
Temporal Profile of the Microbial Community and Volatile Compounds in the Third-Round Fermentation of Sauce-Flavor baijiu in the Beijing Region


Abstract
1. Introduction
2. Materials and Methods
2.1. Samples Collection
2.2. DNA Extraction and Amplicon Sequencing
2.3. Volatile Compounds Analysis
2.4. Fermentation Parameters Analysis
2.5. Data Analysis
3. Results
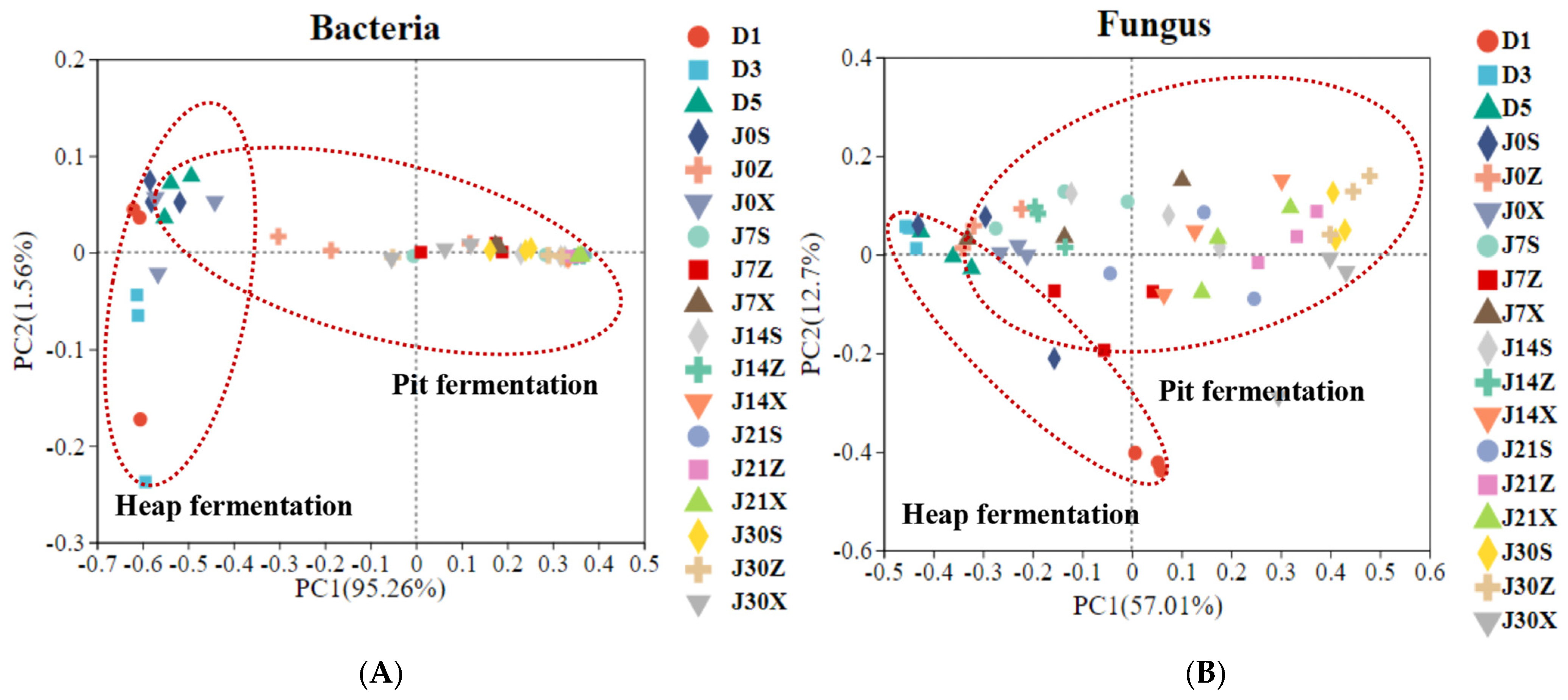
3.1. Temporal Profile of Microbial Community Composition
3.2. Temporal Profile of Volatile Compounds
3.3. Changes in Fermentation Parameters
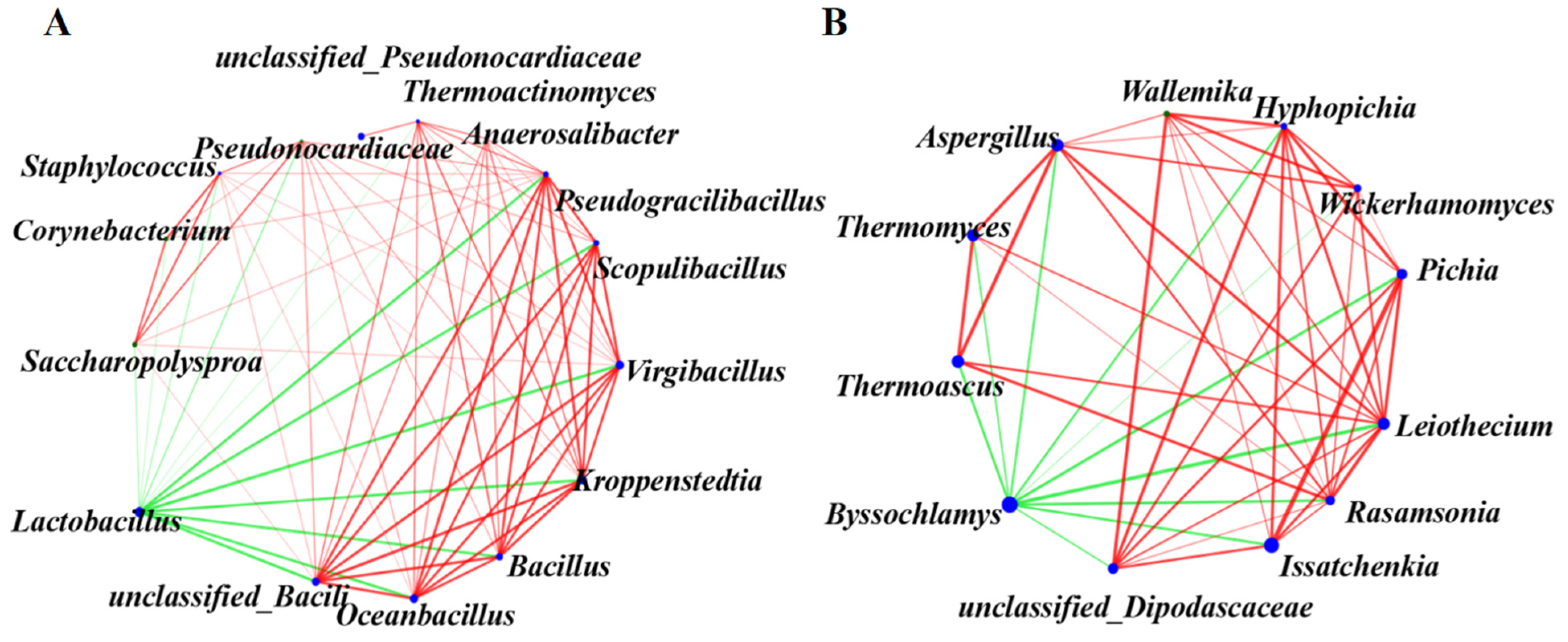
3.4. Correlations between Microbial Community Structure and Fermentation Parameters
3.5. Correlations between Microbial Community Structure and Volatile Compounds
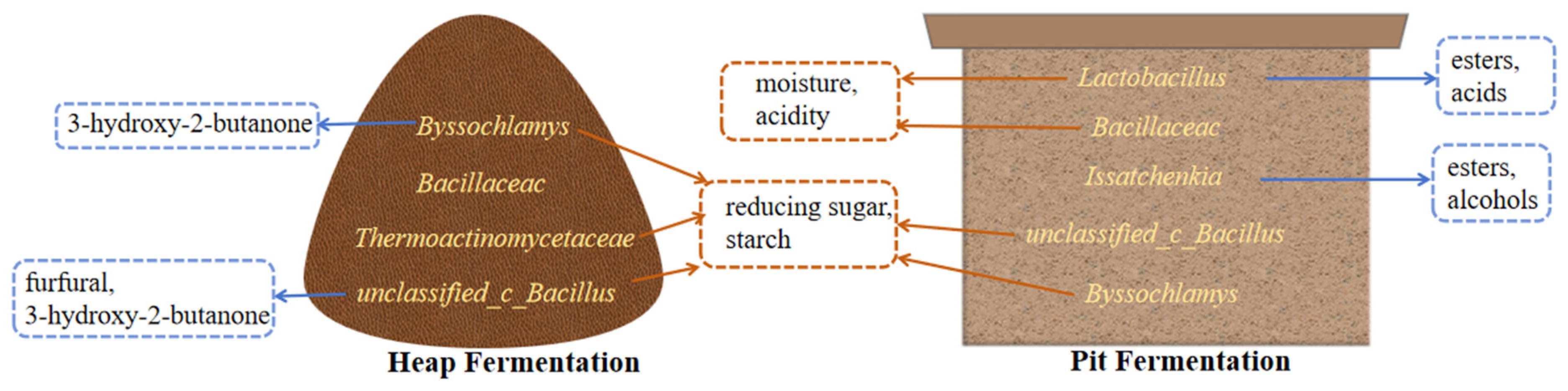
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, Y.H.; Yi, Z.L.; Jin, Y.L.; Huang, M.J.; He, K.Z.; Liu, D.Y.; Zhao, H. Metatranscriptomics Reveals the Functions and Enzyme Profiles of the Microbial Community in Chinese Nong-Flavor Liquor Starter. Front. Microbiol. 2017, 8, 14. [Google Scholar] [CrossRef]
- Sun, J.Y.; Zhao, D.R.; Zhang, F.G.; Sun, B.G.; Zheng, F.P.; Huang, M.Q.; Li, H.H. Joint direct injection and GC-MS chemometric approach for chemical profile and sulfur compounds of sesame-flavor Chinese Baijiu (Chinese liquor). Eur. Food Res. Technol. 2018, 244, 145–160. [Google Scholar] [CrossRef]
- Wu, Y.L.; Hao, F.; Lv, X.B.; Chen, B.; Yang, Y.B.; Zeng, X.L.; Wang, L. Diversity of lactic acid bacteria in Moutai-flavor liquor fermentation process. Food Biotechnol. 2020, 34, 212–227. [Google Scholar] [CrossRef]
- Wang, L. Research trends in Jiang-flavor baijiu fermentation: From fermentation microecology to environmental ecology. J. Food Sci. 2022, 87, 1362–1374. [Google Scholar] [CrossRef]
- Xu, Y.; Zhao, J.; Huang, H.; Guo, X.; Li, X.; Zou, W.; Huang, M. Biodegradation of phthalate esters by Pantoea dispersa BJQ0007 isolated from Baijiu. J. Food Compos. Anal. 2022, 105, 104201. [Google Scholar] [CrossRef]
- Wang, H.; Huang, Y.; Huang, Y. Microbiome diversity and evolution in stacking fermentation during different rounds of Jiang-flavoured Baijiu brewing. LWT-Food Sci. Technol. 2021, 143, 111119. [Google Scholar] [CrossRef]
