
Mesostructured Silica-Coated Magnetic Nanoparticles to Extract Six Opium Alkaloids in Poppy Seeds Prior to Ultra-High-Performance Liquid Chromatography-Tandem Mass Spectrometry Analysis





Abstract
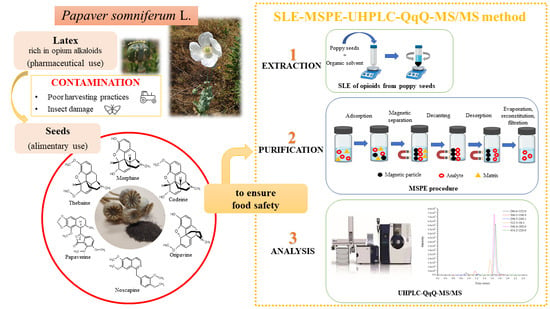
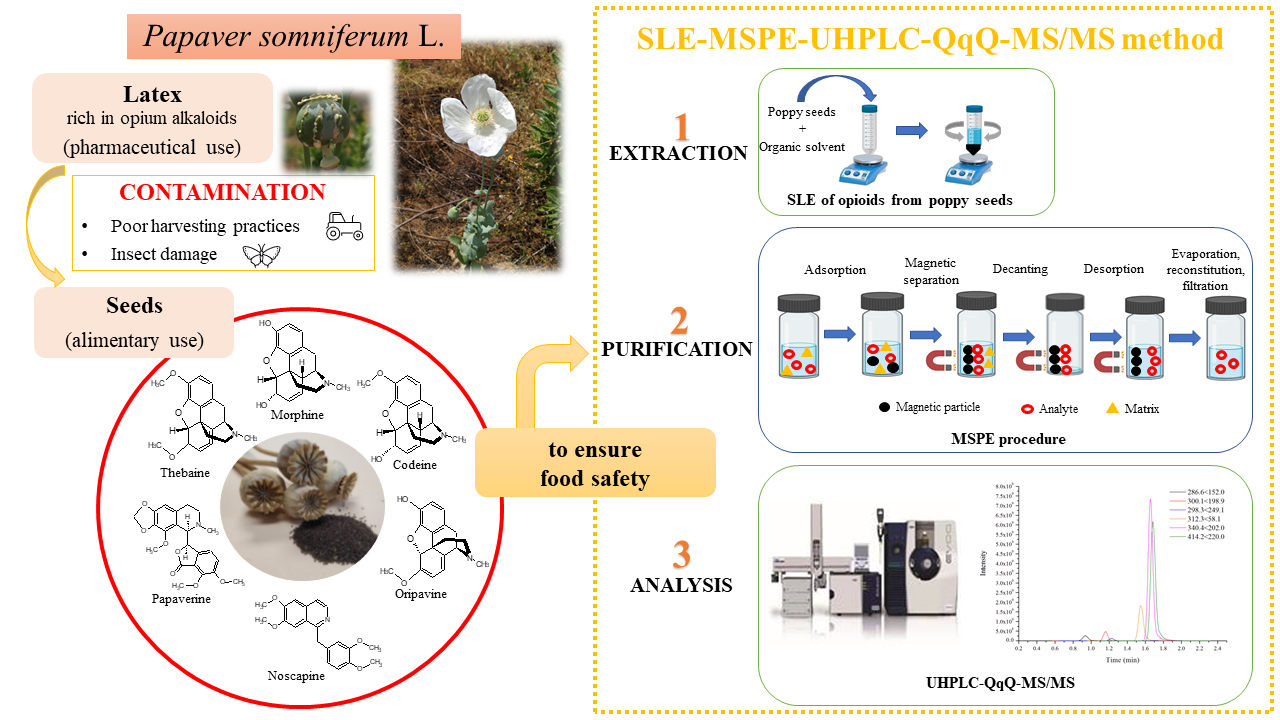
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Sample Collection
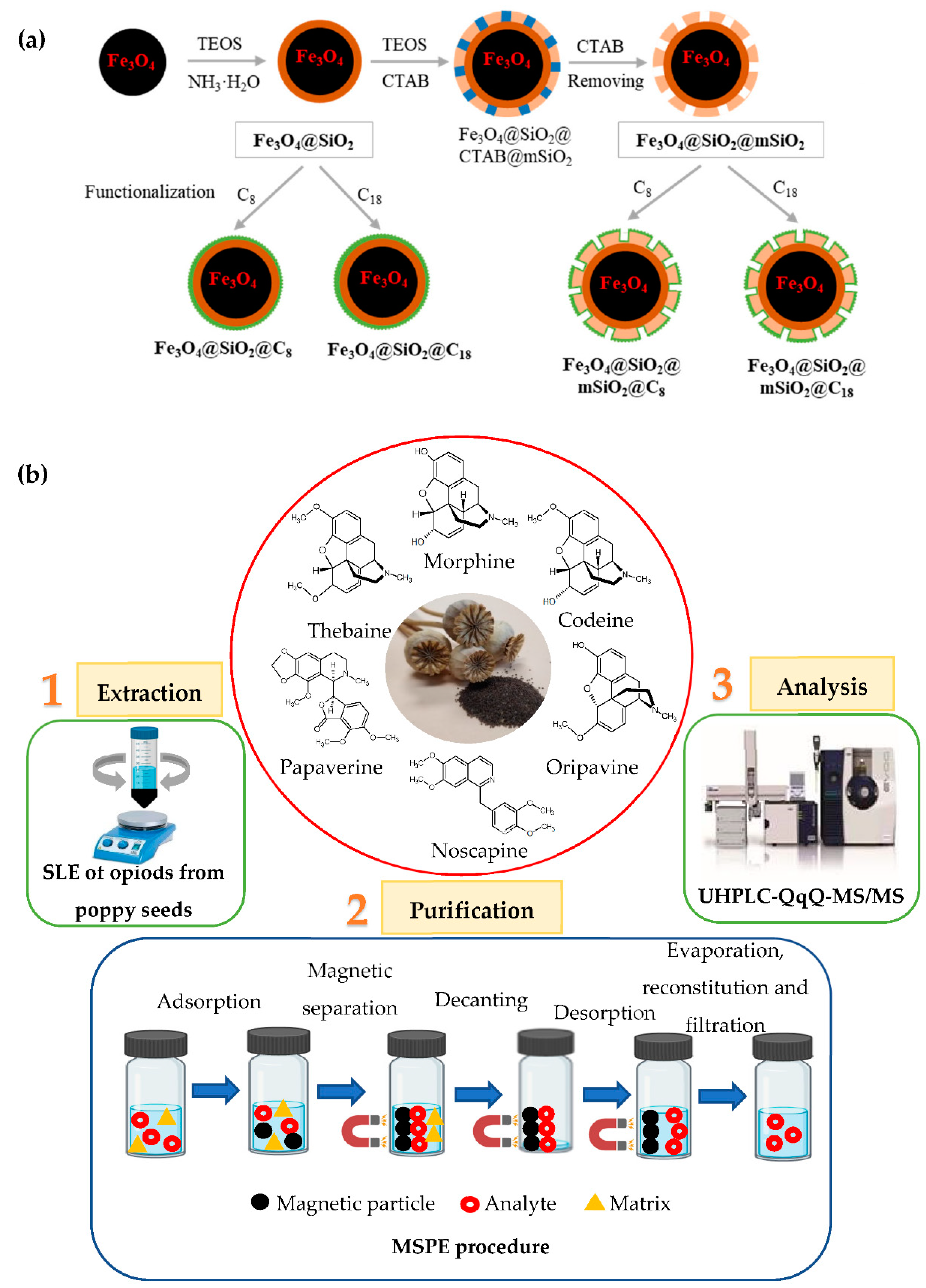
2.3. Synthesis of Organic-Functionalised Magnetic Particles
2.3.1. Preparation of Fe3O4 Particles
2.3.2. Surface Modification of Fe3O4 Particles with Amorphous Silica (Fe3O4@SiO2)
2.3.3. Surface Modification of Fe3O4 Particles with Mesostructurated Silica (Fe3O4@SiO2@mSiO2)
2.3.4. Organic Functionalisation of Fe3O4@SiO2 and Fe3O4@SiO2@mSiO2 Particles with C8 or C18 Groups
