

Food-Borne Chemical Carcinogens and the Evidence for Human Cancer Risk

Abstract
1. Introduction
1.1. Mechanisms of Carcinogenicity of DNA-Reactive Carcinogens
1.2. Mechanisms of Carcinogenicity of Epigenetic Carcinogens
2. Risk Assessment of Food-Derived Carcinogens
2.1. Application of Carcinogenicity Data to Human Risk
2.2. Risk Assessment of DNA-Reactive Rodent Carcinogens
2.3. Risk Assessment of Epigenetic Carcinogens
3. DNA-Reactive Carcinogens and Related Chemicals Present in Food
3.1. Phytotoxins

3.1.1. Alkenylbenzene Derivatives
3.1.1.1. Safrole
3.1.1.2. Estragole
3.1.1.3. Methyl Eugenol
3.1.1.4. α- and β-asarone
3.1.2. Aristolochic Acids
3.1.3. Glucosides
3.1.3.1. Cycasin
3.1.3.2. Ptaquiloside and Bracken Fern
3.1.4. Psoralens
3.1.5. Pyrrolizidine Alkaloids
3.2. Mycotoxins

3.2.1. Aflatoxins
3.2.2. Ochratoxin A
3.3. Carcinogens Formed during Food Processing

3.3.1. Benzene
3.3.2. Chloropropanols
3.3.3. Ethyl Carbamate (Urethane)
3.4. Heat-Generated Carcinogens

3.4.1. Acrylamide
3.4.2. Heterocyclic Amines
3.4.3. Polycyclic Aromatic Hydrocarbons
3.5. Carcinogens Formed Exogenously and Endogenously

3.5.1. Ethylene Oxide
3.5.2. N-Nitroso Compounds
3.5.3. α,β-Unsaturated Aldehydes
3.5.3.1. Malondialdehyde, 4-Hydroxynonenal, Crotonaldehyde, trans,trans-2,4-hexadienal
3.5.3.2. Trans-2 hexenal
3.6. Carcinogenicity of Preserved and Processed Foods
3.6.1. Preserved Vegetables
3.6.2. Red and Processed Meat
3.6.3. Salted Fish
4. Epigenetic Carcinogens or Carcinogens with Uncertain Mode of Action and Related Chemicals Present in Food
4.1. Phytotoxins

4.1.1. β-Myrcene
4.1.2. Pulegone
4.2. Mycotoxins

4.2.1. Fumonisins
4.2.2. Fusarin C
4.3. Environmental, Agricultural and Industrial Contaminants

4.3.1. Agricultural Contaminants
4.3.1.1. p,p′-dichlorodiphenyltrichloroethane
4.3.1.2. Dioxins and Dioxin-Like Compounds
4.3.2. Food Contact Materials
4.3.2.1. Benzophenone
4.3.2.2. Di(2-ethylhexyl) Phthalate
4.3.2.3. 1,4-dioxane
4.3.2.4. Methyl Isobutyl Ketone
4.4. Carcinogens Formed during Processing, Packaging and Storage of Food

4.4.1. Alkylated Imidazoles
4.4.2. Furan
4.5. Food Additives

Monocyclic Phenolics, Synthetic and Natural
5. Food-Borne Chemopreventive Agents
6. Discussion and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- National Research Council Committee, on Comparative Toxicity of Naturally Occurring Carcinogens. Carcinogens and Anticarcinogens in the Human Diet: A Comparison of Naturally Occurring and Synthetic Substances; The National Academies Collection: Reports Funded by National Institutes of Health; National Academies Press (US): Washington, DC, USA, 1996.
- Williams, G.M. Food-borne carcinogens. Prog. Clin. Biol. Res. 1986, 206, 73–81. [Google Scholar]
- Abnet, C.C. Carcinogenic food contaminants. Cancer Investig. 2007, 25, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, T. Nutrition and dietary carcinogens. Carcinogenesis 2000, 21, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.S. Chemical food safety issues in the United States: Past, present, and future. J. Agric. Food Chem. 2009, 57, 8161–8170. [Google Scholar] [CrossRef] [PubMed]
- Rietjens, I.M.C.M.; Michael, A.; Bolt, H.M.; Siméon, B.; Andrea, H.; Nils, H.; Christine, K.; Angela, M.; Gloria, P.; Daniel, R.; et al. The role of endogenous versus exogenous sources in the exposome of putative genotoxins and consequences for risk assessment. Arch. Toxicol. 2022, 96, 1297–1352. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S.; Hoffmann, D. N-nitroso compounds and man: Sources of exposure, endogenous formation and occurrence in body fluids. Eur. J. Cancer Prev. 1998, 7, 165–166. [Google Scholar]
- Tricker, A.R.; Preussmann, R. Carcinogenic N-nitrosamines in the diet: Occurrence, formation, mechanisms and carcinogenic potential. Mutat. Res./Genet. Toxicol. 1991, 259, 277–289. [Google Scholar] [CrossRef]
- Key, T.J.; Bradbury, K.E.; Perez-Cornago, A.; Sinha, R.; Tsilidis, K.K.; Tsugane, S. Diet, nutrition, and cancer risk: What do we know and what is the way forward? BMJ 2020, 368, m511. [Google Scholar] [CrossRef]
- Reddy, B.S.; Cohen, L.A.; David McCoy, G.; Hill, P.; Weisburger, J.H.; Wynder, E.L. Nutrition and its relationship to cancer. Adv. Cancer Res. 1980, 32, 237–345. [Google Scholar] [CrossRef]
- Clapp, R.W.; Howe, G.K.; Jacobs, M.M. Environmental and occupational causes of cancer: A call to act on what we know. Biomed. Pharm. 2007, 61, 631–639. [Google Scholar] [CrossRef]
- Doll, R.; Peto, R. The causes of cancer: Quantitative estimates of avoidable risks of cancer in the United States today. JNCI J. Natl. Cancer Inst. 1981, 66, 1192–1308. [Google Scholar] [CrossRef]
- Rumgay, H.; Shield, K.; Charvat, H.; Ferrari, P.; Sornpaisarn, B.; Obot, I.; Islami, F.; Lemmens, V.; Rehm, J.; Soerjomataram, I. Global burden of cancer in 2020 attributable to alcohol consumption: A population-based study. Lancet Oncol. 2021, 22, 1071–1080. [Google Scholar] [CrossRef]
- Cogliano, V.J.; Baan, R.; Straif, K.; Grosse, Y.; Lauby-Secretan, B.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; et al. Preventable exposures associated with human cancers. J. Natl. Cancer Inst. 2011, 103, 1827–1839. [Google Scholar] [CrossRef] [PubMed]
- IARC, International Agency for Research on Cancer. Pharmaceuticals. Volume 100 A. A review of human carcinogens. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 1–401. [Google Scholar]
- Pflaum, T.; Hausler, T.; Baumung, C.; Ackermann, S.; Kuballa, T.; Rehm, J.; Lachenmeier, D.W. Carcinogenic compounds in alcoholic beverages: An update. Arch. Toxicol. 2016, 90, 2349–2367. [Google Scholar] [CrossRef]
- Kobets, T.; Iatropoulos, M.J.; Williams, G.M. Mechanisms of DNA-reactive and epigenetic chemical carcinogens: Applications to carcinogenicity testing and risk assessment. Toxicol. Res. 2019, 8, 123–145. [Google Scholar] [CrossRef]
- Williams, G.M.; Iatropoulos, M.J.; Enzmann, H.G.; Deschl, U. Carcinogenicity of chemicals: Assessment and human extrapolation. In Hayes’ Principles and Methods of Toxicology, 6th ed.; Hayes, A., Kruger, C.L., Eds.; Taylor and Francis: Philadelphia, PA, USA, 2014; pp. 1251–1303. [Google Scholar]
- Williams, G.M. Mechanisms of chemical carcinogenesis and application to human cancer risk assessment. Toxicology 2001, 166, 3–10. [Google Scholar] [CrossRef]
- Weisburger, J.H.; Williams, G.M. The distinction between genotoxic and epigenetic carcinogens and implication for cancer risk. Toxicol. Sci. 2000, 57, 4–5. [Google Scholar] [CrossRef]
- Williams, G.M. Application of mode-of-action considerations in human cancer risk assessment. Toxicol. Lett. 2008, 180, 75–80. [Google Scholar] [CrossRef]
- Williams, G.M. Chemicals with carcinogenic activity in the rodent liver; mechanistic evaluation of human risk. Cancer Lett. 1997, 117, 175–188. [Google Scholar] [CrossRef]
- Miller, E.C.; Miller, J.A. Biochemical mechanisms of chemical carcinogenesis. Mol. Biol. Cancer 1974, 377–402. [Google Scholar] [CrossRef]
- Preston, R.J.; Williams, G.M. DNA-reactive carcinogens: Mode of action and human cancer hazard. Crit. Rev. Toxicol. 2005, 35, 673–683. [Google Scholar] [CrossRef]
- Hartwig, A.; Arand, M.; Epe, B.; Guth, S.; Jahnke, G.; Lampen, A.; Martus, H.-J.; Monien, B.; Rietjens, I.M.C.M.; Schmitz-Spanke, S.; et al. Mode of action-based risk assessment of genotoxic carcinogens. Arch. Toxicol. 2020, 94, 1787–1877. [Google Scholar] [CrossRef] [PubMed]
- Hanawalt, P. Functional characterization of global genomic DNA repair and its implications for cancer. Mutat. Res./Rev. Mutat. Res. 2003, 544, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.; Fazio, M.; Williams, G.M. Cell cycle-specific mutagenesis at the hypoxanthine phosphoribosyltransferase locus in adult rat liver epithelial cells. Proc. Natl. Acad. Sci. USA 1980, 77, 7377–7379. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, W.K.; Kaufman, D.G. Cell cycle control, DNA repair and initiation of carcinogenesis. FASEB J. 1993, 7, 1188–1191. [Google Scholar] [CrossRef] [PubMed]
- Pagès, V.; Fuchs, R.P. How DNA lesions are turned into mutations within cells? Oncogene 2002, 21, 8957–8966. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Williams, G.M.; Iatropoulos, M.J.; Jeffrey, A.M. Mechanistic basis for nonlinearities and thresholds in rat liver carcinogenesis by the DNA-reactive carcinogens 2-acetylaminofluorene and diethylnitrosamine. Toxicol. Pathol. 2000, 28, 388–395. [Google Scholar] [CrossRef]
- Cohen, S.M.; Arnold, L.L. Chemical carcinogenesis. Toxicol. Sci. 2011, 120 (Suppl. S1), S76–S92. [Google Scholar] [CrossRef]
- Poirier, M.C. Chemical-induced DNA damage and human cancer risk. Discov. Med. 2012, 14, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Paini, A.; Scholz, G.; Marin-Kuan, M.; Schilter, B.; O’Brien, J.; van Bladeren, P.J.; Rietjens, I.M.C.M. Quantitative comparison between in vivo DNA adduct formation from exposure to selected DNA-reactive carcinogens, natural background levels of DNA adduct formation and tumour incidence in rodent bioassays. Mutagenesis 2011, 26, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Hwa Yun, B.; Guo, J.; Bellamri, M.; Turesky, R.J. DNA adducts: Formation, biological effects, and new biospecimens for mass spectrometric measurements in humans. Mass Spectrom. Rev. 2020, 39, 55–82. [Google Scholar] [CrossRef] [PubMed]
- Poirier, M.C.; Beland, F.A. DNA adduct measurements and tumor incidence during chronic carcinogen exposure in rodents. Environ. Health Perspect. 1994, 102 (Suppl. S6), 161–165. [Google Scholar] [CrossRef]
- Doerge, D.R.; Gamboa da Costa, G.; McDaniel, L.P.; Churchwell, M.I.; Twaddle, N.C.; Beland, F.A. DNA adducts derived from administration of acrylamide and glycidamide to mice and rats. Mutat. Res. 2005, 580, 131–141. [Google Scholar] [CrossRef]
- Lafferty, J.S.; Kamendulis, L.M.; Kaster, J.; Jiang, J.; Klaunig, J.E. Subchronic acrylamide treatment induces a tissue-specific increase in DNA synthesis in the rat. Toxicol. Lett. 2004, 154, 95–103. [Google Scholar] [CrossRef]
- Pavanello, S.; Bollati, V.; Pesatori, A.C.; Kapka, L.; Bolognesi, C.; Bertazzi, P.A.; Baccarelli, A. Global and gene-specific promoter methylation changes are related to anti-B[a]PDE-DNA adduct levels and influence micronuclei levels in polycyclic aromatic hydrocarbon-exposed individuals. Int. J. Cancer 2009, 125, 1692–1697. [Google Scholar] [CrossRef]
- Williams, G.M.; Iatropoulos, M.J.; Weisburger, J.H. Chemical carcinogen mechanisms of action and implications for testing methodology. Exp. Toxicol. Pathol. 1996, 48, 101–111. [Google Scholar] [CrossRef]
- Phillips, D.H.; Arlt, V.M. Genotoxicity: Damage to DNA and its consequences. EXS 2009, 99, 87–110. [Google Scholar] [CrossRef]
- Neumann, H.-G. Risk assessment of chemical carcinogens and thresholds. Crit. Rev. Toxicol. 2009, 39, 449–461. [Google Scholar] [CrossRef]
- Kobets, T.; Williams, G.M. Thresholds for hepatocarcinogenicity of DNA-reactive compounds. In Thresholds of Genotoxic Carcinogens; Academic Press: Cambridge, MA, USA, 2016; pp. 19–36. [Google Scholar] [CrossRef]
- Nohmi, T. Thresholds of genotoxic and non-genotoxic carcinogens. Toxicol. Res. 2018, 34, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Kobets, T.; Williams, G.M. Review of the evidence for thresholds for DNA-reactive and epigenetic experimental chemical carcinogens. Chem. Biol. Interact. 2019, 301, 88–111. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.M.; Iatropoulos, M.J.; Jeffrey, A.M. Dose-effect relationships for DNA-reactive liver carcinogens. In Cellular Response to the Genotoxic Insult: The Question of Threshold for Genotoxic Carcinogens; Oxford Academic: Oxford, UK, 2012; pp. 33–51. [Google Scholar] [CrossRef]
- EFSA CONTAM Panel, European Food Safety Authority, Panel on Contaminants in the Food Chain. Scientific Opinion of the Panel on Contaminants in the Food Chain on a Request from the European Commission on Polycyclic Aromatic Hydrocarbons in Food; The EFSA Journal Series, No. 724; European Food Safety Authority: Parma, Italy, 2008; 114p.
- Williams, G.M. DNA reactive and epigenetic carcinogens. Exp. Toxicol. Pathol. 1992, 44, 457–463. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Kamendulis, L.M.; Xu, Y. Epigenetic mechanisms of chemical carcinogenesis. Hum. Exp. Toxicol. 2000, 19, 543–555. [Google Scholar] [CrossRef]
- Sawan, C.; Vaissière, T.; Murr, R.; Herceg, Z. Epigenetic drivers and genetic passengers on the road to cancer. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2008, 642, 1–13. [Google Scholar] [CrossRef]
- Pogribny, I.P.; Rusyn, I.; Beland, F.A. Epigenetic aspects of genotoxic and non-genotoxic hepatocarcinogenesis: Studies in rodents. Environ. Mol. Mutagen. 2008, 49, 9–15. [Google Scholar] [CrossRef]
- Pogribny, I.P.; Rusyn, I. Environmental toxicants, epigenetics, and cancer. Adv. Exp. Med. Biol. 2013, 754, 215–232. [Google Scholar] [CrossRef]
- Williams, G.M.; Jeffrey, A.M. Oxidative DNA damage: Endogenous and chemically induced. Regul. Toxicol. Pharmacol. 2000, 32, 283–292. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Kamendulis, L.M. The role of oxidative stress in carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 239–267. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- IARC, International Agency for Research on Cancer. Preamble to the IARC Monographs (Amended January 2019); International Agency for Research on Cancer: Lyon, France, 2019. [Google Scholar]
- Wiltse, J.; Dellarco, V.L. U.S. Environmental Protection Agency guidelines for carcinogen risk assessment: Past and future. Mutat. Res./Rev. Genet. Toxicol. 1996, 365, 3–15. [Google Scholar] [CrossRef]
- Jeffrey, A.M.; Williams, G.M. Risk assessment of DNA-reactive carcinogens in food. Toxicol. Appl. Pharm. 2005, 207, 628–635. [Google Scholar] [CrossRef]
- Felter, S.P.; Bhat, V.S.; Botham, P.A.; Bussard, D.A.; Casey, W.; Hayes, A.W.; Hilton, G.M.; Magurany, K.A.; Sauer, U.G.; Ohanian, E.V. Assessing chemical carcinogenicity: Hazard identification, classification, and risk assessment. Insight from a Toxicology Forum state-of-the-science workshop. Crit. Rev. Toxicol. 2021; 51, 653–694. [Google Scholar] [CrossRef]
- Barlow, S.; Schlatter, J. Risk assessment of carcinogens in food. Toxicol. Appl. Pharm. 2010, 243, 180–190. [Google Scholar] [CrossRef]
- Raffaele, K.; Vulimiri, S.; Bateson, T. Benefits and barriers to using epidemiology data in environmental risk assessment. Open Epidemiol. J. 2011, 411, 99–105. [Google Scholar] [CrossRef]
- Kobets, T.; Williams, G.M. Chemicals with carcinogenic activity primarily in rodent liver. In Comprehensive Toxicology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 409–442. [Google Scholar] [CrossRef]
- Edler, L.; Hart, A.; Greaves, P.; Carthew, P.; Coulet, M.; Boobis, A.; Williams, G.M.; Smith, B. Selection of appropriate tumour data sets for Benchmark Dose Modelling (BMD) and derivation of a Margin of Exposure (MoE) for substances that are genotoxic and carcinogenic: Considerations of biological relevance of tumour type, data quality and uncertainty assessment. Food Chem. Toxicol. 2014, 70, 264–289. [Google Scholar] [CrossRef]
- Rosenkranz, H.S. SAR modeling of genotoxic phenomena: The consequence on predictive performance of deviation from a unity ratio of genotoxicants/non-genotoxicants. Mutat. Res./Genet. Toxicol. Environ. Mutagen. 2004, 559, 67–71. [Google Scholar] [CrossRef]
- IARC, International Agency for Research on Cancer. Agents Classified by the IARC Monographs; International Agency for Research on Cancer: Lyon, France, 2022; Volume 1–131. [Google Scholar]
- NTP, National Toxicology Program. Report on Carcinogens, (RoC), 15th ed.; National Toxicology Program: Research Triangle, NC, USA, 2021.
- O’Brien, J.; Renwick, A.G.; Constable, A.; Dybing, E.; Müller, D.J.G.; Schlatter, J.; Slob, W.; Tueting, W.; van Benthem, J.; Williams, G.M.; et al. Approaches to the risk assessment of genotoxic carcinogens in food: A critical appraisal. Food Chem. Toxicol. 2006, 44, 1613–1635. [Google Scholar] [CrossRef]
- Benford, D.; Bolger, P.M.; Carthew, P.; Coulet, M.; DiNovi, M.; Leblanc, J.-C.; Renwick, A.G.; Setzer, W.; Schlatter, J.; Smith, B.; et al. Application of the Margin of Exposure (MOE) approach to substances in food that are genotoxic and carcinogenic. Food Chem. Toxicol. 2010, 48, S2–S24. [Google Scholar] [CrossRef]
- Herceg, Z. Epigenetics and cancer: Towards an evaluation of the impact of environmental and dietary factors. Mutagenesis 2007, 22, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Braakhuis, H.M.; Slob, W.; Olthof, E.D.; Wolterink, G.; Zwart, E.P.; Gremmer, E.R.; Rorije, E.; van Benthem, J.; Woutersen, R.; van der Laan, J.W.; et al. Is current risk assessment of non-genotoxic carcinogens protective? Crit. Rev. Toxicol. 2018, 48, 500–511. [Google Scholar] [CrossRef]
- Swenberg, J.A.; Lehman-McKeeman, L.D. alpha 2-Urinary globulin-associated nephropathy as a mechanism of renal tubule cell carcinogenesis in male rats. IARC Sci. Publ. 1999, 147, 95–118. [Google Scholar]
- Williams, G.M.; Whysner, J. Epigenetic carcinogens: Evaluation and risk assessment. Exp. Toxicol. Pathol. 1996, 48, 189–195. [Google Scholar] [CrossRef]
- van den Berg, S.; Restani, P.; Boersma, M.; Delmulle, L.; Rietjens, I. Levels of genotoxic and carcinogenic compounds in plant food supplements and associated risk assessment. Food Nutr. Sci. 2011, 2, 989–1010. [Google Scholar] [CrossRef]
- Rietjens, I.M.; Cohen, S.M.; Fukushima, S.; Gooderham, N.J.; Hecht, S.; Marnett, L.J.; Smith, R.L.; Adams, T.B.; Bastaki, M.; Harman, C.G.; et al. Impact of structural and metabolic variations on the toxicity and carcinogenicity of hydroxy- and alkoxy-substituted allyl- and propenylbenzenes. Chem. Res. Toxicol. 2014, 27, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Eisenreich, A.; Götz, M.E.; Sachse, B.; Monien, B.H.; Herrmann, K.; Schäfer, B. Alkenylbenzenes in foods: Aspects impeding the evaluation of adverse health effects. Foods 2021, 10, 2139. [Google Scholar] [CrossRef]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Safety Evaluation of Certain Food Additives. Prepared by the Sixty-Ninth Meeting of the Joint FAO/WHO Expert Committee on Food Additives; World Health Organization: Geneva, Switzerland, 2009; Volume 952, pp. 9–21. [Google Scholar]
- Smith, R.L.; Adams, T.B.; Doull, J.; Feron, V.J.; Goodman, J.I.; Marnett, L.J.; Portoghese, P.S.; Waddell, W.J.; Wagner, B.M.; Rogers, A.E.; et al. Safety assessment of allylalkoxybenzene derivatives used as flavouring substances—Methyl eugenol and estragole. Food Chem. Toxicol. 2002, 40, 851–870. [Google Scholar] [CrossRef]
- Al-Subeihi, A.A.; Spenkelink, B.; Rachmawati, N.; Boersma, M.G.; Punt, A.; Vervoort, J.; van Bladeren, P.J.; Rietjens, I.M. Physiologically based biokinetic model of bioactivation and detoxification of the alkenylbenzene methyleugenol in rat. Toxicol. Vitr. 2011, 25, 267–285. [Google Scholar] [CrossRef]
- Martati, E.; Boersma, M.G.; Spenkelink, A.; Khadka, D.B.; van Bladeren, P.J.; Rietjens, I.M.; Punt, A. Physiologically based biokinetic (PBBK) modeling of safrole bioactivation and detoxification in humans as compared with rats. Toxicol. Sci. 2012, 128, 301–316. [Google Scholar] [CrossRef]
- Rietjens, I.M.C.M.; Boersma, M.G.; van der Woude, H.; Jeurissen, S.M.F.; Schutte, M.E.; Alink, G.M. Flavonoids and alkenylbenzenes: Mechanisms of mutagenic action and carcinogenic risk. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2005, 574, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Jeurissen, S.M.F.; Punt, A.; Boersma, M.G.; Bogaards, J.J.P.; Fiamegos, Y.C.; Schilter, B.; van Bladeren, P.J.; Cnubben, N.H.P.; Rietjens, I.M.C.M. Human cytochrome P450 enzyme specificity for the bioactivation of estragole and related alkenylbenzenes. Chem. Res. Toxicol. 2007, 20, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Randerath, K.; Putman, K.L.; Randerath, E. Flavor constituents in cola drinks induce hepatic DNA adducts in adult and fetal mice. Biochem. Biophys. Res. Commun. 1993, 192, 61–68. [Google Scholar] [CrossRef]
- Kobets, T.; Duan, J.-D.; Brunnemann, K.D.; Etter, S.; Smith, B.; Williams, G.M. Structure-activity relationships for DNA damage by alkenylbenzenes in turkey egg fetal liver. Toxicol. Sci. 2016, 150, 301–311. [Google Scholar] [CrossRef]
- Kobets, T.; Cartus, A.T.; Fuhlbrueck, J.A.; Brengel, A.; Stegmüller, S.; Duan, J.-D.; Brunnemann, K.D.; Williams, G.M. Assessment and characterization of DNA adducts produced by alkenylbenzenes in fetal turkey and chicken livers. Food Chem. Toxicol. 2019, 129, 424–433. [Google Scholar] [CrossRef]
- IARC, International Agency for Research on Cancer. Overall Evaluations of Carcinogenicity: An Updating of IARC Monographs Volumes 1 to 42; IARC Monographs Supplement 7; World Health Organization: Geneva, Switzerland, 1987. [Google Scholar]
- Kamdem, D.; Gage, D. Chemical composition of essential oil from the root bark of Sassafras albidum. Planta Med. 1995, 61, 574–575. [Google Scholar] [CrossRef] [PubMed]
- IARC, International Agency for Research on Cancer. Some naturally occurring substances. IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Man. Environ. Pollut. 1976, 10, 1–353. [Google Scholar] [CrossRef]
- SCF, Scientific Committee on Food. Opinion of the Scientific Committee on Food on the Safety of the Presence of Safrole (1-allyl-3,4-Methylene Dioxy Benzene) in Flavourings and Other Food Ingredients with Flavouring Properties; SCF/CS/FLAV/FLAVOUR/6; Scientific Committee on Food: Brussels, Belgium, 2002. [Google Scholar]
- Wiseman, R.W.; Miller, E.C.; Miller, J.A.; Liem, A. Structure-activity studies of the hepatocarcinogenicities of alkenylbenzene derivatives related to estragole and safrole on administration to preweanling male C57BL/6J × C3H/HeJ F1 mice. J. Ethnopharmacol. 1987, 22, 319. [Google Scholar] [CrossRef]
- Miller, E.C.; Swanson, A.B.; Phillips, D.H.; Fletcher, T.L.; Liem, A.; Miller, J.A. Structure-activity studies of the carcinogenicities in the mouse and rat of some naturally occurring and synthetic alkenylbenzene derivatives related to safrole and estragole. Cancer Res. 1983, 43, 1124–1134. [Google Scholar]
- Daimon, H.; Sawada, S.; Asakura, S.; Sagami, F. In vivo genotoxicity and DNA adduct levels in the liver of rats treated with safrole. Carcinogenesis 1998, 19, 141–146. [Google Scholar] [CrossRef][Green Version]
- Kevekordes, S.; Spielberger, J.; Burghaus, C.M.; Birkenkamp, P.; Zietz, B.; Paufler, P.; Diez, M.; Bolten, C.; Dunkelberg, H. Micronucleus formation in human lymphocytes and in the metabolically competent human hepatoma cell line Hep-G2: Results with 15 naturally occurring substances. Anticancer Res. 2001, 21, 461–469. [Google Scholar] [PubMed]
- Gupta, K.P.; van Golen, K.L.; Putman, K.L.; Randerath, K. Formation and persistence of safrole-DNA adducts over a 10,000-fold dose range in mouse liver. Carcinogenesis 1993, 14, 1517–1521. [Google Scholar] [CrossRef] [PubMed]
- Randerath, K.; Haglund, R.E.; Phillips, D.H.; Reddy, M.V. 32P-post-labelling analysis of DNA adducts formed in the livers of animals treated with safrole, estragole and other naturally-occurring alkenylbenzenes. I. Adult female CD-1 mice. Carcinogenesis 1984, 5, 1613–1622. [Google Scholar] [CrossRef]
- Lee, J.-M.; Liu, T.-Y.; Wu, D.-C.; Tang, H.-C.; Leh, J.; Wu, M.-T.; Hsu, H.-H.; Huang, P.-M.; Chen, J.-S.; Lee, C.-J.; et al. Safrole–DNA adducts in tissues from esophageal cancer patients: Clues to areca-related esophageal carcinogenesis. Mutat. Res. /Genet. Toxicol. Environ. Mutagen. 2005, 565, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.D.; Moorthy, B.; Bi, J.; Donnelly, K.C.; Randerath, K. DNA adducts from alkoxyallylbenzene herb and spice constituents in cultured human (HepG2) cells. Environ. Mol. Mutagen. 2007, 48, 715–721. [Google Scholar] [CrossRef]
- Boberg, E.W.; Miller, E.C.; Miller, J.A.; Poland, A.; Liem, A. Strong evidence from studies with brachymorphic mice and pentachlorophenol that 1′-sulfoöxysafrole is the major ultimate electrophilic and carcinogenic metabolite of 1′-hydroxysafrole in mouse liver. Cancer Res. 1983, 43, 5163–5173. [Google Scholar]
- Boberg, E.W.; Miller, E.C.; Miller, J.A. The metabolic sulfonation and side-chain oxidation of 3′-hydroxyisosafrole in the mouse and its inactivity as a hepatocarcinogen relative to 1′-hydroxysafrole. Chem. Biol. Interact. 1986, 59, 73–97. [Google Scholar] [CrossRef]
- Daimon, H.; Sawada, S.; Asakura, S.; Sagami, F. Inhibition of sulfotransferase affecting in vivo genotoxicity and DNA adducts induced by safrole in rat liver. Teratog. Carcinog. Mutagen. 1997, 17, 327–337. [Google Scholar] [CrossRef]
- Beyer, J.; Ehlers, D.; Maurer, H.H. Abuse of nutmeg (Myristica Fragrans Houtt.): Studies on the metabolism and the toxicologic detection of its ingredients elemicin, myristicin, and safrole in rat and human urine using gas chromatography/mass spectrometry. Ther. Drug Monit. 2006, 28, 568–575. [Google Scholar] [CrossRef]
- Chen, C.L.; Chi, C.W.; Chang, K.W.; Liu, T.Y. Safrole-like DNA adducts in oral tissue from oral cancer patients with a betel quid chewing history. Carcinogenesis 1999, 20, 2331–2334. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.T.; Chen, C.L.; Wu, C.C.; Chan, S.A.; Chi, C.W.; Liu, T.Y. Safrole-DNA adduct in hepatocellular carcinoma associated with betel quid chewing. Toxicol. Lett. 2008, 183, 21–27. [Google Scholar] [CrossRef] [PubMed]
- FDA, Food and Drug Administration. Title 21-Food and Drugs. Chapter I-Food and Drug Administration Department of Health and Human Services, Subchapter B-Food for Human Consumption (Continued). Part 189-Substances Prohibited from Use in Human Food. Subpart C-Substances Generally Prohibited from Direct Addition or Use as Human Food. Sec. 189.180 Safrole; Food and Drug Administration: Silber Spring, MD, USA, 2017.
- Commission Regulation, (EU). Regulation (EC) No 1334/2008 of the European Parliament and of the Council of 16 December 2008 on flavourings and certain food ingredients with flavouring properties for use in and on foods and amending Council Regulation (EEC) No 1601/91, Regulations (EC) No 2232/96 and (EC) No 110/2008 and Directive 2000/13/EC. Off. J. Eur. Union 2008, 354, 218–234. [Google Scholar]
- Liu, T.Y.; Chung, Y.T.; Wang, P.F.; Chi, C.W.; Hsieh, L.L. Safrole-DNA adducts in human peripheral blood—An association with areca quid chewing and CYP2E1 polymorphisms. Mutat. Res. 2004, 559, 59–66. [Google Scholar] [CrossRef]
- EMA, European Medicines Agency. Public Statement on the Use of Herbal Medicinal Products Containing Estragole; 2nd Draft, Revision 1, MA/HMPC/137212/2005 Rev 1; European Medicines Agency: Amsterdam, The Netherlands, 2019.
- NTP, National Toxicology Program. Technical Report on the 3-Month Toxicity Studies of Estragole (CAS No. 140-67-0) Administered by Gavage to F344/N Rats and B6C3F1 Mice; 1521-4621 (Print); National Toxicology Program: Research Triangle, NC, USA, 2011; pp. 1–111.
- Müller, L.; Kasper, P.; Müller-Tegethoff, K.; Petr, T. The genotoxic potential in vitro and in vivo of the allyl benzene etheric oils estragole, basil oil and trans-anethole. Mutat. Res. Lett. 1994, 325, 129–136. [Google Scholar] [CrossRef]
- Ding, W.; Levy, D.D.; Bishop, M.E.; Pearce, M.G.; Davis, K.J.; Jeffrey, A.M.; Duan, J.D.; Williams, G.M.; White, G.A.; Lyn-Cook, L.E.; et al. In vivo genotoxicity of estragole in male F344 rats. Environ. Mol. Mutagen. 2015, 56, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Suzuki, Y.; Hibi, D.; Jin, M.; Fukuhara, K.; Umemura, T.; Nishikawa, A. Detection and quantification of specific DNA adducts by liquid chromatography-tandem mass spectrometry in the livers of rats given estragole at the carcinogenic dose. Chem. Res. Toxicol. 2011, 24, 532–541. [Google Scholar] [CrossRef]
- Anthony, A.; Caldwell, J.; Hutt, A.J.; Smith, R.L. Metabolism of estragole in rat and mouse and influence of dose size on excretion of the proximate carcinogen 1′-hydroxyestragole. Food Chem. Toxicol. 1987, 25, 799–806. [Google Scholar] [CrossRef]
- Waddell, W.J. Thresholds of carcinogenicity of flavors. Toxicol. Sci. 2002, 68, 275–279. [Google Scholar] [CrossRef]
- IARC, International Agency for Research on Cancer. Some Chemicals Present in Industrial and Consumer Products, Food and Drinking Water; IARC Monograhs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2013; Volume 101. [Google Scholar]
- Moshonas, M.G.; Shaw, P.E. Compounds new to essential orange oil from fruit treated with abscission chemicals. J. Agric. Food Chem. 1978, 26, 1288–1290. [Google Scholar] [CrossRef]
- NTP, National Toxicology Program. Technical Report on the Toxicology and Carcinogenesis Studies of Methyleugenol (CAS NO. 93-15-2) in F344/N Rats and B6C3F1 Mice (Gavage Studies); 0888-8051 (Print); National Toxicology Program: Research Triangle, NC, USA, 2000; pp. 1–412.
- Williams, G.M.; Iatropoulos, M.J.; Jeffrey, A.M.; Duan, J.-D. Methyleugenol hepatocellular cancer initiating effects in rat liver. Food Chem. Toxicol. 2013, 53, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Waddell, W.J. Correlation of tumors with DNA adducts from methyl eugenol and tamoxifen in rats. Toxicol. Sci. 2004, 79, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Al-Subeihi, A.A.; Spenkelink, B.; Punt, A.; Boersma, M.G.; van Bladeren, P.J.; Rietjens, I.M. Physiologically based kinetic modeling of bioactivation and detoxification of the alkenylbenzene methyleugenol in human as compared with rat. Toxicol. Appl. Pharm. 2012, 260, 271–284. [Google Scholar] [CrossRef]
- Lutz, W.; Gaylor, D.; Conolly, R.; Lutz, R. Nonlinearity and thresholds in dose–response relationships for carcinogenicity due to sampling variation, logarithmic dose scaling, or small differences in individual susceptibility. Toxicol. Appl. Pharmacol. 2005, 207, 565–569. [Google Scholar] [CrossRef]
- Smith, B.; Cadby, P.; Leblanc, J.C.; Setzer, R.W. Application of the Margin of Exposure (MoE) approach to substances in food that are genotoxic and carcinogenic: Example: Methyleugenol, CASRN: 93-15-2. Food Chem. Toxicol. 2010, 48 (Suppl. S1), S89–S97. [Google Scholar] [CrossRef]
- Chandra, S.A.; Nolan, M.W.; Malarkey, D.E. Chemical carcinogenesis of the gastrointestinal tract in rodents: An overview with emphasis on NTP carcinogenesis bioassays. Toxicol. Pathol. 2010, 38, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Eisenbrand, G.; Fukushima, S.; Gooderham, N.; Guengerich, F.; Hecht, S.; Rietjens, I.; Harman, C.; Taylor, S. GRAS 28 flavoring substances. Food Technol. 2018, 72, 62–77. [Google Scholar]
- Hermes, L.; Römermann, J.; Cramer, B.; Esselen, M. Quantitative analysis of β-asarone derivatives in Acorus calamus and herbal food products by HPLC-MS/MS. J. Agric. Food Chem. 2021, 69, 776–782. [Google Scholar] [CrossRef]
- Uebel, T.; Hermes, L.; Haupenthal, S.; Müller, L.; Esselen, M. α-Asarone, β-asarone, and γ-asarone: Current status of toxicological evaluation. J. Appl. Toxicol. 2021, 41, 1166–1179. [Google Scholar] [CrossRef]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Toxicological Evaluation of Certain Food Additives; Monograph on β-asarone: WHO Food Additive Series No. 16; World Health Organization: Geneva, Switzerland, 1981; Volume 16. [Google Scholar]
- SCF, Scientific Committee on Food. Opinion of the Scientific Committee on Food on the Presence of β-Asarone in Flavourings and Other Food Ingredients with Flavouring Properties; SCF/CS/FLAV/FLAVOUR/9 ADD1 Final 2002; European Commission Health & Consumer Protection Directorate-General: Brussels, Belgium, 2002. [Google Scholar]
- Kim, S.G.; Liem, A.; Stewart, B.C.; Miller, J.A. New studies on trans-anethole oxide and trans-asarone oxide. Carcinogenesis 1999, 20, 1303–1307. [Google Scholar] [CrossRef]
- Berg, K.; Bischoff, R.; Stegmüller, S.; Cartus, A.; Schrenk, D. Comparative investigation of the mutagenicity of propenylic and allylic asarone isomers in the Ames fluctuation assay. Mutagenesis 2016, 31, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Hasheminejad, G.; Caldwell, J. Genotoxicity of the alkenylbenzenes α− and β-asarone, myristicin and elemicin as determined by the UDS assay in cultured rat hepatocytes. Food Chem. Toxicol. 1994, 32, 223–231. [Google Scholar] [CrossRef]
- Haupenthal, S.; Berg, K.; Gründken, M.; Vallicotti, S.; Hemgesberg, M.; Sak, K.; Schrenk, D.; Esselen, M. In vitro genotoxicity of carcinogenic asarone isomers. Food Funct. 2017, 8, 1227–1234. [Google Scholar] [CrossRef]
- Stegmüller, S.; Schrenk, D.; Cartus, A.T. Formation and fate of DNA adducts of alpha- and beta-asarone in rat hepatocytes. Food Chem. Toxicol. 2018, 116, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Cartus, A.T.; Stegmüller, S.; Simson, N.; Wahl, A.; Neef, S.; Kelm, H.; Schrenk, D. Hepatic metabolism of carcinogenic β-asarone. Chem. Res. Toxicol. 2015, 28, 1760–1773. [Google Scholar] [CrossRef] [PubMed]
- Cartus, A.T.; Schrenk, D. Metabolism of the carcinogen alpha-asarone in liver microsomes. Food Chem. Toxicol. 2016, 87, 103–112. [Google Scholar] [CrossRef]
- Cartus, A.T.; Schrenk, D. Metabolism of carcinogenic alpha-asarone by human cytochrome P450 enzymes. Naunyn Schmiedebergs Arch. Pharm. 2020, 393, 213–223. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, X.X.; Sun, Q.M.; Chen, M.; Liu, S.L.; Zhang, X.; Zhou, J.Y.; Zou, X. β-Asarone inhibits gastric cancer cell proliferation. Oncol. Rep. 2015, 34, 3043–3050. [Google Scholar] [CrossRef]
- Chen, M.; Zhuang, Y.W.; Wu, C.E.; Peng, H.Y.; Qian, J.; Zhou, J.Y. β-Asarone suppresses HCT116 colon cancer cell proliferation and liver metastasis in part by activating the innate immune system. Oncol. Lett. 2021, 21, 435. [Google Scholar] [CrossRef]
- FDA, Food and Drug Administration. Title 21-Food and Drugs. Chapter I-Food and Drug Administration Department of Health and Human Services, Subchapter B-Food for Human Consumption (Continued). Part 189-Substances Prohibited from Use in Human Food. Subpart C-Substances Generally Prohibited from Direct Addition or Use as Human Food. Sec. 189.110 Calamus and Its Derivatives; Food and Drug Administration: Silver Springs, MD, USA, 2013.