- Wang, L.; Huang, Y.; Hu, X.; Li, Y. The impact of environmental factors on the environmental bacterial diversity and composition in the Jiang-flavoured Baijiu production region. LWT-Food Sci. Technol. 2021, 149, 111784. [Google Scholar] [CrossRef]
- Wang, D.; Chen, L.; Yang, F.; Wang, H.; Wang, L. Yeasts and their importance to the flavour of traditional Chinese liquor: A review. J. Inst. Brew. 2019, 125, 214–221. [Google Scholar] [CrossRef]
- Li, Y.; Cheng, Y.; Wang, H.; Hu, X.; Wang, L.; Huang, Y. Diverse structure and characteristics of the fungal community during the different rounds of Jiang-flavoured Baijiu production in Moutai town. LWT-Food Sci. Technol. 2022, 161, 113313. [Google Scholar] [CrossRef]
- Cai, W.; Wang, Y.; Wang, W.; Shu, N.; Hou, Q.; Tang, F.; Shan, C.; Yang, X.; Guo, Z. Insights into the aroma profile of sauce-flavor baijiu by GC-IMS combined with multivariate statistical analysis. J. Anal. Methods Chem. 2022, 2022, 4614330. [Google Scholar] [CrossRef]
- Hao, F.; Tan, Y.; Lv, X.; Chen, L.; Yang, F.; Wang, H.; Xu, Y. Microbial Community Succession and Its Environment Driving Factors During Initial Fermentation of Maotai-Flavor Baijiu. Front. Microbiol. 2021, 12, 669201. [Google Scholar] [CrossRef]
- Wang, X.; Du, H.; Zhang, Y.; Xu, Y. Environmental Microbiota Drives Microbial Succession and Metabolic Profiles during Chinese Liquor Fermentation. Appl. Environ. Microbiol. 2018, 84, e02369-17. [Google Scholar] [CrossRef]
- Wang, W.; Xu, Y.; Huang, H.; Pang, Z.; Fu, Z.; Niu, J.; Sun, B. Correlation between microbial communities and flavor compounds during the fifth and sixth rounds of sauce-flavor baijiu fermentation. Food Res. Int. 2021, 150, 110741. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Tian, Z.; Meng, W.; Li, Z. Microbial Diversity and Physicochemical Characteristics of the Maotai-Flavored Liquor Fermentation Process. J. Nanosci. Nanotechnol. 2020, 20, 4097–4109. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Du, H.; Zhang, H.; Fang, C.; Jin, G.; Chen, S.; Xu, Y. Geographically Associated Fungus-Bacterium Interactions Contribute to the Formation of Geography-Dependent Flavor during High-Complexity Spontaneous Fermentation. Microbiol. Spectr. 2022, 10, e01844-22. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, C.; Zhao, H.; Min, W.; Zhu, H.; Wang, H.; Li, W. Effect of Environmental Microorganisms on Fermentation Microbial Community of Sauce-Flavor baijiu. Foods 2023, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Garcia-Fernandez, D.; Mas, A.; Esteve-Zarzoso, B. Fungal diversity in grape must and wine fermentation assessed by massive sequencing, quantitative PCR and DGGE. Front. Microbiol. 