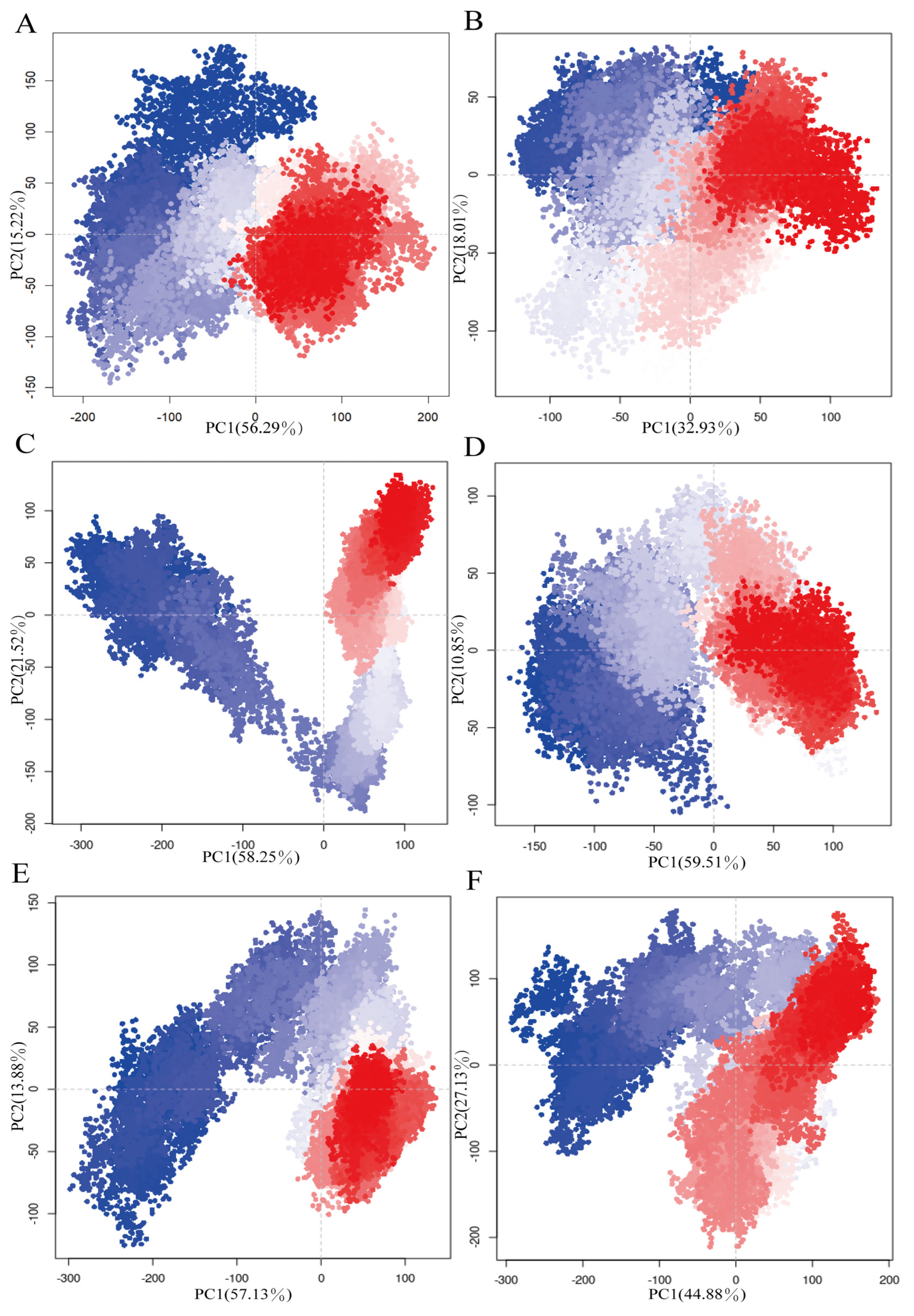
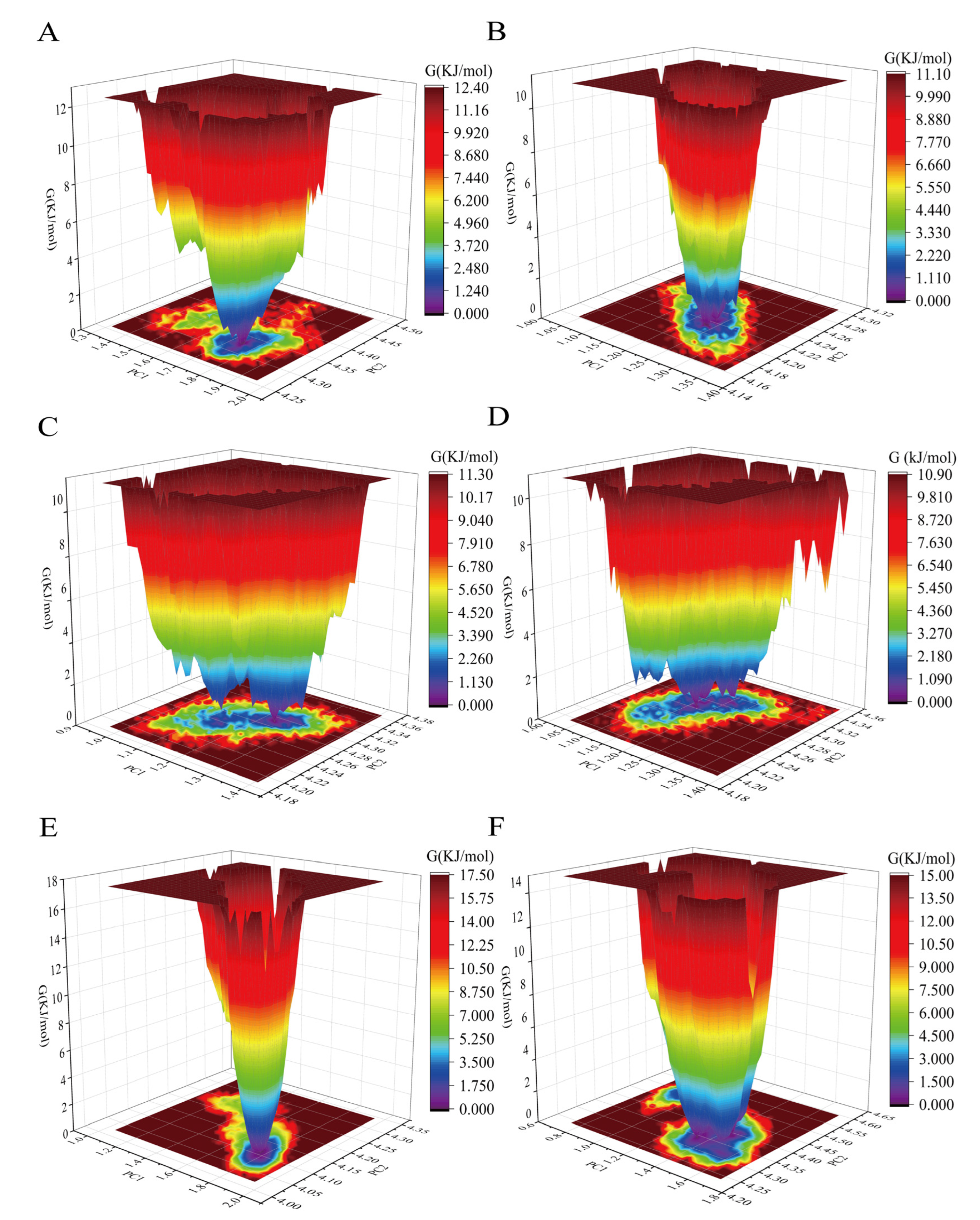
3.3. Influence of Cyclopeptide on Antibody Dynamic Conformational Ensembles
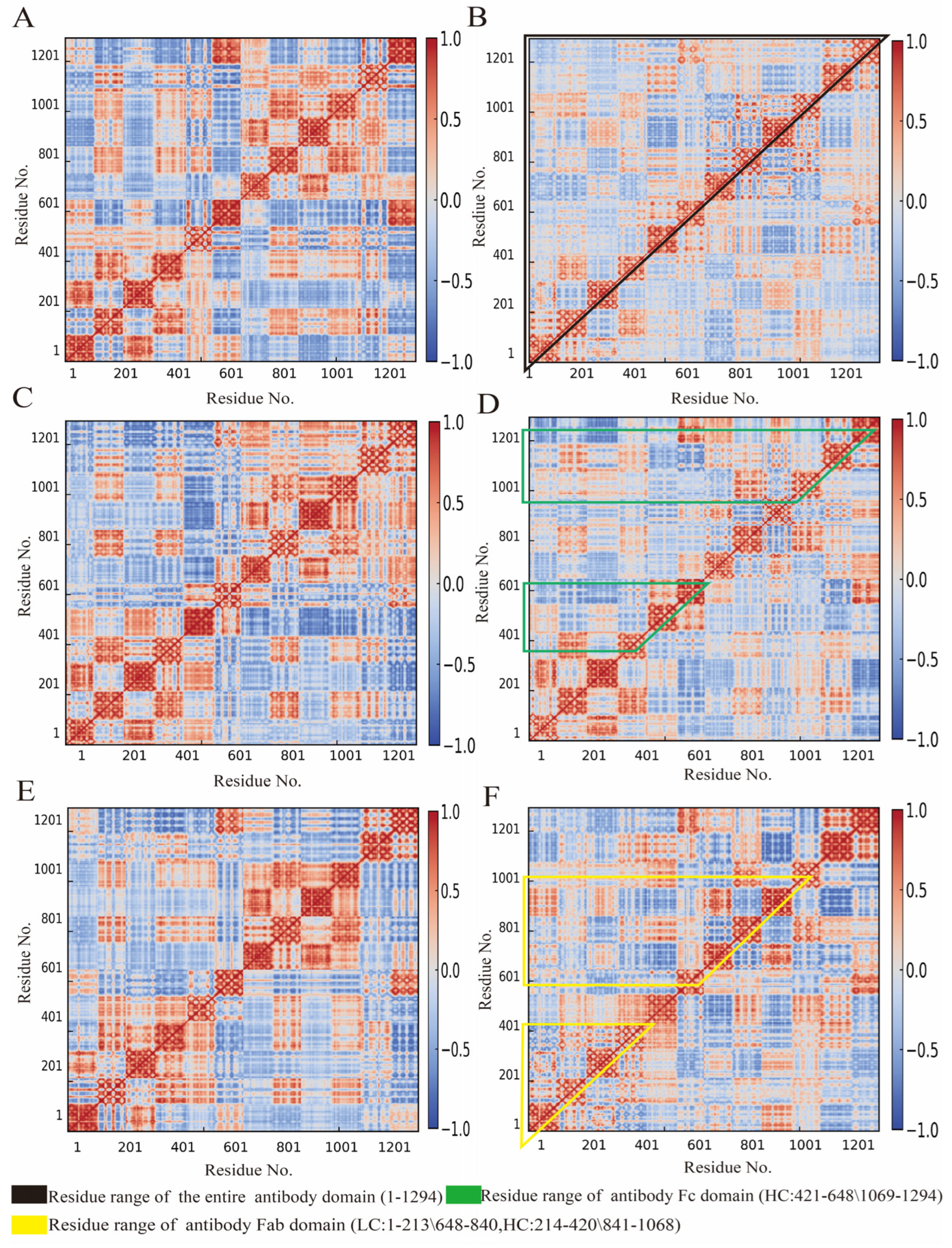
3.4. Influence of Cyclopeptide on Antibody Residue Correlation
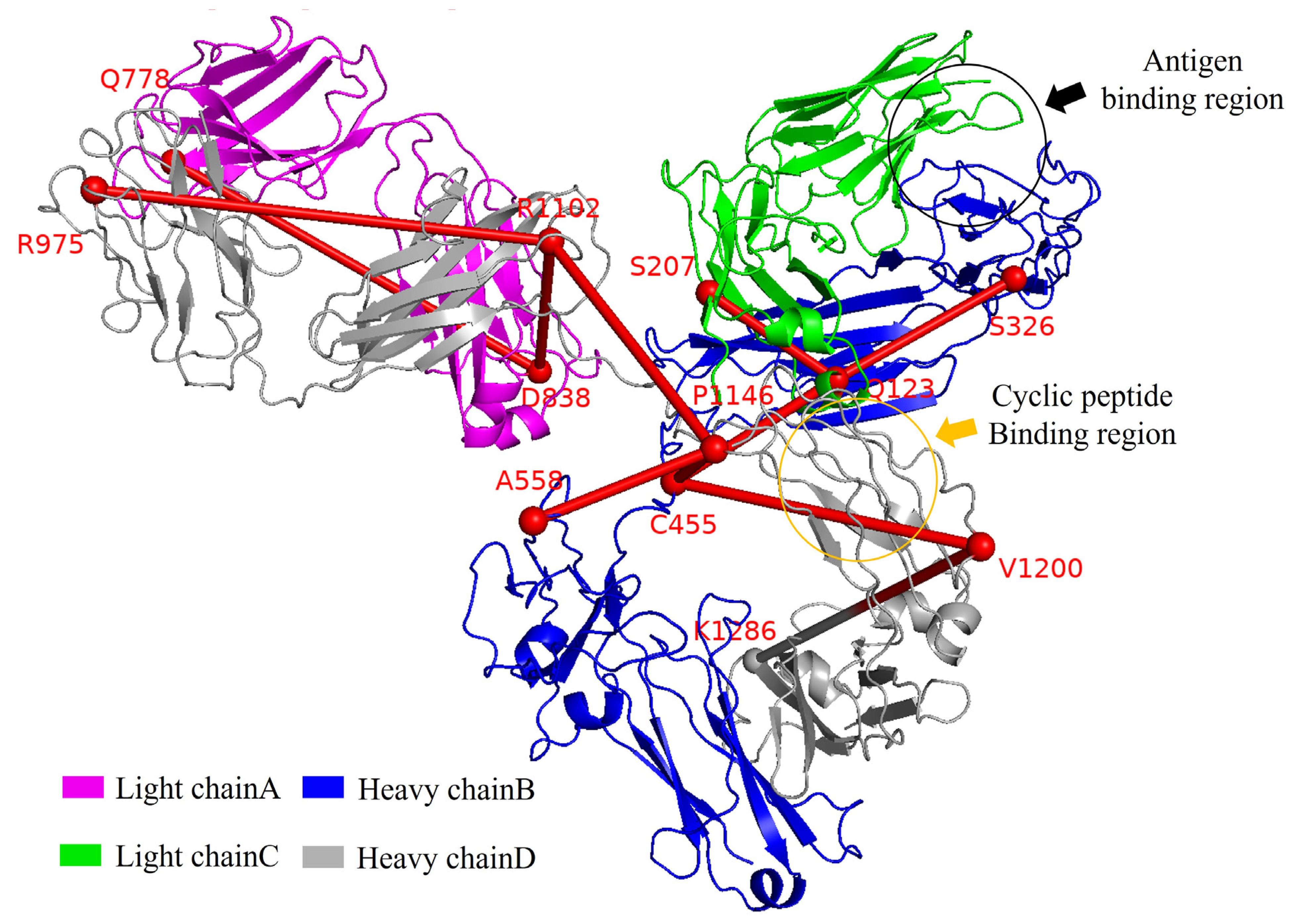
3.5. Allosteric Pathways of Antibody
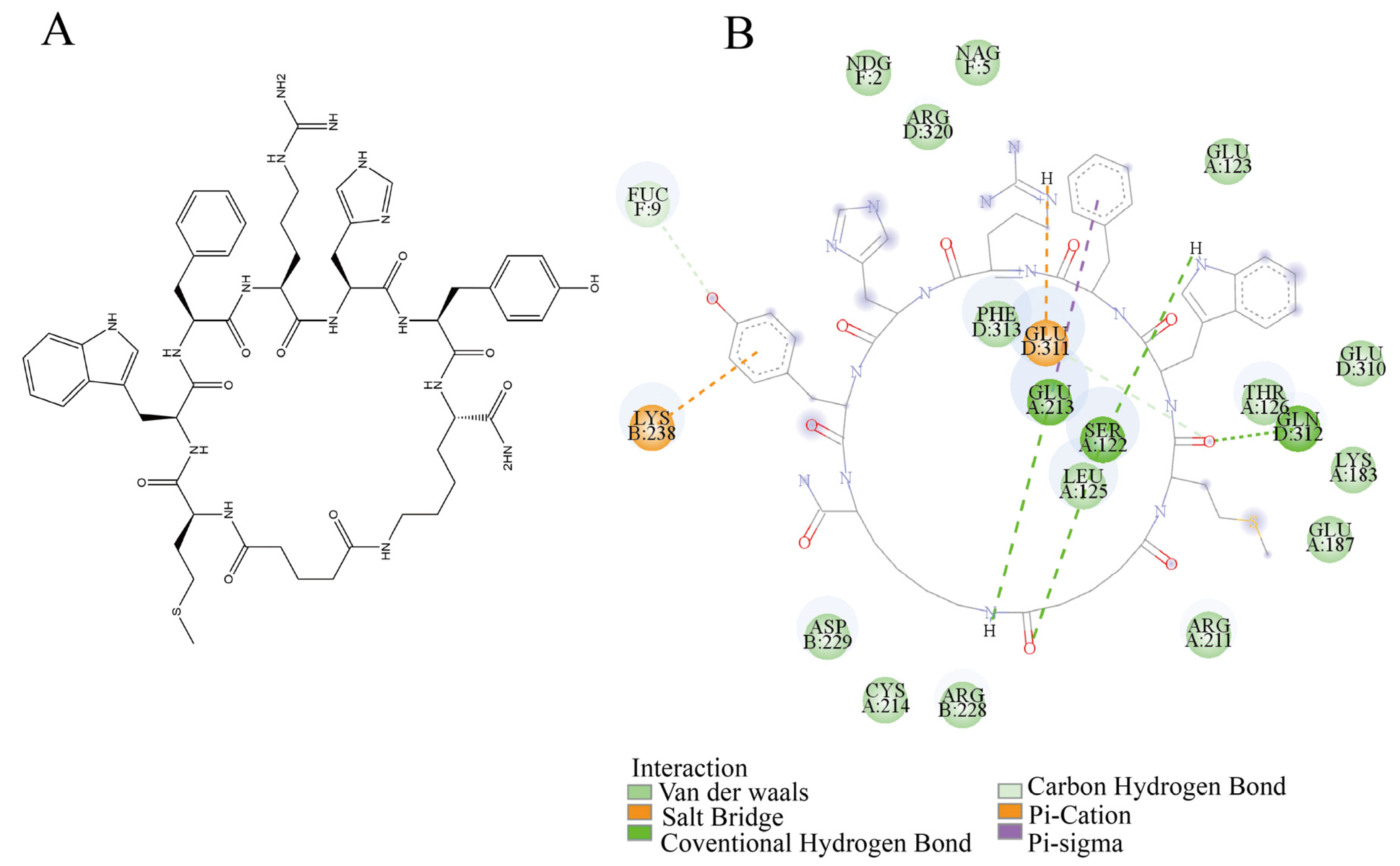
3.6. Key Drivers of Antibody Allostery
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Rossetti, M.; Porchetta, A. Allosterically Regulated DNA-Based Switches: From Design to Bioanalytical Applications. Anal. Chim. Acta 2018, 1012, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Karplus, M. Allostery and Cooperativity Revisited. Protein Sci. 2008, 17, 1295–1307. [Google Scholar] [CrossRef]
- Strickland, D.; Moffat, K.; Sosnick, T.R. Light-Activated DNA Binding in a Designed Allosteric Protein. Proc. Natl. Acad. Sci. USA 2008, 105, 10709–10714. [Google Scholar] [CrossRef] [PubMed]
- Sevcsik, E.; Trexler, A.J.; Dunn, J.M.; Rhoades, E. Allostery in a Disordered Protein: Oxidative Modifications to α-Synuclein Act Distally to Regulate Membrane Binding. J. Am. Chem. Soc. 2011, 133, 7152–7158. [Google Scholar] [CrossRef] [PubMed]