- EMA, European Medicines Agency. Public Statement on the Risks Associated with the Use of Herbal Products Containing Aristolochia Species; EMEA/HMPC/138381/2005; European Medicines Agency: Amsterdam, The Netherlands, 2005.
- Arlt, V.M.; Stiborova, M.; Schmeiser, H.H. Aristolochic acid as a probable human cancer hazard in herbal remedies: A review. Mutagenesis 2002, 17, 265–277. [Google Scholar] [CrossRef]
- Abdullah, R.; Diaz, L.N.; Wesseling, S.; Rietjens, I.M. Risk assessment of plant food supplements and other herbal products containing aristolochic acids using the margin of exposure (MOE) approach. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2017, 34, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Liu, Z.H.; Qiu, Q.; Li, H.; Li, L.S. Tumour induction in rats following exposure to short-term high dose aristolochic acid I. Mutagenesis 2005, 20, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Mengs, U. Tumour induction in mice following exposure to aristolochic acid. Arch. Toxicol. 1988, 61, 504–505. [Google Scholar] [CrossRef]
- Zhang, H.; Cifone, M.A.; Murli, H.; Erexson, G.L.; Mecchi, M.S.; Lawlor, T.E. Application of simplified in vitro screening tests to detect genotoxicity of aristolochic acid. Food Chem. Toxicol. 2004, 42, 2021–2028. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhou, C.; Cao, Y.; Xi, J.; Ohira, T.; He, L.; Huang, P.; You, X.; Liu, W.; Zhang, X.; et al. Assessment of Pig-a, micronucleus, and comet assay endpoints in Tg.RasH2 mice carcinogenicity study of aristolochic acid I. Environ. Mol. Mutagen. 2020, 61, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Bárta, F.; Dedíková, A.; Bebová, M.; Dušková, Š.; Mráz, J.; Schmeiser, H.H.; Arlt, V.M.; Hodek, P.; Stiborová, M. Co-exposure to aristolochic acids I and II increases DNA adduct formation responsible for aristolochic acid I-mediated carcinogenicity in rats. Int. J. Mol. Sci. 2021, 22, 479. [Google Scholar] [CrossRef]
- Abdullah, R.; Wesseling, S.; Spenkelink, B.; Louisse, J.; Punt, A.; Rietjens, I. Defining in vivo dose-response curves for kidney DNA adduct formation of aristolochic acid I in rat, mouse and human by an in vitro and physiologically based kinetic modeling approach. J. Appl. Toxicol. 2020, 40, 1647–1660. [Google Scholar] [CrossRef]
- Schmeiser, H.H.; Schoepe, K.B.; Wiessler, M. DNA adduct formation of aristolochic acid I and II in vitro and in vivo. Carcinogenesis 1988, 9, 297–303. [Google Scholar] [CrossRef]
- McDaniel, L.P.; Elander, E.R.; Guo, X.; Chen, T.; Arlt, V.M.; Mei, N. Mutagenicity and DNA adduct formation by aristolochic acid in the spleen of Big Blue® rats. Environ. Mol. Mutagen. 2012, 53, 358–368. [Google Scholar] [CrossRef]
- Mei, N.; Arlt, V.M.; Phillips, D.H.; Heflich, R.H.; Chen, T. DNA adduct formation and mutation induction by aristolochic acid in rat kidney and liver. Mutat. Res. 2006, 602, 83–91. [Google Scholar] [CrossRef]
- Rebhan, K.; Ertl, I.E.; Shariat, S.F.; Grollman, A.P.; Rosenquist, T. Aristolochic acid and its effect on different cancers in uro-oncology. Curr. Opin. Urol. 2020, 30, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Grollman, A.P. Aristolochic acid nephropathy: Harbinger of a global iatrogenic disease. Environ. Mol. Mutagen. 2013, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. Metabolic activation of carcinogenic aristolochic acid, a risk factor for Balkan endemic nephropathy. Mutat. Res. 2008, 658, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Fernando, R.C.; Schmeiser, H.H.; Scherf, H.R.; Wiessler, M. Formation and persistence of specific purine DNA adducts by 32P-postlabelling in target and non-target organs of rats treated with aristolochic acid I. IARC Sci. Publ. 1993, 124, 167–171. [Google Scholar]
- Feldmeyer, N.; Schmeiser, H.H.; Muehlbauer, K.R.; Belharazem, D.; Knyazev, Y.; Nedelko, T.; Hollstein, M. Further studies with a cell immortalization assay to investigate the mutation signature of aristolochic acid in human p53 sequences. Mutat. Res./Genet. Toxicol. Environ. Mutagen. 2006, 608, 163–168. [Google Scholar] [CrossRef]
- Levi, M.; Guchelaar, H.J.; Woerdenbag, H.J.; Zhu, Y.P. Acute hepatitis in a patient using a Chinese herbal tea—A case report. Pharm. World Sci. 1998, 20, 43–44. [Google Scholar] [CrossRef]
- Schaneberg, B.T.; Applequist, W.L.; Khan, I.A. Determination of aristolochic acid I and II in North American species of Asarum and Aristolochia. Pharmazie 2002, 57, 686–689. [Google Scholar] [PubMed]
- Gökmen, M.R.; Lord, G.M. Aristolochic acid nephropathy. BMJ 2012, 344, e4000. [Google Scholar] [CrossRef]
- Nortier, J.L.; Martinez, M.C.; Schmeiser, H.H.; Arlt, V.M.; Bieler, C.A.; Petein, M.; Depierreux, M.F.; De Pauw, L.; Abramowicz, D.; Vereerstraeten, P.; et al. Urothelial carcinoma associated with the use of a Chinese herb (Aristolochia fangchi). N. Engl. J. Med. 2000, 342, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Nortier, J.L.; Vanherweghem, J.L. Renal interstitial fibrosis and urothelial carcinoma associated with the use of a Chinese herb (Aristolochia fangchi). Toxicology 2002, 181–182, 577–580. [Google Scholar] [CrossRef]
- Martena, M.J.; van der Wielen, J.C.A.; van de Laak, L.F.J.; Konings, E.J.M.; de Groot, H.N.; Rietjens, I.M.C.M. Enforcement of the ban on aristolochic acids in Chinese traditional herbal preparations on the Dutch market. Anal. Bioanal. Chem. 2007, 389, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Dong, Z. Toxic phytochemicals and their potential risks for human cancer. Cancer Prev. Res. 2015, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sieber, S.M.; Correa, P.; Dalgard, D.W.; McIntire, K.R.; Adamson, R.H. Carcinogenicity and hepatotoxicity of cycasin and its aglycone methylazoxymethanol acetate in nonhuman primates. J. Natl. Cancer Inst. 1980, 65, 177–189. [Google Scholar] [PubMed]
- Kuniyasu, T.; Tanaka, T.; Shima, H.; Sugie, S.; Mori, H.; Takahashi, M. Enhancing effect of cholecystectomy on colon carcinogenesis induced by methylazoxymethanol acetate in hamsters. Dis. Colon Rectum 1986, 29, 492–494. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.S.; Maeura, Y. Dose-response studies of the effect of dietary butylated hydroxyanisole on colon carcinogenesis induced by methylazoxymethanol acetate in female CF1 mice. J. Natl. Cancer Inst. 1984, 72, 1181–1187. [Google Scholar] [PubMed]
- Tanaka, T.; Shinoda, T.; Yoshimi, N.; Niwa, K.; Iwata, H.; Mori, H. Inhibitory effect of magnesium hydroxide on methylazoxymethanol acetate-induced large bowel carcinogenesis in male F344 rats. Carcinogenesis 1989, 10, 613–616. [Google Scholar] [CrossRef]
- Lijinsky, W.; Saavedra, J.E.; Reuber, M.D. Organ-specific carcinogenesis in rats by methyl- and ethylazoxyalkanes. Cancer Res. 1985, 45, 76–79. [Google Scholar]
- Hoffmann, G.R.; Morgan, R.W. Review: Putative mutagens and carcinogens in foods. V. Cycad azoxyglycosides. Environ. Mutagen. 1984, 6, 103–116. [Google Scholar] [CrossRef]
- Williams, G.M.; Laspia, M.F.; Mori, H.; Hirono, I. Genotoxicity of cycasin in the hepatocyte primary culture/DNA repair test supplemented with beta-glucosidase. Cancer Lett. 1981, 12, 329–333. [Google Scholar] [CrossRef]
- Kawai, K.; Furukawa, H.; Hirono, I. Genotoxic activity in vivo of the naturally occurring glucoside, cycasin, in the Drosophila wing spot test. Mutat. Res. 1995, 346, 145–149. [Google Scholar] [CrossRef]
- Matsushima, T.; Matsumoto, H.; Shirai, A.; Sawamura, M.; Sugimura, T. Mutagenicity of the naturally occurring carcinogen cycasin and synthetic methylazoxymethanol conjugates in Salmonella typhimurium. Cancer Res. 1979, 39, 3780–3782. [Google Scholar] [PubMed]
- Cavanna, M.; Parodi, S.; Taningher, M.; Bolognesi, C.; Sciabà, L.; Brambilla, G. DNA fragmentation in some organs of rats and mice treated with cycasin. Br. J. Cancer 1979, 39, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Klaus, V.; Bastek, H.; Damme, K.; Collins, L.B.; Frötschl, R.; Benda, N.; Lutter, D.; Ellinger-Ziegelbauer, H.; Swenberg, J.A.; Dietrich, D.R.; et al. Time-matched analysis of DNA adduct formation and early gene expression as predictive tool for renal carcinogenesis in methylazoxymethanol acetate treated Eker rats. Arch. Toxicol. 2017, 91, 3427–3438. [Google Scholar] [CrossRef] [PubMed]
- Kisby, G.E.; Ellison, M.; Spencer, P.S. Content of the neurotoxins cycasin (methylazoxymethanol beta-D-glucoside) and BMAA (beta-N-methylamino-L-alanine) in cycad flour prepared by Guam Chamorros. Neurology 1992, 42, 1336–1340. [Google Scholar] [CrossRef]
- Esclaire, F.; Kisby, G.; Spencer, P.; Milne, J.; Lesort, M.; Hugon, J. The Guam cycad toxin methylazoxymethanol damages neuronal DNA and modulates tau mRNA expression and excitotoxicity. Exp. Neurol. 1999, 155, 11–21. [Google Scholar] [CrossRef]
- Fiala, E.S.; Sohn, O.S.; Hamilton, S.R. Effects of chronic dietary ethanol on in vivo and in vitro metabolism of methylazoxymethanol and on methylazoxymethanol-induced DNA methylation in rat colon and liver. Cancer Res. 1987, 47, 5939–5943. [Google Scholar]
- Sohn, O.S.; Puz, C.; Caswell, N.; Fiala, E.S. Differential susceptibility of rat and guinea pig colon mucosa DNA to methylation by methylazoxymethyl acetate in vivo. Cancer Lett. 1985, 29, 293–300. [Google Scholar] [CrossRef]
- Sohn, O.S.; Fiala, E.S.; Requeijo, S.P.; Weisburger, J.H.; Gonzalez, F.J. Differential effects of CYP2E1 status on the metabolic activation of the colon carcinogens azoxymethane and methylazoxymethanol. Cancer Res. 2001, 61, 8435–8440. [Google Scholar]
- Kisby, G.E.; Fry, R.C.; Lasarev, M.R.; Bammler, T.K.; Beyer, R.P.; Churchwell, M.; Doerge, D.R.; Meira, L.B.; Palmer, V.S.; Ramos-Crawford, A.L.; et al. The cycad genotoxin MAM modulates brain cellular pathways involved in neurodegenerative disease and cancer in a DNA damage-linked manner. PLoS ONE 2011, 6, e20911. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Anderson, D.W.; Mantel, N.; Román, G.C. Motor neuron disease on Guam: Geographic and familial occurrence, 1956–1985. Acta Neurol. Scand. 1996, 94, 51–59. [Google Scholar] [CrossRef]
- Borenstein, A.R.; Mortimer, J.A.; Schofield, E.; Wu, Y.; Salmon, D.P.; Gamst, A.; Olichney, J.; Thal, L.J.; Silbert, L.; Kaye, J.; et al. Cycad exposure and risk of dementia, MCI, and PDC in the Chamorro population of Guam. Neurology 2007, 68, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S.; Chan, Y.L.; Wu, M.L.; Deng, J.F.; Chiu, T.F.; Chen, J.C.; Wang, F.L.; Tseng, C.P. Acute cycas seed poisoning in Taiwan. J. Toxicol. Clin. Toxicol. 2004, 42, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Amelot, M.E.; Avendaño, M. Human carcinogenesis and bracken fern: A review of the evidence. Curr. Med. Chem. 2002, 9, 675–686. [Google Scholar] [CrossRef]
- Gil da Costa, R.M.; Bastos, M.M.; Oliveira, P.A.; Lopes, C. Bracken-associated human and animal health hazards: Chemical, biological and pathological evidence. J. Hazard. Mater. 2012, 203–204, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, L.H. Presence of the carcinogen ptaquiloside in fern-based food products and traditional medicine: Four cases of human exposure. Curr. Res. Food Sci. 2021, 4, 557–564. [Google Scholar] [CrossRef]
- Virgilio, A.; Sinisi, A.; Russo, V.; Gerardo, S.; Santoro, A.; Galeone, A.; Taglialatela-Scafati, O.; Roperto, F. Ptaquiloside, the major carcinogen of bracken fern, in the pooled raw milk of healthy sheep and goats: An underestimated, global concern of food safety. J. Agric. Food Chem. 2015, 63, 4886–4892. [Google Scholar] [CrossRef]
- Aranha, P.C.; Hansen, H.C.; Rasmussen, L.H.; Strobel, B.W.; Friis, C. Determination of ptaquiloside and pterosin B derived from bracken (Pteridium aquilinum) in cattle plasma, urine and milk. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 951–952, 44–51. [Google Scholar] [CrossRef]
- Francesco, B.; Giorgio, B.; Rosario, N.; Saverio, R.F.; Francesco, G.; Romano, M.; Adriano, S.; Cinzia, R.; Antonio, T.; Franco, R.; et al. A new, very sensitive method of assessment of ptaquiloside, the major bracken carcinogen in the milk of farm animals. Food Chem. 2011, 124, 660–665. [Google Scholar] [CrossRef]
- Shahin, M.; Smith, B.L.; Prakash, A.S. Bracken carcinogens in the human diet. Mutat. Res. 1999, 443, 69–79. [Google Scholar] [CrossRef]
- Potter, D.M.; Baird, M.S. Carcinogenic effects of ptaquiloside in bracken fern and related compounds. Br. J. Cancer 2000, 83, 914–920. [Google Scholar] [CrossRef]
- Prakash, A.S.; Pereira, T.N.; Smith, B.L.; Shaw, G.; Seawright, A.A. Mechanism of bracken fern carcinogenesis: Evidence for H-ras activation via initial adenine alkylation by ptaquiloside. Nat. Toxins 1996, 4, 221–227. [Google Scholar] [CrossRef]
- IARC, International Agency for Research on Cancer. Some Naturally Occurring and Synthetic Food Components, Furocoumarins and Ultraviolet Radiation. In IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans; International Agency for Research on Cancer: Lyon, France, 1986; Volume 40. [Google Scholar]
- Pamukcu, A.M.; Yalçiner, S.; Hatcher, J.F.; Bryan, G.T. Quercetin, a rat intestinal and bladder carcinogen present in bracken fern (Pteridium aquilinum). Cancer Res. 1980, 40, 3468–3472. [Google Scholar] [PubMed]
- Hirono, I. Carcinogenic principles isolated from bracken fern. Crit. Rev. Toxicol. 1986, 17, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Hirono, I.; Aiso, S.; Yamaji, T.; Mori, H.; Yamada, K.; Niwa, H.; Ojika, M.; Wakamatsu, K.; Kigoshi, H.; Niiyama, K.; et al. Carcinogenicity in rats of ptaquiloside isolated from bracken. Gan 1984, 75, 833–836. [Google Scholar]
- Hirono, I.; Ogino, H.; Fujimoto, M.; Yamada, K.; Yoshida, Y.; Ikagawa, M.; Okumura, M. Induction of tumors in ACI rats given a diet containing ptaquiloside, a bracken carcinogen. J. Natl. Cancer Inst. 1987, 79, 1143–1149. [Google Scholar]
- Gil da Costa, R.M.; Neto, T.; Estêvão, D.; Moutinho, M.; Félix, A.; Medeiros, R.; Lopes, C.; Bastos, M.; Oliveira, P.A. Ptaquiloside from bracken (Pteridium spp.) promotes oral carcinogenesis initiated by HPV16 in transgenic mice. Food Funct. 2020, 11, 3298–3305. [Google Scholar] [CrossRef]
- Mori, H.; Sugie, S.; Hirono, I.; Yamada, K.; Niwa, H.; Ojika, M. Genotoxicity of ptaquiloside, a bracken carcinogen, in the hepatocyte primary culture/DNA-repair test. Mutat. Res. 1985, 143, 75–78. [Google Scholar] [CrossRef]
- Tourchi-Roudsari, M. Multiple effects of bracken fern under in vivo and in vitro conditions. Asian Pac. J. Cancer Prev. 2014, 15, 7505–7513. [Google Scholar] [CrossRef]
- Nagao, T.; Saito, K.; Hirayama, E.; Uchikoshi, K.; Koyama, K.; Natori, S.; Morisaki, N.; Iwasaki, S.; Matsushima, T. Mutagenicity of ptaquiloside, the carcinogen in bracken, and its related illudane-type sesquiterpenes. I. Mutagenicity in Salmonella typhimurium. Mutat. Res. 1989, 215, 173–178. [Google Scholar] [CrossRef]
- Gil da Costa, R.M.; Coelho, P.; Sousa, R.; Bastos, M.; Porto, B.; Teixeira, J.P.; Malheiro, I.; Lopes, C. Multiple genotoxic activities of ptaquiloside in human lymphocytes: Aneugenesis, clastogenesis and induction of sister chromatid exchange. Mutat. Res. 2012, 747, 77–81. [Google Scholar] [CrossRef]
- Matsuoka, A.; Hirosawa, A.; Natori, S.; Iwasaki, S.; Sofuni, T.; Ishidate, M., Jr. Mutagenicity of ptaquiloside, the carcinogen in bracken, and its related illudane-type sesquiterpenes. II. Chromosomal aberration tests with cultured mammalian cells. Mutat. Res. 1989, 215, 179–185. [Google Scholar] [CrossRef]
- Gomes, J.; Magalhães, A.; Michel, V.; Amado, I.F.; Aranha, P.; Ovesen, R.G.; Hansen, H.C.; Gärtner, F.; Reis, C.A.; Touati, E. Pteridium aquilinum and its ptaquiloside toxin induce DNA damage response in gastric epithelial cells, a link with gastric carcinogenesis. Toxicol. Sci. 2012, 126, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Povey, A.C.; Potter, D.; O’Connor, P.J. 32P-post-labelling analysis of DNA adducts formed in the upper gastrointestinal tissue of mice fed bracken extract or bracken spores. Br. J. Cancer 1996, 74, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Freitas, R.N.; O’Connor, P.J.; Prakash, A.S.; Shahin, M.; Povey, A.C. Bracken (Pteridium aquilinum)-induced DNA adducts in mouse tissues are different from the adduct induced by the activated form of the Bracken carcinogen ptaquiloside. Biochem. Biophys. Res. Commun. 2001, 281, 589–594. [Google Scholar] [CrossRef]
- Shahin, M.; Smith, B.L.; Worral, S.; Moore, M.R.; Seawright, A.A.; Prakash, A.S. Bracken fern carcinogenesis: Multiple intravenous doses of activated ptaquiloside induce DNA adducts, monocytosis, increased TNF alpha levels, and mammary gland carcinoma in rats. Biochem. Biophys. Res. Commun. 1998, 244, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Sardon, D.; de la Fuente, I.; Calonge, E.; Perez-Alenza, M.D.; Castaño, M.; Dunner, S.; Peña, L. H-ras immunohistochemical expression and molecular analysis of urinary bladder lesions in grazing adult cattle exposed to bracken fern. J. Comp. Pathol. 2005, 132, 195–201. [Google Scholar] [CrossRef]
- Shahin, M.; Moore, M.R.; Worrall, S.; Smith, B.L.; Seawright, A.A.; Prakash, A.S. H-ras activation is an early event in the ptaquiloside-induced carcinogenesis: Comparison of acute and chronic toxicity in rats. Biochem. Biophys. Res. Commun. 1998, 250, 491–497. [Google Scholar] [CrossRef]
- Gil da Costa, R.M.; Oliveira, P.A.; Bastos, M.M.; Lopes, C.C.; Lopes, C. Ptaquiloside-induced early-stage urothelial lesions show increased cell proliferation and intact β-catenin and E-cadherin expression. Environ. Toxicol. 2014, 29, 763–769. [Google Scholar] [CrossRef]
- Alonso-amelot, M.E.; Castillo, U.F.; Smith, B.L.; Lauren, D.R. Excretion, through milk, of ptaquiloside in bracken-fed cows. A quantitative assessment. Lait 1998, 78, 413–423. [Google Scholar] [CrossRef]
- Alonso-Amelot, M.E. The link between bracken fern and stomach cancer: Milk. Nutrition 1997, 13, 694–696. [Google Scholar] [CrossRef]
- Alonso-Amelot, M.E.; Avendaño, M. Possible association between gastric cancer and bracken fern in Venezuela: An epidemiologic study. Int. J. Cancer 2001, 91, 252–259. [Google Scholar] [CrossRef]
- Liu, R.; Li, A.; Sun, A.; Kong, L. Preparative isolation and purification of psoralen and isopsoralen from Psoralea corylifolia by high-speed counter-current chromatography. J. Chromatogr. A 2004, 1057, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Siskos, E.P.; Mazomenos, B.E.; Konstantopoulou, M.A. Isolation and identification of insecticidal components from Citrus aurantium fruit peel extract. J. Agric. Food Chem. 2008, 56, 5577–5581. [Google Scholar] [CrossRef]
- Finkelstein, E.V.E.; Afek, U.Z.I.; Gross, E.; Aharoni, N.; Rosenberg, L.; Halevy, S. An outbreak of phytophotodermatitis due to celery. Int. J. Dermatol. 1994, 33, 116–118. [Google Scholar] [CrossRef]
- McCloud, E.S.; Berenbaum, M.R.; Tuveson, R.W. Furanocoumarin content and phototoxicity of rough lemon (Citrus jambhiri) foliage exposed to enhanced ultraviolet-B (UVB) irradiation. J. Chem. Ecol. 1992, 18, 1125–1137. [Google Scholar] [CrossRef]
- Arigò, A.; Rigano, F.; Russo, M.; Trovato, E.; Dugo, P.; Mondello, L. Dietary intake of coumarins and furocoumarins through citrus beverages: A detailed estimation by a HPLC-MS/MS method combined with the Linear Retention Index System. Foods 2021, 10, 1533. [Google Scholar] [CrossRef] [PubMed]
- Melough, M.M.; Chun, O.K. Dietary furocoumarins and skin cancer: A review of current biological evidence. Food Chem. Toxicol. 2018, 122, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Nagayo, K.; Way, B.H.; Tran, R.M.; Song, P.S. Photocarcinogenicity of 8-methoxypsoralen and aflatoxin B1 with longwave ultraviolet light. Cancer Lett. 1983, 18, 191–198. [Google Scholar] [CrossRef]
- Forbes, P.D.; Davies, R.E.; Urbach, F.; Dunnick, J.K. Long-term toxicity of oral 8-methoxypsoralen plus ultraviolet radiation in mice. J. Toxicol. Cutan. Ocul. Toxicol. 1990, 9, 237–250. [Google Scholar] [CrossRef]
- NTP, National Toxicology Program. Toxicology and Carcinogenesis Studies of 8-Methoxypsoralen (CAS No. 298-81-7) in F344/N Rats (Gavage Studies); National Toxicology Program: Research Triangle, NC, USA, 1989; pp. 1–130.
- Müller, L.; Kasper, P.; Kersten, B.; Zhang, J. Photochemical genotoxicity and photochemical carcinogenesis—Two sides of a coin? Toxicol. Lett. 1998, 102–103, 383–387. [Google Scholar] [CrossRef]
- Chételat, A.A.; Albertini, S.; Gocke, E. The photomutagenicity of fluoroquinolones in tests for gene mutation, chromosomal aberration, gene conversion and DNA breakage (Comet assay). Mutagenesis 1996, 11, 497–504. [Google Scholar] [CrossRef]
- Yang, A.; Chen, J.; Ma, Y.; Wang, L.; Fan, Y.; He, X. Studies on the metabolites difference of psoralen/isopsoralen in human and six mammalian liver microsomes in vitro by UHPLC-MS/MS. J. Pharm. Biomed. Anal. 2017, 141, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Lu, D.; Cao, J.; Zheng, L.; Peng, Y.; Zheng, J. Psoralen, a mechanism-based inactivator of CYP2B6. Chem. Biol. Interact. 2015, 240, 346–352. [Google Scholar] [CrossRef]
- Girennavar, B.; Poulose, S.M.; Jayaprakasha, G.K.; Bhat, N.G.; Patil, B.S. Furocoumarins from grapefruit juice and their effect on human CYP 3A4 and CYP 1B1 isoenzymes. Bioorg. Med. Chem. 2006, 14, 2606–2612. [Google Scholar] [CrossRef]
- Santes-Palacios, R.; Romo-Mancillas, A.; Camacho-Carranza, R.; Espinosa-Aguirre, J.J. Inhibition of human and rat CYP1A1 enzyme by grapefruit juice compounds. Toxicol. Lett. 2016, 258, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.M.; Zhong, Y.H.; Xiao, W.B.; Li, H.; Lu, C. Identification and characterization of psoralen and isopsoralen as potent CYP1A2 reversible and time-dependent inhibitors in human and rat preclinical studies. Drug Metab. Dispos. 2013, 41, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Bickers, D.R.; Pathak, M.A. Psoralen pharmacology: Studies on metabolism and enzyme induction. Natl. Cancer Inst. Monogr. 1984, 66, 77–84. [Google Scholar]
- Wagstaff, D.J. Dietary exposure to furocoumarins. Regul. Toxicol. Pharm. 1991, 14, 261–272. [Google Scholar] [CrossRef]
- Gorgus, E.; Lohr, C.; Raquet, N.; Guth, S.; Schrenk, D. Limettin and furocoumarins in beverages containing citrus juices or extracts. Food Chem. Toxicol. 2010, 48, 93–98. [Google Scholar] [CrossRef]
- Ellis, C.R.; Elston, D.M. Psoralen-induced phytophotodermatitis. Dermatitis 2021, 32, 140–143. [Google Scholar] [CrossRef]
- Berkley, S.F. Dermatitis in grocery workers associated with high natural concentrations of furanocoumarins in celery. Ann. Intern. Med. 1986, 105, 351. [Google Scholar] [CrossRef] [PubMed]
- Ljunggren, B. Severe phototoxic burn following celery ingestion. Arch. Derm. 1990, 126, 1334–1336. [Google Scholar] [CrossRef]
- Archier, E.; Devaux, S.; Castela, E.; Gallini, A.; Aubin, F.; Le Maître, M.; Aractingi, S.; Bachelez, H.; Cribier, B.; Joly, P.; et al. Carcinogenic risks of psoralen UV-A therapy and narrowband UV-B therapy in chronic plaque psoriasis: A systematic literature review. J. Eur. Acad. Derm. Venereol. 2012, 26 (Suppl. S3), 22–31. [Google Scholar] [CrossRef] [PubMed]
- Stern, R.S.; Liebman, E.J.; Väkevä, L. Oral psoralen and ultraviolet-A light (PUVA) treatment of psoriasis and persistent risk of nonmelanoma skin cancer. PUVA Follow-up Study. J. Natl. Cancer Inst. 1998, 90, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Marley, A.R.; Li, M.; Champion, V.L.; Song, Y.; Han, J.; Li, X. The association between citrus consumption and melanoma risk in the UK Biobank. Br. J. Derm. 2021, 185, 353–362. [Google Scholar] [CrossRef]
- Moreira, R.; Pereira, D.M.; Valentão, P.; Andrade, P.B. Pyrrolizidine alkaloids: Chemistry, pharmacology, toxicology and food safety. Int. J. Mol. Sci. 2018, 19, 1668. [Google Scholar] [CrossRef]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Safety Evaluation of Certain Food Additives and Contaminants: Prepared by the Eightieth Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA); Supplement 2: Pyrrolizidine alkaloids; Who Food Additives Series: 71-S2; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar] [CrossRef]
- Robertson, J.; Stevens, K. Pyrrolizidine alkaloids: Occurrence, biology, and chemical synthesis. Nat. Prod. Rep. 2017, 34, 62–89. [Google Scholar] [CrossRef]
- EFSA CONTAM Panel, European Food Safety Authority, Panel on Contaminants in the Food Chain. Scientific Opinion on pyrrolizidine alkaloids in food and feed. EFSA J. 2011, 9, 2406. [Google Scholar] [CrossRef]
- Robertson, J.; Stevens, K. Pyrrolizidine alkaloids. Nat. Prod. Rep. 2014, 31, 1721–1788. [Google Scholar] [CrossRef]
- Kopp, T.; Abdel-Tawab, M.; Mizaikoff, B. Extracting and analyzing pyrrolizidine alkaloids in medicinal plants: A review. Toxins 2020, 12, 320. [Google Scholar] [CrossRef]
- Fu, P.P.; Xia, Q.; Lin, G.; Chou, M.W. Pyrrolizidine alkaloids—Genotoxicity, metabolism enzymes, metabolic activation, and mechanisms. Drug Metab. Rev. 2004, 36, 1–55. [Google Scholar] [CrossRef] [PubMed]
- Dusemund, B.; Nowak, N.; Sommerfeld, C.; Lindtner, O.; Schäfer, B.; Lampen, A. Risk assessment of pyrrolizidine alkaloids in food of plant and animal origin. Food Chem. Toxicol. 2018, 115, 63–72. [Google Scholar] [CrossRef] [PubMed]
- IARC, International Agency for Research on Cancer. Some Traditional Herbal Medicines, Some Mycotoxins, Naphthalene and Styrene. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2002; Volume 82. [Google Scholar]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Evaluation of Certain Food Additives and Contaminants; Prepared for the Eightieth Report of the Joint FAO/WHO Expert Committee on Food Additives; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- EMA, European Medicines Agency. Public Statement on the Use of Herbal Medicinal Products Containing Toxic, Unsaturated Pyrrolizidine Alkaloids (PAs) Including Recommendations Regarding Contamination of Herbal Medicinal Products with Pas; EMA/HMPC/893108/2011; European Medicines Agency: Amsterdam, The Netherlands, 2021.
- Fu, P.P. Pyrrolizidine alkaloids: Metabolic activation pathways leading to liver tumor initiation. Chem. Res. Toxicol. 2017, 30, 81–93. [Google Scholar] [CrossRef] [PubMed]
- NTP, National Toxicology Program. Bioassay of Lasiocarpine for Possible Carcinogenicity; 0163-7185 (Print); National Toxicology Program: Research Triangle, NC, USA, 1978; pp. 1–66.
- NTP, National Toxicology Program. Toxicology and Carcinogenesis Studies of Riddelliine (CAS No. 23246-96-0) in F344/N Rats and B6C3F1 Mice (Gavage Studies); 0888-8051 (Print); National Toxicology Program: Research Triangle, NC, USA, 2003; pp. 1–280.