2015, 6, 1156. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, C.; Xu, Q.; Li, Z.; Song, Y.; Zhou, S.; Luo, X. Bacterial Diversity and Lactic Acid Bacteria with High Alcohol Tolerance in the Fermented Grains of Soy Sauce Aroma Type Baijiu in North China. Foods 2022, 11, 1794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Du, H.; Xu, Y. Volatile organic compound-mediated antifungal activity of Pichia spp. and its effect on the metabolic profiles of fermentation communities. Appl. Environ. Microbiol. 2021, 87, e02992-20. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, X.; Liu, X.; Li, X.; Zhang, C.; Li, W.; Sun, B. Discovery and development of a novel short-chain fatty acid ester synthetic biocatalyst under aqueous phase from Monascus purpureus isolated from Baijiu. Food Chem. 2021, 338, 128025. [Google Scholar] [CrossRef]
- Song, Z.; Du, H.; Zhang, Y.; Xu, Y. Unraveling Core Functional Microbiota in Traditional Solid-State Fermentation by High-Throughput Amplicons and Metatranscriptomics Sequencing. Front. Microbiol. 2017, 8, 1294. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Lin, W.; Liu, X.; Wang, X.; Luo, L. Environmental Factors Affecting Microbiota Dynamics during Traditional Solid-state Fermentation of Chinese Daqu Starter. Front. Microbiol. 2016, 7, 1237. [Google Scholar] [CrossRef] [PubMed]
- ISO:5377; Starch Hydrolysis Products. Determination of Reducing Power and Dextrose Equivalent. International Organization for Standardization: Geneva, Switzerland, 1981.
- Chen, X.; Wu, Y.; Zhu, H.; Wang, H.; Lu, H.; Zhang, C.; Wang, Y. Turning over fermented grains elevating heap temperature and driving microbial community succession during the heap fermentation of sauce-flavor baijiu. LWT-Food Sci. Technol. 2022, 172, 114173. [Google Scholar] [CrossRef]
- Liu, C.; Feng, S.; Wu, Q.; Huang, H.; Chen, Z.; Li, S.; Xu, Y. Raw Material Regulates Flavor Formation via Driving Microbiota in Chinese Liquor Fermentation. Front. Microbiol. 2019, 10, 1520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Abdo, A.A.A.; Kaddour, B.; Wu, Q.; Xin, L.; Li, X.; Teng, C. Xylan-oligosaccharides ameliorate high fat diet induced obesity and glucose intolerance and modulate plasma lipid profile and gut microbiota in mice. J. Funct. Foods 2020, 64, 103622. [Google Scholar] [CrossRef]