- Subedi, G.P.; Hanson, Q.M.; Barb, A.W. Restricted Motion of the Conserved Immunoglobulin G1 N-Glycan Is Essential for Efficient FcγRIIIa Binding. Structure 2014, 22, 1478–1488. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, B.; Krüger, A.; Du, X.; Visser, L.; Dömling, A.S.S.; Wrenger, C.; Groves, M.R. Discovery of Small-Molecule Allosteric Inhibitors of PfATC as Antimalarials. J. Am. Chem. Soc. 2022, 144, 19070–19077. [Google Scholar] [CrossRef]
- Berezovsky, I.N.; Nussinov, R. Multiscale Allostery: Basic Mechanisms and Versatility in Diagnostics and Drug Design. J. Mol. Biol. 2022, 434, 167751. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Liu, Y.; Kong, R.; Ni, D.; Yu, Z.; Lu, S.; Zhang, J. Harnessing Reversed Allosteric Communication: A Novel Strategy for Allosteric Drug Discovery. J. Med. Chem. 2021, 64, 17728–17743. [Google Scholar] [CrossRef]
- Gunasekaran, K.; Ma, B.; Nussinov, R. Is Allostery an Intrinsic Property of All Dynamic Proteins? Proteins Struct. Funct. Bioinform. 2004, 57, 433–443. [Google Scholar] [CrossRef]
- Lou, H.; Cao, X. Antibody Variable Region Engineering for Improving Cancer Immunotherapy. Cancer Commun. 2022, 42, 804–827. [Google Scholar] [CrossRef]
- van Erp, E.A.; Luytjes, W.; Ferwerda, G.; van Kasteren, P.B. Fc-Mediated Antibody Effector Functions During Respiratory Syncytial Virus Infection and Disease. Front. Immunol. 2019, 10, 548. [Google Scholar] [CrossRef] [PubMed]