2.4. Characterization of Organic-Functionalised Magnetic Particles
2.5. Optimisation of UHPLC-QqQ-MS/MS Analysis
2.6. Optimisation of Sample Preparation
2.6.1. Optimisation of SLE of Opioids from Poppy Seeds
2.6.2. Discontinuous Adsorption Studies to Select the Best Magnetic Material for MSPE Procedure
2.6.3. Adsorption Kinetic and Isotherm Experiments with Fe3O4@SiO2@mSiO2 Material
2.6.4. Optimisation of MSPE Conditions with Fe3O4@SiO2@mSiO2 Material
2.7. Optimised Sample Preparation Procedure
2.7.1. Optimised SLE of Opioids from Poppy Seeds
2.7.2. Optimised MSPE Procedure
2.8. Instrumental and Method Validation
3. Results and Discussion
3.1. Characterisation of Magnetic Materials Synthetised
3.1.1. SEM
3.1.2. XRD
3.1.3. XRF
3.1.4. FT-IR
3.1.5. BET
3.1.6. Elemental Analysis
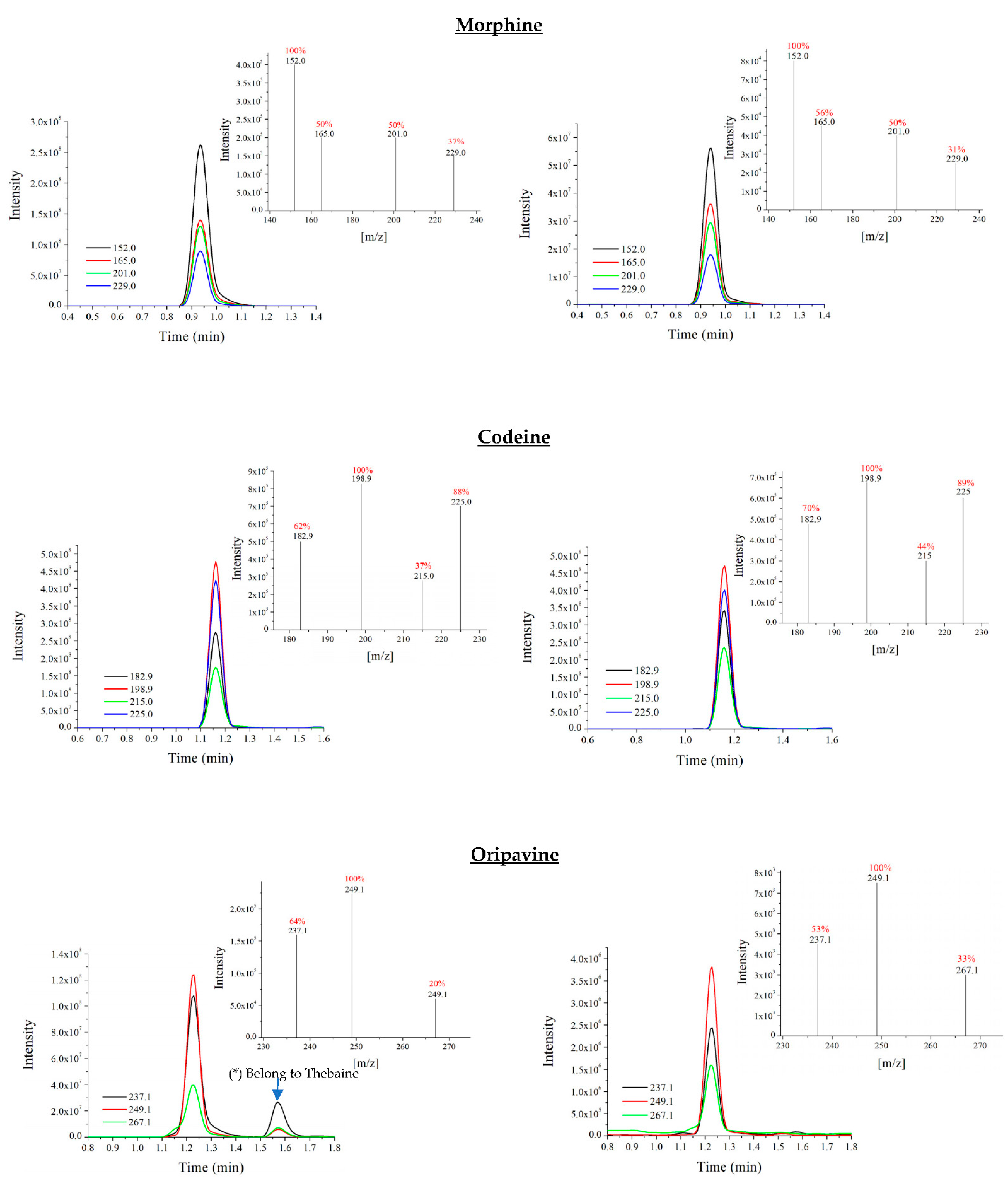
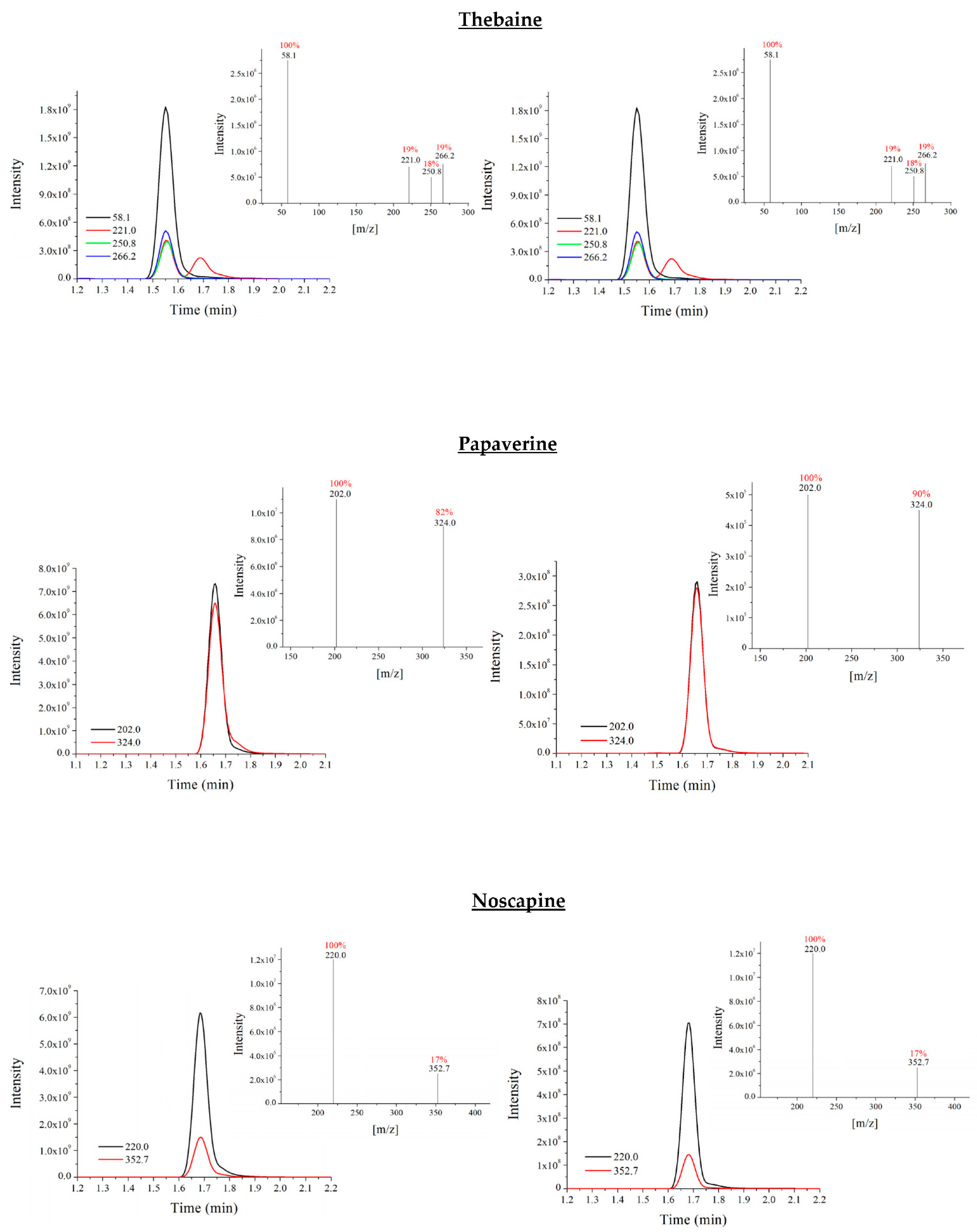
3.2. Optimisation of UHPLC-QqQ-MS/MS Analysis
3.3. Optimisation of Sample Preparation
3.3.1. Optimisation of SLE of Opioids from Poppy Seeds
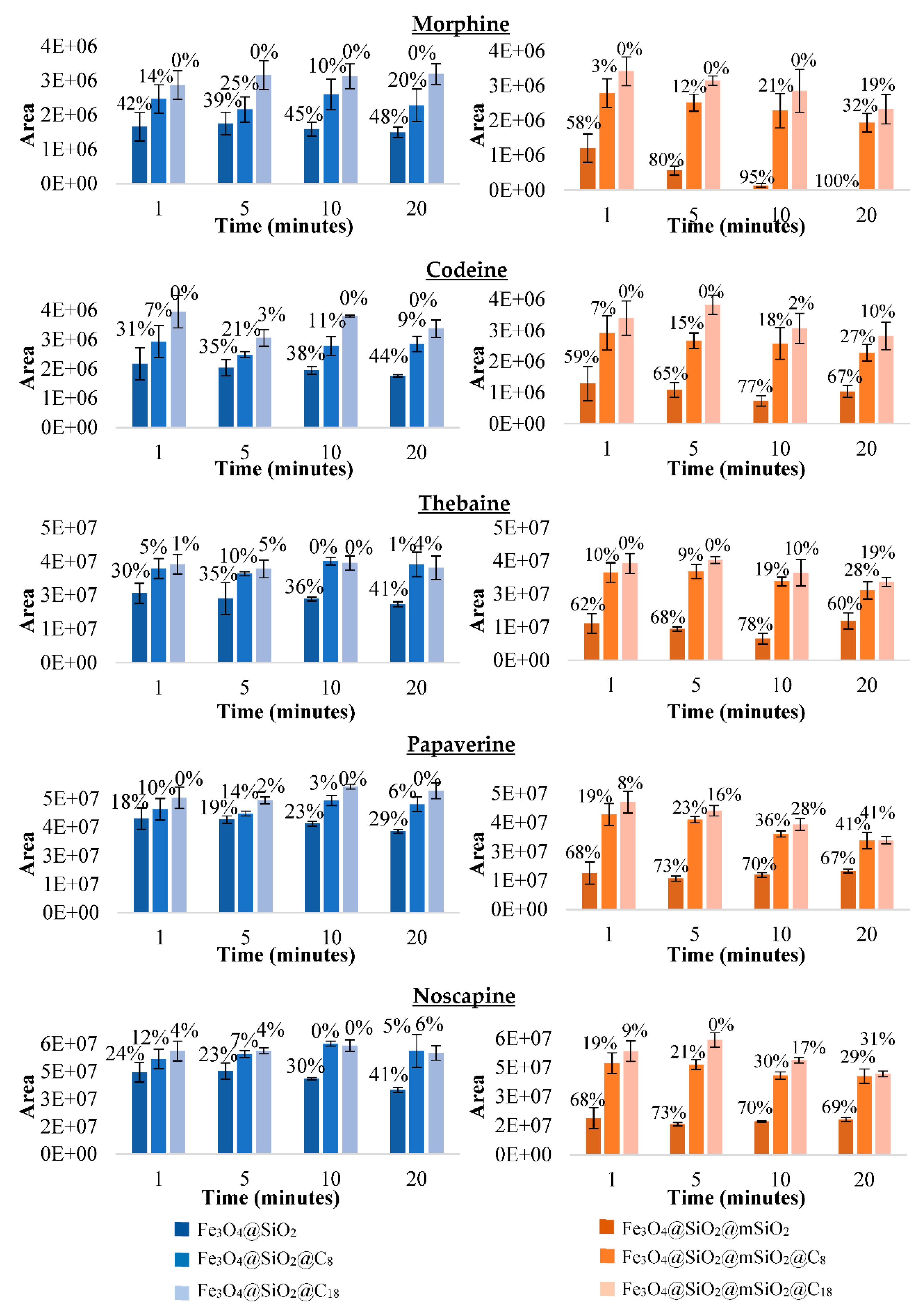
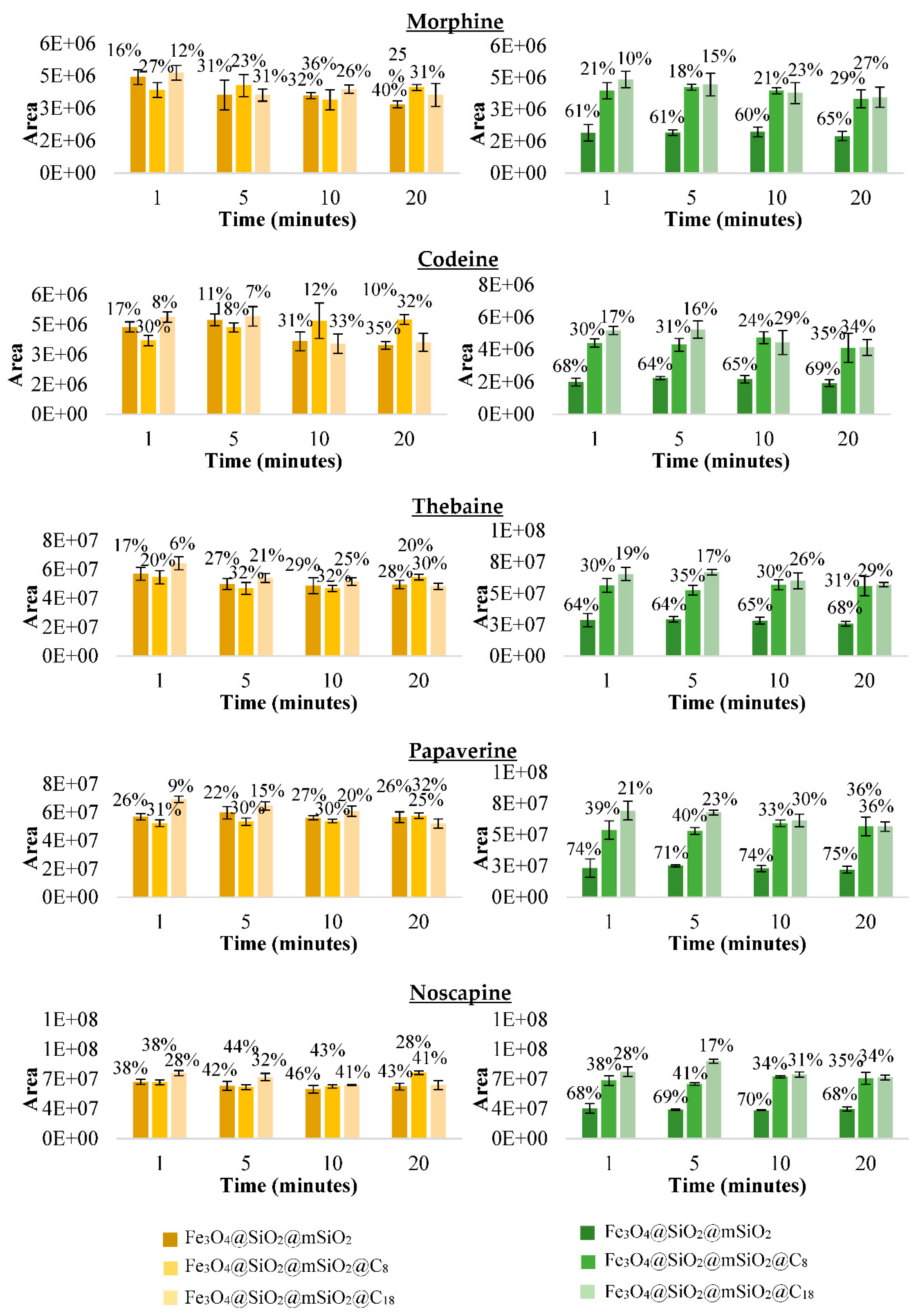
3.3.2. Selecting the Best Material for the MSPE Procedure after Making Discontinuous Adsorption Study with the Synthesised Magnetic Materials
3.3.3. Adsorption Kinetic and Isotherm Experiments of Fe3O4@SiO2@mSiO2 Material
3.3.4. Optimisation of MSPE Procedure with Fe3O4@SiO2@mSiO2 Material
Adsorption Conditions (Time and pH)
Desorption Conditions (Solvent Type, Time, pH and Number of Consecutive Desorptions)