- Jin, Y.; Li, D.; Ai, M.; Tang, Q.; Huang, J.; Ding, X.; Zhou, R. Correlation between volatile profiles and microbial communities: A metabonomic approach to study Jiang-flavor liquor Daqu. Food Res. Int. 2019, 121, 422–432. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, L.; Tan, Y.; Wang, H.; Yang, F.; Chen, L.; Xu, Y. Effect of Pichia on shaping the fermentation microbial community of sauce-flavor Baijiu. Int. J. Food Microbiol. 2021, 336, 108898. [Google Scholar] [CrossRef]
- Shi, X.; Zhao, S.; Chen, S.; Han, X.; Yang, Q.; Zhang, L.; Hu, Y. Tetramethylpyrazine in Chinese baijiu: Presence, analysis, formation, and regulation. Front. Nutr. 2022, 9, 1004435. [Google Scholar] [CrossRef]
- Du, R.; Wu, Q.; Xu, Y. Chinese Liquor Fermentation: Identification of Key Flavor-Producing Lactobacillus spp. by Quantitative Profiling with Indigenous Internal Standards. Appl. Environ. Microbiol. 2020, 86, e00456-20. [Google Scholar] [CrossRef]
- Li, K.; Chen, Y.; Liu, T.; Deng, M.; Xu, Z.; Fu, G.; Zheng, F. Analysis of spatial distribution of bacterial community associated with accumulation of volatile compounds in Jiupei during the brewing of special-flavor liquor. LWT-Food Sci. Technol. 2020, 130, 109620. [Google Scholar] [CrossRef]
- Hu, L.; Wang, J.; Ji, X.; Liu, R.; Chen, F.; Zhang, X. (Selection of non-Saccharomyces yeasts for orange wine fermentation based on their enological traits and volatile compounds formation. J. Food Sci. Technol.-Mysore 2018, 55, 4001–4012. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Y.; Li, R.; Liu, L.; Miao, Y.; Weng, P.; Wu, Z. Metabolic changes of Issatchenkia orientalis under acetic acid stress by transcriptome profile using RNA-sequencing (Nov, 10.1007/s10123-021-00217-6, 2022). Int. Microbiol. 2022, 25, 661. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Miao, L. Multiple Batches of Fermentation Promote the Formation of Functional Microbiota in Chinese Miscellaneous-Flavor Baijiu Fermentation. Front. Microbiol. 2020, 11, 75. [Google Scholar] [CrossRef] [PubMed]