- Soto, J.A.; Gálvez, N.M.S.; Pacheco, G.A.; Bueno, S.M.; Kalergis, A.M. Antibody Development for Preventing the Human Respiratory Syncytial Virus Pathology. Mol. Med. 2020, 26, 35. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhang, H.; Zhou, T.; Pan, K.; Raza, S.H.A.; Shen, X.; Lei, H. A Non-Classical View of Antibody Properties: Allosteric Effect between Variable and Constant Regions. Biotechnol. Adv. 2025, 78, 108482. [Google Scholar] [CrossRef]
- Kennedy, P.J.; Oliveira, C.; Granja, P.L.; Sarmento, B. Monoclonal Antibodies: Technologies for Early Discovery and Engineering. Crit. Rev. Biotechnol. 2018, 38, 394–408. [Google Scholar] [CrossRef]
- Porter, R.R.; Press, E.M. The Fractionation of Bovine γ-Globulin by Partition Chromatography. Biochem. J. 1957, 66, 600–603. [Google Scholar] [CrossRef]
- Feinstein, A.; Rowe, A.J. Molecular Mechanism of Formation of an Antigen–Antibody Complex. Nature 1965, 205, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.R.; Padlan, E.A.; Segal, D.M. Three-Dimensional Structure of Immunoglobulins. Annu. Rev. Biochem. 1975, 44, 639–667. [Google Scholar] [CrossRef]