- Chen, T.; Mei, N.; Fu, P.P. Genotoxicity of pyrrolizidine alkaloids. J. Appl. Toxicol. 2010, 30, 183–196. [Google Scholar] [CrossRef]
- Allemang, A.; Mahony, C.; Lester, C.; Pfuhler, S. Relative potency of fifteen pyrrolizidine alkaloids to induce DNA damage as measured by micronucleus induction in HepaRG human liver cells. Food Chem. Toxicol. 2018, 121, 72–81. [Google Scholar] [CrossRef]
- Williams, G.M.; Mori, H.; Hirono, I.; Nagao, M. Genotoxicity of pyrrolizidine alkaloids in the hepatocyte primary culture/DNA-repair test. Mutat. Res. 1980, 79, 1–5. [Google Scholar] [CrossRef]
- Mori, H.; Sugie, S.; Yoshimi, N.; Asada, Y.; Furuya, T.; Williams, G.M. Genotoxicity of a variety of pyrrolizidine alkaloids in the hepatocyte primary culture-DNA repair test using rat, mouse, and hamster hepatocytes. Cancer Res. 1985, 45, 3125–3129. [Google Scholar]
- Chou, M.W.; Yan, J.; Nichols, J.; Xia, Q.; Beland, F.A.; Chan, P.C.; Fu, P.P. Correlation of DNA adduct formation and riddelliine-induced liver tumorigenesis in F344 rats and B6C3F1 mice. Cancer Lett. 2004, 207, 119–125. [Google Scholar] [CrossRef]
- Wang, Y.P.; Yan, J.; Beger, R.D.; Fu, P.P.; Chou, M.W. Metabolic activation of the tumorigenic pyrrolizidine alkaloid, monocrotaline, leading to DNA adduct formation in vivo. Cancer Lett. 2005, 226, 27–35. [Google Scholar] [CrossRef]
- Xia, Q.; Zhao, Y.; Von Tungeln, L.S.; Doerge, D.R.; Lin, G.; Cai, L.; Fu, P.P. Pyrrolizidine alkaloid-derived DNA adducts as a common biological biomarker of pyrrolizidine alkaloid-induced tumorigenicity. Chem. Res. Toxicol. 2013, 26, 1384–1396. [Google Scholar] [CrossRef]
- Ebmeyer, J.; Braeuning, A.; Glatt, H.; These, A.; Hessel-Pras, S.; Lampen, A. Human CYP3A4-mediated toxification of the pyrrolizidine alkaloid lasiocarpine. Food Chem. Toxicol. 2019, 130, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Schoch, T.K.; Gardner, D.R.; Stegelmeier, B.L. GC/MS/MS detection of pyrrolic metabolites in animals poisoned with the pyrrolizidine alkaloid riddelliine. J. Nat. Toxins 2000, 9, 197–206. [Google Scholar] [PubMed]
- Yang, Y.C.; Yan, J.; Doerge, D.R.; Chan, P.C.; Fu, P.P.; Chou, M.W. Metabolic activation of the tumorigenic pyrrolizidine alkaloid, riddelliine, leading to DNA adduct formation in vivo. Chem. Res. Toxicol. 2001, 14, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Yan, J.; Fu, P.P.; Chou, M.W. Human liver microsomal reduction of pyrrolizidine alkaloid N-oxides to form the corresponding carcinogenic parent alkaloid. Toxicol Lett. 2005, 155, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Geburek, I.; Rutz, L.; Gao, L.; Küpper, J.H.; These, A.; Schrenk, D. Metabolic pattern of hepatotoxic pyrrolizidine alkaloids in liver cells. Chem. Res. Toxicol. 2021, 34, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Fashe, M.M.; Juvonen, R.O.; Petsalo, A.; Räsänen, J.; Pasanen, M. Species-specific differences in the in vitro metabolism of lasiocarpine. Chem. Res. Toxicol. 2015, 28, 2034–2044. [Google Scholar] [CrossRef]
- EFSA, European Food Safety Authority. Dietary exposure assessment to pyrrolizidine alkaloids in the European population. EFSA J. 2016, 14, 4572. [Google Scholar] [CrossRef]
- Rasenack, R.; Müller, C.; Kleinschmidt, M.; Rasenack, J.; Wiedenfeld, H. Veno-occlusive disease in a fetus caused by pyrrolizidine alkaloids of food origin. Fetal Diagn. Ther. 2003, 18, 223–225. [Google Scholar] [CrossRef]
- Neuman, M.G.; Cohen, L.; Opris, M.; Nanau, R.M.; Hyunjin, J. Hepatotoxicity of pyrrolizidine alkaloids. J. Pharm. Pharm. Sci. 2015, 18, 825–843. [Google Scholar] [CrossRef]
- Habs, M.; Binder, K.; Krauss, S.; Müller, K.; Ernst, B.; Valentini, L.; Koller, M. A balanced risk-benefit analysis to determine human risks associated with pyrrolizidine alkaloids (PA)-The case of tea and herbal infusions. Nutrients 2017, 9, 717. [Google Scholar] [CrossRef]
- EFSA CONTAM Panel, European Food Safety Authority, Panel on Contaminants in the Food Chain. Statement on the risks for human health related to the presence of pyrrolizidine alkaloids in honey, tea, herbal infusions and food supplements. EFSA J. 2017, 15, 4908. [Google Scholar] [CrossRef]
- Chen, L.; Mulder, P.P.J.; Louisse, J.; Peijnenburg, A.; Wesseling, S.; Rietjens, I. Risk assessment for pyrrolizidine alkaloids detected in (herbal) teas and plant food supplements. Regul. Toxicol. Pharm. 2017, 86, 292–302. [Google Scholar] [CrossRef]
- Bullerman, L.B.; Bianchini, A. Stability of mycotoxins during food processing. Int. J. Food Microbiol. 2007, 119, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Marin, S.; Ramos, A.J.; Cano-Sancho, G.; Sanchis, V. Mycotoxins: Occurrence, toxicology, and exposure assessment. Food Chem. Toxicol. 2013, 60, 218–237. [Google Scholar] [CrossRef] [PubMed]
- Bryden, W.L. Mycotoxins in the food chain: Human health implications. Asia Pac. J. Clin. Nutr. 2007, 16 (Suppl. S1), 95–101. [Google Scholar] [PubMed]
- IARC, International Agency for Research on Cancer. Chemical Agents and Related Occupations. Review of Human Carcinogens. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2012; Volume 100F. [Google Scholar]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Evaluation of Certain Contaminants in Food; Prepared for the Eighty-Third Report of the Joint FAO/WHO Expert Committee on Food Additives; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Wogan, G.N.; Hecht, S.S.; Felton, J.S.; Conney, A.H.; Loeb, L.A. Environmental and chemical carcinogenesis. Semin. Cancer Biol. 2004, 14, 473–486. [Google Scholar] [CrossRef]
- Rushing, B.R.; Selim, M.I. Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food Chem. Toxicol. 2019, 124, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Ramsdell, H.S.; Eaton, D.L. Species susceptibility to aflatoxin B1 carcinogenesis: Comparative kinetics of microsomal biotransformation. Cancer Res. 1990, 50, 615–620. [Google Scholar]
- Wogan, G.N.; Edwards, G.S.; Newberne, P.M. Structure-activity relationships in toxicity and carcinogenicity of aflatoxins and analogs. Cancer Res. 1971, 31, 1936–1942. [Google Scholar]
- EFSA CONTAM Panel, European Food Safety Authority, Panel on Contaminants in the Food Chain. Scientific Opinion on risk assessment of aflatoxins in food. EFSA J. 2020, 18, 6040. [Google Scholar] [CrossRef]
- Robens, J.F.; Richard, J.L. Aflatoxins in animal and human health. Rev. Environ. Contam. Toxicol. 1992, 127, 69–94. [Google Scholar] [CrossRef] [PubMed]
- Choy, W.N. A review of the dose-response induction of DNA adducts by aflatoxin B1 and its implications to quantitative cancer-risk assessment. Mutat. Res. 1993, 296, 181–198. [Google Scholar] [CrossRef]
- Williams, G.M.; Duan, J.-D.; Brunnemann, K.D.; Iatropoulos, M.J.; Vock, E.; Deschl, U. Chicken fetal liver DNA damage and adduct formation by activation-dependent DNA-reactive carcinogens and related compounds of several structural classes. Toxicol. Sci. 2014, 141, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Chen, C.J.; Haghighi, B.; Yang, G.Y.; Hsieh, L.L.; Wang, L.W.; Santella, R.M. Quantitation of aflatoxin B1-DNA adducts in woodchuck hepatocytes and rat liver tissue by indirect immunofluorescence analysis. Cancer Res. 1991, 51, 1720–1725. [Google Scholar] [PubMed]
- Coskun, E.; Jaruga, P.; Vartanian, V.; Erdem, O.; Egner, P.A.; Groopman, J.D.; Lloyd, R.S.; Dizdaroglu, M. Aflatoxin-guanine DNA adducts and oxidatively induced DNA damage in aflatoxin-treated mice in vivo as measured by Liquid Chromatography-Tandem Mass Spectrometry with Isotope Dilution. Chem. Res. Toxicol. 2019, 32, 80–89. [Google Scholar] [CrossRef]
- Bedard, L.L.; Massey, T.E. Aflatoxin B1-induced DNA damage and its repair. Cancer Lett. 2006, 241, 174–183. [Google Scholar] [CrossRef]
- Wang, J.S.; Groopman, J.D. DNA damage by mycotoxins. Mutat. Res. 1999, 424, 167–181. [Google Scholar] [CrossRef]
- Eaton, D.L.; Gallagher, E.P. Mechanisms of aflatoxin carcinogenesis. Annu. Rev. Pharm. Toxicol. 1994, 34, 135–172. [Google Scholar] [CrossRef]
- McQueen, C.A.; Way, B.M.; Williams, G.M. Genotoxicity of carcinogens in human hepatocytes: Application in hazard assessment. Toxicol. Appl. Pharmacol. 1988, 96, 360–366. [Google Scholar] [CrossRef]
- Theumer, M.G.; Henneb, Y.; Khoury, L.; Snini, S.P.; Tadrist, S.; Canlet, C.; Puel, O.; Oswald, I.P.; Audebert, M. Genotoxicity of aflatoxins and their precursors in human cells. Toxicol. Lett. 2018, 287, 100–107. [Google Scholar] [CrossRef]
- Marchese, S.; Polo, A.; Ariano, A.; Velotto, S.; Costantini, S.; Severino, L. Aflatoxin B1 and M1: Biological properties and their involvement in cancer development. Toxins 2018, 10, 214. [Google Scholar]
- Garner, R.C.; Martin, C.N.; Smith, J.R.; Coles, B.F.; Tolson, M.R. Comparison of aflatoxin B1 and aflatoxin G1 binding to cellular macromolecules in vitro, in vivo and after peracid oxidation; characterisation of the major nucleic acid adducts. Chem. Biol. Interact. 1979, 26, 57–73. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Johnson, W.W.; Shimada, T.; Ueng, Y.F.; Yamazaki, H.; Langouët, S. Activation and detoxication of aflatoxin B1. Mutat. Res. 1998, 402, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Roebuck, B.D.; Siegel, W.G.; Wogan, G.N. In vitro metabolism of aflatoxin B2 by animal and human liver. Cancer Res. 1978, 38, 999–1002. [Google Scholar] [PubMed]
- Roebuck, B.D.; Wogan, G.N. Species comparison of in vitro metabolism of aflatoxin B1. Cancer Res. 1977, 37, 1649–1656. [Google Scholar]
- Benkerroum, N. Chronic and acute toxicities of aflatoxins: Mechanisms of action. Int. J. Environ. Res. Public Health 2020, 17, 423. [Google Scholar] [CrossRef]
- Groopman, J.D.; Wild, C.P.; Hasler, J.; Junshi, C.; Wogan, G.N.; Kensler, T.W. Molecular epidemiology of aflatoxin exposures: Validation of aflatoxin-N7-guanine levels in urine as a biomarker in experimental rat models and humans. Environ. Health Perspect. 1993, 99, 107–113. [Google Scholar] [CrossRef]
- Wogan, G.N.; Kensler, T.W.; Groopman, J.D. Present and future directions of translational research on aflatoxin and hepatocellular carcinoma. A review. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2012, 29, 249–257. [Google Scholar] [CrossRef]
- Kew, M.C. Synergistic interaction between aflatoxin B1 and hepatitis B virus in hepatocarcinogenesis. Liver Int. 2003, 23, 405–409. [Google Scholar] [CrossRef]
- Ross, R.K.; Yuan, J.M.; Yu, M.C.; Wogan, G.N.; Qian, G.S.; Tu, J.T.; Groopman, J.D.; Gao, Y.T.; Henderson, B.E. Urinary aflatoxin biomarkers and risk of hepatocellular carcinoma. Lancet 1992, 339, 943–946. [Google Scholar] [CrossRef]
- Fang, L.; Zhao, B.; Zhang, R.; Wu, P.; Zhao, D.; Chen, J.; Pan, X.; Wang, J.; Wu, X.; Zhang, H.; et al. Occurrence and exposure assessment of aflatoxins in Zhejiang province, China. Environ. Toxicol. Pharmacol. 2022, 92, 103847. [Google Scholar] [CrossRef] [PubMed]
- FDA, Food and Drug Administration. Compliance Policy Guide Sec. 555.400. Aflatoxins in Human Food; Guidance for FDA Staff; Food and Drug Administration: Silver Spring, MD, USA, 2021.
- FDA, Food and Drug Administration. Compliance Policy Guide (CPG) Sec 527.400 Whole Milk, Lowfat Milk, Skim Milk-Aflatoxin M1; Food and Drug Administration: Silver Spring, MD, USA, 2005.
- IARC, International Agency for Research on Cancer. Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Aromatic Amines and Mycotoxins. In IARC Monographs on the Evaluation of Carcinogenic Risk to Humans; International Agency for Research on Cancer: Lyon, France, 1993; Volume 56. [Google Scholar]
- Malir, F.; Ostry, V.; Pfohl-Leszkowicz, A.; Malir, J.; Toman, J. Ochratoxin A: 50 years of research. Toxins 2016, 8, 191. [Google Scholar] [CrossRef] [PubMed]
- EFSA CONTAM Panel, European Food Safety Authority, Panel on Contaminants in the Food Chain. Scientific Opinion on the risk assessment of ochratoxin A in food. EFSA J. 2020, 18, 6113. [Google Scholar] [CrossRef]
- NTP, National Toxicology Program. Toxicology and Carcinogenesis Studies of Ochratoxin A (CAS No. 303-47-9) in F344/N Rats (Gavage Studies); 0888-8051 (Print); National Toxicology Program: Research Triangle, NC, USA, 1989; pp. 1–142.
- Mantle, P.; Kulinskaya, E.; Nestler, S. Renal tumourigenesis in male rats in response to chronic dietary ochratoxin A. Food Addit. Contam. 2005, 22, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Mally, A. Ochratoxin A and mitotic disruption: Mode of action analysis of renal tumor formation by Ochratoxin A. Toxicol. Sci. 2012, 127, 315–330. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Manderville, R.A. Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans. Mol. Nutr. Food Res. 2007, 51, 61–99. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Manderville, R.A. An update on direct genotoxicity as a molecular mechanism of ochratoxin a carcinogenicity. Chem. Res. Toxicol. 2012, 25, 252–262. [Google Scholar] [CrossRef]
- Mally, A.; Dekant, W. DNA adduct formation by ochratoxin A: Review of the available evidence. Food Addit. Contam. 2005, 22, 65–74. [Google Scholar] [CrossRef]
- Tozlovanu, M.; Faucet-Marquis, V.; Pfohl-Leszkowicz, A.; Manderville, R.A. Genotoxicity of the hydroquinone metabolite of ochratoxin A: structure-activity relationships for covalent DNA adduction. Chem. Res. Toxicol. 2006, 19, 1241–1247. [Google Scholar] [CrossRef]
- Mantle, P.G.; Faucet-Marquis, V.; Manderville, R.A.; Squillaci, B.; Pfohl-Leszkowicz, A. Structures of covalent adducts between DNA and ochratoxin a: A new factor in debate about genotoxicity and human risk assessment. Chem. Res. Toxicol. 2010, 23, 89–98. [Google Scholar] [CrossRef]
- Kamp, H.G.; Eisenbrand, G.; Janzowski, C.; Kiossev, J.; Latendresse, J.R.; Schlatter, J.; Turesky, R.J. Ochratoxin A induces oxidative DNA damage in liver and kidney after oral dosing to rats. Mol. Nutr. Food Res. 2005, 49, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Arbillaga, L.; Azqueta, A.; van Delft, J.H.M.; López de Cerain, A. In vitro gene expression data supporting a DNA non-reactive genotoxic mechanism for ochratoxin A. Toxicol. Appl. Pharmacol. 2007, 220, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Czakai, K.; Müller, K.; Mosesso, P.; Pepe, G.; Schulze, M.; Gohla, A.; Patnaik, D.; Dekant, W.; Higgins, J.M.G.; Mally, A. Perturbation of mitosis through inhibition of histone acetyltransferases: The key to Ochratoxin A toxicity and carcinogenicity? Toxicol. Sci. 2011, 122, 317–329. [Google Scholar] [CrossRef]
- Rásonyi, T.; Schlatter, J.; Dietrich, D.R. The role of α2u-globulin in ochratoxin A induced renal toxicity and tumors in F344 rats. Toxicol. Lett. 1999, 104, 83–92. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Pinelli, E.; Bartsch, H.; Mohr, U.; Castegnaro, M. Sex- and strain-specific expression of cytochrome P450s in Ochratoxin A-induced genotoxicity and carcinogenicity in rats. Mol. Carcinog. 1998, 23, 76–85. [Google Scholar] [CrossRef]
- Heussner, A.H.; Bingle, L.E. Comparative ochratoxin toxicity: A review of the available data. Toxins 2015, 7, 4253–4282. [Google Scholar] [CrossRef] [PubMed]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Evaluation of Certain Food Additives and Contaminants; Prepared by the Sixty-Eight Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA); WHO: Geneva, Switzerland, 2007. [Google Scholar]
- McNeal, T.P.; Nyman, P.J.; Diachenko, G.W.; Hollifield, H.C. Survey of benzene in foods by using headspace concentration techniques and capillary gas chromatography. J. AOAC Int. 1993, 76, 1213–1219. [Google Scholar] [CrossRef]
- IARC, International Agency for Research on Cancer. Benzene. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2018; Volume 120. [Google Scholar]
- Salviano Dos Santos, V.P.; Medeiros Salgado, A.; Guedes Torres, A.; Signori Pereira, K. Benzene as a chemical hazard in processed foods. Int. J. Food Sci. 2015, 2015, 545640. [Google Scholar] [CrossRef]
- Smith, B.; Cadby, P.; DiNovi, M.; Setzer, R.W. Application of the Margin of Exposure (MoE) approach to substances in food that are genotoxic and carcinogenic: Example: Benzene, CAS: 71-43-2. Food Chem. Toxicol. 2010, 48 (Suppl. S1), S49–S56. [Google Scholar] [CrossRef]
- Gardner, L.K.; Lawrence, G.D. Benzene production from decarboxylation of benzoic acid in the presence of ascorbic acid and a transition-metal catalyst. J. Agric. Food Chem. 1993, 41, 693–695. [Google Scholar] [CrossRef]
- Jickells, S.M.; Crews, C.; Castle, L.; Gilbert, J. Headspace analysis of benzene in food contact materials and its migration into foods from plastics cookware. Food Addit. Contam. 1990, 7, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Meadows, M. Benzene in beverages. FDA Consum. 2006, 40, 9–10. [Google Scholar] [PubMed]
- Nyman, P.J.; Diachenko, G.W.; Perfetti, G.A.; McNeal, T.P.; Hiatt, M.H.; Morehouse, K.M. Survey results of benzene in soft drinks and other beverages by headspace gas chromatography/mass spectrometry. J. Agric. Food Chem. 2008, 56, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Snyder, R.; Witz, G.; Goldstein, B.D. The toxicology of benzene. Environ. Health Perspect. 1993, 100, 293–306. [Google Scholar] [CrossRef]
- Maltoni, C.; Cotti, G.; Valgimigli, L.; Mandrioli, A. Zymbal gland carcinomas in rats following exposure to benzene by inhalation. Am. J. Ind. Med. 1982, 3, 11–16. [Google Scholar] [CrossRef]
- NTP, National Toxicology Program. Toxicology and Carcinogenesis Studies of Benzene (CAS No. 71-43-2) in F344/N Rats and B6C3F1 Mice (Gavage Studies); 0888-8051 (Print); National Toxicology Program: Research Triangle, NC, USA, 1986; pp. 1–277.
- Whysner, J.; Reddy, M.V.; Ross, P.M.; Mohan, M.; Lax, E.A. Genotoxicity of benzene and its metabolites. Mutat. Res. 2004, 566, 99–130. [Google Scholar] [CrossRef]
- Wetmore, B.A.; Struve, M.F.; Gao, P.; Sharma, S.; Allison, N.; Roberts, K.C.; Letinski, D.J.; Nicolich, M.J.; Bird, M.G.; Dorman, D.C. Genotoxicity of intermittent co-exposure to benzene and toluene in male CD-1 mice. Chem. Biol. Interact. 2008, 173, 166–178. [Google Scholar] [CrossRef]
- Tuo, J.; Loft, S.; Thomsen, M.S.; Poulsen, H.E. Benzene-induced genotoxicity in mice in vivo detected by the alkaline comet assay: Reduction by CYP2E1 inhibition. Mutat. Res. 1996, 368, 213–219. [Google Scholar] [CrossRef]
- Provost, G.S.; Mirsalis, J.C.; Rogers, B.J.; Short, J.M. Mutagenic response to benzene and tris(2,3-dibromopropyl)-phosphate in the lambda lacI transgenic mouse mutation assay: A standardized approach to in vivo mutation analysis. Environ. Mol. Mutagen. 1996, 28, 342–347. [Google Scholar] [CrossRef]
- Salem, E.; El-Garawani, I.; Allam, H.; El-Aal, B.A.; Hegazy, M. Genotoxic effects of occupational exposure to benzene in gasoline station workers. Ind. Health 2018, 56, 132–140. [Google Scholar] [CrossRef]
- Kitamoto, S.; Matsuyama, R.; Uematsu, Y.; Ogata, K.; Ota, M.; Yamada, T.; Miyata, K.; Kimura, J.; Funabashi, H.; Saito, K. Genotoxicity evaluation of benzene, di(2-ethylhexyl) phthalate, and trisodium ethylenediamine tetraacetic acid monohydrate using a combined rat comet/micronucleus assays. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 786–788, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, H.; Edmands, W.M.B.; Lan, Q.; Carlsson, H.; Vermeulen, R.; Zhang, L.; Yin, S.N.; Li, G.L.; Smith, M.T.; Rothman, N.; et al. Adductomic signatures of benzene exposure provide insights into cancer induction. Carcinogenesis 2018, 39, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, C.; Xin, W.; Yin, S. Tissue distribution of DNA adducts and their persistence in blood of mice exposed to benzene. Environ. Health Perspect. 1996, 104 (Suppl. S6), 1337–1338. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bodell, W.J.; Pathak, D.N.; Lévay, G.; Ye, Q.; Pongracz, K. Investigation of the DNA adducts formed in B6C3F1 mice treated with benzene: Implications for molecular dosimetry. Environ. Health Perspect. 1996, 104 (Suppl. S6), 1189–1193. [Google Scholar] [CrossRef]
- Reddy, M.V.; Blackburn, G.R.; Schreiner, C.A.; Mehlman, M.A.; Mackerer, C.R. 32P analysis of DNA adducts in tissues of benzene-treated rats. Environ. Health Perspect. 1989, 82, 253–257. [Google Scholar] [CrossRef]
- Gaskell, M.; McLuckie, K.I.E.; Farmer, P.B. Genotoxicity of the benzene metabolites para-benzoquinone and hydroquinone. Chem. Biol. Interact. 2005, 153–154, 267–270. [Google Scholar] [CrossRef]
- Williams, G.M.; Iatropoulos, M.J.; Jeffrey, A.M.; Duan, J.-D. Inhibition by dietary hydroquinone of acetylaminofluorene induction of initiation of rat liver carcinogenesis. Food Chem. Toxicol. 2007, 45, 1620–1625. [Google Scholar] [CrossRef]
- English, J.C.; Hill, T.; O’Donoghue, J.L.; Reddy, M.V. Measurement of nuclear DNA Modification by 32P-postlabeling in the kidneys of male and female Fischer 344 rats after multiple gavage doses of hydroquinone. Toxicol. Sci. 1994, 23, 391–396. [Google Scholar] [CrossRef]
- Gaskell, M.; McLuckie, K.I.; Farmer, P.B. Comparison of the mutagenic activity of the benzene metabolites, hydroquinone and para-benzoquinone in the supF forward mutation assay: A role for minor DNA adducts formed from hydroquinone in benzene mutagenicity. Mutat. Res. 2004, 554, 387–398. [Google Scholar] [CrossRef]
- Linhart, I.; Mikes, P.; Frantík, E.; Mráz, J. DNA adducts formed from p-benzoquinone, an electrophilic metabolite of benzene, are extensively metabolized in vivo. Chem. Res. Toxicol. 2011, 24, 383–391. [Google Scholar] [CrossRef]
- Xie, Z.; Zhang, Y.; Guliaev, A.B.; Shen, H.; Hang, B.; Singer, B.; Wang, Z. The p-benzoquinone DNA adducts derived from benzene are highly mutagenic. DNA Repair. 2005, 4, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Kolachana, P.; Subrahmanyam, V.V.; Meyer, K.B.; Zhang, L.; Smith, M.T. Benzene and its phenolic metabolites produce oxidative DNA damage in HL60 cells in vitro and in the bone marrow in vivo. Cancer Res. 1993, 53, 1023–1026. [Google Scholar] [PubMed]
- Snyder, R. Leukemia and benzene. Int. J. Environ. Res. Public Health 2012, 9, 2875–2893. [Google Scholar] [CrossRef] [PubMed]
- Wallace, L. Environmental exposure to benzene: An update. Environ. Health Perspect. 1996, 104 (Suppl. S6), 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Weisel, C.P. Benzene exposure: An overview of monitoring methods and their findings. Chem. Biol. Interact. 2010, 184, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Halden, R.U.; Buckley, T.J. Volatile organic compounds in human milk: Methods and measurements. Environ. Sci. Technol. 2007, 41, 1662–1667. [Google Scholar] [CrossRef]
- Medeiros Vinci, R.; Jacxsens, L.; Van Loco, J.; Matsiko, E.; Lachat, C.; de Schaetzen, T.; Canfyn, M.; Van Overmeire, I.; Kolsteren, P.; De Meulenaer, B. Assessment of human exposure to benzene through foods from the Belgian market. Chemosphere 2012, 88, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Cheasley, R.; Keller, C.P.; Setton, E. Lifetime excess cancer risk due to carcinogens in food and beverages: Urban versus rural differences in Canada. Can. J. Public Health 2017, 108, e288–e295. [Google Scholar] [CrossRef]
- Galbraith, D.; Gross, S.A.; Paustenbach, D. Benzene and human health: A historical review and appraisal of associations with various diseases. Crit. Rev. Toxicol. 2010, 40 (Suppl. S2), 1–46. [Google Scholar] [CrossRef]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Evaluation of Certain Food Additives Prepared by the Twenty-third Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA); WHO: Geneva, Switzerland, 1980; Volume 648. [Google Scholar]
- Lachenmeier, D.W.; Reusch, H.; Sproll, C.; Schoeberl, K.; Kuballa, T. Occurrence of benzene as a heat-induced contaminant of carrot juice for babies in a general survey of beverages. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2008, 25, 1216–1224. [Google Scholar] [CrossRef]
- Hamlet, C.G.; Sadd, P.A.; Crews, C.; Velíšek, J.; Baxter, D.E. Occurrence of 3-chloro-propane-1,2-diol (3-MCPD) and related compounds in foods: A review. Food Addit. Contam. 2002, 19, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Andres, S.; Appel, K.E.; Lampen, A. Toxicology, occurrence and risk characterisation of the chloropropanols in food: 2-monochloro-1,3-propanediol, 1,3-dichloro-2-propanol and 2,3-dichloro-1-propanol. Food Chem. Toxicol. 2013, 58, 467–478. [Google Scholar] [CrossRef] [PubMed]
- EFSA CONTAM Panel, European Food Safety Authority, Panel on Contaminants in the Food Chain. Update of the risk assessment on 3-monochloropropanediol and its fatty acid esters. EFSA J. 2018, 16, 5083. [Google Scholar] [CrossRef]
- Crews, C.; Hasnip, S.; Chapman, S.; Hough, P.; Potter, N.; Todd, J.; Brereton, P.; Matthews, W. Survey of chloropropanols in soy sauces and related products purchased in the UK in 2000 and 2002. Food Addit. Contam. 2003, 20, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Nyman, P.J.; Diachenko, G.W.; Perfetti, G.A. Survey of chloropropanols in soy sauces and related products. Food Addit. Contam. 2003, 20, 909–915. [Google Scholar] [CrossRef] [PubMed]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Safety Evaluation of Certain Food Additives and Contaminants; Prepared by the Fifty-Seven Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA); WHO: Geneva, Switzerland, 2002. [Google Scholar]
- Korte, R.; Schulz, S.; Brauer, B. Chloropropanols (3-MCPD, 1,3-DCP) from food contact materials: GC-MS method improvement, market survey and investigations on the effect of hot water extraction. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2021, 38, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Becalski, A.; Zhao, T.; Breton, F.; Kuhlmann, J. 2- and 3-Monochloropropanediols in paper products and their transfer to foods. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2016, 33, 1499–1508. [Google Scholar] [CrossRef]
- NTP, National Toxicology Program. 1,3-Dichloro-2-Propanol [CAS No. 96-23-1]: Review of Toxicological Literature; National Institute of Environmental Health Sciences, National Institutes of Health and U.S. Department of Health and Human Services: Research Triangle Park, NC, USA, 2005.
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. 1,3-Dichloro-2-propanol (addendum). In Safety Evaluation of Certain Food Additives and Contaminants; Prepared by the Fifty-Seven Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA); WHO: Geneva, Switzerland, 2007; pp. 209–238. [Google Scholar]
- El Ramy, R.; Ould Elhkim, M.; Lezmi, S.; Poul, J.M. Evaluation of the genotoxic potential of 3-monochloropropane-1,2-diol (3-MCPD) and its metabolites, glycidol and β-chlorolactic acid, using the single cell gel/comet assay. Food Chem. Toxicol. 2007, 45, 41–48. [Google Scholar] [CrossRef]
- Aasa, J.; Törnqvist, M.; Abramsson-Zetterberg, L. Measurement of micronuclei and internal dose in mice demonstrates that 3-monochloropropane-1,2-diol (3-MCPD) has no genotoxic potency in vivo. Food Chem. Toxicol. 2017, 109, 414–420. [Google Scholar] [CrossRef]
- Lynch, B.S.; Bryant, D.W.; Hook, G.J.; Nestmann, E.R.; Munro, I.C. Carcinogenicity of monochloro-1,2-propanediol (α-chlorohydrin, 3-MCPD). Int. J. Toxicol. 1998, 17, 47–76. [Google Scholar] [CrossRef]
- Buhrke, T.; Voss, L.; Briese, A.; Stephanowitz, H.; Krause, E.; Braeuning, A.; Lampen, A. Oxidative inactivation of the endogenous antioxidant protein DJ-1 by the food contaminants 3-MCPD and 2-MCPD. Arch. Toxicol. 2018, 92, 289–299. [Google Scholar] [CrossRef]
- Abt, E.; Incorvati, V.; Robin, L.P.; Redan, B.W. Occurrence of ethyl carbamate in foods and beverages: Review of the formation mechanisms, advances in analytical methods, and mitigation strategies. J. Food Prot. 2021, 84, 2195–2212. [Google Scholar] [CrossRef] [PubMed]
- Ough, C.S.; Crowell, E.A.; Gutlove, B.R. Carbamyl compound reactions with ethanol. Am. J. Enol. Vitic. 1988, 39, 239–242. [Google Scholar]
- Gowd, V.; Su, H.; Karlovsky, P.; Chen, W. Ethyl carbamate: An emerging food and environmental toxicant. Food Chem. 2018, 248, 312–321. [Google Scholar] [CrossRef]
- IARC, International Agency for Research on Cancer. Alcohol Consumption and Ethyl Carbamate. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2010; Volume 96. [Google Scholar]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Safety Evaluation of Certain Food Contaminants; Prepared by the Sixty-Fourth Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA); FAO Food and Nutrition Paper; World Health Organization: Geneva, Switzerland, 2006; Volume 82, pp. 1–778. [Google Scholar]
- EFSA CONTAM Panel, European Food Safety Authority, Panel on Contaminants in the Food Chain. Opinion of the Scientific Panel on Contaminants in the Food chain on a request from the European Commission on ethyl carbamate and hydrocyanic acid in food and beverages. EFSA J. 2007, 551, 1–44. [Google Scholar]
- Choi, B.; Koh, E. Effect of fruit thermal processing on ethyl carbamate content in maesil (Prunus mume) liqueur. Food Sci. Biotechnol. 2021, 30, 1427–1434. [Google Scholar] [CrossRef]
- NTP, National Toxicology Program. Toxicology and Carcinogenesis Studies of Urethane, Ethanol and Urethane/Ethanol (Urethane, CAS No. 51-79-6; Ethanol CAS No. 64-17-5) in B6C3F1 Mice (Drinking Water Studies); National Toxicology Program: Research Triangle, NC, USA, 2004.
- Chan, P.C. NTP Technical Report on Toxicity Studies of Urethane in Drinking Water and Urethane in 5% Ethanol Administered to F344/N Rats and B6C3F1 Mice; Toxicity Report Series; United States Department of Health and Human Services Public Health Service National Institutes of Health: Research Triangle Park, NC, USA, 1996; pp. 1–91. [Google Scholar]
- Hübner, P.; Groux, P.M.; Weibel, B.; Sengstag, C.; Horlbeck, J.; Leong-Morgenthaler, P.M.; Lüthy, J. Genotoxicity of ethyl carbamate (urethane) in Salmonella, yeast and human lymphoblastoid cells. Mutat. Res. 1997, 390, 11–19. [Google Scholar] [CrossRef]
- Allen, J.W.; Stoner, G.D.; Pereira, M.A.; Backer, L.C.; Sharief, Y.; Hatch, G.G.; Campbell, J.A.; Stead, A.G.; Nesnow, S. Tumorigenesis and genotoxicity of ethyl carbamate and vinyl carbamate in rodent cells. Cancer Res. 1986, 46, 4911–4915. [Google Scholar]
- Sotomayor, R.E.; Washington, M.C. Formation of etheno and oxoethyl adducts in liver DNA from rats exposed subchronically to urethane in drinking water and ethanol. Cancer Lett. 1996, 100, 155–161. [Google Scholar] [CrossRef]
- Fernando, R.C.; Nair, J.; Barbin, A.; Miller, J.A.; Bartsch, H. Detection of 1,N6-ethenodeoxyadenosine and 3,N4-ethenodeoxycytidine by immunoaffinity/32P-postlabelling in liver and lung DNA of mice treated with ethyl carbamate (urethane) or its metabolites. Carcinogenesis 1996, 17, 1711–1718. [Google Scholar] [CrossRef] [PubMed]
- Barbin, A. Formation of DNA etheno adducts in rodents and humans and their role in carcinogenesis. Acta Biochim. Pol. 1998, 45, 145–161. [Google Scholar] [CrossRef]
- Forkert, P.G. Mechanisms of lung tumorigenesis by ethyl carbamate and vinyl carbamate. Drug Metab. Rev. 2010, 42, 355–378. [Google Scholar] [CrossRef] [PubMed]
- Park, K.K.; Liem, A.; Stewart, B.C.; Miller, J.A. Vinyl carbamate epoxide, a major strong electrophilic, mutagenic and carcinogenic metabolite of vinyl carbamate and ethyl carbamate (urethane). Carcinogenesis 1993, 14, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Hoffler, U.; El-Masri, H.A.; Ghanayem, B.I. Cytochrome P450 2E1 (CYP2E1) is the principal enzyme responsible for urethane metabolism: Comparative studies using CYP2E1-null and wild-type mice. J. Pharm. Exp. 2003, 305, 557–564. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Kim, D.H. Enzymatic oxidation of ethyl carbamate to vinyl carbamate and its role as an intermediate in the formation of 1,N6-ethenoadenosine. Chem. Res. Toxicol. 1991, 4, 413–421. [Google Scholar] [CrossRef]
- Forkert, P.G.; Lee, R.P.; Reid, K. Involvement of CYP2E1 and carboxylesterase enzymes in vinyl carbamate metabolism in human lung microsomes. Drug Metab. Dispos. 2001, 29, 258–263. [Google Scholar]
- Sozio, F.; Schioppa, T.; Sozzani, S.; Del Prete, A. Urethane-induced lung carcinogenesis. Methods Cell Biol. 2021, 163, 45–57. [Google Scholar] [CrossRef]
- Zimmerli, B.; Schlatter, J. Ethyl carbamate: Analytical methodology, occurrence, formation, biological activity and risk assessment. Mutat. Res. 1991, 259, 325–350. [Google Scholar] [CrossRef]
- Schlatter, J.; Lutz, W.K. The carcinogenic potential of ethyl carbamate (urethane): Risk assessment at human dietary exposure levels. Food Chem. Toxicol. 1990, 28, 205–211. [Google Scholar] [CrossRef]
- Jägerstad, M.; Skog, K. Genotoxicity of heat-processed foods. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2005, 574, 156–172. [Google Scholar] [CrossRef]
- Mottram, D.S.; Wedzicha, B.L.; Dodson, A.T. Acrylamide is formed in the Maillard reaction. Nature 2002, 419, 448–449. [Google Scholar] [CrossRef]
- Stadler, R.H.; Blank, I.; Varga, N.; Robert, F.; Hau, J.; Guy, P.A.; Robert, M.-C.; Riediker, S. Acrylamide from Maillard reaction products. Nature 2002, 419, 449–450. [Google Scholar] [CrossRef]
- Friedman, M. Chemistry, biochemistry, and safety of acrylamide. A review. J. Agric. Food Chem. 2003, 51, 4504–4526. [Google Scholar] [CrossRef]
- Tritscher, A. Human health risk assessment of processing-related compounds in food. Toxicol. Lett. 2004, 149, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Dybing, E.; Farmer, P.B.; Andersen, M.; Fennell, T.R.; Lalljie, S.P.D.; Müller, D.J.G.; Olin, S.; Petersen, B.J.; Schlatter, J.; Scholz, G.; et al. Human exposure and internal dose assessments of acrylamide in food. Food Chem. Toxicol. 2005, 43, 365–410. [Google Scholar] [CrossRef] [PubMed]
- IARC, International Agency for Research on Cancer. Some Industrial Chemicals. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 1994; Volume 60. [Google Scholar]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Safety Evaluation of Certain Contaminants in Food; Prepared by the Seventy-Second Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA); FAO JECFA Monographs 8; WHO: Geneva, Switzerland, 2011. [Google Scholar]
- Commission Regulation (EU). Commission Regulation (EU) 2017/2158 of 20 November 2017 Establishing Mitigation Measures and Benchmark Levels for the Reduction of the Presence of Acrylamide in Food; Commission Regulation (EU): Brussels, Belgium, 2017; pp. 24–44. [Google Scholar]
- Xu, F.; Oruna-Concha, M.J.; Elmore, J.S. The use of asparaginase to reduce acrylamide levels in cooked food. Food Chem. 2016, 210, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.M. The carcinogenicity of acrylamide. Mutat. Res./Genet. Toxicol. Environ. Mutagen. 2005, 580, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E. Acrylamide carcinogenicity. J. Agric. Food Chem. 2008, 56, 5984–5988. [Google Scholar] [CrossRef]
- NTP, National Toxicology Program. Toxicology and Carcinogenesis Studies of Acrylamide (CAS No. 79-06-1) in F344/N Rats and B6C3F1 Mice (Feed and Drinking Water Studies); 1551-8272; National Toxicology Program, National Institutes of Health: Research Triangle Park, NC, USA, 2011; pp. 25–28.