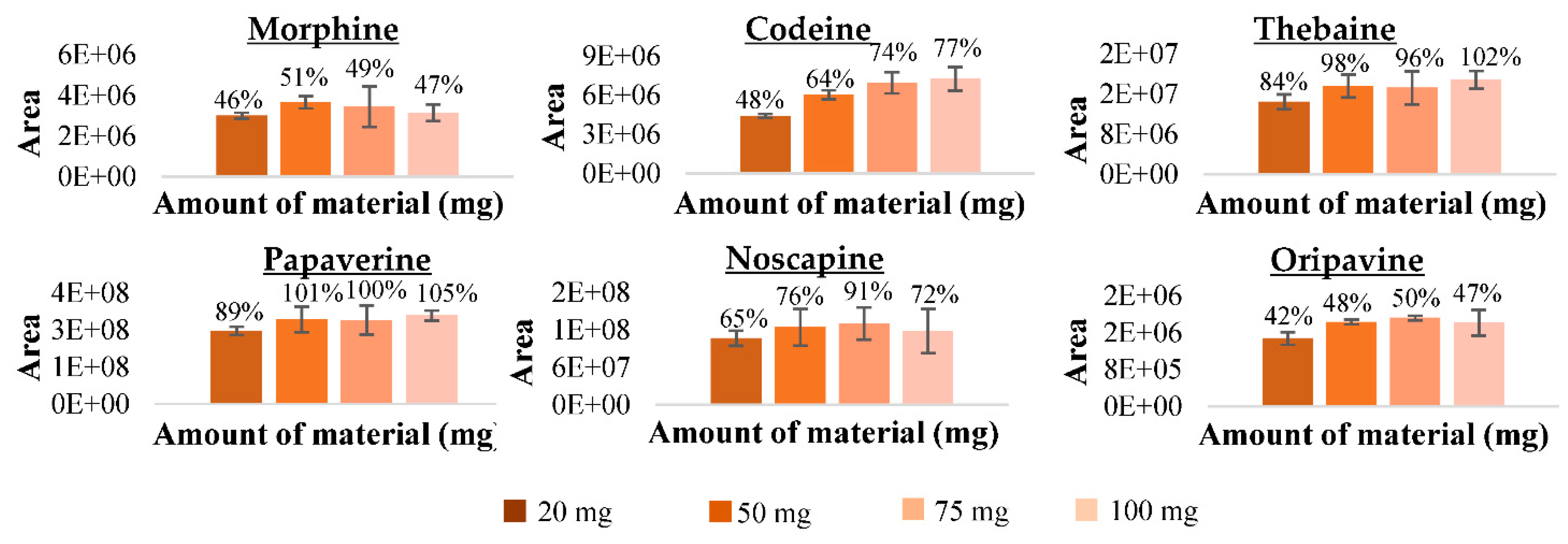
Amount of Fe3O4@SiO2@mSiO2 Material
3.4. Instrumental and Method Validation
3.5. Comparison of the Proposed Methodology with Others Reported Methods
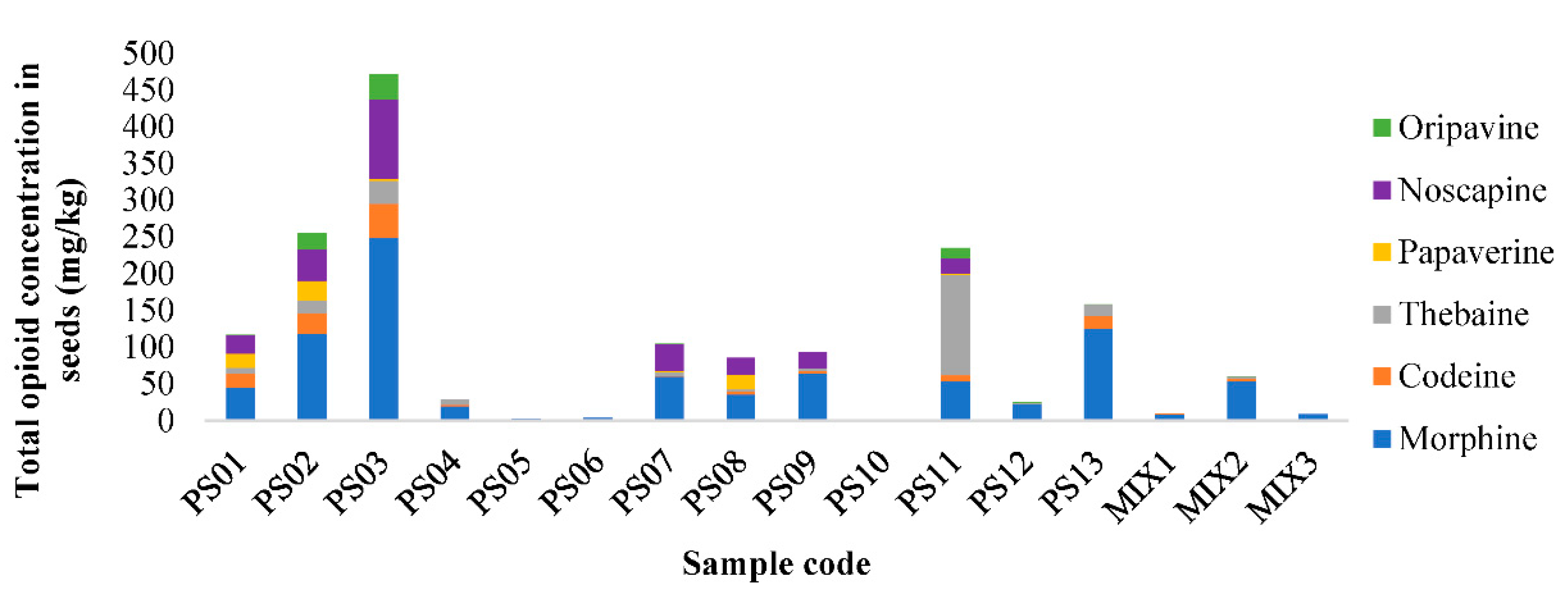
3.6. Application of the Method SLE-MSPE-UHPLC-QqQ-MS/MS to Real Samples of Poppy Seeds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- López, P.; Fauw, D.P.P.-D.; Mulder, P.P.; Spanjer, M.; de Stoppelaar, J.; Mol, H.G.; de Nijs, M. Straightforward analytical method to determine opium alkaloids in poppy seeds and bakery products. Food Chem. 2018, 242, 443–450. [Google Scholar] [CrossRef]
- Meos, A.; Saks, L.; Raal, A. Content of alkaloids in ornamental Papaver somniferum L. cultivars growing in Estonia. Proc. Est. Acad. Sci. 2017, 66, 34. [Google Scholar] [CrossRef]
- Powers, D.; Erickson, S.; Swortwood, M.J. Quantification of Morphine, Codeine, and Thebaine in Home-Brewed Poppy Seed Tea by LC-MS/MS. J. Forensic Sci. 2018, 63, 1229–1235. [Google Scholar] [CrossRef]
- Casado-Hidalgo, G.; Morante-Zarcero, S.; Pérez-Quintanilla, D.; Sierra, I. Opium alkaloids in food products: Current and future perspectives. Trends Food Sci. Technol. 2021, 108, 92–102. [Google Scholar] [CrossRef]
- AESAN. Spanish Food Safety and Nutrition Agency. Opium Alkaloids in Poppy Seeds. 2020. Available online: http://www.aecosan.msssi.gob.es/AECOSAN/docs/documentos/seguridad_alimentaria/gestion_riesgos/opio_semillas_adormidera.pdf (accessed on 23 January 2020).