- Sun, Y.; Estevez, A.; Schlothauer, T.; Wecksler, A.T. Antigen Physiochemical Properties Allosterically Effect the IgG Fc-Region and Fc Neonatal Receptor Affinity. MAbs 2020, 12, 1802135. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Cutler, J.E. Hybridoma Passage In Vitro May Result in Reduced Ability of Antimannan Antibody To Protect against Disseminated Candidiasis. Infect. Immun. 2006, 74, 4310–4321. [Google Scholar] [CrossRef]
- Zhao, J.; Nussinov, R.; Ma, B. Antigen Binding Allosterically Promotes Fc Receptor Recognition. MAbs 2018, 11, 58–74. [Google Scholar] [CrossRef]
- Lindberg, M.C.; Cunningham, A.; Lindberg, N.H. Acute Phenobarbital Intoxication. S. Med. J. 1992, 85, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Food Safety Sampling, Inspection and Testing Department State Administration for Market Regulation People’s Republic of China. Detection of 75 Kinds of Illegally Added Chemical Drugs in Health Foods; BJS 201710; State Administration for Market Regulation: Beijing, China, 2017. Available online: https://www.samr.gov.cn/spcjs/bz/cs/art/2017/art_0365495da2fa4ee782b888f0cacd0b5b.html (accessed on 11 April 2025).
- Krausz, L.M.; Hitz, J.B.; Buckler, R.T.; Burd, J.F. Substrate-Labeled Fluorescent Immunoassay for Phenobarbital. Ther. Drug Monit. 1980, 2, 261–272. [Google Scholar] [CrossRef]