- Besaratinia, A.; Pfeifer, G.P. A review of mechanisms of acrylamide carcinogenicity. Carcinogenesis 2007, 28, 519–528. [Google Scholar] [CrossRef]
- Watzek, N.; Böhm, N.; Feld, J.; Scherbl, D.; Berger, F.; Merz, K.H.; Lampen, A.; Reemtsma, T.; Tannenbaum, S.R.; Skipper, P.L.; et al. N7-glycidamide-guanine DNA adduct formation by orally ingested acrylamide in rats: A dose-response study encompassing human diet-related exposure levels. Chem. Res. Toxicol. 2012, 25, 381–390. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Kamendulis, L.M. Mechanisms of acrylamide induced rodent carcinogenesis. Adv. Exp. Med. Biol. 2005, 561, 49–62. [Google Scholar] [CrossRef]
- Hagio, S.; Tsuji, N.; Furukawa, S.; Takeuchi, K.; Hayashi, S.; Kuroda, Y.; Honma, M.; Masumura, K. Effect of sampling time on somatic and germ cell mutations induced by acrylamide in gpt delta mice. Genes Environ. 2021, 43, 4. [Google Scholar] [CrossRef]
- Shimamura, Y.; Iio, M.; Urahira, T.; Masuda, S. Inhibitory effects of Japanese horseradish (Wasabia japonica) on the formation and genotoxicity of a potent carcinogen, acrylamide. J. Sci. Food Agric. 2017, 97, 2419–2425. [Google Scholar] [CrossRef] [PubMed]
- Katen, A.L.; Stanger, S.J.; Anderson, A.L.; Nixon, B.; Roman, S.D. Chronic acrylamide exposure in male mice induces DNA damage to spermatozoa; Potential for amelioration by resveratrol. Reprod. Toxicol. 2016, 63, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolsky, V.N.; Pacheco-Martinez, M.M.; McDaniel, L.P.; Pearce, M.G.; Ding, W. In vivo genotoxicity assessment of acrylamide and glycidyl methacrylate. Food Chem. Toxicol. 2016, 87, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Calleman, C.J. The metabolism and pharmacokinetics of acrylamide: Implications for mechanisms of toxicity and human risk estimation. Drug Metab. Rev. 1996, 28, 527–590. [Google Scholar] [CrossRef]
- Exon, J.H. A review of the toxicology of acrylamide. J. Toxicol. Environ. Health Part B 2006, 9, 397–412. [Google Scholar] [CrossRef]
- Gamboa da Costa, G.; Churchwell, M.I.; Hamilton, L.P.; Von Tungeln, L.S.; Beland, F.A.; Marques, M.M.; Doerge, D.R. DNA adduct formation from acrylamide via conversion To glycidamide in adult and neonatal mice. Chem. Res. Toxicol. 2003, 16, 1328–1337. [Google Scholar] [CrossRef]
- Dearfield, K.L.; Douglas, G.R.; Ehling, U.H.; Moore, M.M.; Sega, G.A.; Brusick, D.J. Acrylamide: A review of its genotoxicity and an assessment of heritable genetic risk. Mutat. Res./Fundam. Mol. Mech. Mutagen. 1995, 330, 71–99. [Google Scholar] [CrossRef]
- Besaratinia, A.; Pfeifer, G.P. Genotoxicity of acrylamide and glycidamide. J. Natl. Cancer Inst. 2004, 96, 1023–1029. [Google Scholar] [CrossRef]
- Jones, D.J.L.; Singh, R.; Emms, V.; Farmer, P.B.; Grant, D.; Quinn, P.; Maxwell, C.; Mina, A.; Ng, L.L.; Schumacher, S.; et al. Determination of N7-glycidamide guanine adducts in human blood DNA following exposure to dietary acrylamide using liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2022, 36, e9245. [Google Scholar] [CrossRef]
- Fennell, T.R.; Friedman, M.A. Comparison of acrylamide metabolism in humans and rodents. Adv. Exp. Med. Biol. 2005, 561, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.; Zakłos-Szyda, M.; Żyżelewicz, D.; Koszucka, A.; Motyl, I. Acrylamide decreases cell viability, and provides oxidative stress, DNA damage, and apoptosis in human colon adenocarcinoma cell line Caco-2. Molecules 2020, 25, 368. [Google Scholar] [CrossRef] [PubMed]
- De Conti, A.; Tryndyak, V.; VonTungeln, L.S.; Churchwell, M.I.; Beland, F.A.; Antunes, A.M.M.; Pogribny, I.P. Genotoxic and epigenotoxic alterations in the lung and liver of mice induced by acrylamide: A 28 day drinking water study. Chem. Res. Toxicol. 2019, 32, 869–877. [Google Scholar] [CrossRef] [PubMed]
- EFSA CONTAM Panel, European Food Safety Authority, Panel on Contaminants in the Food Chain. Scientific Opinion on acrylamide in food. EFSA J. 2015, 13, 4104.
- Mucci, L.A.; Wilson, K.M. Acrylamide intake through diet and human cancer risk. J. Agric. Food Chem. 2008, 56, 6013–6019. [Google Scholar] [CrossRef]
- Pelucchi, C.; Bosetti, C.; Galeone, C.; La Vecchia, C. Dietary acrylamide and cancer risk: An updated meta-analysis. Int. J. Cancer 2015, 136, 2912–2922. [Google Scholar] [CrossRef]
- Duale, N.; Bjellaas, T.; Alexander, J.; Becher, G.; Haugen, M.; Paulsen, J.E.; Frandsen, H.; Olesen, P.T.; Brunborg, G. Biomarkers of human exposure to acrylamide and relation to polymorphisms in metabolizing genes. Toxicol. Sci. 2009, 108, 90–99. [Google Scholar] [CrossRef]
- Hogervorst, J.G.; Schouten, L.J.; Konings, E.J.; Goldbohm, R.A.; van den Brandt, P.A. A prospective study of dietary acrylamide intake and the risk of endometrial, ovarian, and breast cancer. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2304–2313. [Google Scholar] [CrossRef]
- Bonneck, S. Acrylamide Risk Governance in Germany. In Global Risk Governance; Springer: Dordrecht, The Netherlands, 2008; pp. 231–274. [Google Scholar]
- Baum, M.; Fauth, E.; Fritzen, S.; Herrmann, A.; Mertes, P.; Merz, K.; Rudolphi, M.; Zankl, H.; Eisenbrand, G. Acrylamide and glycidamide: Genotoxic effects in V79-cells and human blood. Mutat. Res. 2005, 580, 61–69. [Google Scholar] [CrossRef]
- Sugimura, T.; Wakabayashi, K.; Nakagama, H.; Nagao, M. Heterocyclic amines: Mutagens/carcinogens produced during cooking of meat and fish. Cancer Sci. 2004, 95, 290–299. [Google Scholar] [CrossRef]
- Nagao, M.; Ushijima, T.; Watanabe, N.; Okochi, E.; Ochiai, M.; Nakagama, H.; Sugimura, T. Studies on mammary carcinogenesis induced by a heterocyclic amine, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine, in mice and rats. Environ. Mol. Mutagen. 2002, 39, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Weisburger, J.H.; Rivenson, A.; Kingston, D.G.; Wilkins, T.D.; Van Tassell, R.L.; Nagao, M.; Sugimura, T.; Hara, Y. Dietary modulation of the carcinogenicity of the heterocyclic amines. Princess Takamatsu Symp. 1995, 23, 240–250. [Google Scholar] [PubMed]
- Sasaki, Y.F.; Saga, A.; Akasaka, M.; Nishidate, E.; Watanabe-Akanuma, M.; Ohta, T.; Matsusaka, N.; Tsuda, S. In vivo genotoxicity of heterocyclic amines detected by a modified alkaline single cell gel electrophoresis assay in a multiple organ study in the mouse. Mutat. Res. 1997, 395, 57–73. [Google Scholar] [CrossRef]
- Bellamri, M.; Walmsley, S.J.; Turesky, R.J. Metabolism and biomarkers of heterocyclic aromatic amines in humans. Genes Environ. 2021, 43, 29. [Google Scholar] [CrossRef] [PubMed]
- Fuccelli, R.; Rosignoli, P.; Servili, M.; Veneziani, G.; Taticchi, A.; Fabiani, R. Genotoxicity of heterocyclic amines (HCAs) on freshly isolated human peripheral blood mononuclear cells (PBMC) and prevention by phenolic extracts derived from olive, olive oil and olive leaves. Food Chem. Toxicol. 2018, 122, 234–241. [Google Scholar] [CrossRef]
- Bellamri, M.; Le Hegarat, L.; Vernhet, L.; Baffet, G.; Turesky, R.J.; Langouët, S. Human T lymphocytes bioactivate heterocyclic aromatic amines by forming DNA adducts. Environ. Mol. Mutagen. 2016, 57, 656–667. [Google Scholar] [CrossRef]
- Dingley, K.H.; Curtis, K.D.; Nowell, S.; Felton, J.S.; Lang, N.P.; Turteltaub, K.W. DNA and protein adduct formation in the colon and blood of humans after exposure to a dietary-relevant dose of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine. Cancer Epidemiol. Biomark. Prev. 1999, 8, 507–512. [Google Scholar]
- Reistad, R.; Rossland, O.J.; Latva-Kala, K.J.; Rasmussen, T.; Vikse, R.; Becher, G.; Alexander, J. Heterocyclic aromatic amines in human urine following a fried meat meal. Food Chem. Toxicol. 1997, 35, 945–955. [Google Scholar] [CrossRef]
- Friesen, M.D.; Kaderlik, K.; Lin, D.; Garren, L.; Bartsch, H.; Lang, N.P.; Kadlubar, F.F. Analysis of DNA adducts of 2-amino-1-methyl-6-phenylimidazo[4,5- b]pyridine in rat and human tissues by alkaline hydrolysis and gas chromatography/electron capture mass spectrometry: Validation by comparison with 32P-postlabeling. Chem. Res. Toxicol. 1994, 7, 733–739. [Google Scholar] [CrossRef]
- Totsuka, Y.; Fukutome, K.; Takahashi, M.; Takahashi, S.; Tada, A.; Sugimura, T.; Wakabayashi, K. Presence of N2-(deoxyguanosin-8-yl)-2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline (dG-C8-MeIQx) in human tissues. Carcinogenesis 1996, 17, 1029–1034. [Google Scholar] [CrossRef]
- Pathak, K.V.; Chiu, T.L.; Amin, E.A.; Turesky, R.J. Methemoglobin formation and characterization of hemoglobin adducts of carcinogenic aromatic amines and heterocyclic aromatic amines. Chem. Res. Toxicol. 2016, 29, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Guengerich, F.P. Cytochrome P450 activation of arylamines and heterocyclic amines. Annu. Rev. Pharm. Toxicol. 2005, 45, 27–49. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Stampfer, M.J.; Hough, H.L.; Garcia-Closas, M.; Willett, W.C.; Hennekens, C.H.; Kelsey, K.T.; Hunter, D.J. A prospective study of N-acetyltransferase genotype, red meat intake, and risk of colorectal cancer. Cancer Res. 1998, 58, 3307–3311. [Google Scholar] [PubMed]
- Gooderham, N.J.; Murray, S.; Lynch, A.M.; Yadollahi-Farsani, M.; Zhao, K.; Boobis, A.R.; Davies, D.S. Food-derived heterocyclic amine mutagens: Variable metabolism and significance to humans. Drug Metab. Dispos. 2001, 29, 529–534. [Google Scholar] [PubMed]
- McQueen, C.A.; Maslansky, C.J.; Williams, G.M. Role of the acetylation polymorphism in determining susceptibility of cultured rabbit hepatocytes to DNA damage by aromatic amines. Cancer Res. 1983, 43, 3120–3123. [Google Scholar] [PubMed]
- Layton, D.W.; Bogen, K.T.; Knize, M.G.; Hatch, F.T.; Johnson, V.M.; Felton, J.S. Cancer risk of heterocyclic amines in cooked foods: An analysis and implications for research. Carcinogenesis 1995, 16, 39–52. [Google Scholar] [CrossRef]
- Augustsson, K.; Skog, K.; Jägerstad, M.; Dickman, P.W.; Steineck, G. Dietary heterocyclic amines and cancer of the colon, rectum, bladder, and kidney: A population-based study. Lancet 1999, 353, 703–707. [Google Scholar] [CrossRef]
- Bogen, K.T.; Keating, G.A. U.S. dietary exposures to heterocyclic amines. J. Expo. Anal. Environ. Epidemiol. 2001, 11, 155–168. [Google Scholar] [CrossRef]
- Snyderwine, E.G. Some perspectives on the nutritional aspects of breast cancer research. Food-derived heterocyclic amines as etiologic agents in human mammary cancer. Cancer 1994, 74, 1070–1077. [Google Scholar] [CrossRef]
- Ward, M.H.; Sinha, R.; Heineman, E.F.; Rothman, N.; Markin, R.; Weisenburger, D.D.; Correa, P.; Zahm, S.H. Risk of adenocarcinoma of the stomach and esophagus with meat cooking method and doneness preference. Int. J. Cancer 1997, 71, 14–19. [Google Scholar] [CrossRef]
- Sinha, R.; Kulldorff, M.; Swanson, C.A.; Curtin, J.; Brownson, R.C.; Alavanja, M.C. Dietary heterocyclic amines and the risk of lung cancer among Missouri women. Cancer Res. 2000, 60, 3753–3756. [Google Scholar] [PubMed]
- Zhu, J.; Chang, P.; Bondy, M.L.; Sahin, A.A.; Singletary, S.E.; Takahashi, S.; Shirai, T.; Li, D. Detection of 2-amino-1-methyl-6-phenylimidazo[4,5-b]-pyridine-DNA adducts in normal breast tissues and risk of breast cancer. Cancer Epidemiol. Biomark. Prev. 2003, 12, 830–837. [Google Scholar]
- Tang, D.; Kryvenko, O.N.; Wang, Y.; Trudeau, S.; Rundle, A.; Takahashi, S.; Shirai, T.; Rybicki, B.A. 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-DNA adducts in benign prostate and subsequent risk for prostate cancer. Int. J. Cancer 2013, 133, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Jakszyn, P.; Agudo, A.; Ibáñez, R.; García-Closas, R.; Pera, G.; Amiano, P.; González, C.A. Development of a food database of nitrosamines, heterocyclic amines, and polycyclic aromatic hydrocarbons. J. Nutr. 2004, 134, 2011–2014. [Google Scholar] [CrossRef]
- Zelinkova, Z.; Wenzl, T. The occurrence of 16 EPA PAHs in food—A review. Polycycl. Aromat. Compd. 2015, 35, 248–284. [Google Scholar] [CrossRef]
- Lijinsky, W. The formation and occurrence of polynuclear aromatic hydrocarbons associated with food. Mutat. Res. 1991, 259, 251–261. [Google Scholar] [CrossRef]
- Park, K.C.; Pyo, H.; Kim, W.; Yoon, K.S. Effects of cooking methods and tea marinades on the formation of benzo[a]pyrene in grilled pork belly (Samgyeopsal). Meat Sci. 2017, 129, 1–8. [Google Scholar] [CrossRef]
- Kazerouni, N.; Sinha, R.; Hsu, C.H.; Greenberg, A.; Rothman, N. Analysis of 200 food items for benzo[a]pyrene and estimation of its intake in an epidemiologic study. Food Chem. Toxicol. 2001, 39, 423–436. [Google Scholar] [CrossRef]
- IARC, International Agency for Research on Cancer. Some non-heterocyclic polycyclic aromatic hydrocarbons and some related exposures. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2010; Volume 92. [Google Scholar]
- Kroese, E.D.; Muller, J.J.A.; Mohn, G.R.; Dortant, P.M.; Wester, P.W. Tumorigenic Effects in Wistar Rats Orally Administered benzo[a]pyrene for Two Years (Gavage Studies): Implications for Human Cancer Risks Associated with Oral Exposure to Polycyclic Aromatic Hydrocarbons; 658603 010; National Institute for Public Health and the Environment (RIVM): Bilthoven, The Netherlands, 2001. [Google Scholar]
- IARC, International Agency for Research on Cancer. Bitumens and Bitumen Emissions, and some N- and S-Heterocyclic Polycyclic Aromatic Hydrocarbons. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2013; Volume 103. [Google Scholar]
- Moorthy, B.; Chu, C.; Carlin, D.J. Polycyclic aromatic hydrocarbons: From metabolism to lung cancer. Toxicol. Sci. 2015, 145, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Gelboin, H.V. Benzo[alpha]pyrene metabolism, activation and carcinogenesis: Role and regulation of mixed-function oxidases and related enzymes. Physiol. Rev. 1980, 60, 1107–1166. [Google Scholar] [CrossRef]
- Jerina, D.M.; Sayer, J.M.; Agarwal, S.K.; Yagi, H.; Levin, W.; Wood, A.W.; Conney, A.H.; Pruess-Schwartz, D.; Baird, W.M.; Pigott, M.A.; et al. Reactivity and tumorigenicity of bay-region diol epoxides derived from polycyclic aromatic hydrocarbons. Adv. Exp. Med. Biol. 1986, 197, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Lee, B.M. Oxidative stress to DNA, protein, and antioxidant enzymes (superoxide dismutase and catalase) in rats treated with benzo(a)pyrene. Cancer Lett. 1997, 113, 205–212. [Google Scholar] [CrossRef]
- Kawabata, T.T.; White, K.L., Jr. Suppression of the vitro humoral immune response of mouse splenocytes by benzo(a)pyrene metabolites and inhibition of benzo(a)pyrene-induced immunosuppression by alpha-naphthoflavone. Cancer Res. 1987, 47, 2317–2322. [Google Scholar] [PubMed]
- Myers, J.N.; Harris, K.L.; Rekhadevi, P.V.; Pratap, S.; Ramesh, A. Benzo(a)pyrene-induced cytotoxicity, cell proliferation, DNA damage, and altered gene expression profiles in HT-29 human colon cancer cells. Cell Biol. Toxicol. 2021, 37, 891–913. [Google Scholar] [CrossRef] [PubMed]
- Damiani, L.A.; Yingling, C.M.; Leng, S.; Romo, P.E.; Nakamura, J.; Belinsky, S.A. Carcinogen-induced gene promoter hypermethylation is mediated by DNMT1 and causal for transformation of immortalized bronchial epithelial cells. Cancer Res. 2008, 68, 9005–9014. [Google Scholar] [CrossRef]
- Commission Regulation (EU). Commission Regulation (EU) No 835/2011 of 19 August 2011 Amending Regulation (EC) No 1881/2006 as Regards Maximum Levels for Polycyclic Aromatic Hydrocarbons in Foodstuffs; Commission Regulation (EU): Brussels, Belgium, 2011; pp. 4–8. [Google Scholar]
- Commission Regulation (EU). Commission Regulation (EU) No 2020/1255 of 7 September 2020 Amending Regulation (EC) No 1881/2006 as Regards Maximum Levels of Polycyclic Aromatic Hydrocarbons (PAHs) in Traditionally Smoked Meat and Smoked Meat Products and Traditionally Smoked Fish and Smoked Fishery Products and Establishing a Maximum Level of PAHs in Powders of Food of Plant Origin Used for the Preparation of Beverages; Commission Regulation (EU): Brussels, Belgium, 2020. [Google Scholar]
- Sinha, R.; Kulldorff, M.; Gunter, M.J.; Strickland, P.; Rothman, N. Dietary benzo[a]pyrene intake and risk of colorectal adenoma. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2030–2034. [Google Scholar] [CrossRef]
- Yoon, E.; Park, K.; Lee, H.; Yang, J.-H.; Lee, C. Estimation of excess cancer risk on time-weighted lifetime average daily intake of PAHs from food ingestion. Hum. Ecol. Risk Assess. Int. J. 2007, 13, 669–680. [Google Scholar] [CrossRef]
- Bononi, M.; Quaglia, G.; Tateo, F. Identification of ethylene oxide in herbs, spices and other dried vegetables imported into Italy. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2014, 31, 271–275. [Google Scholar] [CrossRef]
- Heuser, S.G.; Scudamore, K.A. Fumigant residues in wheat and flour: Solvent extraction and gas-chromatographic determination of free methyl bromide and ethylene oxide. Analyst 1968, 93, 252–258. [Google Scholar] [CrossRef]
- Jensen, K.G. Determination of ethylene oxide residues in processed food products by gas-liquid chromatography after derivatization. Z. Lebensm. Unters. 1988, 187, 535–540. [Google Scholar] [CrossRef]
- Tthe European Parliament and the Council of the European Union. Regulation (EC) No 1107/2009 of the European Parliament and of the Council of 21 October 2009 Concerning the Placing of Plant Protection Products on the Market and Repealing Council Directives 79/117/EEC and 91/414/EEC; The European Parliament: Brussels, Belgium, 2009. [Google Scholar]
- The European Parliament. Regulation (EC) No 396/2005 of the European Parliament and of the Council of 23 February 2005 on Maximum Residue Levels of Pesticides in or on Food and Feed of Plant and Animal Origin and Amending Council Directive 91/414/EEC; The European Parliament: Brussels, Belgium, 2005. [Google Scholar]
- Kowalska, A.; Manning, L. Food Safety Governance and Guardianship: The role of the private sector in addressing the EU ehylene oxide incident. Foods 2022, 11, 204. [Google Scholar] [CrossRef] [PubMed]
- Bessaire, T.; Stroheker, T.; Eriksen, B.; Mujahid, C.; Hammel, Y.A.; Varela, J.; Delatour, T.; Panchaud, A.; Mottier, P.; Stadler, R.H. Analysis of ethylene oxide in ice creams manufactured with contaminated carob bean gum (E410). Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2021, 38, 2116–2127. [Google Scholar] [CrossRef] [PubMed]
- Kirman, C.R.; Li, A.A.; Sheehan, P.J.; Bus, J.S.; Lewis, R.C.; Hays, S.M. Ethylene oxide review: Characterization of total exposure via endogenous and exogenous pathways and their implications to risk assessment and risk management. J. Toxicol. Environ. Health B Crit. Rev. 2021, 24, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Bolt, H.M. Quantification of endogenous carcinogens. The ethylene oxide paradox. Biochem. Pharm. 1996, 52, 1–5. [Google Scholar] [CrossRef]
- Törnqvist, M.; Gustafsson, B.; Kautiainen, A.; Harms-Ringdahl, M.; Granath, F.; Ehrenberg, L. Unsaturated lipids and intestinal bacteria as sources of endogenous production of ethene and ethylene oxide. Carcinogenesis 1989, 10, 39–41. [Google Scholar] [CrossRef]
- Kirman, C.R.; Hays, S.M. Derivation of endogenous equivalent values to support risk assessment and risk management decisions for an endogenous carcinogen: Ethylene oxide. Regul. Toxicol. Pharm. 2017, 91, 165–172. [Google Scholar] [CrossRef]
- NTP, National Toxicology Program. Toxicology and carcinogenesis studies of ethylene oxide (CAS No. 75-21-8) in B6C3F1 mice (inhalation studies). Natl. Toxicol. Program. Tech. Rep. Ser. 1987, 326, 1–114. [Google Scholar]
- Bolt, H.M. Carcinogenicity and genotoxicity of ethylene oxide: New aspects and recent advances. Crit. Rev. Toxicol. 2000, 30, 595–608. [Google Scholar] [CrossRef]
- ATSDR, Agency for Toxic Substances and Disease Registry. Toxicological Profile for Ethylene Oxide; Draft for Public Comment; Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2020.
- Lynch, H.; Kozal, J.S.; Russell, A.J.; Thompson, W.J.; Divis, H.R.; Freid, R.D.; Calabrese, E.J.; Mundt, K.A. Systematic review of the scientific evidence on ethylene oxide as a human carcinogen. Chem. Biol. Interact. 2022, 364, 110031. [Google Scholar] [CrossRef]
- Dunkelberg, H. Carcinogenicity of ethylene oxide and 1,2-propylene oxide upon intragastric administration to rats. Br. J. Cancer 1982, 46, 924–933. [Google Scholar] [CrossRef]
- Natarajan, A.T.; Preston, R.J.; Dellarco, V.; Ehrenberg, L.; Generoso, W.; Lewis, S.; Tates, A.D. Ethylene oxide: Evaluation of genotoxicity data and an exploratory assessment of genetic risk. Mutat. Res. 1995, 330, 55–70. [Google Scholar] [CrossRef]
- Farooqi, Z.; Törnqvist, M.; Ehrenberg, L.; Natarajan, A.T. Genotoxic effects of ethylene oxide and propylene oxide in mouse bone marrow cells. Mutat. Res. 1993, 288, 223–228. [Google Scholar] [CrossRef]
- Ghosh, M.; Godderis, L. Genotoxicity of ethylene oxide: A review of micronucleus assay results in human population. Mutat. Res. Rev. Mutat. Res. 2016, 770, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Kolman, A.; Chovanec, M.; Osterman-Golkar, S. Genotoxic effects of ethylene oxide, propylene oxide and epichlorohydrin in humans: Update review (1990–2001). Mutat. Res. 2002, 512, 173–194. [Google Scholar] [CrossRef]
- Recio, L.; Donner, M.; Abernethy, D.; Pluta, L.; Steen, A.M.; Wong, B.A.; James, A.; Preston, R.J. In vivo mutagenicity and mutation spectrum in the bone marrow and testes of B6C3F1 lacI transgenic mice following inhalation exposure to ethylene oxide. Mutagenesis 2004, 19, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Bolt, H.M.; Peter, H.; Föst, U. Analysis of macromolecular ethylene oxide adducts. Int. Arch. Occup. Environ. Health 1988, 60, 141–144. [Google Scholar] [CrossRef]
- Rusyn, I.; Asakura, S.; Li, Y.; Kosyk, O.; Koc, H.; Nakamura, J.; Upton, P.B.; Swenberg, J.A. Effects of ethylene oxide and ethylene inhalation on DNA adducts, apurinic/apyrimidinic sites and expression of base excision DNA repair genes in rat brain, spleen, and liver. DNA Repair. 2005, 4, 1099–1110. [Google Scholar] [CrossRef]
- Marsden, D.A.; Jones, D.J.; Lamb, J.H.; Tompkins, E.M.; Farmer, P.B.; Brown, K. Determination of endogenous and exogenously derived N7-(2-hydroxyethyl)guanine adducts in ethylene oxide-treated rats. Chem. Res. Toxicol. 2007, 20, 290–299. [Google Scholar] [CrossRef]
- Walker, V.E.; Fennell, T.R.; Upton, P.B.; Skopek, T.R.; Prevost, V.; Shuker, D.E.; Swenberg, J.A. Molecular dosimetry of ethylene oxide: Formation and persistence of 7-(2-hydroxyethyl)guanine in DNA following repeated exposures of rats and mice. Cancer Res. 1992, 52, 4328–4334. [Google Scholar]
- Fennell, T.R.; Brown, C.D. A physiologically based pharmacokinetic model for ethylene oxide in mouse, rat, and human. Toxicol. Appl. Pharm. 2001, 173, 161–175. [Google Scholar] [CrossRef]
- Müller, M.; Krämer, A.; Angerer, J.; Hallier, E. Ethylene oxide-protein adduct formation in humans: Influence of glutathione-S-transferase polymorphisms. Int. Arch. Occup. Environ. Health 1998, 71, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Yong, L.C.; Schulte, P.A.; Wiencke, J.K.; Boeniger, M.F.; Connally, L.B.; Walker, J.T.; Whelan, E.A.; Ward, E.M. Hemoglobin adducts and sister chromatid exchanges in hospital workers exposed to ethylene oxide: Effects of glutathione S-transferase T1 and M1 genotypes. Cancer Epidemiol. Biomark. Prev. 2001, 10, 539–550. [Google Scholar]
- Fennell, T.R.; MacNeela, J.P.; Morris, R.W.; Watson, M.; Thompson, C.L.; Bell, D.A. Hemoglobin adducts from acrylonitrile and ethylene oxide in cigarette smokers: Effects of glutathione S-transferase T1-null and M1-null genotypes. Cancer Epidemiol. Biomark. Prev. 2000, 9, 705–712. [Google Scholar]
- Haufroid, V.; Merz, B.; Hofmann, A.; Tschopp, A.; Lison, D.; Hotz, P. Exposure to ethylene oxide in hospitals: Biological monitoring and influence of glutathione S-transferase and epoxide hydrolase polymorphisms. Cancer Epidemiol. Biomark. Prev. 2007, 16, 796–802. [Google Scholar] [CrossRef]
- Philippin, G.; Cadet, J.; Gasparutto, D.; Mazon, G.; Fuchs, R.P. Ethylene oxide and propylene oxide derived N7-alkylguanine adducts are bypassed accurately in vivo. DNA Repairr. 2014, 22, 133–136. [Google Scholar] [CrossRef]
- Tompkins, E.M.; McLuckie, K.I.; Jones, D.J.; Farmer, P.B.; Brown, K. Mutagenicity of DNA adducts derived from ethylene oxide exposure in the pSP189 shuttle vector replicated in human Ad293 cells. Mutat. Res. 2009, 678, 129–137. [Google Scholar] [CrossRef]
- Pottenger, L.H.; Boysen, G.; Brown, K.; Cadet, J.; Fuchs, R.P.; Johnson, G.E.; Swenberg, J.A. Understanding the importance of low-molecular weight (ethylene oxide- and propylene oxide-induced) DNA adducts and mutations in risk assessment: Insights from 15 years of research and collaborative discussions. Environ. Mol. Mutagen. 2019, 60, 100–121. [Google Scholar] [CrossRef]
- Gollapudi, B.B.; Su, S.; Li, A.A.; Johnson, G.E.; Reiss, R.; Albertini, R.J. Genotoxicity as a toxicologically relevant endpoint to inform risk assessment: A case study with ethylene oxide. Environ. Mol. Mutagen. 2020, 61, 852–871. [Google Scholar] [CrossRef]
- van Sittert, N.J.; Boogaard, P.J.; Natarajan, A.T.; Tates, A.D.; Ehrenberg, L.G.; Törnqvist, M.A. Formation of DNA adducts and induction of mutagenic effects in rats following 4 weeks inhalation exposure to ethylene oxide as a basis for cancer risk assessment. Mutat. Res. 2000, 447, 27–48. [Google Scholar] [CrossRef]
- SCF, Scientific Committee on Food. Opinion of the Scientific Committee on Food on Impurities of Ethylene Oxide in Food Additives; SCF/CS/ADD/EMU/186 Final; Scientific Committee on Food: Brussels, Belgium, 2002. [Google Scholar]
- Fowles, J.; Mitchell, J.; McGrath, H. Assessment of cancer risk from ethylene oxide residues in spices imported into New Zealand. Food Chem. Toxicol. 2001, 39, 1055–1062. [Google Scholar] [CrossRef]
- Bisanti, L.; Maggini, M.; Raschetti, R.; Alegiani, S.S.; Ippolito, F.M.; Caffari, B.; Segnan, N.; Ponti, A. Cancer mortality in ethylene oxide workers. Br. J. Ind. Med. 1993, 50, 317–324. [Google Scholar] [CrossRef]
- Steenland, K.; Whelan, E.; Deddens, J.; Stayner, L.; Ward, E. Ethylene oxide and breast cancer incidence in a cohort study of 7576 women (United States). Cancer Causes Control 2003, 14, 531–539. [Google Scholar] [CrossRef]
- Jinot, J.; Fritz, J.M.; Vulimiri, S.V.; Keshava, N. Carcinogenicity of ethylene oxide: Key findings and scientific issues. Toxicol. Mech. Methods 2018, 28, 386–396. [Google Scholar] [CrossRef]
- Marsh, G.M.; Keeton, K.A.; Riordan, A.S.; Best, E.A.; Benson, S.M. Ethylene oxide and risk of lympho-hematopoietic cancer and breast cancer: A systematic literature review and meta-analysis. Int. Arch. Occup. Environ. Health 2019, 92, 919–939. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.J.; Kozal, J.S.; Thompson, W.J.; Maier, A.; Dotson, G.S.; Best, E.A.; Mundt, K.A. Ethylene oxide: Cancer evidence integration and dose-response implications. Dose Response 2019, 17, 1559325819888317. [Google Scholar] [CrossRef]
- EPA, US Environmental Protection Agency. Evaluation of the Inhalation Carcinogenicity of Ethylene Oxide (CASRN 75-21-8). In Support of Summary Information on the Integrated Risk Information System (IRIS); EPA/635/R-16/350Fa; US Environmental Protection Agency: Washington, DC, USA, 2016. [Google Scholar]
- German Federal Institute for Risk Assessment, Bundesinstitut für Risikobewertung (BfR). Updated BfR Opinion on Health Risk Assessment of Ethylene Oxide Residues in Sesame Seeds. Opinion no 024/2021 Issued 1 September 2021; German Federal Institute for Risk Assessment: Berlin, Germany, 2021; p. 9.
- Lijinsky, W. N-Nitroso compounds in the diet. Mutat. Res. 1999, 443, 129–138. [Google Scholar] [CrossRef]
- Loeppky, R.N.; Yu, H. Amidine nitrosation. J. Org. Chem. 2004, 69, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- EMA, European Medicines Agency. Assessment Report. Procedure under Article 5(3) of Regulation EC (No) 726/2004. Nitrosamine Impurities in Human Medicinal Products. Procedure Number: EMEA/H/A-5(3)/1490; EMA/369136/2020; European Medicines Agency: Amsterdam, The Netherlands, 2020.
- Leaf, C.D.; Wishnok, J.S.; Tannenbaum, S.R. Mechanisms of endogenous nitrosation. Cancer Surv. 1989, 8, 323–334. [Google Scholar]
- Bartsch, H. N-nitroso compounds and human cancer: Where do we stand? IARC Sci. Publ. 1991, 105, 1–10. [Google Scholar]
- Bartsch, H.; Ohshima, H.; Pignatelli, B.; Calmels, S. Endogenously formed N-nitroso compounds and nitrosating agents in human cancer etiology. Pharmacogenetics 1992, 2, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Griesenbeck, J.S.; Steck, M.D.; Huber, J.C., Jr.; Sharkey, J.R.; Rene, A.A.; Brender, J.D. Development of estimates of dietary nitrates, nitrites, and nitrosamines for use with the Short Willet Food Frequency Questionnaire. Nutr. J. 2009, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Haorah, J.; Zhou, L.; Wang, X.; Xu, G.; Mirvish, S.S. Determination of total N-nitroso compounds and their precursors in frankfurters, fresh meat, dried salted fish, sauces, tobacco, and tobacco smoke particulates. J. Agric. Food Chem. 2001, 49, 6068–6078. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, P.; Xu, X.; Zhou, G. Influence of various cooking methods on the concentrations of volatile N-nitrosamines and biogenic amines in dry-cured sausages. J. Food Sci. 2012, 77, C560–C565. [Google Scholar] [CrossRef] [PubMed]
- Havery, D.C.; Kline, D.A.; Miletta, E.M.; Joe, F.L., Jr.; Fazio, T. Survey of food products for volatile N-nitrosamines. J. Assoc. Off. Anal. Chem. 1976, 59, 540–546. [Google Scholar] [CrossRef]
- IARC, International Agency for Research on Cancer. Some industrial chemicals. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2000; Volume 77. [Google Scholar]
- IARC, International Agency for Research on Cancer. Ingested nitrate and nitrite, and cyanobacterial peptide toxins. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2010; Volume 94. [Google Scholar]
- Jeffrey, A.M.; Iatropoulos, M.J.; Williams, G.M. Nasal cytotoxic and carcinogenic activities of systemically distributed organic chemicals. Toxicol. Pathol. 2006, 34, 827–852. [Google Scholar] [CrossRef]
- Pour, P.; Gingell, R.; Langenbach, R.; Nagel, D.; Grandjean, C.; Lawson, T.; Salmasi, S. Carcinogenicity of N-nitrosomethyl(2-oxopropyl)amine in Syrian hamsters. Cancer Res. 1980, 40, 3585–3590. [Google Scholar] [PubMed]
- Thresher, A.; Foster, R.; Ponting, D.J.; Stalford, S.A.; Tennant, R.E.; Thomas, R. Are all nitrosamines concerning? A review of mutagenicity and carcinogenicity data. Regul. Toxicol. Pharm. 2020, 116, 104749. [Google Scholar] [CrossRef]
- Lijinsky, W. Mutagenesis, carcinogenesis and alkylating properties of nitrosamines and related compounds. Prog. Clin. Biol. Res. 1986, 209, 141–148. [Google Scholar]
- Tsuda, S.; Matsusaka, N.; Madarame, H.; Miyamae, Y.; Ishida, K.; Satoh, M.; Sekihashi, K.; Sasaki, Y.F. The alkaline single cell electrophoresis assay with eight mouse organs: Results with 22 mono-functional alkylating agents (including 9 dialkyl N-nitrosoamines) and 10 DNA crosslinkers. Mutat. Res. 2000, 467, 83–98. [Google Scholar] [CrossRef]
- Verna, L. N-Nitrosodiethylamine mechanistic data and risk assessment: Bioactivation, DNA-adduct formation, mutagenicity, and tumor initiation. Pharmacol. Ther. 1996, 71, 57–81. [Google Scholar] [CrossRef]
- Arimoto-Kobayashi, S.; Kaji, K.; Sweetman, G.M.; Hayatsu, H. Mutation and formation of methyl- and hydroxylguanine adducts in DNA caused by N-nitrosodimethylamine and N-nitrosodiethylamine with UVA irradiation. Carcinogenesis 1997, 18, 2429–2433. [Google Scholar] [CrossRef] [PubMed]
- Otteneder, M.; Lutz, W.K. Correlation of DNA adduct levels with tumor incidence: Carcinogenic potency of DNA adducts. Mutat. Res. 1999, 424, 237–247. [Google Scholar] [CrossRef]
- Loeppky, R.N.; Ye, Q.; Goelzer, P.; Chen, Y. DNA adducts from N-nitrosodiethanolamine and related beta-oxidized nitrosamines in vivo: (32)P-postlabeling methods for glyoxal- and O(6)-hydroxyethyldeoxyguanosine adducts. Chem. Res. Toxicol. 2002, 15, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Rajewsky, M.F.; Engelbergs, J.; Thomale, J.; Schweer, T. Relevance of DNA repair to carcinogenesis and cancer therapy. Recent Results Cancer Res. 1998, 154, 127–146. [Google Scholar]
- Kamataki, T.; Fujita, K.-i.; Nakayama, K.; Yamazaki, Y.; Miyamoto, M.; Ariyoshi, N. Role of human cytochrome P450 (CYP) in the metabolic activation of nitrosamine derivatives: Application of genetically engineered Salmonella expressing human CYP. Drug Metab. Rev. 2002, 34, 667–676. [Google Scholar] [CrossRef]
- Yang, C.S.; Smith, T.; Ishizaki, H.; Hong, J.Y. Enzyme mechanisms in the metabolism of nitrosamines. IARC Sci. Publ. 1991, 105, 265–274. [Google Scholar]
- Johnson, G.E.; Dobo, K.; Gollapudi, B.; Harvey, J.; Kenny, J.; Kenyon, M.; Lynch, A.; Minocherhomji, S.; Nicolette, J.; Thybaud, V.; et al. Permitted daily exposure limits for noteworthy N-nitrosamines. Environ. Mol. Mutagen. 2021, 62, 293–305. [Google Scholar] [CrossRef]
- Gushgari, A.J.; Halden, R.U. Critical review of major sources of human exposure to N-nitrosamines. Chemosphere 2018, 210, 1124–1136. [Google Scholar] [CrossRef]
- Poirier, S.; Hubert, A.; de-Thé, G.; Ohshima, H.; Bourgade, M.C.; Bartsch, H. Occurrence of volatile nitrosamines in food samples collected in three high-risk areas for nasopharyngeal carcinoma. IARC Sci. Publ. 1987, 84, 415–419. [Google Scholar]
- O’Brien, P.J.; Siraki, A.G.; Shangari, N. Aldehyde sources, metabolism, molecular toxicity mechanisms, and possible effects on human health. Crit. Rev. Toxicol. 2005, 35, 609–662. [Google Scholar] [CrossRef]
- Sousa, B.C.; Pitt, A.R.; Spickett, C.M. Chemistry and analysis of HNE and other prominent carbonyl-containing lipid oxidation compounds. Free Radic. Biol. Med. 2017, 111, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.B.; Gavin, C.L.; Taylor, S.V.; Waddell, W.J.; Cohen, S.M.; Feron, V.J.; Goodman, J.; Rietjens, I.M.; Marnett, L.J.; Portoghese, P.S.; et al. The FEMA GRAS assessment of alpha,beta-unsaturated aldehydes and related substances used as flavor ingredients. Food Chem. Toxicol. 2008, 46, 2935–2967. [Google Scholar] [CrossRef] [PubMed]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Evaluation of Certain Food Additives and Contaminants; Prepared by the Sixty-First Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA); WHO: Geneva, Switzerland, 2004. [Google Scholar]
- Gasc, N.; Taché, S.; Rathahao, E.; Bertrand-Michel, J.; Roques, V.; Guéraud, F. 4-Hydroxynonenal in foodstuffs: Heme concentration, fatty acid composition and freeze-drying are determining factors. Redox Rep. 2007, 12, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Feron, V.J.; Til, H.P.; de Vrijer, F.; Woutersen, R.A.; Cassee, F.R.; van Bladeren, P.J. Aldehydes: Occurrence, carcinogenic potential, mechanism of action and risk assessment. Mutat. Res./Genet. Toxicol. 1991, 259, 363–385. [Google Scholar] [CrossRef]
- IARC, International Agency for Research on Cancer. Acrolein, crotonaldehyde, and arecoline. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2021; Volume 128. [Google Scholar]
- EFSA CONTAM Panel, European Food Safety Authority, Panel on Contaminants in the Food Chain. Scientific Opinion on Flavouring Group Evaluation 200, Revision 1 (FGE.200 Rev.1): 74 a,b-unsaturated aliphatic aldehydes and precursors from chemical subgroup 1.1.1 of FGE.19. EFSA J. 2018, 16, 5422. [Google Scholar] [CrossRef]
- Chung, F.L.; Tanaka, T.; Hecht, S.S. Induction of liver tumors in F344 rats by crotonaldehyde. Cancer Res. 1986, 46, 1285–1289. [Google Scholar]
- NTP, National Toxicology Program. Toxicology and Carcinogenesis Studies of Malonaldehyde, Sodium Salt (3-hydroxy-2-propenal, Sodium Salt) (CAS No. 24382-04-5) in F344/N Rats and B6C3F1 Mice (Gavage Studies); 0888-8051 (Print); National Toxicology Program: Research Triangle, NC, USA, 1988; pp. 1–182.