- Houbraken, J.; Samson, R.A.; Frisvad, J.C. Byssochlamys: Significance of heat resistance and mycotoxin production. In Advances in Food Mycology; Hocking, A.D., Pitt, J.I., Samson, R.A., Thrane, U., Eds.; Springer: Boston, MA, USA, 2006; Volume 571, pp. 211–224. [Google Scholar]
- Utami, A.S.; Sunarti, T.; Isono, N.; Hisamatsu, M.; Ehara, H. Preparation of biodegradable foam from sago residue. Sago Palm 2014, 22, 1–5. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Sampling Location | Sampling Time |
|---|---|---|
| D1 | heap fermentation mixing the upper and lower layers | 1 days |
| D3 | heap fermentation mixing the upper and lower layers | 3 days |
| D5 | heap fermentation mixing the upper and lower layers | 5 days |
| J0S | pit fermentation upper layers | 0 days |
| J0Z | pit fermentation middle layers | 0 days |
| J0X | pit fermentation lower layers | 0 days |
| J7S | pit fermentation upper layers | 7 days |
| J7Z | pit fermentation middle layers | 7 days |
| J7X | pit fermentation lower layers | 7 days |
| J14S | pit fermentation upper layers | 14 days |
| J14Z | pit fermentation middle layers | 14 days |
| J14X | pit fermentation lower layers | 14 days |
| J21S | pit fermentation upper layers | 21 days |
| J21Z | pit fermentation middle layers | 21 days |
| J21X | pit fermentation lower layers | 21 days |
| J30S | pit fermentation upper layers | 30 days |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Zhang, H.; Wang, R.; Zhang, C.; Li, X. Temporal Profile of the Microbial Community and Volatile Compounds in the Third-Round Fermentation of Sauce-Flavor baijiu in the Beijing Region. Foods 2024, 13, 670. https://doi.org/10.3390/foods13050670
Li W, Zhang H, Wang R, Zhang C, Li X. Temporal Profile of the Microbial Community and Volatile Compounds in the Third-Round Fermentation of Sauce-Flavor baijiu in the Beijing Region. Foods. 2024; 13(5):670. https://doi.org/10.3390/foods13050670
Chicago/Turabian StyleLi, Weiwei, Hui Zhang, Runnan Wang, Chengnan Zhang, and Xiuting Li. 2024. "Temporal Profile of the Microbial Community and Volatile Compounds in the Third-Round Fermentation of Sauce-Flavor baijiu in the Beijing Region" Foods 13, no. 5: 670. https://doi.org/10.3390/foods13050670
APA StyleLi, W., Zhang, H., Wang, R., Zhang, C., & Li, X. (2024). Temporal Profile of the Microbial Community and Volatile Compounds in the Third-Round Fermentation of Sauce-Flavor baijiu in the Beijing Region. Foods, 13(5), 670. https://doi.org/10.3390/foods13050670