- Harris, L.J.; Skaletsky, E.; McPherson, A. Crystallographic Structure of an Intact IgG1 Monoclonal Antibody1. J. Mol. Biol. 1998, 275, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Menegatti, S.; Hussain, M.; Naik, A.D.; Carbonell, R.G.; Rao, B.M. mRNA Display Selection and Solid-Phase Synthesis of Fc-Binding Cyclic Peptide Affinity Ligands. Biotechnol. Bioeng. 2013, 110, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Xiao, S.; Jiang, X.; Tao, P. PASSer: Fast and Accurate Prediction of Protein Allosteric Sites. Nucleic Acids Res. 2023, 51, W427–W431. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, S.; Hu, Q.; Gao, S.; Ma, X.; Zhang, W.; Shen, Y.; Chen, F.; Lai, L.; Pei, J. CavityPlus: A Web Server for Protein Cavity Detection with Pharmacophore Modelling, Allosteric Site Identification and Covalent Ligand Binding Ability Prediction. Nucleic Acids Res. 2018, 46, W374–W379. [Google Scholar] [CrossRef]
- Huang, W.; Nussinov, R.; Zhang, J. Computational Tools for Allosteric Drug Discovery: Site Identification and Focus Library Design. In Computational Protein Design; Samish, I., Ed.; Springer: New York, NY, USA, 2017; pp. 439–446. ISBN 978-1-4939-6637-0. [Google Scholar]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Biovia, D.S. Discovery Studio Visualizer Software, Version 2019, Dassault Systèmes: San Diego, CA, USA, 2019. Available online: https://www.3ds.com/products-services/biovia/ (accessed on 11 April 2025).