- NTP, National Toxicology Program. Toxicology and Carcinogensis Studies of 2,4-Hexadienal (89% trans,trans Isomer, CAS No. 142-83-6; 11% cis, trans Isomer) (Gavage Studies); 0888-8051 (Print); National Toxicology Program: Research Triangle, NC, USA, 2003; pp. 1–290.
- EFSA CONTAM Panel, European Food Safety Authority, Panel on Contaminants in the Food Chain. Scientific Opinion on Flavouring Group Evaluation 203, Revision 2 (FGE.203Rev2): a,b-unsaturated aliphatic aldehydes and precursors from chemical subgroup 1.1.4 of FGE.19 with two or more conjugated double-bonds and with or without additional non-conjugated double-bonds. EFSA J. 2018, 16, 5322. [Google Scholar] [CrossRef]
- IARC, International Agency for Research on Cancer. Re-evaluation of some organic chemicals, hydrazine and hydrogen Peroxide. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 1999; Volume 71. [Google Scholar]
- Von Tungeln, L.S.; Yi, P.; Bucci, T.J.; Samokyszyn, V.M.; Chou, M.W.; Kadlubar, F.F.; Fu, P.P. Tumorigenicity of chloral hydrate, trichloroacetic acid, trichloroethanol, malondialdehyde, 4-hydroxy-2-nonenal, crotonaldehyde, and acrolein in the B6C3F(1) neonatal mouse. Cancer Lett. 2002, 185, 13–19. [Google Scholar] [CrossRef]
- Eder, E.; Deininger, C.; Neudecker, T.; Deininger, D. Mutagenicity of β-alkyl substituted acrolein congeners in the Salmonella typhimurium strain TA100 and genotoxicity testing in the SOS chromotest. Environ. Mol. Mutagen. 1992, 19, 338–345. [Google Scholar] [CrossRef]
- Eder, E.; Scheckenbach, S.; Deininger, C.; Hoffman, C. The possible role of alpha, beta-unsaturated carbonyl compounds in mutagenesis and carcinogenesis. Toxicol Lett. 1993, 67, 87–103. [Google Scholar] [CrossRef]
- Brambilla, G.; Sciabà, L.; Faggin, P.; Maura, A.; Marinari, U.M.; Ferro, M.; Esterbauer, H. Cytotoxicity, DNA fragmentation and sister-chromatid exchange in Chinese hamster ovary cells exposed to the lipid peroxidation product 4-hydroxynonenal and homologous aldehydes. Mutat. Res. 1986, 171, 169–176. [Google Scholar] [CrossRef]
- Esterbauer, H. Cytotoxicity and genotoxicity of lipid-oxidation products. Am. J. Clin. Nutr. 1993, 57, 779S–785S, discussion 785S–786S. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Villalta, P.W.; Wang, M.; Hecht, S.S. Analysis of crotonaldehyde- and acetaldehyde-derived 1,n(2)-propanodeoxyguanosine adducts in DNA from human tissues using liquid chromatography electrospray ionization tandem mass spectrometry. Chem. Res. Toxicol. 2006, 19, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Guéraud, F. 4-Hydroxynonenal metabolites and adducts in pre-carcinogenic conditions and cancer. Free Radic. Biol. Med. 2017, 111, 196–208. [Google Scholar] [CrossRef]
- Hu, W.; Feng, Z.; Eveleigh, J.; Iyer, G.; Pan, J.; Amin, S.; Chung, F.L.; Tang, M.S. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis 2002, 23, 1781–1789. [Google Scholar] [CrossRef]
- Singh, S.P.; Chen, T.; Chen, L.; Mei, N.; McLain, E.; Samokyszyn, V.; Thaden, J.J.; Moore, M.M.; Zimniak, P. Mutagenic effects of 4-hydroxynonenal triacetate, a chemically protected form of the lipid peroxidation product 4-hydroxynonenal, as assayed in L5178Y/Tk+/– mouse lymphoma cells. J. Pharmacol. Exp. Ther. 2005, 313, 855–861. [Google Scholar] [CrossRef]
- Gutteridge, J.M.C. Aspects to consider when detecting and measuring lipid peroxidation. Free Radic. Res. Commun. 1986, 1, 173–184. [Google Scholar] [CrossRef]
- Tsikas, D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: Analytical and biological challenges. Anal. Biochem. 2017, 524, 13–30. [Google Scholar] [CrossRef]
- Nielsen, N.S.; Timm-Heinrich, M.; Jacobsen, C. Comparison of wet-chemical methods for determination of lipid hydroperoxides. J. Food Lipids 2003, 10, 35–50. [Google Scholar] [CrossRef]
- Calingasan, N.Y.; Uchida, K.; Gibson, G.E. Protein-bound acrolein. J. Neurochem. 1999, 72, 751–756. [Google Scholar] [CrossRef]
- Yuan, J.M.; Gao, Y.T.; Wang, R.; Chen, M.; Carmella, S.G.; Hecht, S.S. Urinary levels of volatile organic carcinogen and toxicant biomarkers in relation to lung cancer development in smokers. Carcinogenesis 2012, 33, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.M.; Butler, L.M.; Gao, Y.T.; Murphy, S.E.; Carmella, S.G.; Wang, R.; Nelson, H.H.; Hecht, S.S. Urinary metabolites of a polycyclic aromatic hydrocarbon and volatile organic compounds in relation to lung cancer development in lifelong never smokers in the Shanghai Cohort Study. Carcinogenesis 2014, 35, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, H.; Schiffman, C.; Gunter, M.J.; Naccarati, A.; Polidoro, S.; Dagnino, S.; Dudoit, S.; Vineis, P.; Rappaport, S.M. Cys34 adductomics links colorectal cancer with the gut microbiota and redox biology. Cancer Res. 2019, 79, 6024–6031. [Google Scholar] [CrossRef] [PubMed]
- Eder, E.; Schuler, D.; Budiawan. Cancer risk assessment for crotonaldehyde and 2-hexenal: An approach. IARC Sci. Publ. 1999, 150, 219–232. [Google Scholar]
- Kunishima, M.; Yamauchi, Y.; Mizutani, M.; Kuse, M.; Takikawa, H.; Sugimoto, Y. Identification of (Z)-3:(E)-2-hexenal isomerases essential to the production of the leaf aldehyde in plants. J. Biol Chem. 2016, 291, 14023–14033. [Google Scholar] [CrossRef]
- Jordán, M.J.; Tandon, K.; Shaw, P.E.; Goodner, K.L. Aromatic profile of aqueous banana essence and banana fruit by Gas Chromatography−Mass Spectrometry (GC-MS) and Gas Chromatography−Olfactometry (GC-O). J. Agric. Food Chem. 2001, 49, 4813–4817. [Google Scholar] [CrossRef]
- EFSA CONTAM Panel, European Food Safety Authority, Panel on Contaminants in the Food Chain. Scientific Opinion on Flavouring Group Evaluation 71 Revision 1 (FGE.71Rev1): Consideration of aliphatic, linear, a,b-unsaturated alcohols, aldehydes, carboxylic acids, and related esters evaluated by JECFA (63rd and 69th meeting) structurally related to flavouring substances evaluated in FGE.05Rev3. EFSA J. 2020, 18, 5924. [Google Scholar] [CrossRef]
- Nádasi, E.; Varjas, T.; Pajor, L.; Ember, I. Carcinogenic potential of trans-2-hexenal is based on epigenetic effect. In Vivo 2005, 19, 559–562. [Google Scholar]
- Dittberner, U.; Eisenbrand, G.; Zankl, H. Genotoxic effects of the α, β-unsaturated aldehydes 2-trans-butenal,2-trans-hexenal and 2-trans, 6-cis-rmnonadienal. Mutat. Res./Environ. Mutagen. Relat. Subj. 1995, 335, 259–265. [Google Scholar] [CrossRef]
- Dittberner, U.; Schmetzer, B.; Gölzer, P.; Eisenbrand, G.; Zankl, H. Genotoxic effects of 2-trans-hexenal in human buccal mucosa cells in vivo. Mutat. Res. 1997, 390, 161–165. [Google Scholar] [CrossRef]
- Eder, E.; Schuler, D. An approach to cancer risk assessment for the food constituent 2-hexenal on the basis of 1,N2-propanodeoxyguanosine adducts of 2-hexenal in vivo. Arch. Toxicol. 2000, 74, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Schuler, D.; Eder, E. Detection of 1,N2-propanodeoxyguanosine adducts of 2-hexenal in organs of Fischer 344 rats by a 32P-post-labeling technique. Carcinogenesis 1999, 20, 1345–1350. [Google Scholar] [CrossRef][Green Version]
- Gölzer, P.; Janzowski, C.; Pool-Zobel, B.L.; Eisenbrand, G. (E)-2-hexenal-induced DNA damage and formation of cyclic 1,N2-(1,3-propano)-2’-deoxyguanosine adducts in mammalian cells. Chem. Res. Toxicol. 1996, 9, 1207–1213. [Google Scholar] [CrossRef]
- Stout, M.D.; Jeong, Y.C.; Boysen, G.; Li, Y.; Sangaiah, R.; Ball, L.M.; Gold, A.; Swenberg, J.A. LC/MS/MS method for the quantitation of trans-2-hexenal-derived exocyclic 1,N(2)-propanodeoxyguanosine in DNA. Chem. Res. Toxicol. 2006, 19, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Stout, M.D.; Bodes, E.; Schoonhoven, R.; Upton, P.B.; Travlos, G.S.; Swenberg, J.A. Toxicity, DNA binding, and cell proliferation in male F344 rats following short-term gavage exposures to trans-2-hexenal. Toxicol. Pathol. 2008, 36, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Kiwamoto, R.; Rietjens, I.M.; Punt, A. A physiologically based in silico model for trans-2-hexenal detoxification and DNA adduct formation in rat. Chem. Res. Toxicol. 2012, 25, 2630–2641. [Google Scholar] [CrossRef]
- Janzowski, C.; Glaab, V.; Mueller, C.; Straesser, U.; Kamp, H.G.; Eisenbrand, G. Alpha,beta-unsaturated carbonyl compounds: Induction of oxidative DNA damage in mammalian cells. Mutagenesis 2003, 18, 465–470. [Google Scholar] [CrossRef]
- Palmer, S. Diet, nutrition, and cancer. Prog. Food Nutr. Sci. 1985, 9, 283–341. [Google Scholar]
- Weisburger, J.H. Carcinogenesis in our food and cancer prevention. Adv. Exp. Med. Biol. 1991, 289, 137–151. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Cho, H.J.; Moon, S.; Choi, J.; Lee, S.; Ahn, C.; Yoo, K.Y.; Kim, I.; Ko, K.P.; Lee, J.E.; et al. Pickled vegetable and salted fish intake and the risk of gastric cancer: Two prospective cohort studies and a meta-analysis. Cancers 2020, 12, 996. [Google Scholar] [CrossRef]
- Gallicchio, L.; Matanoski, G.; Tao, X.; Chen, L.; Lam, T.K.; Boyd, K.; Robinson, K.A.; Balick, L.; Mickelson, S.; Caulfield, L.E.; et al. Adulthood consumption of preserved and nonpreserved vegetables and the risk of nasopharyngeal carcinoma: A systematic review. Int. J. Cancer 2006, 119, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- IARC, International Agency for Research on Cancer. Red meat and processed meat. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2015; Volume 114. [Google Scholar]
- Farvid, M.S.; Sidahmed, E.; Spence, N.D.; Mante Angua, K.; Rosner, B.A.; Barnett, J.B. Consumption of red meat and processed meat and cancer incidence: A systematic review and meta-analysis of prospective studies. Eur. J. Epidemiol. 2021, 36, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.D.; Lloyd, S.K. Association between red meat consumption and colon cancer: A systematic review of experimental results. Exp. Biol. Med. 2017, 242, 813–839. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.J.; Darwis, N.D.M.; Mackay, D.F.; Celis-Morales, C.A.; Lyall, D.M.; Sattar, N.; Gill, J.M.R.; Pell, J.P. Red and processed meat consumption and breast cancer: UK Biobank cohort study and meta-analysis. Eur. J. Cancer 2018, 90, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Turesky, R.J. Mechanistic evidence for red meat and processed meat intake and cancer risk: A follow-up on the International Agency for Research on Cancer evaluation of 2015. Chimia 2018, 72, 718–724. [Google Scholar] [CrossRef]
- Tannenbaum, S.R.; Bishop, W.; Yu, M.C.; Henderson, B.E. Attempts to isolate N-nitroso compounds from Chinese-style salted fish. Natl. Cancer Inst. Monogr. 1985, 69, 209–211. [Google Scholar]
- IARC, International Agency for Research on Cancer. Review of human carcinogens. Personal habits and indoor combustions. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2012; Volume 100E. [Google Scholar]
- Qiu, Y.; Chen, J.H.; Yu, W.; Wang, P.; Rong, M.; Deng, H. Contamination of Chinese salted fish with volatile N-nitrosamines as determined by QuEChERS and gas chromatography-tandem mass spectrometry. Food Chem. 2017, 232, 763–769. [Google Scholar] [CrossRef]
- Poirier, S.; Bouvier, G.; Malaveille, C.; Ohshima, H.; Shao, Y.M.; Hubert, A.; Zeng, Y.; de Thé, G.; Bartsch, H. Volatile nitrosamine levels and genotoxicity of food samples from high-risk areas for nasopharyngeal carcinoma before and after nitrosation. Int. J. Cancer 1989, 44, 1088–1094. [Google Scholar] [CrossRef]
- Zheng, X.; Luo, Y.; Christensson, B.; Drettner, B. Induction of nasal and nasopharyngeal tumours in Sprague-Dawley rats fed with Chinese salted fish. Acta Otolaryngol. 1994, 114, 98–104. [Google Scholar] [CrossRef]
- Yu, M.C.; Nichols, P.W.; Zou, X.N.; Estes, J.; Henderson, B.E. Induction of malignant nasal cavity tumours in Wistar rats fed Chinese salted fish. Br. J. Cancer 1989, 60, 198–201. [Google Scholar] [CrossRef][Green Version]
- Huang, D.P.; Ho, J.H.; Saw, D.; Teoh, T.B. Carcinoma of the nasal and paranasal regions in rats fed Cantonese salted marine fish. IARC Sci. Publ. 1978, 20, 315–328. [Google Scholar]
- Weisburger, J.H.; Marquardt, H.; Hirota, N.; Mori, H.; Williams, G.M. Induction of cancer of the glandular stomach in rats by an extract of nitrite-treated fish. J. Natl. Cancer Inst. 1980, 64, 163–167. [Google Scholar] [PubMed]
- Widlak, P.; Zheng, X.; Osterdahl, B.G.; Drettner, B.; Christensson, B.; Kumar, R.; Hemminki, K. N-nitrosodimethylamine and 7-methylguanine DNA adducts in tissues of rats fed Chinese salted fish. Cancer Lett. 1995, 94, 85–90. [Google Scholar] [CrossRef]
- Yuan, J.M.; Wang, X.L.; Xiang, Y.B.; Gao, Y.T.; Ross, R.K.; Yu, M.C. Preserved foods in relation to risk of nasopharyngeal carcinoma in Shanghai, China. Int. J. Cancer 2000, 85, 358–363. [Google Scholar] [CrossRef]
- Lian, M. Salted fish and processed foods intake and nasopharyngeal carcinoma risk: A dose-response meta-analysis of observational studies. Eur. Arch. Otorhinolaryngol. 2022, 279, 2501–2509. [Google Scholar] [CrossRef]
- Hara, N.; Sakata, K.; Nagai, M.; Fujita, Y.; Hashimoto, T.; Yanagawa, H. Statistical analyses on the pattern of food consumption and digestive-tract cancers in Japan. Nutr. Cancer 1984, 6, 220–228. [Google Scholar] [CrossRef]
- FDA, Food and Drug Administration. Food Additive Regulations. Synthetic Flavoring Agents and Adjuvants. A Rule by the Food and Drug Administration on 10/09/2018 83 FR 50490. 21 CFR 172. 21 CFR 177. Document Number: 2018-21807. Final Rule; Notif. Part Denial Petition; Food and Drug Administration: Rome, Italy, 2018; pp. 50490–50503.
- IARC, International Agency for Research on Cancer. Some chemicals that cause tumours of the urinary tract in rodents. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2019; Volume 119. [Google Scholar]
- Felter, S.P.; Llewelyn, C.; Navarro, L.; Zhang, X. How the 62-year old Delaney Clause continues to thwart science: Case study of the flavor substance β-myrcene. Regul. Toxicol. Pharm. 2020, 115, 104708. [Google Scholar] [CrossRef]
- Api, A.M.; Belmonte, F.; Belsito, D.; Biserta, S.; Botelho, D.; Bruze, M.; Burton, G.A., Jr.; Buschmann, J.; Cancellieri, M.A.; Dagli, M.L.; et al. RIFM fragrance ingredient safety assessment, myrcene, CAS Registry Number 123-35-3. Food Chem. Toxicol. 2020, 144 (Suppl. S1), 111631. [Google Scholar] [CrossRef]
- NTP, National Toxicology Program. Technical report on the toxicology and carcinogenesis studies of beta-myrcene (CAS No. 123-35-3) in F344/N rats and B6C3F1 mice (gavage studies). Natl. Toxicol. Program Tech. Rep. Ser. 2010, 557, 1–163. [Google Scholar]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Safety Evaluation of Certain Food Additives; Prepared for the Seventy-Ninth Meeting of the Joint FAO/WHO Expert Committee on Food Additives; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- EFSA CEF Panel, European Food Safety Authority, Panel on Food Contact Materials, Enzymes, Flavourings and Processing Aids. Scientific Opinion on Flavouring Group Evaluation 78, Revision 2 (FGE.78Rev2): Consideration of aliphatic and alicyclic and aromatic hydrocarbons evaluated by JECFA (63rd meeting) structurally related to aliphatic hydrocarbonsevaluated by EFSA in FGE.25Rev3. EFSA J. 2015, 13, 4067. [Google Scholar] [CrossRef]
- Kauderer, B.; Zamith, H.; Paumgartten, F.J.; Speit, G. Evaluation of the mutagenicity of beta-myrcene in mammalian cells in vitro. Environ. Mol. Mutagen. 1991, 18, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Zamith, H.P.; Vidal, M.N.; Speit, G.; Paumgartten, F.J. Absence of genotoxic activity of beta-myrcene in the in vivo cytogenetic bone marrow assay. Braz. J. Med. Biol. Res. 1993, 26, 93–98. [Google Scholar] [PubMed]
- Cohen, S.M.; Eisenbrand, G.; Fukushima, S.; Gooderham, N.J.; Guengerich, F.P.; Hecht, S.S.; Rietjens, I.; Bastaki, M.; Davidsen, J.M.; Harman, C.L.; et al. FEMA GRAS assessment of natural flavor complexes: Citrus-derived flavoring ingredients. Food Chem. Toxicol. 2019, 124, 192–218. [Google Scholar] [CrossRef]
- Madyastha, K.M.; Srivatsan, V. Metabolism of beta-myrcene in vivo and in vitro: Its effects on rat-liver microsomal enzymes. Xenobiotica 1987, 17, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Asakawa, Y.; Takemoto, T.; Aratani, T. Terpenoids biotransformation in mammals III: Biotransformation of alpha-pinene, beta-pinene, pinane, 3-carene, carane, myrcene, and p-cymene in rabbits. J. Pharm. Sci. 1981, 70, 406–415. [Google Scholar] [CrossRef]
- Adams, T.B.; Gavin, C.L.; McGowen, M.M.; Waddell, W.J.; Cohen, S.M.; Feron, V.J.; Marnett, L.J.; Munro, I.C.; Portoghese, P.S.; Rietjens, I.M.; et al. The FEMA GRAS assessment of aliphatic and aromatic terpene hydrocarbons used as flavor ingredients. Food Chem. Toxicol. 2011, 49, 2471–2494. [Google Scholar] [CrossRef]
- De-Oliveira, A.C.; Ribeiro-Pinto, L.F.; Paumgartten, J.R. In vitro inhibition of CYP2B1 monooxygenase by beta-myrcene and other monoterpenoid compounds. Toxicol. Lett. 1997, 92, 39–46. [Google Scholar] [CrossRef]
- De-Oliveira, A.C.; Ribeiro-Pinto, L.F.; Otto, S.S.; Gonçalves, A.; Paumgartten, F.J. Induction of liver monooxygenases by beta-myrcene. Toxicology 1997, 124, 135–140. [Google Scholar] [CrossRef]
- Cesta, M.F.; Hard, G.C.; Boyce, J.T.; Ryan, M.J.; Chan, P.C.; Sills, R.C. Complex histopathologic response in rat kidney to oral β-myrcene: An unusual dose-related nephrosis and low-dose alpha2u-globulin nephropathy. Toxicol. Pathol. 2013, 41, 1068–1077. [Google Scholar] [CrossRef]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Evaluation of Certain Food Additives; Prepared by the Sixty-third meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA); WHO: Geneva, Switzerland, 2005. [Google Scholar]
- Mog, S.R.; Zang, Y.J. Safety assessment of food additives: Case example with myrcene, a synthetic flavoring agent. Toxicol. Pathol. 2019, 47, 1035–1037. [Google Scholar] [CrossRef]
- IARC, International Agency for Research on Cancer. Some drugs and herbal products. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2016; Volume 108. [Google Scholar]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Evaluation of Certain Food Additives and Contaminants; Prepared by the Fifty-Fifth Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA); WHO: Geneva, Switzerland, 2001. [Google Scholar]
- EFSA AFC Panel, European Food Safety Authority, Panel on Food Additives, Flavourings, Processing Aids and Materials in contact with Food. Opinion of the Scientific Panel on Food Additives, Flavourings, Processing Aids and Materials in contact with Foods on a request from the Commission on Pulegone and Menthofuran in flavourings and other food ingredients with flavouring properties. Question number EFSA-Q-2003-119. EFSA J. 2005, 298, 1–32. [Google Scholar] [CrossRef]
- EMA, European Medicines Agency. Public Statement on the Use of Herbal Medicinal Products Containing Pulegone and Menthofuran; EMA/HMPC/138386/2005 Rev. 1; European Medicines Agency: Amsterdam, The Netherlands, 2016.
- NTP, National Toxicology Program. Toxicology and carcinogenesis studies of pulegone (CAS No. 89-82-7) in F344/N rats and B6C3F1 mice (gavage studies). Natl. Toxicol. Program. Tech. Rep. Ser. 2011, 563, 1–201. [Google Scholar]
- Api, A.M.; Belsito, D.; Biserta, S.; Botelho, D.; Bruze, M.; Burton, G.A., Jr.; Buschmann, J.; Cancellieri, M.A.; Dagli, M.L.; Date, M.; et al. RIFM fragrance ingredient safety assessment, pulegone, CAS Registry Number 89-82-7. Food Chem. Toxicol. 2021, 149 (Suppl. S1), 112092. [Google Scholar] [CrossRef] [PubMed]
- Khojasteh, S.C.; Hartley, D.P.; Ford, K.A.; Uppal, H.; Oishi, S.; Nelson, S.D. Characterization of rat liver proteins adducted by reactive metabolites of menthofuran. Chem. Res. Toxicol. 2012, 25, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.J.; Lebetkin, E.H.; Burka, L.T. Metabolism of (R)-(+)-pulegone in F344 rats. Drug Metab. Dispos. 2001, 29, 1567–1577. [Google Scholar]
- Chen, L.J.; Lebetkin, E.H.; Burka, L.T. Comparative disposition of (R)-(+)-pulegone in B6C3F1 mice and F344 rats. Drug Metab. Dispos. 2003, 31, 892–899. [Google Scholar] [CrossRef]
- Khojasteh-Bakht, S.C.; Chen, W.; Koenigs, L.L.; Peter, R.M.; Nelson, S.D. Metabolism of (R)-(+)-pulegone and (R)-(+)-menthofuran by human liver cytochrome P-450s: Evidence for formation of a furan epoxide. Drug Metab. Dispos. 1999, 27, 574–580. [Google Scholar]
- Gordon, W.P.; Huitric, A.C.; Seth, C.L.; McClanahan, R.H.; Nelson, S.D. The metabolism of the abortifacient terpene, (R)-(+)-pulegone, to a proximate toxin, menthofuran. Drug Metab. Dispos. 1987, 15, 589–594. [Google Scholar]
- Nelson, S.D.; McClanahan, R.H.; Thomassen, D.; Gordon, W.P.; Knebel, N. Investigations of mechanisms of reactive metabolite formation from (R)-(+)-pulegone. Xenobiotica 1992, 22, 1157–1164. [Google Scholar] [CrossRef]
- Engel, W. In vivo studies on the metabolism of the monoterpene pulegone in humans using the metabolism of ingestion-correlated amounts (MICA) approach: Explanation for the toxicity differences between (S)-(-)- and (R)-(+)-pulegone. J. Agric. Food Chem. 2003, 51, 6589–6597. [Google Scholar] [CrossRef]
- McClanahan, R.H.; Thomassen, D.; Slattery, J.T.; Nelson, S.D. Metabolic activation of (R)-(+)-pulegone to a reactive enonal that covalently binds to mouse liver proteins. Chem. Res. Toxicol. 1989, 2, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, M.S.; Dodmane, P.R.; Arnold, L.L.; Pennington, K.L.; Anwar, M.M.; Adams, B.R.; Taylor, S.V.; Wermes, C.; Adams, T.B.; Cohen, S.M. Mode of action of pulegone on the urinary bladder of F344 rats. Toxicol. Sci. 2012, 128, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Alshannaq, A.; Yu, J.H. Occurrence, toxicity, and analysis of major mycotoxins in food. Int. J. Environ. Res. Public Health 2017, 14, 632. [Google Scholar] [CrossRef]
- Riley, R.T.; Merrill, A.H., Jr. Ceramide synthase inhibition by fumonisins: A perfect storm of perturbed sphingolipid metabolism, signaling, and disease. J. Lipid Res. 2019, 60, 1183–1189. [Google Scholar] [CrossRef]
- Schroeder, J.J.; Crane, H.M.; Xia, J.; Liotta, D.C.; Merrill, A.H., Jr. Disruption of sphingolipid metabolism and stimulation of DNA synthesis by fumonisin B1. A molecular mechanism for carcinogenesis associated with Fusarium moniliforme. J. Biol. Chem. 1994, 269, 3475–3481. [Google Scholar] [CrossRef]
- NTP, National Toxicology Program. Toxicology and carcinogenesis studies of fumonisin B1 (cas no. 116355-83-0) in F344/N rats and B6C3F1 mice (feed studies). Natl. Toxicol. Program. Tech. Rep. Ser. 2001, 496, 1–352. [Google Scholar]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Safety Evaluation of Certain Mycotoxins in Food; Prepared by the Fifty-Sixth Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA); WHO: Geneva, Switzerland, 2001. [Google Scholar]
- Gelderblom, W.C.A.; Marasas, W.F.O.; Lebepe-Mazur, S.; Swanevelder, S.; Abel, S. Cancer initiating properties of fumonisin B1 in a short-term rat liver carcinogenesis assay. Toxicology 2008, 250, 89–95. [Google Scholar] [CrossRef]
- Gelderblom, W.C.A.; Snyman, S.D.; Lebepe-Mazur, S.; van der Westhuizen, L.; Kriek, N.P.J.; Marasas, W.F.O. The cancer-promoting potential of fumonisin B1 in rat liver using diethylnitrosamine as a cancer initiator. Cancer Lett. 1996, 109, 101–108. [Google Scholar] [CrossRef]
- Sakai, A.; Suzuki, C.; Masui, Y.; Kuramashi, A.; Takatori, K.; Tanaka, N. The activities of mycotoxins derived from Fusarium and related substances in a short-term transformation assay using v-Ha-ras-transfected BALB/3T3 cells (Bhas 42 cells). Mutat. Res. 2007, 630, 103–111. [Google Scholar] [CrossRef]
- Carlson, D.B.; Williams, D.E.; Spitsbergen, J.M.; Ross, P.F.; Bacon, C.W.; Meredith, F.I.; Riley, R.T. Fumonisin B1 promotes aflatoxin B1 and N-methyl-N’-nitro-nitrosoguanidine-initiated liver tumors in rainbow trout. Toxicol. Appl. Pharm. 2001, 172, 29–36. [Google Scholar] [CrossRef]
- Mobio, T.A.; Tavan, E.; Baudrimont, I.; Anane, R.; Carratú, M.R.; Sanni, A.; Gbeassor, M.F.; Shier, T.W.; Narbonne, J.F.; Creppy, E.E. Comparative study of the toxic effects of fumonisin B1 in rat C6 glioma cells and p53-null mouse embryo fibroblasts. Toxicology 2003, 183, 65–75. [Google Scholar] [CrossRef]
- Galvano, F.; Russo, A.; Cardile, V.; Galvano, G.; Vanella, A.; Renis, M. DNA damage in human fibroblasts exposed to fumonisin B(1). Food Chem. Toxicol. 2002, 40, 25–31. [Google Scholar] [CrossRef]
- Domijan, A.M.; Zeljezić, D.; Milić, M.; Peraica, M. Fumonisin B(1): Oxidative status and DNA damage in rats. Toxicology 2007, 232, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.M.; Abdel-Aziem, S.H.; El-Nekeety, A.A.; Abdel-Wahhab, M.A. Panax ginseng extract modulates oxidative stress, DNA fragmentation and up-regulate gene expression in rats sub chronically treated with aflatoxin B1 and fumonisin B 1. Cytotechnology 2015, 67, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, V.; Darroudi, F.; Uhl, M.; Steinkellner, H.; Zsivkovits, M.; Knasmueller, S. Fumonisin B(1) is genotoxic in human derived hepatoma (HepG2) cells. Mutagenesis 2002, 17, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Aranda, M.; Pérez-Alzola, L.P.; Ellahueñe, M.F.; Sepúlveda, C. Assessment of in vitro mutagenicity in Salmonella and in vivo genotoxicity in mice of the mycotoxin fumonisin B(1). Mutagenesis 2000, 15, 469–471. [Google Scholar] [CrossRef]
- Mary, V.S.; Valdehita, A.; Navas, J.M.; Rubinstein, H.R.; Fernández-Cruz, M.L. Effects of aflatoxin B₁, fumonisin B₁ and their mixture on the aryl hydrocarbon receptor and cytochrome P450 1A induction. Food Chem. Toxicol. 2015, 75, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Spotti, M.; Maas, R.F.; de Nijs, C.M.; Fink-Gremmels, J. Effect of fumonisin B(1) on rat hepatic P450 system. Environ. Toxicol. Pharm. 2000, 8, 197–204. [Google Scholar] [CrossRef]
- Müller, S.; Dekant, W.; Mally, A. Fumonisin B1 and the kidney: Modes of action for renal tumor formation by fumonisin B1 in rodents. Food Chem. Toxicol. 2012, 50, 3833–3846. [Google Scholar] [CrossRef]
- Harrer, H.; Humpf, H.U.; Voss, K.A. In vivo formation of N-acyl-fumonisin B1. Mycotoxin Res. 2015, 31, 33–40. [Google Scholar] [CrossRef]
- Wang, E.; Norred, W.P.; Bacon, C.W.; Riley, R.T.; Merrill, A.H., Jr. Inhibition of sphingolipid biosynthesis by fumonisins. Implications for diseases associated with Fusarium moniliforme. J. Biol. Chem. 1991, 266, 14486–14490. [Google Scholar] [CrossRef]
- Voss, K.A.; Howard, P.C.; Riley, R.T.; Sharma, R.P.; Bucci, T.J.; Lorentzen, R.J. Carcinogenicity and mechanism of action of fumonisin B1: A mycotoxin produced by Fusarium moniliforme (F. verticillioides). Cancer Detect. Prev. 2002, 26, 1–9. [Google Scholar] [CrossRef]
- Riley, R.T.; Enongene, E.; Voss, K.A.; Norred, W.P.; Meredith, F.I.; Sharma, R.P.; Spitsbergen, J.; Williams, D.E.; Carlson, D.B.; Merrill, A.H., Jr. Sphingolipid perturbations as mechanisms for fumonisin carcinogenesis. Environ. Health Perspect. 2001, 109 (Suppl. S2), 301–308. [Google Scholar] [CrossRef] [PubMed]
- Merrill, A.H., Jr.; Sullards, M.C.; Wang, E.; Voss, K.A.; Riley, R.T. Sphingolipid metabolism: Roles in signal transduction and disruption by fumonisins. Environ. Health Perspect. 2001, 109 (Suppl. S2), 283–289. [Google Scholar] [CrossRef]
- Gelderblom, W.C.; Abel, S.; Smuts, C.M.; Marnewick, J.; Marasas, W.F.; Lemmer, E.R.; Ramljak, D. Fumonisin-induced hepatocarcinogenesis: Mechanisms related to cancer initiation and promotion. Environ. Health Perspect. 2001, 109 (Suppl. S2), 291–300. [Google Scholar] [CrossRef]
- Rumora, L.; Domijan, A.M.; Grubisić, T.Z.; Peraica, M. Mycotoxin fumonisin B1 alters cellular redox balance and signalling pathways in rat liver and kidney. Toxicology 2007, 242, 31–38. [Google Scholar] [CrossRef]
- Yin, J.J.; Smith, M.J.; Eppley, R.M.; Page, S.W.; Sphon, J.A. Effects of fumonisin B1 on lipid peroxidation in membranes. Biochim. Biophys. Acta 1998, 1371, 134–142. [Google Scholar] [CrossRef]
- Sun, G.; Wang, S.; Hu, X.; Su, J.; Huang, T.; Yu, J.; Tang, L.; Gao, W.; Wang, J.S. Fumonisin B1 contamination of home-grown corn in high-risk areas for esophageal and liver cancer in China. Food Addit. Contam. 2007, 24, 181–185. [Google Scholar] [CrossRef]
- Wang, H.; Wei, H.; Ma, J.; Luo, X. The fumonisin B1 content in corn from North China, a high-risk area of esophageal cancer. J. Environ. Pathol. Toxicol. Oncol. 2000, 19, 139–141. [Google Scholar]
- Isaacson, C. The change of the staple diet of black South Africans from sorghum to maize (corn) is the cause of the epidemic of squamous carcinoma of the oesophagus. Med. Hypotheses 2005, 64, 658–660. [Google Scholar] [CrossRef]
- van der Westhuizen, L.; Shephard, G.S.; Scussel, V.M.; Costa, L.L.; Vismer, H.F.; Rheeder, J.P.; Marasas, W.F. Fumonisin contamination and fusarium incidence in corn from Santa Catarina, Brazil. J. Agric. Food Chem. 2003, 51, 5574–5578. [Google Scholar] [CrossRef] [PubMed]
- Claeys, L.; Romano, C.; De Ruyck, K.; Wilson, H.; Fervers, B.; Korenjak, M.; Zavadil, J.; Gunter, M.J.; De Saeger, S.; De Boevre, M.; et al. Mycotoxin exposure and human cancer risk: A systematic review of epidemiological studies. Compr. Rev. Food Sci. Food Saf. 2020, 19, 1449–1464. [Google Scholar] [CrossRef] [PubMed]
- Persson, E.C.; Sewram, V.; Evans, A.A.; London, W.T.; Volkwyn, Y.; Shen, Y.J.; Van Zyl, J.A.; Chen, G.; Lin, W.; Shephard, G.S.; et al. Fumonisin B1 and risk of hepatocellular carcinoma in two Chinese cohorts. Food Chem. Toxicol. 2012, 50, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Cantalejo, M.J.; Torondel, P.; Amate, L.; Carrasco, J.M.; Hernández, E. Detection of fusarin C and trichothecenes in Fusarium strains from Spain. J. Basic Microbiol. 1999, 39, 143–153. [Google Scholar] [CrossRef]
- Kleigrewe, K.; Söhnel, A.C.; Humpf, H.U. A new high-performance liquid chromatography-tandem mass spectrometry method based on dispersive solid phase extraction for the determination of the mycotoxin fusarin C in corn ears and processed corn samples. J. Agric. Food Chem. 2011, 59, 10470–10476. [Google Scholar] [CrossRef]
- Han, Z.; Tangni, E.K.; Huybrechts, B.; Munaut, F.; Scauflaire, J.; Wu, A.; Callebaut, A. Screening survey of co-production of fusaric acid, fusarin C, and fumonisins B₁, B₂ and B₃ by Fusarium strains grown in maize grains. Mycotoxin Res. 2014, 30, 231–240. [Google Scholar] [CrossRef]
- Zhu, B.; Jeffrey, A.M. Stability of Fusarin C: Effects of the normal cooking procedure used in china and ph. Nutr. Cancer 1992, 18, 53–58. [Google Scholar] [CrossRef]
- Gelderblom, W.C.; Thiel, P.G.; Jaskiewicz, K.; Marasas, W.F. Investigations on the carcinogenicity of fusarin C--a mutagenic metabolite of Fusarium moniliforme. Carcinogenesis 1986, 7, 1899–1901. [Google Scholar] [CrossRef]
- Cheng, S.J.; Jiang, Y.Z.; Li, M.H.; Lo, H.Z. A mutagenic metabolite produced by Fusarium moniliforme isolated from Linxian county, China. Carcinogenesis 1985, 6, 903–905. [Google Scholar] [CrossRef]
- Gelderblom, W.C.; Snyman, S.D. Mutagenicity of potentially carcinogenic mycotoxins produced by Fusarium moniliforme. Mycotoxin Res. 1991, 7, 46–52. [Google Scholar] [CrossRef]
- Norred, W.P.; Plattner, R.D.; Vesonder, R.F.; Bacon, C.W.; Voss, K.A. Effects of selected secondary metabolites of Fusarium moniliforme on unscheduled synthesis of DNA by rat primary hepatocytes. Food Chem. Toxicol. 1992, 30, 233–237. [Google Scholar] [CrossRef]
- Bever, R.J., Jr.; Couch, L.H.; Sutherland, J.B.; Williams, A.J.; Beger, R.D.; Churchwell, M.I.; Doerge, D.R.; Howard, P.C. DNA adduct formation by Fusarium culture extracts: Lack of role of fusarin C. Chem. Biol. Interact. 2000, 128, 141–157. [Google Scholar] [CrossRef]
- Lu, S.J.; Ronai, Z.A.; Li, M.H.; Jeffrey, A.M. Fusarium moniliforme metabolites: Genotoxicity of culture extracts. Carcinogenesis 1988, 9, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, W.C.; Swart, P.; Kramer, P.S. Investigations on the spectral interactions of fusarin C with rat liver microsomal cytochrome P-450. Xenobiotica 1988, 18, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.J.; Li, M.H.; Park, S.S.; Gelboin, H.V.; Jeffrey, A.M. Metabolism of fusarin C by rat liver microsomes. Role of esterase and cytochrome P-450 enzymes with respect to the mutagenicity of fusarin C in Salmonella typhimurium. Biochem. Pharm. 1989, 38, 3811–3817. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Jeffrey, A.M. Fusarin C: Isolation and identification of two microsomal metabolites. Chem. Res. Toxicol. 1993, 6, 97–101. [Google Scholar] [CrossRef]
- Sondergaard, T.E.; Hansen, F.T.; Purup, S.; Nielsen, A.K.; Bonefeld-Jørgensen, E.C.; Giese, H.; Sørensen, J.L. Fusarin C acts like an estrogenic agonist and stimulates breast cancer cells in vitro. Toxicol. Lett. 2011, 205, 116–121. [Google Scholar] [CrossRef]
- Marasas, W.F.; Jaskiewicz, K.; Venter, F.S.; Van Schalkwyk, D.J. Fusarium moniliforme contamination of maize in oesophageal cancer areas in Transkei. S. Afr. Med. J. 1988, 74, 110–114. [Google Scholar]
- IARC, International Agency for Research on Cancer. Polychlorinated biphenyls and polybrominted biphenyls. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2016; Volume 107. [Google Scholar]
- IARC, International Agency for Research on Cancer. DDT, lindane, and 2,4-D. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2018; Volume 113. [Google Scholar]
- Walker, K. Cost-comparison of DDT and alternative insecticides for malaria control. Med. Vet. Entomol. 2000, 14, 345–354. [Google Scholar] [CrossRef]
- Pedercini, M.; Movilla Blanco, S.; Kopainsky, B. Application of the malaria management model to the analysis of costs and benefits of DDT versus non-DDT malaria control. PLoS ONE 2011, 6, e27771. [Google Scholar] [CrossRef]
- ATSDR, Agency for Toxic Substances and Disease Registry. Toxicological Profile for DDT, DDE, and DDD; Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2022.