- Sproll, C.; Perz, R.C.; Lachenmeier, D.W. Optimized LC/MS/MS analysis of morphine and codeine in poppy seed and evaluation of their fate during food processing as a basis for risk analysis. J. Agric. Food Chem. 2006, 54, 5292–5298. [Google Scholar] [CrossRef] [PubMed]
- Moeller, M.R.; Hammer, K.; Engel, O. Poppy seed consumption and toxicological analysis of blood and urine samples. Forensic Sci. Int. 2004, 143, 183–186. [Google Scholar] [CrossRef]
- Newmeyer, M.; Concheiro, M.; da Costa, J.L.; LoDico, C.; Gorelick, D.A.; Huestis, M.A. Simultaneous plasma and oral fluid morphine and codeine concentrations after controlled administration of poppy seeds with known opiate content. Forensic Toxicol. 2015, 33, 235–243. [Google Scholar] [CrossRef]
- Özbunar, E.; Aydoğdu, M.; Döğer, R.; Bostancı, H.I.; Koruyucu, M.; Akgür, S.A. Morphine concentrations in human urine following poppy seed paste consumption. Forensic Sci. Int. 2019, 295, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Rohrig, T.P.; Moore, C. The determination of morphine in urine and oral fluid following ingestion of poppy seeds. J. Anal. Toxicol. 2003, 27, 449–452. [Google Scholar] [CrossRef]
- European Food Safety Authority. Scientific Opinion on the risks for public health related to the presence of opium alkaloids in poppy seeds, EFSA Panel on Contaminants in the Food Chain (CONTAM). EFSA J. 2011, 9, 150. [Google Scholar] [CrossRef]
- BfR Recommends Provisional Daily Upper Intake Level and Guidance Value for Morphine in Poppy Seeds, BfR Heath Assessment No. 0.12/2006. 27 December 2005. Available online: https://mobil.bfr.bund.de/cm/349/bfr_recommends_provisional_daily_upper_intake_level_and_a_guidance_value_for_morphine_in_poppy_seeds.pdf (accessed on 23 January 2020).
- European Commision, Health and Food Safety Directorate-General, Summary Report of the Standing Committee on Plants, Animals, Food and Feed Held in Brussels on 25 November 2016. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/reg-com_toxic_20161125_sum.pdf (accessed on 23 January 2020).
- Commission Recommendation 2014/662/EU of 10 September 2014 on Good Practices to Prevent and to Reduce the Presence of Opium Alkaloids in Poppy Seeds and Poppy Seed Products, Brussels, Belgium, Official Journal of the European Union, L 271/96 2014. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32014H0662&from=SK (accessed on 23 January 2020).
- European Food Safety Authority. Update of the Scientific Opinion on opium alkaloids in poppy seeds, EFSA Panel on Contaminants in the Food Chain (CONTAM). EFSA J. 2018, 16, 5243. [Google Scholar] [CrossRef]
- Thevis, M.; Opfermann, G.; Schänzer, W. Urinary concentrations of morphine and codeine after consumption of poppy seeds. J. Anal. Toxicol. 2003, 27, 53–56. [Google Scholar] [CrossRef][Green Version]
- Acevska, J.; Dimitrovska, A.; Stefkov, G.; Brezovska, K.; Karapandzova, M.; Kulevanova, S. Development and validation of a reversed-phase HPLC method for determination of alkaloids from Papaver somniferum L. (Papaveraceae). J. AOAC Int. 2012, 95, 399–405. [Google Scholar] [CrossRef]
- Cao, Q.; Li, S.; He, C.; Li, K.; Liu, F. Extraction and determination of papaverin in pericarpium papaveris using aqueous two-phase system of poly(ethyleneglycol)–(NH4)2SO4 coupled with high-performance liquid chromatography. Anal. Chim. Acta 2007, 590, 187–194. [Google Scholar] [CrossRef]
- Guo, Q.Z.; Zhang, J.; Zhao, S.; Shao, B. Determination of five alkaloids of pericarpium papaveris in hot pot broth using ultra-performance liquid chromatography coupled to triple quadruple mass spectrometry. Food Anal. Methods 2013, 6, 698–704. [Google Scholar] [CrossRef]
- Stranska, I.; Skalicky, M.; Novak, J.; Matyasova, E.; Hejnák, V. Analysis of selected poppy (Papaver somniferum L.) cultivars: Pharmaceutically important alkaloids. Ind. Crop. Prod. 2013, 41, 120–126. [Google Scholar] [CrossRef]
- Casado, N.; Gañán, J.; Morante-Zarcero, S.; Sierra, I. New advanced materials and sorbent-based microextraction techniques as strategies in sample preparation to improve the determination of natural toxins in food samples. Molecules 2020, 25, 702. [Google Scholar] [CrossRef]
- Xu, F.; Liu, F.; Wang, C.; Wei, Y. Amantadine-functionalized magnetic microspheres and stable isotope labeled internal standards for reducing matrix effect in determination of five opium alkaloids by liquid chromatography-quadrupole linear ion trap mass spectrometry. J. Chin. Chem. Soc. 2019, 66, 484–492. [Google Scholar] [CrossRef]
- Tang, T.; Cao, S.; Xi, C.; Li, X.; Zhang, L.; Wang, G.; Chen, Z. Chitosan functionalized magnetic graphene oxide nanocomposite for the sensitive and effective determination of alkaloids in hotpot. Int. J. Biol. Macromol. 2020, 146, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, A.L.; Cavaliere, C.; LA Barbera, G.; Montone, C.M.; Piovesana, S.; Laganà, A. Recent Applications of Magnetic Solid-phase Extraction for Sample Preparation. Chromatographia 2019, 82, 1251–1274. [Google Scholar] [CrossRef]
- Tajik, M.; Yamini, Y.; Baheri, T.; Safari, M.; Asiabi, H. Supercritical fluid extraction of papaverine and noscapine from poppy capsules followed by preconcentration with magnetic nano Fe3O4@Cu@diphenylthiocarbazone particles. New J. Chem. 2017, 41, 7028–7037. [Google Scholar] [CrossRef]
- Deng, Y.; Qi, D.; Deng, C.; Zhang, A.X.; Zhao, D. Superparamagnetic high-magnetization microspheres with an Fe3O4@SiO2 core and perpendicularly aligned mesoporous SiO2 shell for removal of microcystins. J. Am. Chem. Soc. 2008, 130, 28–29. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Liu, F.; Wang, C.; Wei, Y. Reversed-phase/weak anion Exchange magnetic mesoporous microspheres for removal of matrix effects in lipophilic marine biotoxins analysis by ultrahigh-performance liquid chromatography coupled to tandem mass spectrometry. Food Chem. 2019, 294, 104–111. [Google Scholar] [CrossRef]
- Zhang, H.-F.; Shi, Y.-P. Magnetic retrieval of chitosan: Extraction of bioactive constituents from green tea beverage samples. Analyst 2011, 137, 910–916. [Google Scholar] [CrossRef]
- Zeng, H.; Wang, Y.; Nie, C.; Kong, J.; Liu, X. Preparation of magnetic molecularly imprinted polymers for separating rutin from Chinese medicinal plants. Analyst 2012, 137, 2503–2512. [Google Scholar] [CrossRef]
- Lagergren, S. About the theory of so-called adsorption of soluble substances. K. Sven. Vetenskapsakad. Handl. 1898, 24, 1–39. [Google Scholar]
- Ho, Y.; McKay, G. Pseudo-second order model for sorption processes. Process. Biochem. 1999, 34, 451–465. [Google Scholar] [CrossRef]
- Weber, W.J.; Morris, J.C. Kinetics of adsorption on carbon from solution. J. San. Eng. Div. 1963, 89, 31–60. [Google Scholar] [CrossRef]
- Langmuir, I. The adsorption of gases on plane surfaces of glass, mica and platinum. J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar] [CrossRef]
- Freundlich, H.M.F. Over the adsorption in solution. Am. J. Phys. Chem. 1906, 57, 385–471. [Google Scholar]
- European Commission SANTE/11813/2017. Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticide Residues and Analysis in Food and Feed. 2017. Available online: https://www.eurl-pesticides.eu/userfiles/file/EurlALL/AqcGuidance_SANTE_2019_12682.pdf (accessed on 9 April 2021).