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Haddad, M.; Gaudreault, R.; Sasseville, G.; Nguyen, P.T.; Wiebe, H.; Van De Ven, T.; Bourgault, S.; Mousseau, N.; Ramassamy, C. Molecular Interactions of Tannic Acid with Proteins Associated with SARS-CoV-2 Infectivity. Int. J. Mol. Sci. 2022, 23, 2643. [Google Scholar] [CrossRef]
- Li, L.; Zhang, R.; Xu, Y.; Zhang, W. Comprehensive Screening Strategy Coupled with Structure-Guided Engineering of l-Threonine Aldolase from Pseudomonas Putida for Enhanced Catalytic Efficiency towards l-Threo-4-Methylsulfonylphenylserine. Front. Bioeng. Biotechnol. 2023, 11, 1117890. [Google Scholar] [CrossRef]
- Petersen, B.M.; Ulmer, S.A.; Rhodes, E.R.; Gutierrez-Gonzalez, M.F.; Dekosky, B.J.; Sprenger, K.G.; Whitehead, T.A. Regulatory Approved Monoclonal Antibodies Contain Framework Mutations Predicted From Human Antibody Repertoires. Front. Immunol. 2021, 12, 728694. [Google Scholar] [CrossRef] [PubMed]
- Ngo, S.T.; Nguyen, T.H.; Pham, D.-H.; Tung, N.T.; Nam, P.C. Thermodynamics and Kinetics in Antibody Resistance of the 501Y.V2 SARS-CoV-2 Variant. RSC Adv. 2021, 11, 33438–33446. [Google Scholar] [CrossRef]
- Grant, B.J.; Skjærven, L.; Yao, X.-Q. The Bio3D Packages for Structural Bioinformatics. Protein Sci. 2021, 30, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Chang, S.; Hu, J.; Tian, Y.; Tian, X. Molecular Dynamics Simulations of Ternary Complexes: Comparisons of LEAFY Protein Binding to Different DNA Motifs. J. Chem. Inf. Model. 2015, 55, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Chen, Z.; Li, Q.; Liu, R.; Song, W.; Kong, R.; Chang, S. Molecular Dynamics Simulations of Wild Type and Mutants of SAPAP in Complexed with Shank3. Int. J. Mol. Sci. 2019, 20, 224. [Google Scholar] [CrossRef]
- Li, M.; Liu, X.; Zhang, S.; Liang, S.; Zhang, Q.; Chen, J. Deciphering the Binding Mechanism of Inhibitors of the SARS-CoV-2 Main Protease through Multiple Replica Accelerated Molecular Dynamics Simulations and Free Energy Landscapes. Phys. Chem. Chem. Phys. 2022, 24, 22129–22143. [Google Scholar] [CrossRef]
- Yan, F.; Liu, X.; Zhang, S.; Su, J.; Zhang, Q.; Chen, J. Effect of Double Mutations T790M/L858R on Conformation and Drug-Resistant Mechanism of Epidermal Growth Factor Receptor Explored by Molecular Dynamics Simulations. RSC Adv. 2018, 8, 39797–39810. [Google Scholar] [CrossRef]
- Bao, H.; Wang, W.; Sun, H.; Chen, J. Probing Mutation-Induced Conformational Transformation of the GTP/M-RAS Complex through Gaussian Accelerated Molecular Dynamics Simulations. J. Enzym. Inhib. Med. Chem. 2023, 38, 2195995. [Google Scholar] [CrossRef]
- Almeleebia, T.M.; Ahamad, S.; Ahmad, I.; Alshehri, A.; Alkhathami, A.G.; Alshahrani, M.Y.; Asiri, M.A.; Saeed, A.; Siddiqui, J.A.; Yadav, D.K.; et al. Identification of PARP12 Inhibitors By Virtual Screening and Molecular Dynamics Simulations. Front. Pharmacol. 2022, 13, 847499. [Google Scholar] [CrossRef]
- Belhassan, A.; Chtita, S.; Zaki, H.; Alaqarbeh, M.; Alsakhen, N.; Almohtaseb, F.; Lakhlifi, T.; Bouachrine, M. In Silico Detection of Potential Inhibitors from Vitamins and Their Derivatives Compounds against SARS-CoV-2 Main Protease by Using Molecular Docking, Molecular Dynamic Simulation and ADMET Profiling. J. Mol. Struct. 2022, 1258, 132652. [Google Scholar] [CrossRef]
- Ricci, C.G.; Silveira, R.L.; Rivalta, I.; Batista, V.S.; Skaf, M.S. Allosteric Pathways in the PPARγ-RXRα Nuclear Receptor Complex. Sci. Rep. 2016, 6, 19940. [Google Scholar] [CrossRef]
- Liu, M.; Wang, L.; Sun, X.; Zhao, X. Investigating the Impact of Asp181 Point Mutations on Interactions between PTP1B and Phosphotyrosine Substrate. Sci. Rep. 2014, 4, 5095. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J.; Zhu, W. Binding Modes of Three Inhibitors 8CA, F8A and I4A to A-FABP Studied Based on Molecular Dynamics Simulation. PLoS ONE 2014, 9, e99862. [Google Scholar] [CrossRef] [PubMed]