- WHO, World Health Organization. The Use of DDT in Malaria Vector Control; WHO Position Statement; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Smith, D. Worldwide trends in DDT levels in human breast milk. Int. J. Epidemiol. 1999, 28, 179–188. [Google Scholar] [CrossRef]
- van den Berg, M.; Kypke, K.; Kotz, A.; Tritscher, A.; Lee, S.Y.; Magulova, K.; Fiedler, H.; Malisch, R. WHO/UNEP global surveys of PCDDs, PCDFs, PCBs and DDTs in human milk and benefit-risk evaluation of breastfeeding. Arch. Toxicol. 2017, 91, 83–96. [Google Scholar] [CrossRef] [PubMed]
- NCI, National Cancer Institute. Bioassay of DDT, TDE, and p,p′-DDE for Possible Carcinogenicity; National Institutes of Health: Bethesda, MD, USA, 1978.
- IARC, International Agency for Research on Cancer. DDT and associated compounds. Occupational exposures in insecticide application, and some pesticides. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 1991; Volume 53. [Google Scholar]
- EFSA CONTAM Panel, European Food Safety Authority, Panel on Contaminants in the Food Chain. Opinion of the Scientific Panel on contaminants in the food chain [CONTAM] related to DDT as an undesirable substance in animal feed. EFSA J. 2006, 433, 1–69. [Google Scholar] [CrossRef]
- Tebourbi, O.; Driss, M.R.; Sakly, M.; Rhouma, K.B. Metabolism of DDT in different tissues of young rats. J. Environ. Sci. Health B 2006, 41, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.P.; Roan, C.C. Absorption, storage, and metabolic conversion of ingested DDT and DDT metabolites in man. Arch. Environ. Health 1971, 22, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Nims, R.W.; Lubet, R.A.; Fox, S.D.; Jones, C.R.; Thomas, P.E.; Reddy, A.B.; Kocarek, T.A. Comparative pharmacodynamics of CYP2B induction by DDT, DDE, and DDD in male rat liver and cultured rat hepatocytes. J. Toxicol. Environ. Health A 1998, 53, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Lund, B.O.; Bergman, A.; Brandt, I. Metabolic activation and toxicity of a DDT-metabolite, 3-methylsulphonyl-DDE, in the adrenal zona fasciculata in mice. Chem. Biol. Interact. 1988, 65, 25–40. [Google Scholar] [CrossRef]
- Williams, G.M.; Numoto, S. Promotion of mouse liver neoplasms by the organochlorine pesticides chlordane and heptachlor in comparison to dichlorodiphenyltrichlorpethane. Carcinogenesis 1984, 5, 1689–1696. [Google Scholar] [CrossRef]
- Williams, G.M.; Telang, S.; Tong, C. Inhibition of intercellular communication between liver cells by the liver tumor promoter 1,1,1-trichloro-2,2-bis(p-chlorophenyl)ethane. Cancer Lett. 1981, 11, 339–344. [Google Scholar] [CrossRef]
- López-Cervantes, M.; Torres-Sánchez, L.; Tobías, A.; López-Carrillo, L. Dichlorodiphenyldichloroethane burden and breast cancer risk: A meta-analysis of the epidemiologic evidence. Environ. Health Perspect. 2003, 112, 207–214. [Google Scholar] [CrossRef]
- Gatto, N.M.; Longnecker, M.P.; Press, M.F.; Sullivan-Halley, J.; McKean-Cowdin, R.; Bernstein, L. Serum organochlorines and breast cancer: A case–control study among African-American women. Cancer Causes Control 2007, 18, 29–39. [Google Scholar] [CrossRef]
- Mouly, T.A.; Toms, L.L. Breast cancer and persistent organic pollutants (excluding DDT): A systematic literature review. Environ. Sci. Pollut. Res. Int. 2016, 23, 22385–22407. [Google Scholar] [CrossRef] [PubMed]
- VoPham, T.; Bertrand, K.A.; Hart, J.E.; Laden, F.; Brooks, M.M.; Yuan, J.M.; Talbott, E.O.; Ruddell, D.; Chang, C.H.; Weissfeld, J.L. Pesticide exposure and liver cancer: A review. Cancer Causes Control 2017, 28, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Baris, D.; Zahm, S.H.; Cantor, K.P.; Blair, A. Agricultural use of DDT and risk of non-Hodgkin’s lymphoma: Pooled analysis of three case-control studies in the United States. Occup. Environ. Med. 1998, 55, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.S.; Crespo, J.G.; Afonso, C.A. Dioxins sources and current remediation technologies—A review. Environ. Int. 2008, 34, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine, (US) Committee on the Implications of Dioxin in the Food Supply. Dioxins and Dioxin-like Compounds in the Food Supply: Strategies to Decrease Exposure; National Academies Press (US): Washington, DC, USA, 2003.
- Van den Berg, M.; Birnbaum, L.; Bosveld, A.T.; Brunström, B.; Cook, P.; Feeley, M.; Giesy, J.P.; Hanberg, A.; Hasegawa, R.; Kennedy, S.W.; et al. Toxic equivalency factors (TEFs) for PCBs, PCDDs, PCDFs for humans and wildlife. Environ. Health Perspect. 1998, 106, 775–792. [Google Scholar] [CrossRef]
- Van den Berg, M.; Birnbaum, L.S.; Denison, M.; De Vito, M.; Farland, W.; Feeley, M.; Fiedler, H.; Hakansson, H.; Hanberg, A.; Haws, L.; et al. The 2005 World Health Organization reevaluation of human and Mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicol. Sci. 2006, 93, 223–241. [Google Scholar] [CrossRef]
- Schecter, A.; Birnbaum, L.; Ryan, J.J.; Constable, J.D. Dioxins: An overview. Environ. Res. 2006, 101, 419–428. [Google Scholar] [CrossRef]
- Charnley, G.; Kimbrough, R.D. Overview of exposure, toxicity, and risks to children from current levels of 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds in the USA. Food Chem. Toxicol. 2006, 44, 601–615. [Google Scholar] [CrossRef]
- Scialli, A.R.; Watkins, D.K.; Ginevan, M.E. Agent Orange eposure and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in human milk. Birth Defects Res. B Dev. Reprod. Toxicol. 2015, 104, 129–139. [Google Scholar] [CrossRef]
- Ulaszewska, M.M.; Zuccato, E.; Davoli, E. PCDD/Fs and dioxin-like PCBs in human milk and estimation of infants’ daily intake: A review. Chemosphere 2011, 83, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Arisawa, K. Recent decreasing trends of exposure to PCDDs/PCDFs/dioxin-like PCBs in general populations, and associations with diabetes, metabolic syndrome, and gout/hyperuricemia. J. Med. Investig. 2018, 65, 151–161. [Google Scholar] [CrossRef] [PubMed]
- NTP, National Toxicology Program. Toxicology and carcinogenesis studies of a mixture of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) (Cas No. 1746-01-6), 2,3,4,7,8-pentachlorodibenzofuran (PeCDF) (Cas No. 57117-31-4), and 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126) (Cas No. 57465-28-8) in female Harlan Sprague-Dawley rats (gavage studies). Natl. Toxicol. Program. Tech. Rep. Ser. 2006, 526, 1–180. [Google Scholar]
- NTP, National Toxicology Program. Toxicology and carcinogenesis studies of 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126) (CAS No. 57465-28-8) in female Harlan Sprague-Dawley rats (Gavage Studies). Natl. Toxicol. Program. Tech. Rep. Ser. 2006, 520, 4–246. [Google Scholar]
- NTP, National Toxicology Program. Technical report on the toxicology and carcinogenesis studies of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) (CAS No. 1746-01-6) in female Harlan Sprague-Dawley rats (Gavage Studies). Natl. Toxicol. Program. Tech. Rep. Ser. 2006, 521, 4–232.
- Knerr, S.; Schrenk, D. Carcinogenicity of “non-dioxinlike” polychlorinated biphenyls. Crit. Rev. Toxicol. 2006, 36, 663–694. [Google Scholar] [CrossRef]
- Huff, J.E.; Salmon, A.G.; Hooper, N.K.; Zeise, L. Long-term carcinogenesis studies on 2,3,7,8-tetrachlorodibenzo-p-dioxin and hexachlorodibenzo-p-dioxins. Cell Biol. Toxicol. 1991, 7, 67–94. [Google Scholar] [CrossRef] [PubMed]
- Turteltaub, K.W.; Felton, J.S.; Gledhill, B.L.; Vogel, J.S.; Southon, J.R.; Caffee, M.W.; Finkel, R.C.; Nelson, D.E.; Proctor, I.D.; Davis, J.C. Accelerator mass spectrometry in biomedical dosimetry: Relationship between low-level exposure and covalent binding of heterocyclic amine carcinogens to DNA. Proc. Natl. Acad. Sci. USA 1990, 87, 5288–5292. [Google Scholar] [CrossRef]
- Randerath, K.; Putman, K.L.; Randerath, E.; Mason, G.; Kelley, M.; Safe, S. Organ-specific effects of long term feeding of 2,3,7,8-tetrachlorodibenzo-p-dioxin and 1,2,3,7,8-pentachlorodibenzo-p-dioxin on I-compounds in hepatic and renal DNA of female Sprague-Dawley rats. Carcinogenesis 1988, 9, 2285–2289. [Google Scholar] [CrossRef]
- Dragan, Y.P.; Schrenk, D. Animal studies addressing the carcinogenicity of TCDD (or related compounds) with an emphasis on tumour promotion. Food Addit. Contam. 2000, 17, 289–302. [Google Scholar] [CrossRef]
- Whysner, J.; Montandon, F.; McClain, R.M.; Downing, J.; Verna, L.K.; Steward, R.E.; Williams, G.M. Absence of DNA adduct formation by phenobarbital, polychlorinated biphenyls, and chlordane in mouse liver using the 32P-postlabeling assay. Toxicol. Appl. Pharmacol. 1998, 148, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Schilderman, P.A.; Maas, L.M.; Pachen, D.M.; de Kok, T.M.; Kleinjans, J.C.; van Schooten, F.J. Induction of DNA adducts by several polychlorinated biphenyls. Environ. Mol. Mutagen. 2000, 36, 79–86. [Google Scholar] [CrossRef]
- Pohjanvirta, R.; Vartiainen, T.; Uusi-Rauva, A.; Mönkkönen, J.; Tuomisto, J. Tissue distribution, metabolism, and excretion of 14C-TCDD in a TCDD-susceptible and a TCDD-resistant rat strain. Pharm. Toxicol. 1990, 66, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.R. Metabolism and disposition of 2,3,7,8-tetrachlorodibenzo-p-dioxin in guinea pigs. Toxicol. Appl. Pharm. 1986, 85, 263–273. [Google Scholar] [CrossRef]
- Kahn, P.C.; Gochfeld, M.; Nygren, M.; Hansson, M.; Rappe, C.; Velez, H.; Ghent-Guenther, T.; Wilson, W.P. Dioxins and dibenzofurans in blood and adipose tissue of Agent Orange-exposed Vietnam veterans and matched controls. JAMA 1988, 259, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Diliberto, J.J.; DeVito, M.J.; Ross, D.G.; Birnbaum, L.S. Subchronic exposure of [3H]- 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in female B6C3F1 mice: Relationship of steady-state levels to disposition and metabolism. Toxicol. Sci. 2001, 61, 241–255. [Google Scholar] [CrossRef]
- Gasiewicz, T.A.; Geiger, L.E.; Rucci, G.; Neal, R.A. Distribution, excretion, and metabolism of 2,3,7,8-tetrachlorodibenzo-p-dioxin in C57BL/6J, DBA/2J, and B6D2F1/J mice. Drug Metab. Dispos. 1983, 11, 397–403. [Google Scholar]
- Inui, H.; Itoh, T.; Yamamoto, K.; Ikushiro, S.; Sakaki, T. Mammalian cytochrome P450-dependent metabolism of polychlorinated dibenzo-p-dioxins and coplanar polychlorinated biphenyls. Int. J. Mol. Sci. 2014, 15, 14044–14057. [Google Scholar] [CrossRef]
- Wroblewski, V.J.; Olson, J.R. Hepatic metabolism of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the rat and guinea pig. Toxicol. Appl. Pharm. 1985, 81, 231–240. [Google Scholar] [CrossRef]
- Whysner, J.; Williams, G.M. 2,3,7,8-Tetrachlorodibenzo-p-dioxin mechanistic data and risk assessment: Gene regulation, cytotoxicity, enhanced cell proliferation, and tumor promotion. Pharmacol. Ther. 1996, 71, 193–223. [Google Scholar] [CrossRef]
- Patrizi, B.; Siciliani de Cumis, M. TCDD toxicity mediated by epigenetic mechanisms. Int. J. Mol. Sci. 2018, 19, 4101. [Google Scholar] [CrossRef]
- Knerr, S.; Schrenk, D. Carcinogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in experimental models. Mol. Nutr. Food Res. 2006, 50, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Lucier, G.; Clark, G.; Hiermath, C.; Tritscher, A.; Sewall, C.; Huff, J. Carcinogenicity of TCDD in laboratory animals: Implications for risk assessment. Toxicol. Ind. Health 1993, 9, 631–668. [Google Scholar] [CrossRef]
- Schecter, A.; Startin, J.; Wright, C.; Kelly, M.; Päpke, O.; Lis, A.; Ball, M.; Olson, J. Dioxins in U.S. food and estimated daily intake. Chemosphere 1994, 29, 2261–2265. [Google Scholar] [CrossRef]
- González, N.; Domingo, J.L. Polychlorinated dibenzo-p-dioxins and dibenzofurans (PCDD/Fs) in food and human dietary intake: An update of the scientific literature. Food Chem. Toxicol. 2021, 157, 112585. [Google Scholar] [CrossRef] [PubMed]
- Danjou, A.M.; Fervers, B.; Boutron-Ruault, M.C.; Philip, T.; Clavel-Chapelon, F.; Dossus, L. Estimated dietary dioxin exposure and breast cancer risk among women from the French E3N prospective cohort. Breast Cancer Res. 2015, 17, 39. [Google Scholar] [CrossRef]
- Boffetta, P.; Mundt, K.A.; Adami, H.O.; Cole, P.; Mandel, J.S. TCDD and cancer: A critical review of epidemiologic studies. Crit Rev. Toxicol. 2011, 41, 622–636. [Google Scholar] [CrossRef]
- Cole, P.; Trichopoulos, D.; Pastides, H.; Starr, T.; Mandel, J.S. Dioxin and cancer: A critical review. Regul. Toxicol. Pharm. 2003, 38, 378–388. [Google Scholar] [CrossRef]
- WHO, World Health Organization. Assessment of the Health Risk of Dioxins: Re-Evaluation of the Tolerable Daily Intake (TDI); Executive Summary; WHO Regional Office for Europe: Copenhagen, Denmark, 1998. [Google Scholar]
- van Leeuwen, F.X.; Feeley, M.; Schrenk, D.; Larsen, J.C.; Farland, W.; Younes, M. Dioxins: WHO’s tolerable daily intake (TDI) revisited. Chemosphere 2000, 40, 1095–1101. [Google Scholar] [CrossRef]
- SCF, Scientific Committee on Food. Opinion of the Scientific Committee on Food on the Risk Assessment of Dioxins and Dioxin-like PCBs in Food; Adopted on 30 May 2001; Scientific Committee on Food: Brussels, Belgium, 2001. [Google Scholar]
- EPA, US Environmental Protection Agency. Dioxin Reassessment—An SAB Review of the Office of Research and Development’s Reassessment of Dioxin: Review of the Revised Sections (Dose Response Modeling, Integrated Summary, Risk Characterization, and Toxicity Equivalence Factors) of the EPA’s Reassessment of Dioxin by the Dioxin Reassessment Review Subcommittee of the EPA Science Advisory Board (SAB); EPA-SAB-EC-01–006; US Environmental Protection Agency: Washington, DC, USA, 2001.
- Starr, T.B. Significant shortcomings of the U.S. Environmental Protection Agency’s latest draft risk characterization for dioxin-like compounds. Toxicol. Sci. 2001, 64, 7–13. [Google Scholar] [CrossRef][Green Version]
- Paustenbach, D.J. The U.S. EPA Science Advisory Board Evaluation (2001) of the EPA dioxin reassessment. Regul. Toxicol. Pharm. 2002, 36, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Popp, J.A.; Crouch, E.; McConnell, E.E. A Weight-of-evidence analysis of the cancer dose-response characteristics of 2,3,7,8-tetrachlorodibenzodioxin (TCDD). Toxicol. Sci. 2006, 89, 361–369. [Google Scholar] [CrossRef] [PubMed][Green Version]
- NRC, National Research Council. Health Risks from Dioxin and Related Compounds; Evaluation of EPA Reassessment; The National Academies Press: Washington, DC, USA, 2006. [CrossRef]
- EFSA CEP Panel, European Food Safety Authority, Panel on Food Contact Materials, Enzymes and Processing Aids. Scientific Opinion on safety of benzophenone to be used as flavouring. EFSA J. 2017, 15, 5013. [Google Scholar]
- Adams, T.B.; McGowen, M.M.; Williams, M.C.; Cohen, S.M.; Feron, V.J.; Goodman, J.I.; Marnett, L.J.; Munro, I.C.; Portoghese, P.S.; Smith, R.L.; et al. The FEMA GRAS assessment of aromatic substituted secondary alcohols, ketones, and related esters used as flavor ingredients. Food Chem. Toxicol. 2007, 45, 171–201. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Tian, M.; Feng, W.; He, H.; Wang, Y.; Yang, L. Sensitive detection of benzophenone-type ultraviolet filters in plastic food packaging materials by sheathless capillary electrophoresis-electrospray ionization-tandem mass spectrometry. J. Chromatogr. A 2019, 1604, 460469. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.A.; Castle, L. Benzophenone in cartonboard packaging materials and the factors that influence its migration into food. Food Addit. Contam. 2003, 20, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Castle, L.; Damant, A.P.; Honeybone, C.A.; Johns, S.M.; Jickells, S.M.; Sharman, M.; Gilbert, J. Migration studies from paper and board food packaging materials. Part 2. Survey for residues of dialkylamino benzophenone UV-cure ink photoinitiators. Food Addit. Contam. 1997, 14, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Simat, T.J.; Altkofer, W.; Fügel, D. Survey on the occurrence of photo-initiators and amine synergists in cartonboard packaging on the German market and their migration into the packaged foodstuffs. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2013, 30, 1993–2016. [Google Scholar] [CrossRef]
- Rhodes, M.C.; Bucher, J.R.; Peckham, J.C.; Kissling, G.E.; Hejtmancik, M.R.; Chhabra, R.S. Carcinogenesis studies of benzophenone in rats and mice. Food Chem. Toxicol. 2007, 45, 843–851. [Google Scholar] [CrossRef]
- NTP, National Toxicology Program. Technical Report on the Toxicity Studies of Benzophenone (CAS No. 119-61-9) Administered in Feed to F344/N Rats and B6C3F1 Mice 0888-8051 (Print); National Toxicology Program: Research Triangle, NC, USA, 2000; pp. 1–412.
- NTP, National Toxicology Program. Toxicology and carcinogenesis studies of benzophenone (CAS No. 119-61-9) in F344/N rats and B6C3F1 mice (feed studies). Natl. Toxicol. Program. Tech. Rep. Ser. 2006, 533, 1–264. [Google Scholar]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Safety Evaluation of Certain Food Additives and Contaminants; Prepared by the Seventy-Third Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA); WHO: Geneva, Switzerland, 2011. [Google Scholar]
- Abramsson-Zetterberg, L.; Svensson, K. 4-Methylbenzophenone and benzophenone are inactive in the micronucleus assay. Toxicol. Lett. 2011, 201, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, K.; Yamazaki, H.; Nakajima, M.; Yokoi, T. Genotoxic activation of benzophenone and its two metabolites by human cytochrome P450s in SOS/umu assay. Mutat. Res. 2002, 519, 199–204. [Google Scholar] [CrossRef]
- Cuquerella, M.C.; Lhiaubet-Vallet, V.; Cadet, J.; Miranda, M.A. Benzophenone photosensitized DNA damage. Acc. Chem. Res. 2012, 45, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Dumont, E.; Wibowo, M.; Roca-Sanjuán, D.; Garavelli, M.; Assfeld, X.; Monari, A. Resolving the benzophenone DNA-photosensitization mechanism at QM/MM level. J. Phys. Chem. Lett. 2015, 6, 576–580. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Suzuki, T.; Tayama, S. Metabolism and toxicity of benzophenone in isolated rat hepatocytes and estrogenic activity of its metabolites in MCF-7 cells. Toxicology 2000, 156, 27–36. [Google Scholar] [CrossRef]
- Jeon, H.K.; Sarma, S.N.; Kim, Y.J.; Ryu, J.C. Toxicokinetics and metabolisms of benzophenone-type UV filters in rats. Toxicology 2008, 248, 89–95. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Tayama, K. Benzophenone-induced estrogenic potency in ovariectomized rats. Arch. Toxicol. 2002, 76, 727–731. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Tayama, K. Estrogenic potency of benzophenone and its metabolites in juvenile female rats. Arch. Toxicol. 2001, 75, 74–79. [Google Scholar] [CrossRef]
- Suzuki, T.; Kitamura, S.; Khota, R.; Sugihara, K.; Fujimoto, N.; Ohta, S. Estrogenic and antiandrogenic activities of 17 benzophenone derivatives used as UV stabilizers and sunscreens. Toxicol. Appl. Pharm. 2005, 203, 9–17. [Google Scholar] [CrossRef]
- Chen, M.L.; Chen, C.H.; Huang, Y.F.; Chen, H.C.; Chang, J.W. Cumulative dietary risk assessment of benzophenone-type photoinitiators from packaged foodstuffs. Foods 2022, 11, 152. [Google Scholar] [CrossRef]
- Erythropel, H.C.; Maric, M.; Nicell, J.A.; Leask, R.L.; Yargeau, V. Leaching of the plasticizer di(2-ethylhexyl)phthalate (DEHP) from plastic containers and the question of human exposure. Appl. Microbiol. Biotechnol. 2014, 98, 9967–9981. [Google Scholar] [CrossRef] [PubMed]
- Serrano, S.E.; Braun, J.; Trasande, L.; Dills, R.; Sathyanarayana, S. Phthalates and diet: A review of the food monitoring and epidemiology data. Environ. Health 2014, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- ATSDR, Agency for Toxic Substances and Disease Registry. Toxicological Profile for di(2-Ethylhexyl)Phthalate (DEHP); Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2022.
- Fierens, T.; Servaes, K.; Van Holderbeke, M.; Geerts, L.; De Henauw, S.; Sioen, I.; Vanermen, G. Analysis of phthalates in food products and packaging materials sold on the Belgian market. Food Chem. Toxicol. 2012, 50, 2575–2583. [Google Scholar] [CrossRef] [PubMed]
- Sharman, M.; Read, W.A.; Castle, L.; Gilbert, J. Levels of di-(2-ethylhexyl)phthalate and total phthalate esters in milk, cream, butter and cheese. Food Addit. Contam. 1994, 11, 375–385. [Google Scholar] [CrossRef] [PubMed]
- EFSA CEP Panel, European Food Safety Authority, Panel on Food Contact Materials, Enzymes and Processing Aids. Scientific Opinion on the update of the risk assessment of di-butylphthalate(DBP), butyl-benzyl-phthalate (BBP), bis(2-ethylhexyl)phthalate (DEHP), di-isononylphthalate (DINP) anddi-isodecylphthalate (DIDP) for use in food contact materials. EFSA J. 2019, 17, 5838. [Google Scholar]
- Bosgra, S.; Bos, P.M.; Vermeire, T.G.; Luit, R.J.; Slob, W. Probabilistic risk characterization: An example with di(2-ethylhexyl) phthalate. Regul. Toxicol. Pharm. 2005, 43, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Khedr, A. Optimized extraction method for LC-MS determination of bisphenol A, melamine and di(2-ethylhexyl) phthalate in selected soft drinks, syringes, and milk powder. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 930, 98–103. [Google Scholar] [CrossRef]
- NTP, National Toxicology Program. Carcinogenesis bioassay of di(2-ethylhexyl)phthalate (CAS No. 117-81-7) in F344 rats and B6C3F1 mice (feed studies). Natl. Toxicol. Program. Tech. Rep. Ser. 1982, 217, 1–127. [Google Scholar]
- NTP, National Toxicology Program. Toxicology and carcinogenesis studies of di(2-ethylhexyl) phthalate administered in feed to Sprague Dawley (Hsd:Sprague Dawley SD) rats. Natl. Toxicol. Program. Tech. Rep. Ser. 2021, 601, NTP-TR-601. [Google Scholar] [CrossRef]
- Voss, C.; Zerban, H.; Bannasch, P.; Berger, M.R. Lifelong exposure to di-(2-ethylhexyl)-phthalate induces tumors in liver and testes of Sprague-Dawley rats. Toxicology 2005, 206, 359–371. [Google Scholar] [CrossRef]
- Ito, Y.; Yamanoshita, O.; Asaeda, N.; Tagawa, Y.; Lee, C.H.; Aoyama, T.; Ichihara, G.; Furuhashi, K.; Kamijima, M.; Gonzalez, F.J.; et al. Di(2-ethylhexyl)phthalate induces hepatic tumorigenesis through a peroxisome proliferator-activated receptor alpha-independent pathway. J. Occup. Health 2007, 49, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Doull, J.; Cattley, R.; Elcombe, C.; Lake, B.G.; Swenberg, J.; Wilkinson, C.; Williams, G.; van Gemert, M. A cancer risk assessment of di(2-ethylhexyl)phthalate: Application of the new U.S. EPA Risk Assessment Guidelines. Regul. Toxicol. Pharm. 1999, 29, 327–357. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.M.; Ohshima, M.; Lynch, P.; Riggs, C. Di(2-ethylhexyl)phthalate but not phenobarbital promotes N-nitrosodiethylamine-initiated hepatocellular proliferative lesions after short-term exposure in male B6C3F1 mice. Cancer Lett. 1984, 24, 49–55. [Google Scholar] [CrossRef]
- Diwan, B.A.; Ward, J.M.; Rice, J.M.; Colburn, N.H.; Spangler, E.F. Tumor-promoting effects of di(2-ethylhexyl)phthalate in JB6 mouse epidermal cells and mouse skin. Carcinogenesis 1985, 6, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, B.E.; Bermudez, E.; Smith-Oliver, T.; Earle, L.; Cattley, R.; Martin, J.; Popp, J.A.; Strom, S.; Jirtle, R.; Michalopoulos, G. Lack of genotoxic activity of di(2-ethylhexyl)phthalate (DEHP) in rat and human hepatocytes. Carcinogenesis 1984, 5, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, J.C. DEHP: Genotoxicity and potential carcinogenic mechanisms-a review. Mutat. Res. 2012, 751, 82–157. [Google Scholar] [CrossRef]
- Erkekoglu, P.; Kocer-Gumusel, B. Genotoxicity of phthalates. Toxicol. Mech. Methods 2014, 24, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Karabulut, G.; Barlas, N. Genotoxic, histologic, immunohistochemical, morphometric and hormonal effects of di-(2-ethylhexyl)-phthalate (DEHP) on reproductive systems in pre-pubertal male rats. Toxicol. Res. 2018, 7, 859–873. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, L.; Ge, L.; Chen, M.; Yang, G.; Ji, F.; Zhong, L.; Guan, Y.; Liu, X. Oxidative DNA damage induced by di-(2-ethylhexyl) phthalate in HEK-293 cell line. Environ. Toxicol. Pharm. 2015, 39, 1099–1106. [Google Scholar] [CrossRef]
- Erkekoglu, P.; Rachidi, W.; Yuzugullu, O.G.; Giray, B.; Favier, A.; Ozturk, M.; Hincal, F. Evaluation of cytotoxicity and oxidative DNA damaging effects of di(2-ethylhexyl)-phthalate (DEHP) and mono(2-ethylhexyl)-phthalate (MEHP) on MA-10 Leydig cells and protection by selenium. Toxicol. Appl. Pharm. 2010, 248, 52–62. [Google Scholar] [CrossRef]
- She, Y.; Jiang, L.; Zheng, L.; Zuo, H.; Chen, M.; Sun, X.; Li, Q.; Geng, C.; Yang, G.; Jiang, L.; et al. The role of oxidative stress in DNA damage in pancreatic β cells induced by di-(2-ethylhexyl) phthalate. Chem. Biol. Interact. 2017, 265, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Takagi, A.; Sai, K.; Umemura, T.; Hasegawa, R.; Kurokawa, Y. Significant increase of 8-hydroxydeoxyguanosine in liver DNA of rats following short-term exposure to the peroxisome proliferators di(2-ethylhexyl)phthalate and di(2-ethylhexyl)adipate. Jpn. J. Cancer Res. 1990, 81, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Takagi, A.; Sai, K.; Umemura, T.; Hasegawa, R.; Kurokawa, Y. Relationship between hepatic peroxisome proliferation and 8-hydroxydeoxyguanosine formation in liver DNA of rats following long-term exposure to three peroxisome proliferators; di(2-ethylhexyl) phthalate, aluminium clofibrate and simfibrate. Cancer Lett. 1990, 53, 33–38. [Google Scholar] [CrossRef]
- von Däniken, A.; Lutz, W.K.; Jäckh, R.; Schlatter, C. Investigation of the potential for binding of Di(2-ethylhexyl) phthalate (DEHP) and Di(2-ethylhexyl) adipate (DEHA) to liver DNA in vivo. Toxicol. Appl. Pharm. 1984, 73, 373–387. [Google Scholar] [CrossRef]
- Lutz, W.K. Investigation of the potential for binding of di(2-ethylhexyl) phthalate (DEHP) to rat liver DNA in vivo. Environ. Health Perspect. 1986, 65, 267–269. [Google Scholar] [CrossRef]
- Gupta, R.C.; Goel, S.K.; Earley, K.; Singh, B.; Reddy, J.K. 32P-postlabeling analysis of peroxisome proliferator-DNA adduct formation in rat liver in vivo and hepatocytes in vitro. Carcinogenesis 1985, 6, 933–936. [Google Scholar] [CrossRef]
- Koch, H.M.; Preuss, R.; Angerer, J. Di(2-ethylhexyl)phthalate (DEHP): Human metabolism and internal exposure—An update and latest results. Int. J. 2006, 29, 155–165, discussion 181-155. [Google Scholar] [CrossRef]
- Silva, M.J.; Samandar, E.; Preau, J.L., Jr.; Needham, L.L.; Calafat, A.M. Urinary oxidative metabolites of di(2-ethylhexyl) phthalate in humans. Toxicology 2006, 219, 22–32. [Google Scholar] [CrossRef]
- Frederiksen, H.; Skakkebaek, N.E.; Andersson, A.M. Metabolism of phthalates in humans. Mol. Nutr. Food Res. 2007, 51, 899–911. [Google Scholar] [CrossRef]
- Ito, Y.; Yokota, H.; Wang, R.; Yamanoshita, O.; Ichihara, G.; Wang, H.; Kurata, Y.; Takagi, K.; Nakajima, T. Species differences in the metabolism of di(2-ethylhexyl) phthalate (DEHP) in several organs of mice, rats, and marmosets. Arch. Toxicol. 2005, 79, 147–154. [Google Scholar] [CrossRef]
- Ito, Y.; Kamijima, M.; Hasegawa, C.; Tagawa, M.; Kawai, T.; Miyake, M.; Hayashi, Y.; Naito, H.; Nakajima, T. Species and inter-individual differences in metabolic capacity of di(2-ethylhexyl)phthalate (DEHP) between human and mouse livers. Environ. Health Prev. Med. 2014, 19, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Kamijima, M.; Nakajima, T. Di(2-ethylhexyl) phthalate-induced toxicity and peroxisome proliferator-activated receptor alpha: A review. Environ. Health Prev. Med. 2019, 24, 47. [Google Scholar] [CrossRef] [PubMed]
- Melnick, R.L. Is peroxisome proliferation an obligatory precursor step in the carcinogenicity of di(2-ethylhexyl)phthalate (DEHP)? Environ. Health Perspect. 2001, 109, 437–442. [Google Scholar] [CrossRef]
- Corton, J.C.; Peters, J.M.; Klaunig, J.E. The PPARα-dependent rodent liver tumor response is not relevant to humans: Addressing misconceptions. Arch. Toxicol. 2018, 92, 83–119. [Google Scholar] [CrossRef]
- Rusyn, I.; Corton, J.C. Mechanistic considerations for human relevance of cancer hazard of di(2-ethylhexyl) phthalate. Mutat. Res. 2012, 750, 141–158. [Google Scholar] [CrossRef]
- Shelby, M.D. NTP-CERHR monograph on the potential human reproductive and developmental effects of di (2-ethylhexyl) phthalate (DEHP). Ntp. Cerhr. Mon. 2006, 5, 1–13. [Google Scholar]
- Schecter, A.; Lorber, M.; Guo, Y.; Wu, Q.; Yun, S.H.; Kannan, K.; Hommel, M.; Imran, N.; Hynan, L.S.; Cheng, D.; et al. Phthalate concentrations and dietary exposure from food purchased in New York State. Environ. Health Perspect. 2013, 121, 473–494. [Google Scholar] [CrossRef]
- Qu, J.; Xia, W.; Qian, X.; Wu, Y.; Li, J.; Wen, S.; Xu, S. Geographic distribution and time trend of human exposure of Di(2-ethylhexyl) phthalate among different age groups based on global biomonitoring data. Chemosphere 2022, 287, 132115. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.; Deoraj, A.; Felty, Q.; Roy, D. Environmental estrogen-like endocrine disrupting chemicals and breast cancer. Mol. Cell Endocrinol. 2017, 457, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Stickney, J.A.; Sager, S.L.; Clarkson, J.R.; Smith, L.A.; Locey, B.J.; Bock, M.J.; Hartung, R.; Olp, S.F. An updated evaluation of the carcinogenic potential of 1,4-dioxane. Regul. Toxicol. Pharm. 2003, 38, 183–195. [Google Scholar] [CrossRef]
- ATSDR, Agency for Toxic Substances and Disease Registry. Toxicological Profile for 1,4-Dioxane; Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2012.