- Commission Regulation (EC) No 401/2006. Laying Down the Methods of Sampling and Analysis for the Official Control of the Levels of Mycotoxins in Foodstuffs. 2006. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32006R0401&from=ES (accessed on 9 April 2021).
- Chen, F.F.; Xie, X.Y.; Shi, Y.P. Preparation of magnetic imprinted polymer for selective recognition of resveratrol in wine. J. Chromatogr. A 2013, 1300, 112–118. [Google Scholar] [CrossRef]
- Zhang, M.; Mei, J.; Lv, S.; Lai, J.; Zheng, X.; Yang, J.; Cui, S. Simultaneous extraction of permethrin diastereomers and deltamethrin in environmental water samples based on aperture regulated magnetic mesoporous silica. New J. Chem. 2020, 44, 16152–16162. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquérol, J.; Siemieniewska, T. Reporting physisorption data for gas/solid systems with special reference to the determination of Surface area and porosity. IUPAC Pure Appl. Chem. 1984, 57, 603–619. [Google Scholar] [CrossRef]
- Cassella, G.; Wu, A.; Shaw, B.; Hill, D. The analysis of thebaine in urine for the detection of poppy seed consumption. J. Anal. Toxicol. 1997, 21, 376–383. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Acevska, J.; Stefkov, G.; Petkovska, R.; Kulevanova, S.; Dimitrovska, A. Chemometric approach for development, optimization, and validation of different chromatographic methods for separation of opium alkaloids. Anal. Bioanal. Chem. 2012, 403, 1117–1129. [Google Scholar] [CrossRef]
- Skopikova, M.; Hashimoto, M.; Richomme, P.; Schinkovitz, A. Matrix free laser desorption ionization mass spectrometry as an efficient tool for the rapid detection of opiates in crude extracts of Papaver somniferum. J. Agric. Food Chem. 2020, 68, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Zentai, A.; Sali, J.; Szeitznė-Szabó, M.; Szabó, I.; Ambrus, A. Exposure of consumers to morphine from poppy seeds in Hungary. Food Addit. Contam. Part A 2011, 29, 1–12. [Google Scholar] [CrossRef]
- Carlin, M.G.; Dean, J.R.; Ames, J.M. Opium alkaloids in harvested and thermally processed poppy seeds. Front. Chem. 2020, 8, 737. [Google Scholar] [CrossRef]
- Bjerver, K.; Jonsson, J.; Nilsson, A.; Schuberth, J.; Schuberth, J. Morphine intake from poppy seed food. J. Pharm. Pharmacol. 2011, 34, 798–801. [Google Scholar] [CrossRef]
- Hayes, L.W.; Krasselt, W.G.; A Mueggler, P. Concentrations of morphine and codeine in serum and urine after ingestion of poppy seeds. Clin. Chem. 1987, 33, 806–808. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | SBET (m2/g) a | Pore Volume (cm3/g) b | Pore Diameter (Å) c | Elemental Analysis (%) | |||
|---|---|---|---|---|---|---|---|
| C | N | H | (mmol ligand/g) d | ||||
| Fe3O4 | - | - | - | - | - | - | - |
| Fe3O4@SiO2 | 147 | 0.18 | 125.8 | - | - | - | - |
| Fe3O4@SiO2@C8 | 26 | 0.09 | 124.4 | 3.432 | 0.000 | 1.070 | 0.286 |
| Fe3O4@SiO2@C18 | 24 | 0.10 | 124.9 | 3.227 | 0.000 | 0.928 | 0.134 |
| Fe3O4@SiO2@mSiO2 | 355 | 0.23 | 38.9 | - | - | - | - |
| Fe3O4@SiO2@mSiO2@C8 | 191 | 0.14 | 39.0 | 10.426 | 0.000 | 2.174 | 0.869 |
| Fe3O4@SiO2@mSiO2@C18 | 14 | 0.04 | 36.4 | 11.310 | 0.000 | 2.178 | 0.471 |
| Analytes | Linear Range (µg/L) | Matrix-Matched Calibration (R2) | Cm | ME | MDLn (µg/kg) | MQL (µg/kg) | Accuracy | Precision | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Recovery (% ± SD) | Mean Recovery (% ± SD) | Intra-Day Precision (RSD %) | Inter-Day Precision (RSD %) | |||||||
| Morphine | 0.01–5000 | y = 3269x + 32,452 (0.999) | 92 | 64 | 0.07 | 0.24 | 50 ± 1 a | 46 ± 2 | 4 a | 11 a |
| 42 ± 2 b | 2 b | 5 b | ||||||||
| Codeine | 0.01–5000 | y = 2554x + 29,642 (0.999) | 96 | 86 | 0.07 | 0.24 | 64 ± 2 a | 68 ± 4 | 3 a | 8 a |
| 71 ± 6 b | 2 b | 4 b | ||||||||
| Thebaine | 0.1–5000 | y = 10,093x + 485,953 (0.999) | 96 | 109 | 0.72 | 2.40 | 72 ± 3 a | 74 ± 4 | 5 a | 7 a |
| 76 ± 4 b | 0 b | 4 b | ||||||||
| Papaverine | 0.01–5000 | y = 33,441x + 2,814,320 (0.999) | 92 | 80 | 0.07 | 0.24 | 116 ± 5 a | 109 ± 3 | 5 a | 9 a |
| 101 ± 1 b | 2 b | 7 b | ||||||||
| Noscapine | 0.01–5000 | y = 42,914x + 1,631,091 (0.999) | 94 | 65 | 0.07 | 0.24 | 109 ± 1 a | 103 ± 1 | 4 a | 10 a |
| 97 ± 1 b | 3 b | 8 b | ||||||||