- Mercadante, D.; Gräter, F.; Daday, C. CONAN: A Tool to Decode Dynamical Information from Molecular Interaction Maps. Biophys. J. 2018, 114, 1267–1273. [Google Scholar] [CrossRef]
- Pellegrini, M. Tandem Repeats in Proteins: Prediction Algorithms and Biological Role. Front. Bioeng. Biotechnol. 2015, 3, 143. [Google Scholar] [CrossRef] [PubMed]
- Kayikci, M.; Venkatakrishnan, A.J.; Scott-Brown, J.; Ravarani, C.N.J.; Flock, T.; Babu, M.M. Visualization and Analysis of Non-Covalent Contacts Using the Protein Contacts Atlas. Nat. Struct. Mol. Biol. 2018, 25, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, G.; Casadevall, J.; Duran, C.; Osuna, S. The Shortest Path Method (SPM) Webserver for Computational Enzyme Design. Protein Eng. Des. Sel. 2024, 37, gzae005. [Google Scholar] [CrossRef]
- Maria-Solano, M.A.; Kinateder, T.; Iglesias-Fernández, J.; Sterner, R.; Osuna, S. In Silico Identification and Experimental Validation of Distal Activity-Enhancing Mutations in Tryptophan Synthase. ACS Catal. 2021, 11, 13733–13743. [Google Scholar] [CrossRef]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domański, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, USA, 11–17 July 2016. [Google Scholar] [CrossRef]
- CharlesHahn. CharlesHahn/DuIvyProcedures_Docs: DuIvyProcedures (v1.0.0). Zenodo. 2024. Available online: https://zenodo.org/records/10646114 (accessed on 11 April 2025).
- Kluska, K.; Chorążewska, A.; Peris-Díaz, M.D.; Adamczyk, J.; Krężel, A. Non-Conserved Amino Acid Residues Modulate the Thermodynamics of Zn(II) Binding to Classical Ββα Zinc Finger Domains. Int. J. Mol. Sci. 2022, 23, 14602. [Google Scholar] [CrossRef]
- Lechner, B.-D.; Smith, P.; McGill, B.; Marshall, S.; Trick, J.L.; Chumakov, A.P.; Winlove, C.P.; Konovalov, O.V.; Lorenz, C.D.; Petrov, P.G. The Effects of Cholesterol Oxidation on Erythrocyte Plasma Membranes: A Monolayer Study. Membranes 2022, 12, 828. [Google Scholar] [CrossRef]
- Tsigelny, I.F.; Sharikov, Y.; Wrasidlo, W.; Gonzalez, T.; Desplats, P.A.; Crews, L.; Spencer, B.; Masliah, E. Role of α-Synuclein Penetration into the Membrane in the Mechanisms of Oligomer Pore Formation. FEBS J. 2012, 279, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Łątka, K.; Bajda, M. Analysis of Binding Determinants for Different Classes of Competitive and Noncompetitive Inhibitors of Glycine Transporters. Int. J. Mol. Sci. 2022, 23, 8050. [Google Scholar] [CrossRef]
- Ikeuchi, E.; Kuroda, D.; Nakakido, M.; Murakami, A.; Tsumoto, K. Delicate Balance among Thermal Stability, Binding Affinity, and Conformational Space Explored by Single-Domain VHH Antibodies. Sci. Rep. 2021, 11, 20624. [Google Scholar] [CrossRef] [PubMed]
- Pathak, R.K.; Kim, J.-M. Identification of Histidine Kinase Inhibitors through Screening of Natural Compounds to Combat Mastitis Caused by Streptococcus Agalactiae in Dairy Cattle. J. Biol. Eng. 2023, 17, 59. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, J.W.; Bajardi-Taccioli, A.; Houde, D.; Feschenko, M.; Carlage, T.; Kaltashov, I.A. Influence of Glycan Modification on IgG1 Biochemical and Biophysical Properties. J. Pharm. Biomed. Anal. 2018, 151, 133–144. [Google Scholar] [CrossRef]
- Pathak, R.K.; Kim, W.-I.; Kim, J.-M. Targeting the PEDV 3CL Protease for Identification of Small Molecule Inhibitors: An Insight from Virtual Screening, ADMET Prediction, Molecular Dynamics, Free Energy Landscape, and Binding Energy Calculations. J. Biol. Eng. 2023, 17, 29. [Google Scholar] [CrossRef]