- SCF, Scientific Committee on Food. Opinion of the Scientific Committee on Food on Impurities of 1,4-Dioxane, 2-Chloroethanol and Mono- and Diethylene Glycol in Currently Permitted Food Additives and in Proposed Use of Ethyl Hydroxyethyl Cellulose in Gluten-Free Bread; SCF/CS/ADD/EMU/198 Final; Scientific Committee on Food: Brussels, Belgium, 2002. [Google Scholar]
- Nishimura, T.; Iizuka, S.; Kibune, N.; Ando, M. Study of 1,4-dioxane intake in the total diet using the market-basket method. J. Health Sci. 2004, 50, 101–107. [Google Scholar] [CrossRef]
- NCI, National Cancer Institute. Bioassay of 1,4-dioxane for possible carcinogenicity. Natl. Cancer Inst. Carcinog. Tech. Rep. Ser. 1978, 80, 1–123. [Google Scholar]
- Kano, H.; Umeda, Y.; Kasai, T.; Sasaki, T.; Matsumoto, M.; Yamazaki, K.; Nagano, K.; Arito, H.; Fukushima, S. Carcinogenicity studies of 1,4-dioxane administered in drinking-water to rats and mice for 2 years. Food Chem. Toxicol. 2009, 47, 2776–2784. [Google Scholar] [CrossRef]
- Kano, H.; Umeda, Y.; Saito, M.; Senoh, H.; Ohbayashi, H.; Aiso, S.; Yamazaki, K.; Nagano, K.; Fukushima, S. Thirteen-week oral toxicity of 1,4-dioxane in rats and mice. J. Toxicol. Sci. 2008, 33, 141–153. [Google Scholar] [CrossRef]
- Lundberg, I.; Högberg, J.; Kronevi, T.; Holmberg, B. Three industrial solvents investigated for tumor promoting activity in the rat liver. Cancer Lett. 1987, 36, 29–33. [Google Scholar] [CrossRef]
- Rosenkranz, H.S.; Klopman, G. 1,4-Dioxane: Prediction of in vivo clastogenicity. Mutat. Res. 1992, 280, 245–251. [Google Scholar] [CrossRef]
- Mirkova, E.T. Activity of the rodent carcinogen 1,4-dioxane in the mouse bone marrow micronucleus assay. Mutat. Res. 1994, 322, 142–144. [Google Scholar] [CrossRef]
- Kitchin, K.T.; Brown, J.L. Is 1,4-dioxane a genotoxic carcinogen? Cancer Lett. 1990, 53, 67–71. [Google Scholar] [CrossRef]
- Morita, T.; Hayashi, M. 1,4-Dioxane is not mutagenic in five in vitro assays and mouse peripheral blood micronucleus assay, but is in mouse liver micronucleus assay. Environ. Mol. Mutagen. 1998, 32, 269–280. [Google Scholar] [CrossRef]
- Itoh, S.; Hattori, C. In vivo genotoxicity of 1,4-dioxane evaluated by liver and bone marrow micronucleus tests and Pig-a assay in rats. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2019, 837, 8–14. [Google Scholar] [CrossRef]
- Gi, M.; Fujioka, M.; Kakehashi, A.; Okuno, T.; Masumura, K.; Nohmi, T.; Matsumoto, M.; Omori, M.; Wanibuchi, H.; Fukushima, S. In vivo positive mutagenicity of 1,4-dioxane and quantitative analysis of its mutagenicity and carcinogenicity in rats. Arch. Toxicol. 2018, 92, 3207–3221. [Google Scholar] [CrossRef]
- Charkoftaki, G.; Golla, J.P.; Santos-Neto, A.; Orlicky, D.J.; Garcia-Milian, R.; Chen, Y.; Rattray, N.J.W.; Cai, Y.; Wang, Y.; Shearn, C.T.; et al. Identification of dose-dependent DNA damage and repair responses from subchronic exposure to 1,4-dioxane in mice using a systems analysis approach. Toxicol. Sci. 2021, 183, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.K.; Thilagar, A.K.; Eastmond, D.A. Chromosome breakage is primarily responsible for the micronuclei induced by 1,4-dioxane in the bone marrow and liver of young CD-1 mice. Mutat. Res. 2005, 586, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Totsuka, Y.; Maesako, Y.; Ono, H.; Nagai, M.; Kato, M.; Gi, M.; Wanibuchi, H.; Fukushima, S.; Shiizaki, K.; Nakagama, H. Comprehensive analysis of DNA adducts (DNA adductome analysis) in the liver of rats treated with 1,4-dioxane. Proc. Jpn. Acad. Ser. B 2020, 96, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Göen, T.; von Helden, F.; Eckert, E.; Knecht, U.; Drexler, H.; Walter, D. Metabolism and toxicokinetics of 1,4-dioxane in humans after inhalational exposure at rest and under physical stress. Arch. Toxicol. 2016, 90, 1315–1324. [Google Scholar] [CrossRef]
- Nannelli, A.; De Rubertis, A.; Longo, V.; Gervasi, P.G. Effects of dioxane on cytochrome P450 enzymes in liver, kidney, lung and nasal mucosa of rat. Arch. Toxicol. 2005, 79, 74–82. [Google Scholar] [CrossRef]
- Lafranconi, M.; Budinsky, R.; Corey, L.; Klapacz, J.; Crissman, J.; LeBaron, M.; Golden, R.; Pleus, R. A 90-day drinking water study in mice to characterize early events in the cancer mode of action of 1,4-dioxane. Regul. Toxicol. Pharm. 2021, 119, 104819. [Google Scholar] [CrossRef]
- Dourson, M.L.; Higginbotham, J.; Crum, J.; Burleigh-Flayer, H.; Nance, P.; Forsberg, N.D.; Lafranconi, M.; Reichard, J. Update: Mode of action (MOA) for liver tumors induced by oral exposure to 1,4-dioxane. Regul Toxicol Pharm. 2017, 88, 45–55. [Google Scholar] [CrossRef]
- Dourson, M.; Reichard, J.; Nance, P.; Burleigh-Flayer, H.; Parker, A.; Vincent, M.; McConnell, E.E. Mode of action analysis for liver tumors from oral 1,4-dioxane exposures and evidence-based dose response assessment. Regul. Toxicol. Pharm. 2014, 68, 387–401. [Google Scholar] [CrossRef]
- Chappell, G.A.; Heintz, M.M.; Haws, L.C. Transcriptomic analyses of livers from mice exposed to 1,4-dioxane for up to 90 days to assess potential mode(s) of action underlying liver tumor development. Curr. Res. Toxicol. 2021, 2, 30–41. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Y.; Charkoftaki, G.; Orlicky, D.J.; Davidson, E.; Wan, F.; Ginsberg, G.; Thompson, D.C.; Vasiliou, V. Oxidative stress and genotoxicity in 1,4-dioxane liver toxicity as evidenced in a mouse model of glutathione deficiency. Sci. Total Environ. 2022, 806, 150703. [Google Scholar] [CrossRef]
- Goldsworthy, T.L.; Monticello, T.M.; Morgan, K.T.; Bermudez, E.; Wilson, D.M.; Jäckh, R.; Butterworth, B.E. Examination of potential mechanisms of carcinogenicity of 1,4-dioxane in rat nasal epithelial cells and hepatocytes. Arch. Toxicol. 1991, 65, 1–9. [Google Scholar] [CrossRef]
- FDA, Food and Drug Administration. Indirect Food Additives: Adhesives and Components of Coatings; Food and Drug Administration: Silver Spring, MD, USA, 1998; pp. 350–351.
- Nishimura, T.; Iizuka, S.; Kibune, N.; Ando, M.; Magara, Y. Study of 1,4-dioxane intake in the total diet. J. Health Sci. 2005, 51, 514–517. [Google Scholar] [CrossRef]
- EFSA CONTAM Panel, European Food Safety Authority, Panel on Contaminants in the Food Chain. Scientific Opinion on the evaluation of the substances currently on the list in the annex to Commission Directive 96/3/EC as acceptable previous cargoes for edible fats and oils—Part II of III. EFSA J. 2012, 10, 2703. [Google Scholar] [CrossRef]
- Johnson, W., Jr. Safety assessment of MIBK (methyl isobutyl ketone). Int. J. Toxicol. 2004, 23 (Suppl. S1), 29–57. [Google Scholar] [CrossRef] [PubMed]
- Api, A.M.; Belsito, D.; Botelho, D.; Bruze, M.; Burton, G.A., Jr.; Buschmann, J.; Dagli, M.L.; Date, M.; Dekant, W.; Deodhar, C.; et al. RIFM fragrance ingredient safety assessment, 4-methyl-2-pentanone, CAS Registry Number 108-10-1. Food Chem. Toxicol. 2019, 130 (Suppl. S1), 110587. [Google Scholar] [CrossRef]
- Stout, M.D.; Herbert, R.A.; Kissling, G.E.; Suarez, F.; Roycroft, J.H.; Chhabra, R.S.; Bucher, J.R. Toxicity and carcinogenicity of methyl isobutyl ketone in F344N rats and B6C3F1 mice following 2-year inhalation exposure. Toxicology 2008, 244, 209–219. [Google Scholar] [CrossRef]
- NTP, National Toxicology Program. Toxicology and carcinogenesis studies of methyl isobutyl ketone (Cas No. 108-10-1) in F344/N rats and B6C3F1 mice (inhalation studies). Natl. Toxicol. Program. Tech. Rep. Ser. 2007, 538, 1–236. [Google Scholar]
- Brooks, T.M.; Meyer, A.L.; Hutson, D.H. The genetic toxicology of some hydrocarbon and oxygenated solvents. Mutagenesis 1988, 3, 227–232. [Google Scholar] [CrossRef]
- O’Donoghue, J.L.; Haworth, S.R.; Curren, R.D.; Kirby, P.E.; Lawlor, T.; Moran, E.J.; Phillips, R.D.; Putnam, D.L.; Rogers-Back, A.M.; Slesinski, R.S.; et al. Mutagenicity studies on ketone solvents: Methyl ethyl ketone, methyl isobutyl ketone, and isophorone. Mutat. Res. 1988, 206, 149–161. [Google Scholar] [CrossRef]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Safety Evaluation of Certain Food Additives; Prepared by the Fifty-First Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA); WHO: Geneva, Switzerland, 1999. [Google Scholar]
- Granvil, C.P.; Sharkawi, M.; Plaa, G.L. Metabolic fate of methyl n-butyl ketone, methyl isobutyl ketone and their metabolites in mice. Toxicol. Lett. 1994, 70, 263–267. [Google Scholar] [CrossRef]
- Duguay, A.B.; Plaa, G.L. Plasma concentrations in methyl isobutyl ketone-potentiated experimental cholestasis after inhalation or oral administration. Fundam. Appl. Toxicol. 1993, 21, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Duguay, A.B.; Plaa, G.L. Tissue concentrations of methyl isobutyl ketone, methyl n-butyl ketone and their metabolites after oral or inhalation exposure. Toxicol. Lett. 1995, 75, 51–58. [Google Scholar] [CrossRef]
- Vézina, M.; Kobusch, A.B.; du Souich, P.; Greselin, E.; Plaa, G.L. Potentiation of chloroform-induced hepatotoxicity by methyl isobutyl ketone and two metabolites. Can. J. Physiol. Pharm. 1990, 68, 1055–1061. [Google Scholar] [CrossRef]
- Gingell, R.; Régnier, J.F.; Wilson, D.M.; Guillaumat, P.O.; Appelqvist, T. Comparative metabolism of methyl isobutyl carbinol and methyl isobutyl ketone in male rats. Toxicol. Lett. 2003, 136, 199–204. [Google Scholar] [CrossRef]
- Raymond, P.; Plaa, G.L. Ketone potentiation of haloalkane-induced hepato- and nephrotoxicity. I. Dose-response relationships. J. Toxicol. Environ. Health 1995, 45, 465–480. [Google Scholar] [CrossRef]
- Raymond, P.; Plaa, G.L. Ketone potentiation of haloalkane-induced hepato- and nephrotoxicity. II. Implication of monooxygenases. J. Toxicol. Environ. Health 1995, 46, 317–328. [Google Scholar] [CrossRef]
- Borghoff, S.J.; Poet, T.S.; Green, S.; Davis, J.; Hughes, B.; Mensing, T.; Sarang, S.S.; Lynch, A.M.; Hard, G.C. Methyl isobutyl ketone exposure-related increases in specific measures of α2u-globulin (α2u) nephropathy in male rats along with in vitro evidence of reversible protein binding. Toxicology 2015, 333, 1–13. [Google Scholar] [CrossRef]
- Borghoff, S.J.; Hard, G.C.; Berdasco, N.M.; Gingell, R.; Green, S.M.; Gulledge, W. Methyl isobutyl ketone (MIBK) induction of alpha2u-globulin nephropathy in male, but not female rats. Toxicology 2009, 258, 131–138. [Google Scholar] [CrossRef]
- Vézina, M.; Plaa, G.L. Potentiation by methyl isobutyl ketone of the cholestasis induced in rats by a manganese-bilirubin combination or manganese alone. Toxicol. Appl. Pharm. 1987, 91, 477–483. [Google Scholar] [CrossRef]
- Joseph, L.D.; Yousef, I.M.; Plaa, G.L.; Sharkawi, M. Potentiation of lithocholic-acid-induced cholestasis by methyl isobutyl ketone. Toxicol. Lett. 1992, 61, 39–47. [Google Scholar] [CrossRef]
- Hughes, B.J.; Thomas, J.; Lynch, A.M.; Borghoff, S.J.; Green, S.; Mensing, T.; Sarang, S.S.; LeBaron, M.J. Methyl isobutyl ketone-induced hepatocellular carcinogenesis in B6C3F(1) mice: A constitutive androstane receptor (CAR)-mediated mode of action. Regul. Toxicol. Pharm. 2016, 81, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Grober, E.; Schaumburg, H.H. Occupational exposure to methyl isobutyl ketone causes lasting impairment in working memory. Neurology 2000, 54, 1853–1855. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.M.; Bernhard, R.A. Effect of nitrogen source on pyrazine formation. J. Agric. Food Chem. 1988, 36, 123–129. [Google Scholar] [CrossRef]
- Wu, X.; Huang, M.; Kong, F.; Yu, S. Short communication: Study on the formation of 2-methylimidazole and 4-methylimidazole in the Maillard reaction. J. Dairy Sci. 2015, 98, 8565–8571. [Google Scholar] [CrossRef]
- Chan, P.C. NTP technical report on the toxicity studies of 2- and 4-Methylimidazole (CAS No. 693-98-1 and 822-36-6) administered in feed to F344/N rats and B6C3F1 mice. Toxic Rep. Ser. 2004, 67, 1-g12. [Google Scholar]
- Schlee, C.; Markova, M.; Schrank, J.; Laplagne, F.; Schneider, R.; Lachenmeier, D.W. Determination of 2-methylimidazole, 4-methylimidazole and 2-acetyl-4-(1,2,3,4-tetrahydroxybutyl)imidazole in caramel colours and cola using LC/MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 927, 223–226. [Google Scholar] [CrossRef]
- Jacobs, G.; Voorspoels, S.; Vloemans, P.; Fierens, T.; Van Holderbeke, M.; Cornelis, C.; Sioen, I.; De Maeyer, M.; Vinkx, C.; Vanermen, G. Caramel colour and process by-products in foods and beverages: Part I—Development of a UPLC-MS/MS isotope dilution method for determination of 2-acetyl-4-(1,2,3,4-tetrahydroxybutyl)imidazole (THI), 4-methylimidazole (4-MEI) and 2-methylimidazol (2-MEI). Food Chem. 2018, 255, 348–356. [Google Scholar] [CrossRef]
- Choi, S.J.; Jung, M.Y. Simple and fast sample preparation followed by Gas Chromatography-Tandem Mass Spectrometry (GC-MS/MS) for the analysis of 2- and 4-methylimidazole in cola and dark beer. J. Food Sci. 2017, 82, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Toxicological Evaluation of Certain Food Additives and Contaminants; Prepared by the Twenty-Ninth Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA); WHO: Geneva, Switzerland, 1985. [Google Scholar]
- EFSA ANS Panel, European Food Safety Authority, Panel on Food Additives and Nutrient Sources added to Food. Scientific Opinion on the re-evaluation of caramel colours (E 150a,b,c,d) as food additives. EFSA J. 2011, 9, 2004. [Google Scholar] [CrossRef]
- Vollmuth, T.A. Caramel color safety—An update. Food Chem. Toxicol. 2018, 111, 578–596. [Google Scholar] [CrossRef]
- Morgan, S.E.; Edwards, W.C. Pilot studies in cattle and mice to determine the presence of 4-methylimidazole in milk after oral ingestion. Vet. Hum. Toxicol. 1986, 28, 240–242. [Google Scholar]
- Müller, L.; Sivertsen, T.; Langseth, W. Ammoniated forage poisoning: Concentrations of alkylimidazoles in ammoniated forage and in milk, plasma and urine in sheep and cow. Acta Vet. Scand. 1998, 39, 511–514. [Google Scholar] [CrossRef]
- Mottier, P.; Mujahid, C.; Tarres, A.; Bessaire, T.; Stadler, R.H. Process-induced formation of imidazoles in selected foods. Food Chem. 2017, 228, 381–387. [Google Scholar] [CrossRef] [PubMed]
- NTP, National Toxicology Program. Toxicology and carcinogenesis studies of 4-methylimidazole (Cas No. 822-36-6) in F344/N rats and B6C3F1 mice (feed studies). Natl. Toxicol. Program. Tech. Rep. Ser. 2007, 535, 1–274. [Google Scholar]
- Chan, P.C.; Sills, R.C.; Kissling, G.E.; Nyska, A.; Richter, W. Induction of thyroid and liver tumors by chronic exposure to 2-methylimidazole in F344/N rats and B6C3F1 mice. Arch. Toxicol. 2008, 82, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.C.; Hill, G.D.; Kissling, G.E.; Nyska, A. Toxicity and carcinogenicity studies of 4-methylimidazole in F344/N rats and B6C3F1 mice. Arch. Toxicol. 2008, 82, 45–53. [Google Scholar] [CrossRef]
- NTP, National Toxicology Program. Toxicology and carcinogensis studies of 2-methylimidazole (Cas No. 693-98-1) in B6C3F1 mice (feed studies). Natl. Toxicol. Program. Tech. Rep. Ser. 2004, 516, 1–292. [Google Scholar]
- Chan, P.; Mahler, J.; Travlos, G.; Nyska, A.; Wenk, M. Induction of thyroid lesions in 14-week toxicity studies of 2 and 4-methylimidazole in Fischer 344/N rats and B6C3F1 mice. Arch. Toxicol. 2006, 80, 169–180. [Google Scholar] [CrossRef]
- Beevers, C.; Adamson, R.H. Evaluation of 4-methylimidazole, in the Ames/Salmonella test using induced rodent liver and lung S9. Environ. Mol. Mutagen. 2016, 57, 51–57. [Google Scholar] [CrossRef]
- Morita, T.; Uneyama, C. Genotoxicity assessment of 4-methylimidazole: Regulatory perspectives. Genes Environ. 2016, 38, 20. [Google Scholar] [CrossRef]
- Celik, R.; Topaktas, M. Genotoxic effects of 4-methylimidazole on human peripheral lymphocytes in vitro. Drug Chem. Toxicol. 2018, 41, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.D.; Reichelderfer, D.; Zutshi, A.; Graves, S.; Walters, D.; Smith, C. Toxicokinetics of 2-methylimidazole in male and female F344 rats. J. Toxicol. Environ. Health Part A 2002, 65, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Fennell, T.R.; Watson, S.L.; Dhungana, S.; Snyder, R.W. Metabolism of 4-methylimidazole in Fischer 344 rats and B6C3F1 mice. Food Chem. Toxicol 2019, 123, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.M.; Griffin, R.J.; Burka, L.T.; Matthews, H.B. Disposition of 2-methylimidazole in rats. J. Toxicol. Environ. Health A 1998, 54, 121–132. [Google Scholar] [CrossRef]
- Dalvie, D.K.; Kalgutkar, A.S.; Khojasteh-Bakht, S.C.; Obach, R.S.; O’Donnell, J.P. Biotransformation reactions of five-membered aromatic heterocyclic rings. Chem. Res. Toxicol. 2002, 15, 269–299. [Google Scholar] [CrossRef] [PubMed]
- Ohta, K.; Fukasawa, Y.; Yamaguchi, J.; Kohno, Y.; Fukushima, K.; Suwa, T.; Awazu, S. Retention mechanism of imidazoles in connective tissue. IV. Identification of a nucleophilic imidazolone metabolite in rats. Biol. Pharm. Bull. 1998, 21, 1334–1337. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Capen, C.C. Mechanisms of chemical injury of thyroid gland. Prog. Clin. Biol Res. 1994, 387, 173–191. [Google Scholar]
- Williams, G.M.; Iatropoulos, M.J. Alteration of liver cell function and proliferation: Differentiation between adaptation and toxicity. Toxicol. Pathol. 2002, 30, 41–53. [Google Scholar] [CrossRef]
- Borghoff, S.J.; Fitch, S.E.; Black, M.B.; McMullen, P.D.; Andersen, M.E.; Chappell, G.A. A systematic approach to evaluate plausible modes of actions for mouse lung tumors in mice exposed to 4-methylimidozole. Regul. Toxicol. Pharm. 2021, 124, 104977. [Google Scholar] [CrossRef]
- Brusick, D.; Aardema, M.J.; Allaben, W.T.; Kirkland, D.J.; Williams, G.; Llewellyn, G.C.; Parker, J.M.; Rihner, M.O. A weight of evidence assessment of the genotoxic potential of 4-methylimidazole as a possible mode of action for the formation of lung tumors in exposed mice. Food Chem. Toxicol. 2020, 145, 111652. [Google Scholar] [CrossRef] [PubMed]
- Folmer, D.E.; Doell, D.L.; Lee, H.S.; Noonan, G.O.; Carberry, S.E. A U.S. population dietary exposure assessment for 4-methylimidazole (4-MEI) from foods containing caramel colour and from formation of 4-MEI through the thermal treatment of food. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2018, 35, 1890–1910. [Google Scholar] [CrossRef] [PubMed]
- Fierens, T.; Van Holderbeke, M.; Cornelis, C.; Jacobs, G.; Sioen, I.; De Maeyer, M.; Vinkx, C.; Vanermen, G. Caramel colour and process contaminants in foods and beverages: Part II—Occurrence data and exposure assessment of 2-acetyl-4-(1,2,3,4-tetrahydroxybutyl)imidazole (THI) and 4-methylimidazole (4-MEI) in Belgium. Food Chem. 2018, 255, 372–379. [Google Scholar] [CrossRef] [PubMed]
- JECFA, Joint FAO/WHO Expert Committee on Food Additives. Combined Compendium of Food Additive Specification; Caramel Colours; Monograph 11; WHO: Geneva, Switzerland, 2011. [Google Scholar]
- Yaylayan, V.A. Precursors, formation and determination of furan in food. J. Verbrauch. Lebensm. 2006, 1, 5–9. [Google Scholar] [CrossRef]
- EFSA CONTAM Panel, European Food Safety Authority, Panel on Contaminants in the Food Chain. Scientific opinion on the risks for publichealth related to the presence of furan and methylfurans in food. EFSA J. 2017, 15, 5005. [Google Scholar] [CrossRef]
- Moro, S.; Chipman, J.K.; Wegener, J.W.; Hamberger, C.; Dekant, W.; Mally, A. Furan in heat-treated foods: Formation, exposure, toxicity, and aspects of risk assessment. Mol. Nutr. Food Res. 2012, 56, 1197–1211. [Google Scholar] [CrossRef]
- FDA, Food and Drug Administration. Exploratory Data on Furan in Food: Individual Food Products; Food and Drug Administration: Silver Spring, MD, USA, 2009.
- Van Lancker, F.; Adams, A.; Owczarek, A.; De Meulenaer, B.; De Kimpe, N. Impact of various food ingredients on the retention of furan in foods. Mol. Nutr. Food Res. 2009, 53, 1505–1511. [Google Scholar] [CrossRef]
- NTP, National Toxicology Program. Toxicology and Carcinogenesis Studies of Furan (CAS No. 110-00-9) in F344 Rats and B6C3F1 Mice(Gavage Studies); 0888-8051 (Print); National Toxicology Program: Research Triangle, NC, USA, 1993; pp. 1–286.
- IARC, International Agency for Research on Cancer. Dry cleaning, some chlorinated solvents and other industrial chemicals. In IARC Monographs on the Evaluation of Carcinogenic Risk to Humans; International Agency for Research on Cancer: Lyon, France, 1995; Volume 63. [Google Scholar]
- Von Tungeln, L.S.; Walker, N.J.; Olson, G.R.; Mendoza, M.C.; Felton, R.P.; Thorn, B.T.; Marques, M.M.; Pogribny, I.P.; Doerge, D.R.; Beland, F.A. Low dose assessment of the carcinogenicity of furan in male F344/N Nctr rats in a 2-year gavage study. Food Chem. Toxicol. 2017, 99, 170–181. [Google Scholar] [CrossRef]
- Moser, G.J.; Foley, J.; Burnett, M.; Goldsworthy, T.L.; Maronpot, R. Furan-induced dose-response relationships for liver cytotoxicity, cell proliferation, and tumorigenicity (furan-induced liver tumorigenicity). Exp. Toxicol. Pathol. 2009, 61, 101–111. [Google Scholar] [CrossRef]
- Durling, L.; Svensson, K.; Abramssonzetterberg, L. Furan is not genotoxic in the micronucleus assay in vivo or in vitro. Toxicol. Lett. 2007, 169, 43–50. [Google Scholar] [CrossRef]
- Wilson, D.M.; Goldsworthy, T.L.; Popp, J.A.; Butterworth, B.E. Evaluation of genotoxicity, pathological lesions, and cell proliferation in livers of rats and mice treated with furan. Environ. Mol. Mutagen. 1992, 19, 209–222. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, L.P.; Ding, W.; Dobrovolsky, V.N.; Shaddock, J.G., Jr.; Mittelstaedt, R.A.; Doerge, D.R.; Heflich, R.H. Genotoxicity of furan in Big Blue rats. Mutat. Res. 2012, 742, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Petibone, D.M.; Latendresse, J.R.; Pearce, M.G.; Muskhelishvili, L.; White, G.A.; Chang, C.W.; Mittelstaedt, R.A.; Shaddock, J.G.; McDaniel, L.P.; et al. In vivo genotoxicity of furan in F344 rats at cancer bioassay doses. Toxicol. Appl. Pharm. 2012, 261, 164–171. [Google Scholar] [CrossRef]
- Jeffrey, A.M.; Brunnemann, K.D.; Duan, J.D.; Schlatter, J.; Williams, G.M. Furan induction of DNA cross-linking and strand breaks in turkey fetal liver in comparison to 1,3-propanediol. Food Chem. Toxicol. 2012, 50, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.A.; Naruko, K.C.; Predecki, D.P. A reactive metabolite of furan, cis-2-butene-1,4-dial, is mutagenic in the Ames assay. Chem. Res. Toxicol. 2000, 13, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Byrns, M.C.; Vu, C.C.; Neidigh, J.W.; Abad, J.-L.; Jones, R.A.; Peterson, L.A. Detection of DNA adducts derived from the reactive metabolite of furan, cis-2-butene-1,4-dial. Chem. Res. Toxicol. 2006, 19, 414–420. [Google Scholar] [CrossRef]
- Churchwell, M.I.; Scheri, R.C.; Von Tungeln, L.S.; Gamboa da Costa, G.; Beland, F.A.; Doerge, D.R. Evaluation of serum and liver toxicokinetics for furan and liver DNA adduct formation in male Fischer 344 rats. Food Chem. Toxicol. 2015, 86, 1–8. [Google Scholar] [CrossRef]
- Neuwirth, C.; Mosesso, P.; Pepe, G.; Fiore, M.; Malfatti, M.; Turteltaub, K.; Dekant, W.; Mally, A. Furan carcinogenicity: DNA binding and genotoxicity of furan in rats in vivo. Mol. Nutr. Food Res. 2012, 56, 1363–1374. [Google Scholar] [CrossRef]
- Kedderis, G.L.; Carfagna, M.A.; Held, S.D.; Batra, R.; Murphy, J.E.; Gargas, M.L. Kinetic analysis of furan biotransformation by F-344 rats in vivo and in vitro. Toxicol. Appl. Pharm. 1993, 123, 274–282. [Google Scholar] [CrossRef]
- Chen, L.J.; Hecht, S.S.; Peterson, L.A. Identification of cis-2-butene-1,4-dial as a microsomal metabolite of furan. Chem. Res. Toxicol. 1995, 8, 903–906. [Google Scholar] [CrossRef]
- Byrns, M.C.; Predecki, D.P.; Peterson, L.A. Characterization of nucleoside adducts of cis-2-butene-1,4-dial, a reactive metabolite of furan. Chem. Res. Toxicol. 2002, 15, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.T.; De Luca, G.; Palma, N.; Leopardi, P.; Degan, P.; Cinelli, S.; Pepe, G.; Mosesso, P.; Di Carlo, E.; Sorrentino, C.; et al. Oxidative stress, mutations and chromosomal aberrations induced by in vitro and in vivo exposure to furan. Int. J. Mol. Sci. 2021, 22, 9687. [Google Scholar] [CrossRef] [PubMed]
- De Conti, A.; Kobets, T.; Escudero-Lourdes, C.; Montgomery, B.; Tryndyak, V.; Beland, F.A.; Doerge, D.R.; Pogribny, I.P. Dose- and time-dependent epigenetic changes in the livers of Fisher 344 rats exposed to furan. Toxicol. Sci. 2014, 139, 371–380. [Google Scholar] [CrossRef] [PubMed]
- de Conti, A.; Kobets, T.; Tryndyak, V.; Burnett, S.D.; Han, T.; Fuscoe, J.C.; Beland, F.A.; Doerge, D.R.; Pogribny, I.P. Persistence of furan-induced epigenetic aberrations in the livers of F344 rats. Toxicol. Sci. 2015, 144, 217–226. [Google Scholar] [CrossRef]
- FDA, Food and Drug Administration. An Updated Exposure Assessment for Furan from the Consumption of Adult and Baby Foods; 18 April 2007; Food and Drug Administration: Silver Spring, MD, USA, 2007.
- Lachenmeier, D.W.; Maser, E.; Kuballa, T.; Reusch, H.; Kersting, M.; Alexy, U. Detailed exposure assessment of dietary furan for infants consuming commercially jarred complementary food based on data from the DONALD study. Matern Child. Nutr. 2012, 8, 390–403. [Google Scholar] [CrossRef] [PubMed]
- IFIC, International Food Information Council; FDA, US Food and Drug Administration. Overview of Food Ingredients, Additives & Colors; US Food and Drug Administration: Washington, DC, USA, 2010.