| Oripavine | 10–5000 | y = 1080x − 31,137 (0.999) | 94 | 31 | 72.07 | 240 | 51 ± 3 a | 52 ± 3 | 3 a | 5 a |
| 53 ± 3 b | 3 b | 5 b | ||||||||
| Sample | Analytes | Sample Treatment | Analysis Technique | MDL (µg/kg) | MQL (µg/kg) | Recovery (%) | RSD (%) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| Extraction | Purification | ||||||||
| Poppy seeds | MOR, COD, THEB, NOS, PAP | AcN/water/formic acid, 80/19/1, v/v/v (100 mL, 30 min, ×2) | − | UHPLC-QqQ-MS/MS | − | 100 | 77–172 | <20.0 | [1] |
| Poppy seeds, cake, buns | MOR, COD, PAP, NOS | MeOH 0.1% acetic acid (30 mL, 60 min) | − | HPLC-QqQ-MS/MS | 70–300 | 200–1000 | − | 7.4–9.0 | [6] |
| Poppy straw | MOR, COD, THEB, PAP | MeOH (5 mL, 20 min, ×2) | − | HPLC-DAD | 200–1800 | 600–5400 | 97–99 | 0.2–0.4 | [17] |
| Hot pot | MOR, COD, THEB, PAP, NOS | HCl 0.1 M (20 mL, 10 min) and PE (10 mL) | SPE (Oasis MCX 60 mg) | UHPLC-QqQ-MS/MS | 0.003–0.04 | 0.01–0.1 | 72–124 | 7.9–23.7 | [19] |
| Poppy straw | MOR, COD, THEB, PAP | Water 5% acetic acid | SPE (Oasis MCX) | HPLC- Ion trap-MS/MS | 400–17,500 | 1100–52,200 | − | − | [20] |
| Hot pot | MOR, COD, THEB, PAP, NOS | Water/AcN 50% (20 mg, 5 min) | MSPE (Fe3O4@SiO2@ADME 50 mg) | HPLC-QqQLIT-MS/MS | 0.05–0.8 | 0.25–2.5 | 80–115 | 4.3–10.7 | [22] |
| Hot pot | MOR, COD, THEB, PAP, NAR | AcN 0.1% formic acid and n-hexane | MSPE (Fe3O4@SiO2@CS/GO 15 mg) | UHPLC- QqQLIT-MS/MS | 0.016–0.092 | 0.036–0.31 | 75–104 | 0.7–9.5 | [23] |
| Poppy capsules | NAR, PAP | SFE | MSPE (Fe3O4@Cu@DPTC 50 mg) | HPLC-UV | 1–100 | − | 88–99 | 5.8–7.7 | [25] |
| Poppy seeds | MOR, COD, THEB, PAP, NOS, ORIP | MeOH/water, 50/50 (v/v) | MSPE (Fe3O4@SiO2@mSiO2 50 mg) | UHPLC-QqQ-MS/MS | 0.07–72.01 | 0.24–240 | 46–109 | 0.4–11 | This work |
| Code | Morphine | Codeine | Thebaine | Papaverine | Noscapine | Oripavine |
|---|---|---|---|---|---|---|
| PS01 | 20.6–45.4 | 5.7–19.2 | <MQL–7.3 | 7.3–19.5 | 9.4–24.8 | <MQL–0.6 |
| PS02 | 23.2–118.7 | 3.2–26.7 | 0.9–17.7 | 2.1–27.1 | 6.5–42.6 | 3.8–22.9 |
| PS03 | 154.9–249.0 | 21.3–45.8 | 12.4–31.5 | 0.8–2.9 | 0.3–108.7 | 9.8–33.4 |
| PS04 | 17.0–19.5 | 1.0–1.9 | 3.4–6.5 | <MQL | <MQL | <MQL |
| PS05 | 1.9–2.2 | <MQL | <MQL | <MQL | <MQL | <MQL |
| PS06 | 1.5–3.7 | <MQL | <MQL | <MQL | <MQL | <MQL |
| PS07 | 6.9–59.3 | 0.4–0.9 | <MQL–5.8 | <MQL–1.1 | 0.7–36.3 | <MQL–0.8 |
| PS08 | 28.8–35.1 | 3.4–4.0 | 0.8–3.4 | 10.5–19.9 | 18.1–22.7 | <MQL |
| PS09 | 58.6–64.3 | 2.9–3.6 | 1.3–3.2 | <MQL | 19.0–21.7 | <MQL |
| PS10 | ND | ND | ND | ND | ND | ND |
| PS11 | 33.7–53.0 | 3.7–8.7 | 37.4–136.2 | <MQL–1.9 | 2.0–21.1 | 2.3–13.8 |
| PS12 | 4.1–22.3 | <MQL-0.3 | <MQL-1.5 | <MQL | <MQL | <MQL-0.5 |
| PS13 | 49.6–125.4 | 2.3–16.5 | 3.9–15.6 | <MQL | <MQL | <MQL-0.5 |
| MIX1 | 6.5–8.7 | <MQL-0.04 | <MQL | <MQL | <MQL | <MQL |
| MIX2 | 44.0–54.0 | 0.7–3.0 | 0.68–2.6 | <MQL | <MQL | <MQL-0.3 |
| MIX3 | 7.0–8.6 | <MQL | <MQL | <MQL | <MQL | <MQL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casado-Hidalgo, G.; Pérez-Quintanilla, D.; Morante-Zarcero, S.; Sierra, I. Mesostructured Silica-Coated Magnetic Nanoparticles to Extract Six Opium Alkaloids in Poppy Seeds Prior to Ultra-High-Performance Liquid Chromatography-Tandem Mass Spectrometry Analysis. Foods 2021, 10, 1587. https://doi.org/10.3390/foods10071587
Casado-Hidalgo G, Pérez-Quintanilla D, Morante-Zarcero S, Sierra I. Mesostructured Silica-Coated Magnetic Nanoparticles to Extract Six Opium Alkaloids in Poppy Seeds Prior to Ultra-High-Performance Liquid Chromatography-Tandem Mass Spectrometry Analysis. Foods. 2021; 10(7):1587. https://doi.org/10.3390/foods10071587
Chicago/Turabian StyleCasado-Hidalgo, Gema, Damián Pérez-Quintanilla, Sonia Morante-Zarcero, and Isabel Sierra. 2021. "Mesostructured Silica-Coated Magnetic Nanoparticles to Extract Six Opium Alkaloids in Poppy Seeds Prior to Ultra-High-Performance Liquid Chromatography-Tandem Mass Spectrometry Analysis" Foods 10, no. 7: 1587. https://doi.org/10.3390/foods10071587
APA StyleCasado-Hidalgo, G., Pérez-Quintanilla, D., Morante-Zarcero, S., & Sierra, I. (2021). Mesostructured Silica-Coated Magnetic Nanoparticles to Extract Six Opium Alkaloids in Poppy Seeds Prior to Ultra-High-Performance Liquid Chromatography-Tandem Mass Spectrometry Analysis. Foods, 10(7), 1587. https://doi.org/10.3390/foods10071587