- KS, S.; Nair, A.S. Insights on the Interaction of SARS-CoV-2 Variant B.1.617.2 with Antibody CR3022 and Analysis of Antibody Resistance. J. Genet. Eng. Biotechnol. 2023, 21, 35. [Google Scholar] [CrossRef]
- Ma, N.; Nivedha, A.K.; Vaidehi, N. Allosteric Communication Regulates Ligand-Specific GPCR Activity. FEBS J. 2021, 288, 2502–2512. [Google Scholar] [CrossRef]
- Chong, W.L.; Saparpakorn, P.; Sangma, C.; Lee, V.S.; Hannongbua, S. Insight into Free Energy and Dynamic Cross-Correlations of Residue for Binding Affinity of Antibody and Receptor Binding Domain SARS-CoV-2. Heliyon 2023, 9, e12667. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody System | Signal Transduction Pathway |
|---|---|
| nGAb | T113, Q123, Y185, A223, Y305, T321, S333, M350, Y360, C455, P483, V512, R554, KG633, N779, F824, Y896, T952, C970, N1007, T1036, G1057, V1070, V1102, V1151, E1201, K1286 |
| nGAb-pep | A4, S12, T20, V30, K45, R61, K106, Y139, S170, S200, A226, A327, G359, S410, T480, S659, F759, I831, T1057, G1070, I1154 |
| GAb | C133, P203, S207, R210, T323, L356, C455, S557, D640, F759, G777, A817, L838, A971, G1005, R1102, T1108, S1152, N1199, K1286 |
| GAb-pep | F98, I105, F134, T171, E186, N209, V406, A558, V766, S840, D857, R973, G1007, S1152, E1201, K1286 |
| GAb-Ag | S14, R107, T113, K206, A230, V324, L353, C453, Q525, E662, S722, Q778, S818, G839, E872, R975, W1004, T1108, P1146 |
| GAb-Ag-pep | Q123, K182, R210, S333, I540, E729, Y780, V820, L838, R1102, S1153, K1286 |
| Common residues | Q123, S207, S326, C455, A558, Q778, D838, R975, R1102, P1146, V1200, K1286 |
| Ligand Bind Site | Interaction Type | Antibody System | Occupancy (%) |
|---|---|---|---|
| Antigen binding site (orthosteric site) | Hydrogen bond | GAb-Ag | 4.5 |
| GAb-Ag-pep | 16.7 | ||
| Hydrophobic | Gab-Ag | 23.7 | |
| Gab-Ag-pep | 22.2 | ||
| Cyclopeptide binding site (allosteric site) | Hydrogen bond | GAb-pep | 17.5 |
| GAb-Ag-pep | 67.1 | ||
| Hydrophobic | GAb-pep | 35.8 | |
| GAb-Ag-pep | 46.8 |
| Interaction Type | Antibody System | Min | Max | Average |
|---|---|---|---|---|
| Hydrogen bond count (pair) | nGAb | 146 | 249 | 208 |
| NGAb-pep | 162 | 245 | 208 | |
| GAb | 169 | 252 | 213 | |
| Gab-pep | 158 | 260 | 212 | |
| Gab-Ag | 153 | 251 | 206 | |
| GAb-Ag-pep | 159 | 248 | 209 | |
| Hydrophobic count (pair) | nGAb | 40,544 | 41,530 | 41,096 |
| nGAb-pep | 40,604 | 41,588 | 41,153 | |
| GAb | 40,588 | 41,590 | 41,144 | |
| GAb-pep | 40,650 | 41,800 | 41,256 | |
| GAb-Ag | 40,568 | 41,660 | 41,126 | |
| GAb-Ag-pep | 40,632 | 41,616 | 41,142 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, T.; Zhang, H.; Yu, X.; Pan, K.; Yao, X.; Shen, X.; Lei, H. Fc-Binding Cyclopeptide Induces Allostery from Fc to Fab: Revealed Through in Silico Structural Analysis to Anti-Phenobarbital Antibody. Foods 2025, 14, 1360. https://doi.org/10.3390/foods14081360
Zhou T, Zhang H, Yu X, Pan K, Yao X, Shen X, Lei H. Fc-Binding Cyclopeptide Induces Allostery from Fc to Fab: Revealed Through in Silico Structural Analysis to Anti-Phenobarbital Antibody. Foods. 2025; 14(8):1360. https://doi.org/10.3390/foods14081360
Chicago/Turabian StyleZhou, Tao, Huiling Zhang, Xiaoting Yu, Kangliang Pan, Xiaojun Yao, Xing Shen, and Hongtao Lei. 2025. "Fc-Binding Cyclopeptide Induces Allostery from Fc to Fab: Revealed Through in Silico Structural Analysis to Anti-Phenobarbital Antibody" Foods 14, no. 8: 1360. https://doi.org/10.3390/foods14081360
APA StyleZhou, T., Zhang, H., Yu, X., Pan, K., Yao, X., Shen, X., & Lei, H. (2025). Fc-Binding Cyclopeptide Induces Allostery from Fc to Fab: Revealed Through in Silico Structural Analysis to Anti-Phenobarbital Antibody. Foods, 14(8), 1360. https://doi.org/10.3390/foods14081360