- Krishan, M.; Navarro, L.; Beck, B.; Carvajal, R.; Dourson, M. A regulatory relic: After 60 years of research on cancer risk, the Delaney Clause continues to keep us in the past. Toxicol. Appl. Pharm. 2021, 433, 115779. [Google Scholar] [CrossRef]
- Williams, G.M.; Karbe, E.; Fenner-Crisp, P.; Iatropoulos, M.J.; Weisburger, J.H. Risk assessment of carcinogens in food with special consideration of non-genotoxic carcinogens. Exp. Toxicol. Pathol. 1996, 48, 209–215. [Google Scholar] [CrossRef]
- Felter, S.P.; Zhang, X.; Thompson, C. Butylated hydroxyanisole: Carcinogenic food additive to be avoided or harmless antioxidant important to protect food supply? Regul. Toxicol. Pharm. 2021, 121, 104887. [Google Scholar] [CrossRef]
- Xu, X.; Liu, A.; Hu, S.; Ares, I.; Martínez-Larrañaga, M.R.; Wang, X.; Martínez, M.; Anadón, A.; Martínez, M.A. Synthetic phenolic antioxidants: Metabolism, hazards and mechanism of action. Food Chem. 2021, 353, 129488. [Google Scholar] [CrossRef]
- EFSA ANS Panel, European Food Safety Authority, Panel on Food Additives and Nutrient Sources added to Food. Scientific Opinion on the reevaluation of butylated hydroxyanisole-BHA (E 320) as a food additive. EFSA J. 2011, 9, 2392. [Google Scholar] [CrossRef]
- EFSA ANS Panel, European Food Safety Authority, Panel on Food Additives and Nutrient Sources added to Food. Scientific Opinion on the reevaluation of butylated hydroxytoluene BHT (E 321) as a food additive. EFSA J. 2012, 10, 2588. [Google Scholar]
- EFSA ANS Panel, European Food Safety Authority, Panel on Food Additives and Nutrient Sources added to Food. Statement on the refined exposure assessment of tertiary-butyl hydroquinone (E 319). EFSA J. 2016, 14, 4363. [Google Scholar]
- Deisinger, P.J. Human exposure to naturally occurring hydroquinone. J. Toxicol. Environ. Health 1996, 47, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Whysner, J. Butylated hydroxyanisole mechanistic data and risk assessment: Conditional species-specific cytotoxicity, enhanced cell proliferation, and tumor promotion. Pharmacol. Ther. 1996, 71, 137–151. [Google Scholar] [CrossRef]
- Williams, G.M.; Iatropoulos, M.J.; Whysner, J. Safety assessment of butylated hydroxyanisole and butylated hydroxytoluene as antioxidant food additives. Food Chem. Toxicol. 1999, 37, 1027–1038. [Google Scholar] [CrossRef]
- Hirose, M.; Fukushima, S.; Sakata, T.; Inui, M.; Ito, N. Effect of quercetin on two-stage carcinogenesis of the rat urinary bladder. Cancer Lett. 1983, 21, 23–27. [Google Scholar] [CrossRef]
- Gharavi, N.; Haggarty, S.; El-Kadi, A.S. Chemoprotective and carcinogenic effects of tert-butylhydroquinone and its metabolites. Curr. Drug Metab. 2007, 8, 1–7. [Google Scholar] [CrossRef]
- NTP, National Toxicology Program. Toxicology and carcinogenesis studies of hydroquinone (CAS No. 123-31-9) in F344/N rats and B6C3F1 mice (gavage studies). Natl. Toxicol. Program. Tech. Rep. Ser. 1989, 366, 1–248. [Google Scholar]
- Lanigan, R.S.; Yamarik, T.A. Final report on the safety assessment of BHT(1). Int. J. Toxicol. 2002, 21 (Suppl. S2), 19–94. [Google Scholar] [CrossRef]
- Williams, G.M.; McQueen, C.A.; Tong, C. Toxicity studies of butylated hydroxyanisole and butylated hydroxytoluene. I. Genetic and cellular effects. Food Chem. Toxicol. 1990, 28, 793–798. [Google Scholar] [CrossRef]
- van Esch, G.J. Toxicology of tert-butylhydroquinone (TBHQ). Food Chem. Toxicol. 1986, 24, 1063–1065. [Google Scholar] [CrossRef]
- Eskandani, M.; Hamishehkar, H.; Ezzati Nazhad Dolatabadi, J. Cytotoxicity and DNA damage properties of tert-butylhydroquinone (TBHQ) food additive. Food Chem. 2014, 153, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Nakagawa, S.; Yoshitake, A.; Miyamoto, J.; Hirose, M.; Ito, N. DNA-adduct formation in the forestomach of rats treated with 3-tert-butyl-4-hydroxyanisole and its metabolites as assessed by an enzymatic 32P-postlabeling method. Cancer Lett. 1989, 48, 189–195. [Google Scholar] [CrossRef]
- Jagetia, G.C.; Menon, K.S.L.; Jain, V. Genotoxic effect of hydroquinone on the cultured mouse spleenocytes. Toxicol. Lett. 2001, 121, 15–20. [Google Scholar] [CrossRef]
- Peters, M.M.; Lau, S.S.; Dulik, D.; Murphy, D.; van Ommen, B.; van Bladeren, P.J.; Monks, T.J. Metabolism of tert-butylhydroquinone to S-substituted conjugates in the male Fischer 344 rat. Chem. Res. Toxicol. 1996, 9, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Malkinson, A.; Radcliffe, R.; Bauer, A.; Dwyer-Nield, L.; Kleeberger, S. Quantitative trait loci that regulate susceptibility to both butylated hydroxytoluene-induced pulmonary inflammation and lung tumor promotion in CXB recombinant inbred mice. Chest 2002, 121, 82s. [Google Scholar] [CrossRef]
- Whysner, J.; Wang, C.X.; Zang, E.; Iatropoulos, M.J.; Williams, G.M. Dose response of promotion by butylated hydroxyanisole in chemically initiated tumours of the rat forestomach. Food Chem. Toxicol. 1994, 32, 215–222. [Google Scholar] [CrossRef]
- Bauer, A.K.; Dwyer-Nield, L.D.; Keil, K.; Koski, K.; Malkinson, A.M. Butylated hydroxytoluene (BHT) induction of pulmonary inflammation: A role in tumor promotion. Exp. Lung Res. 2001, 27, 197–216. [Google Scholar] [CrossRef]
- Malkinson, A.M. Role of inflammation in mouse lung tumorigenesis: A review. Exp. Lung Res. 2005, 31, 57–82. [Google Scholar] [CrossRef]
- English, J.C.; Perry, L.G.; Vlaovic, M.; Moyer, C.; O’Donoghue, J.L. Measurement of cell proliferation in the kidneys of Fischer 344 and Sprague-Dawley rats after gavage administration of hydroquinone. Fundam. Appl. Toxicol. 1994, 23, 397–406. [Google Scholar] [CrossRef]
- Whysner, J.; Verna, L.; English, J.C.; Williams, G.M. Analysis of studies related to tumorigenicity induced by hydroquinone. Regul. Toxicol. Pharmacol. 1995, 21, 158–176. [Google Scholar] [CrossRef] [PubMed]
- Peters, M. Cytotoxicity and cell-proliferation induced by the nephrocarcinogen hydroquinone and its nephrotoxic metabolite 2,3,5-(tris-glutathion-S- yl)hydroquinone. Carcinogenesis 1997, 18, 2393–2401. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hard, G.C.; Whysner, J.; English, J.C.; Zang, E.; Williams, G.M. Relationship of hydroquinone-associated rat renal tumors with spontaneous chronic progressive nephropathy. Toxicol. Pathol. 1997, 25, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Botterweck, A.A.; Verhagen, H.; Goldbohm, R.A.; Kleinjans, J.; van den Brandt, P.A. Intake of butylated hydroxyanisole and butylated hydroxytoluene and stomach cancer risk: Results from analyses in the Netherlands Cohort Study. Food Chem. Toxicol. 2000, 38, 599–605. [Google Scholar] [CrossRef]
- IARC, International Agency for Research on Cancer. Cruciferous Vegetables, Isothiocyanates and Indoles; IARC Handbooks of Cancer Prevention; International Agency for Research on Cancer: Lyon, France, 2004; Volume 9. [Google Scholar]
- Williams, G.M.; Iatropoulos, M.J.; Jeffrey, A.M. Anticarcinogenicity of monocyclic phenolic compounds. Eur. J. Cancer Prev. 2002, 11 (Suppl. S2), S101–S107. [Google Scholar]
- O’Donoghue, J.L. Hydroquinone and its analogues in dermatology—A risk-benefit viewpoint. J. Cosmet. Dermatol. 2006, 5, 196–203. [Google Scholar] [CrossRef]
- Suh, H.J.; Chung, M.S.; Cho, Y.H.; Kim, J.W.; Kim, D.H.; Han, K.W.; Kim, C.J. Estimated daily intakes of butylated hydroxyanisole (BHA), butylated hydroxytoluene (BHT) andtert-butyl hydroquinone (TBHQ) antioxidants in Korea. Food Addit. Contam. 2005, 22, 1176–1188. [Google Scholar] [CrossRef]
- Haque, A.; Brazeau, D.; Amin, A.R. Perspectives on natural compounds in chemoprevention and treatment of cancer: An update with new promising compounds. Eur. J. Cancer 2021, 149, 165–183. [Google Scholar] [CrossRef]
- Langner, E.; Rzeski, W. Dietary derived compounds in cancer chemoprevention. Contemp. Oncol. 2012, 16, 394–400. [Google Scholar] [CrossRef]
- Murakami, A.; Ohigashi, H.; Koshimizu, K. Anti-tumor promotion with food phytochemicals: A strategy for cancer chemoprevention. Biosci. Biotechnol. Biochem. 1996, 60, 1–8. [Google Scholar] [CrossRef]
- Ranjan, A.; Ramachandran, S.; Gupta, N.; Kaushik, I.; Wright, S.; Srivastava, S.; Das, H.; Srivastava, S.; Prasad, S.; Srivastava, S.K. Role of phytochemicals in cancer prevention. Int. J. Mol. Sci. 2019, 20, 4981. [Google Scholar] [CrossRef]
- Wattenberg, L.W. Inhibition of carcinogenesis by naturally-occurring and synthetic compounds. Basic Life Sci. 1990, 52, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Gescher, A.J.; Sharma, R.A.; Steward, W.P. Cancer chemoprevention by dietary constituents: A tale of failure and promise. Lancet Oncol. 2001, 2, 371–379. [Google Scholar] [CrossRef]
- Potter, J.D. The failure of cancer chemoprevention. Carcinogenesis 2014, 35, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Steward, W.P.; Brown, K. Cancer chemoprevention: A rapidly evolving field. Br. J. Cancer 2013, 109, 1–7. [Google Scholar] [CrossRef]
- Patterson, S.L.; Colbert Maresso, K.; Hawk, E. Cancer chemoprevention: Successes and failures. Clin. Chem. 2013, 59, 94–101. [Google Scholar] [CrossRef]
- Johnson, I.T. Phytochemicals and cancer. Proc. Nutr. Soc. 2007, 66, 207–215. [Google Scholar] [CrossRef]
- Kleiner, H.E. Oral administration of naturally occurring coumarins leads to altered phase I and II enzyme activities and reduced DNA adduct formation by polycyclic aromatic hydrocarbons in various tissues of SENCAR mice. Carcinogenesis 2001, 22, 73–82. [Google Scholar] [CrossRef][Green Version]
- Prince, M.; Campbell, C.T.; Robertson, T.A.; Wells, A.J.; Kleiner, H.E. Naturally occurring coumarins inhibit 7,12-dimethylbenz[a]anthracene DNA adduct formation in mouse mammary gland. Carcinogenesis 2005, 27, 1204–1213. [Google Scholar] [CrossRef]
- Huber, W.W.; McDaniel, L.P.; Kaderlik, K.R.; Teitel, C.H.; Lang, N.P.; Kadlubar, F.F. Chemoprotection against the formation of colon DNA adducts from the food-borne carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in the rat. Mutat. Res./Fundam. Mol. Mech. Mutagen. 1997, 376, 115–122. [Google Scholar] [CrossRef]
- Cavin, C. The coffee-specific diterpenes cafestol and kahweol protect against aflatoxin B1-induced genotoxicity through a dual mechanism. Carcinogenesis 1998, 19, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.G.; McWhorter, K.; Rivera-Hidalgo, F.; Wright, J.M.; Hirsbrunner, P.; Sunahara, G.I. Kahweol and cafestol: Inhibitors of hamster buccal pouch carcinogenesis. Nutr. Cancer 1991, 15, 41–46. [Google Scholar] [CrossRef]
- Alhusainy, W.; Williams, G.M.; Jeffrey, A.M.; Iatropoulos, M.J.; Taylor, S.; Adams, T.B.; Rietjens, I.M.C.M. The natural basil flavonoid nevadensin protects against a methyleugenol-induced marker of hepatocarcinogenicity in male F344 rat. Food Chem. Toxicol. 2014, 74, 28–34. [Google Scholar] [CrossRef]
- Azuine, M.A.; Bhide, S.V. Chemopreventive effect of turmeric against stomach and skin tumors induced by chemical carcinogens in Swiss mice. Nutr. Cancer 1992, 17, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Ikezaki, S.; Nishikawa, A.; Furukawa, F.; Kudo, K.; Nakamura, H.; Tamura, K.; Mori, H. Chemopreventive effects of curcumin on glandular stomach carcinogenesis induced by N-methyl-N’-nitro-N-nitrosoguanidine and sodium chloride in rats. Anticancer Res. 2001, 21, 3407–3411. [Google Scholar] [PubMed]
- Singletary, K.W.; Nelshoppen, J.M.; Scardefield, S.; Wallig, M. Inhibition by butylated hydroxytoluene and its oxidative metabolites of DMBA-induced mammary tumorigenesis and of mammary DMBA-DNA adduct formation in vivo in the female rat. Food Chem. Toxicol. 1992, 30, 455–465. [Google Scholar] [CrossRef]
- Williams, G.M.; Tanaka, T.; Maeura, Y. Dose-related inhibition of aflatoxin B1 induced hepatocarcinogenesis by the phenolic antioxidants, butylated hydroxyanisole and butylated hydroxytoluene. Carcinogenesis 1986, 7, 1043–1050. [Google Scholar] [CrossRef]
- Williams, G.M.; Iatropoulos, M.J. Inhibition of the hepatocarcinogenicity of aflatoxin B1 in rats by low levels of the phenolic antioxidants butylated hydroxyanisole and butylated hydroxytoluene. Cancer Lett. 1996, 104, 49–53. [Google Scholar] [CrossRef]
- Simonich, M.T.; Egner, P.A.; Roebuck, B.D.; Orner, G.A.; Jubert, C.; Pereira, C.; Groopman, J.D.; Kensler, T.W.; Dashwood, R.H.; Williams, D.E.; et al. Natural chlorophyll inhibits aflatoxin B1-induced multi-organ carcinogenesis in the rat. Carcinogenesis 2007, 28, 1294–1302. [Google Scholar] [CrossRef]
- Kensler, T.W.; Egner, P.A.; Dolan, P.M.; Groopman, J.D.; Roebuck, B.D. Mechanism of protection against aflatoxin tumorigenicity in rats fed 5-(2-pyrazinyl)-4-methyl-1,2-dithiol-3-thione (oltipraz) and related 1,2-dithiol-3-thiones and 1,2-dithiol-3-ones. Cancer Res. 1987, 47, 4271–4277. [Google Scholar] [PubMed]
- Wattenberg, L.W.; Loub, W.D. Inhibition of polycyclic aromatic hydrocarbon-induced neoplasia by naturally occurring indoles. Cancer Res. 1978, 38, 1410–1413. [Google Scholar] [PubMed]
- Rauscher, R.; Edenharder, R.; Platt, K.L. In vitro antimutagenic and in vivo anticlastogenic effects of carotenoids and solvent extracts from fruits and vegetables rich in carotenoids. Mutat. Res./Genet. Toxicol. Environ. Mutagen. 1998, 413, 129–142. [Google Scholar] [CrossRef]
- Tanaka, T.; Shnimizu, M.; Moriwaki, H. Cancer chemoprevention by carotenoids. Molecules 2012, 17, 3202–3242. [Google Scholar] [CrossRef]
- Hecht, S.S. Chemoprevention of cancer by isothiocyanates, modifiers of carcinogen metabolism. J. Nutr. 1999, 129, 768S–774S. [Google Scholar] [CrossRef]
- Wattenberg, L.W. Inhibition of carcinogen-induced neoplasia by sodium cyanate, tert-butyl isocyanate, and benzyl isothiocyanate administered subsequent to carcinogen exposure. Cancer Res. 1981, 41, 2991–2994. [Google Scholar]
- Zhang, Y.; Kensler, T.W.; Cho, C.G.; Posner, G.H.; Talalay, P. Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates. Proc. Natl. Acad. Sci. USA 1994, 91, 3147–3150. [Google Scholar] [CrossRef]
- Walters, D.G. Cruciferous vegetable consumption alters the metabolism of the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in humans. Carcinogenesis 2004, 25, 1659–1669. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Baluch, N.; Homayouni, T.S.; Morgatskaya, E.; Kumar, S.; Kazemi, P.; Yeger, H. The role of Sulforaphane in cancer chemoprevention and health benefits: A mini-review. J. Cell Commun. Signal. 2018, 12, 91–101. [Google Scholar] [CrossRef]
- Cömert, E.D.; Gökmen, V. Evolution of food antioxidants as a core topic of food science for a century. Food Res. Int. 2018, 105, 76–93. [Google Scholar] [CrossRef]
- Liu, R.H. Potential synergy of phytochemicals in cancer prevention: Mechanism of action. J. Nutr. 2004, 134, 3479S–3485S. [Google Scholar] [CrossRef] [PubMed]
- Racchi, M.L. Antioxidant defenses in plants with attention to Prunus and Citrus spp. Antioxidants 2013, 2, 340–369. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A. Cancer prevention and treatment with resveratrol: From rodent studies to clinical trials. Cancer Prev. Res. 2009, 2, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Heidor, R.; de Conti, A.; Ortega, J.F.; Furtado, K.S.; Silva, R.C.; Tavares, P.E.L.M.; Purgatto, E.; Ract, J.N.R.; de Paiva, S.A.R.; Gioielli, L.A.; et al. The chemopreventive activity of butyrate-containing structured lipids in experimental rat hepatocarcinogenesis. Mol. Nutr. Food Res. 2015, 60, 420–429. [Google Scholar] [CrossRef]
- Guariento, A.H.; Furtado, K.S.; de Conti, A.; Campos, A.; Purgatto, E.; Carrilho, J.; Shinohara, E.M.G.; Tryndyak, V.; Han, T.; Fuscoe, J.C.; et al. Transcriptomic responses provide a new mechanistic basis for the chemopreventive effects of folic acid and tributyrin in rat liver carcinogenesis. Int. J. Cancer 2014, 135, 7–18. [Google Scholar] [CrossRef]
- Ulrich, S.; Wolter, F.; Stein, J.M. Molecular mechanisms of the chemopreventive effects of resveratrol and its analogs in carcinogenesis. Mol. Nutr. Food Res. 2005, 49, 452–461. [Google Scholar] [CrossRef]
- Dashwood, R.H. Indole-3-carbinol: Anticarcinogen or tumor promoter in brassica vegetables? Chem. Biol. Interact. 1998, 110, 1–5. [Google Scholar] [CrossRef]
- Lake, B.G. Coumarin metabolism, toxicity and carcinogenicity: Relevance for human risk assessment. Food Chem. Toxicol. 1999, 37, 423–453. [Google Scholar] [CrossRef]

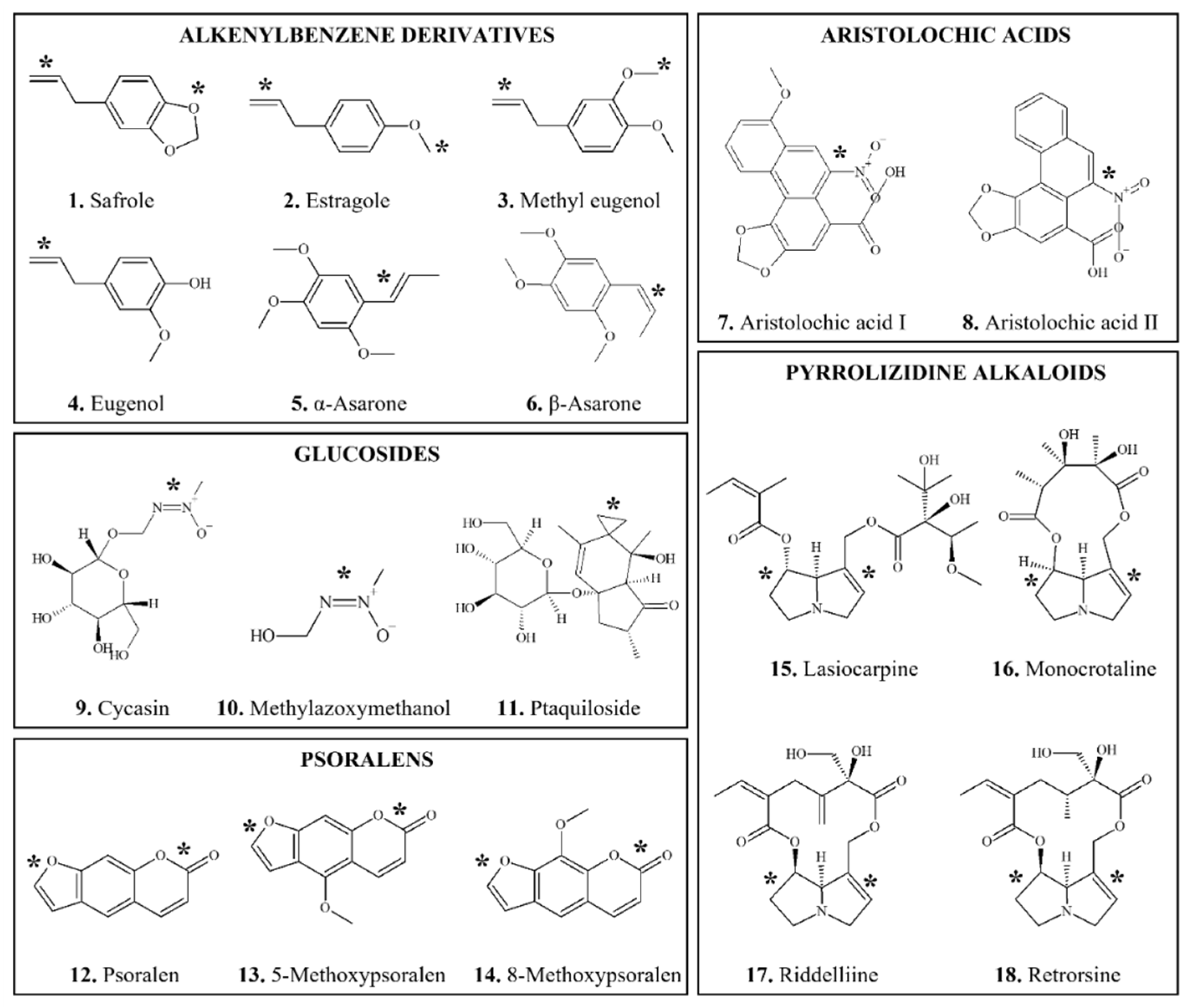{kind=link}
{kind=link}
{kind=link}
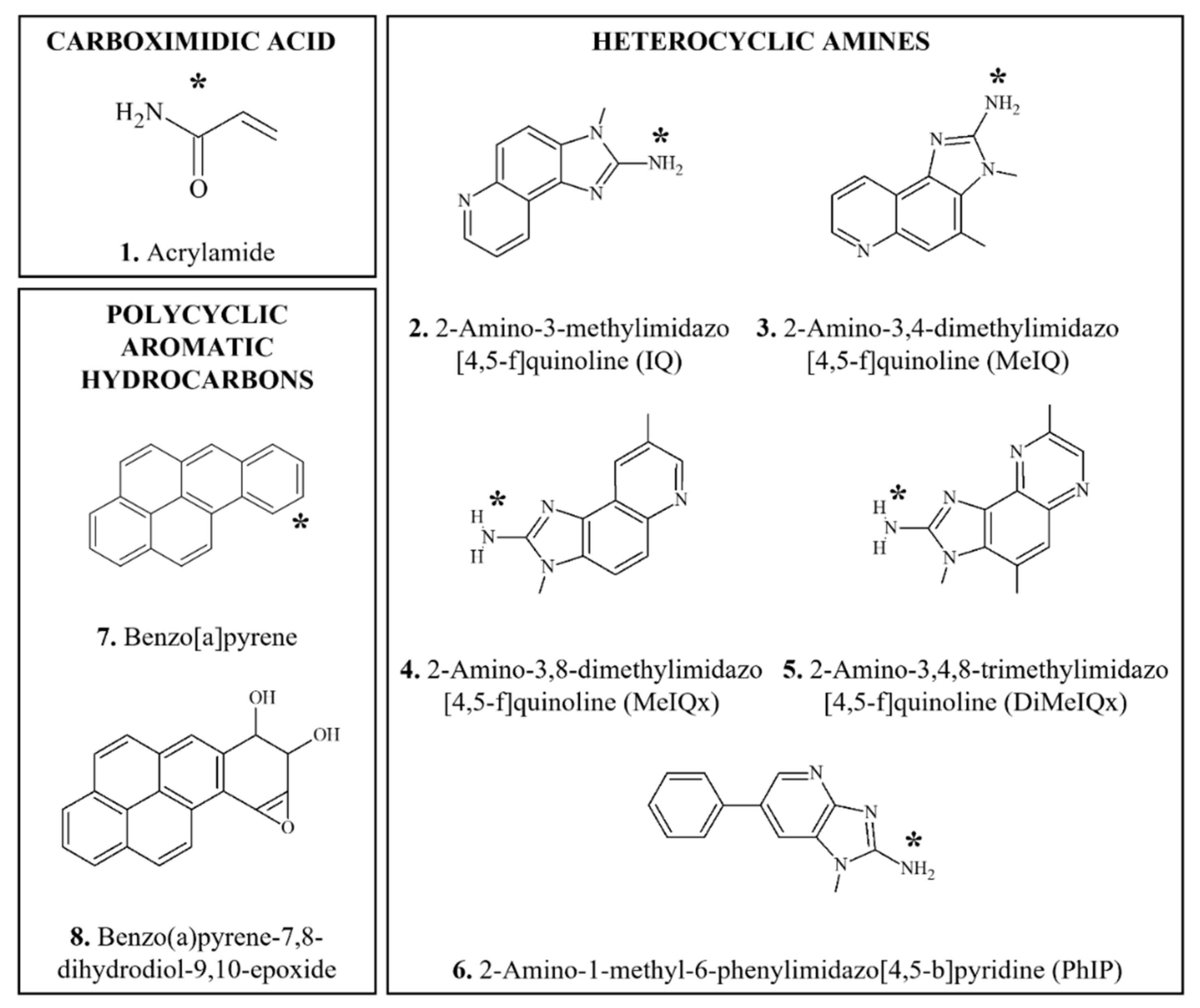{kind=link}
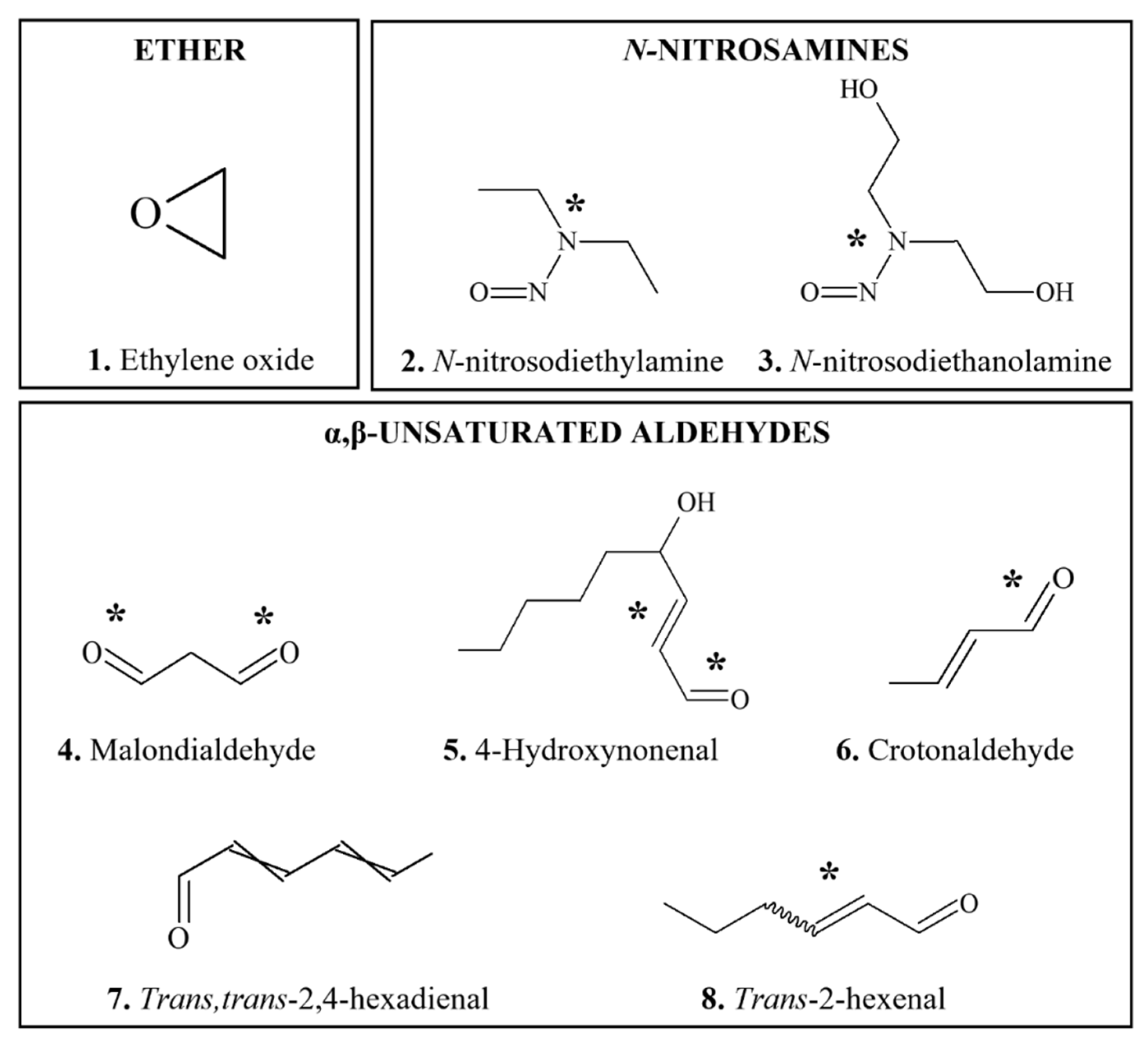{kind=link}
{kind=link}
{kind=link}
{kind=link}
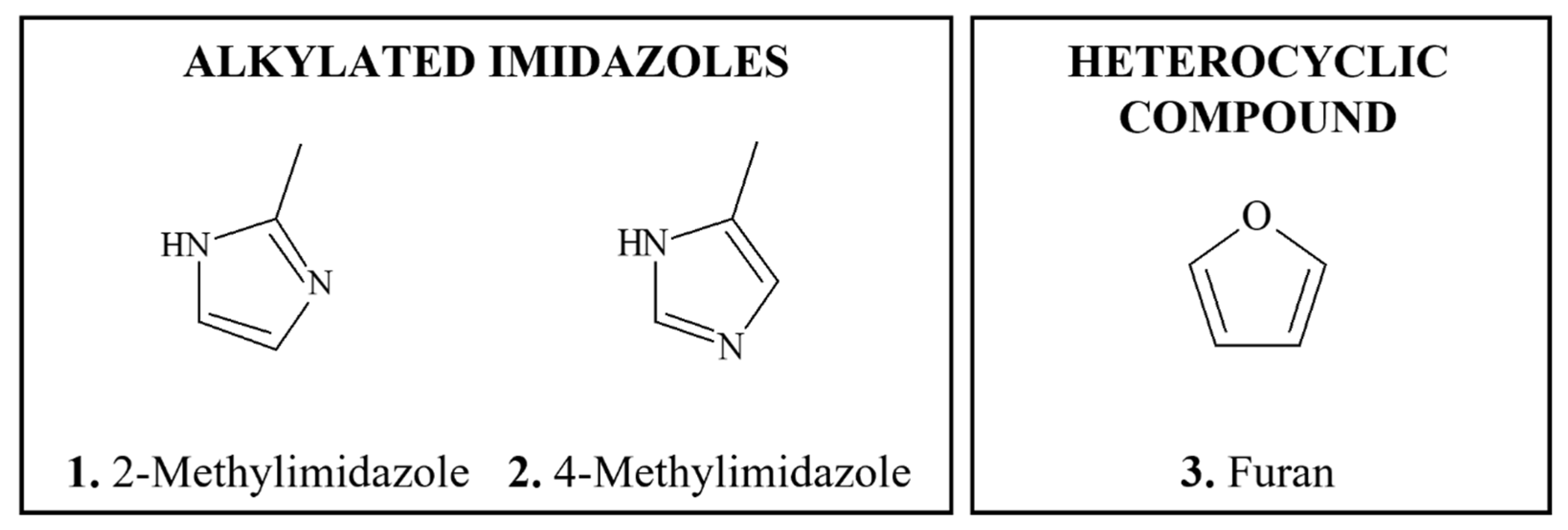{kind=link}
{kind=link}
| Source | Examples a | |
|---|---|---|
| 1. Naturally occurring | ||
| Plant: | alkenylbenzene derivatives aristolochic acid cycasin ptaquiloside d-limonene | psoralen pyrrolizidine alkaloids pulegone β-myrcene |
| Microbial/Fungal: | various mycotoxins | |
| 2. Contaminants | ||
| Introduced before processing: | daminozide dioxins | DDT flumequine |
| Introduced during processing: | trichloroethylene | methylene chloride |
| Food contact materials: | plastics (polyolefins, polyesters, polystyrene, polyamides, etc.) polymeric coatings | monomers (vinyl chloride, styrene, acrylonitrile) |
| 3. Additives | ||
| Anthropogenic: | α,β-aldehydes butylated hydroxyanisole and butylated hydroxytoluene | hexenal saccharin |
| 4. Formed from food components | ||
| During processing: | acrylamide chloropropanols ethyl carbamate (urethane) | furan various nitrosamines alkylated imidazoles |
| During packaging: | bisphenol A furan | phthalates |
| During storage: | benzene | |
| During cooking: | acrylamide benzo[a]pyrene | various heterocyclic amines |
| In the body: | nitrosamines and nitrosamides α,β-aldehydes | ethylene oxide |
| Chemical Name | CAS Registry Number | Classification | Carcinogenic Potency (TD50, mg/kg/d) c | MoA | |
|---|---|---|---|---|---|
| IARC a | NTP b | ||||
| 1. Human carcinogens | |||||
| Aflatoxins | 1 | 1 | 0.343 (mouse) 0.0032 (rat) | GTX | |
| Aristolochic acid I | 313-67-7 | 1 | 1 | N/A | GTX |
| Benzene | 71-43-2 | 1 | 1 | 77.5 (mouse) 169 (rat) | GTX |
| Benzo[a]pyrene | 50-32-8 | 1 | 2 | 3.47 (mouse) 0.956 (rat) | GTX |
| Dioxin (TCDD) | 1746-01-6 | 1 | 1 | 0.000156 (mouse) 0.0000235 (rat) | EPI |
| Dioxin-like compounds (PBCs) | 1 | N/L | N/A | EPI | |
| Ethylene oxide | 75-21-8 | 1 | 1 | 63.7 (mouse) 21.3 (rat) | GTX |
| Methoxsalen with UV A radiation | 298-81-7 | 1 | 1 | 32.4 (rat) | GTX |
| Processed meat | 1 | N/L | N/A | GTX | |
| Salted fish | 1 | N/L | N/A | GTX | |
| 2. Likely to be human carcinogens | |||||
| Acrylamide | 79-06-1 | 2A | 2 | 3.75 (rat) | GTX |
| 2-Amino-3-methylimidazo[4,5-f]quinoline | 76180-96-6 | 2A | 2 | 19.6 (mouse) 0.812 (rat) | GTX |
| p,p′-Dichlorodiphenyl-trichloroethane (DDT) | 50-29-3 | 2A | 2 | 12.8 (mouse) 84.7 (rat) | EPI |
| Ethyl carbamate (urethane) | 51-79-6 | 2A | 2 | 16.9 (mouse) 41.3 (rat) | GTX |
| 5-Methoxypsoralen | 484-20-8 | 2A | N/L | N/A | GTX |
| N-nitrosodiethylamine | 55-18-5 | 2A | 2 | 0.0265 (rat) | GTX |
| Red meat | 2A | N/L | N/A | GTX | |
| 2-Amino-3,4-dimethylimidazo[4,5-f]quinoline | 77094-11-2 | 2B | 2 | 15.5 (mouse) | GTX |
| 2-Amino-3,8-dimethylimidazo[4,5-f]quinoline | 77500-04-0 | 2B | 2 | 24.3 (mouse) 1.66 (rat) | GTX |
| 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine | 105650-23-5 | 2B | 2 | 33.2 (mouse) 1.78 (rat) | GTX |
| Benzophenone | 119-61-9 | 2B | N/L | 152 (rat) 379 (mouse) | EPI |
| Bracken fern | 2B | N/L | N/A | GTX | |
| Butylated hydroxyanisole | 25013-16-5 | 2B | 2 | 5530 (mouse) 405 (rat) | EPI |
| 3-Chloro-1,2-propanediol | 96-24-2 | 2B | N/L | 117 (rat) | Uncertain |
| Crotonaldehyde | 4170-30-3 | 2B | N/L | 4.2 (rat) | GTX |
| Cycasin | 14901-08-7 | 2B | N/L | N/A | GTX |
| 1,3-Dichloro-2-propanol | 96-23-1 | 2B | N/L | 46.4 (rat) | GTX |
| Di(2-ethylhexyl) phthalate | 117-81-7 | 2B | 2 | 476 (rat) 484 (mouse) | EPI |
| 1,4-Dioxane | 123-91-1 | 2B | 2 | 204 (mouse) 267 (rat) | Uncertain/EPI |
| Fumonisin B1 | 116355-83-0 | 2B | N/L | 6.79 (mouse) 5.75 (rat) | Uncertain/EPI |
| Fusarin C | 79748-81-5 | 2B | N/L | N/A | Uncertain/EPI |
| Furan | 110-00-9 | 2B | 2 | 2.72 (mouse) 0.396 (rat) | EPI |
| Lasiocarpine | 303-34-4 | 2B | N/L | 0.389 (rat) | GTX |
| Methyl eugenol | 93-15-2 | 2B | 2 | 19.3 (mouse) 19.7 (rat) | GTX |
| Methylazoxymethanol | 592-62-1 | 2B | N/L | N/A | GTX |
| 2-Methylimidazole | 693-98-1 | 2B | N/L | 782 (mouse) 868 (rat) | EPI |
| 4-Methylimidazole | 822-36-6 | 2B | N/L | 387 (mouse) 317 (rat) | EPI |
| Methyl isobutyl ketone | 108-10-1 | 2B | N/L | 612 (rat) | EPI |
| Monocrotaline | 315-22-0 | 2B | N/L | 0.94 (rat) | GTX |
| β-Myrcene | 123-35-3 | 2B | N/L | 15,400 (rat) | EPI |
| N-nitrosodiethanolamine | 1116-54-7 | 2B | 2 | 3.17 (rat) | GTX |
| Ochratoxin A | 303-47-9 | 2B | 2 | 6.41 (mouse) 0.136 (rat) | GTX/EPI |
| Pickled vegetables | 2B | N/L | N/A | GTX | |
| Pulegone | 89-82-7 | 2B | N/L | 232 (mouse) 156 (rat) | EPI |
| Riddelliine | 23246-96-0 | 2B | 2 | 1.97 (mouse) 0.119 (rat) | GTX |
| Safrole | 94-59-7 | 2B | 2 | 51.3 (mouse) 441 (rat) | GTX |
| trans,trans-2,4-Hexadienal | 142-83-6 | 2B | N/L | 176 (mouse) 62.2 (rat) | GTX |
| 3. Unknown carcinogenic potential | |||||
| Agaritine d | 2757-90-6 | 3 | N/L | N/A | GTX |
| Butylated hydroxytoluene | 128-37-0 | 3 | N/L | 653 (mouse) | EPI |
| Carrageenan (native) d | 9000-07-1 | 3 | N/L | N/A | |
| Chlorate (sodium salt) d | 7775-09-9 | 3 | N/L | 69.1 (mouse) 0.865 (rat) | EPI |
| Eugenol d | 97-53-0 | 3 | N/L | N/A | |
| Furfural d | 98-01-1 | 3 | N/L | 197 (mouse) 683 (rat) | Uncertain |
| Hydroquinone | 123-31-9 | 3 | N/L | 225 (mouse) 82.8 (rat) | EPI |
| Isatidine d | 15503-86-3 | 3 | N/L | 0.716 (rat) | GTX |
| d-Limonene d | 5989-27-5 | 3 | N/L | 204 (rat) | EPI |
| Malondialdehyde | 24382-04-5 | 3 | N/L | 14.1 (mouse) 122 (rat) | GTX |
| Patulin d | 149-29-1 | 3 | N/L | N/A | Uncertain |
| Ptaquiloside | 87625-62-5 | 3 | N/L | N/A | GTX |
| Quercetin d | 117-39-5 | 3 | N/L | 10.1 (rat) | EPI |
| Retrorsine d | 480-54-6 | 3 | N/L | 0.862 (rat) | GTX |
| Senkirkine d | 2318-18-5 | 3 | N/L | 1.7 (rat) | GTX |
| Sodium saccharin d | 128-44-9 | 3 | N/L | 2140 (rat) | EPI |
| Symphytine d | 22571-95-5 | 3 | N/L | 1.91 | GTX |
| Zearalenone d | 17924-92-4 | 3 | N/L | 39 (mouse) | EPI |
| 4. Not classified by IARC/NTP | |||||
| Daminozide d | 1596-84-5 | N/L | N/L | 1030 (mouse) 2500 (rat) | EPI |
| Estragole | 140-67-0 | N/L | N/L | 51.8 (mouse) | GTX |
| Genistein d | 446-72-0 | N/L | N/L | 27.1 (rat) | EPI |
| N-methyl-N-formylhydrazine d | 758-17-8 | N/L | N/L | 1.37 (mouse) | GTX |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobets, T.; Smith, B.P.C.; Williams, G.M. Food-Borne Chemical Carcinogens and the Evidence for Human Cancer Risk. Foods 2022, 11, 2828. https://doi.org/10.3390/foods11182828
Kobets T, Smith BPC, Williams GM. Food-Borne Chemical Carcinogens and the Evidence for Human Cancer Risk. Foods. 2022; 11(18):2828. https://doi.org/10.3390/foods11182828
Chicago/Turabian StyleKobets, Tetyana, Benjamin P. C. Smith, and Gary M. Williams. 2022. "Food-Borne Chemical Carcinogens and the Evidence for Human Cancer Risk" Foods 11, no. 18: 2828. https://doi.org/10.3390/foods11182828
APA StyleKobets, T., Smith, B. P. C., & Williams, G. M. (2022). Food-Borne Chemical Carcinogens and the Evidence for Human Cancer Risk. Foods, 11(18), 2828. https://doi.org/10.3390/foods11182828