
iPS Cells—The Triumphs and Tribulations

Abstract
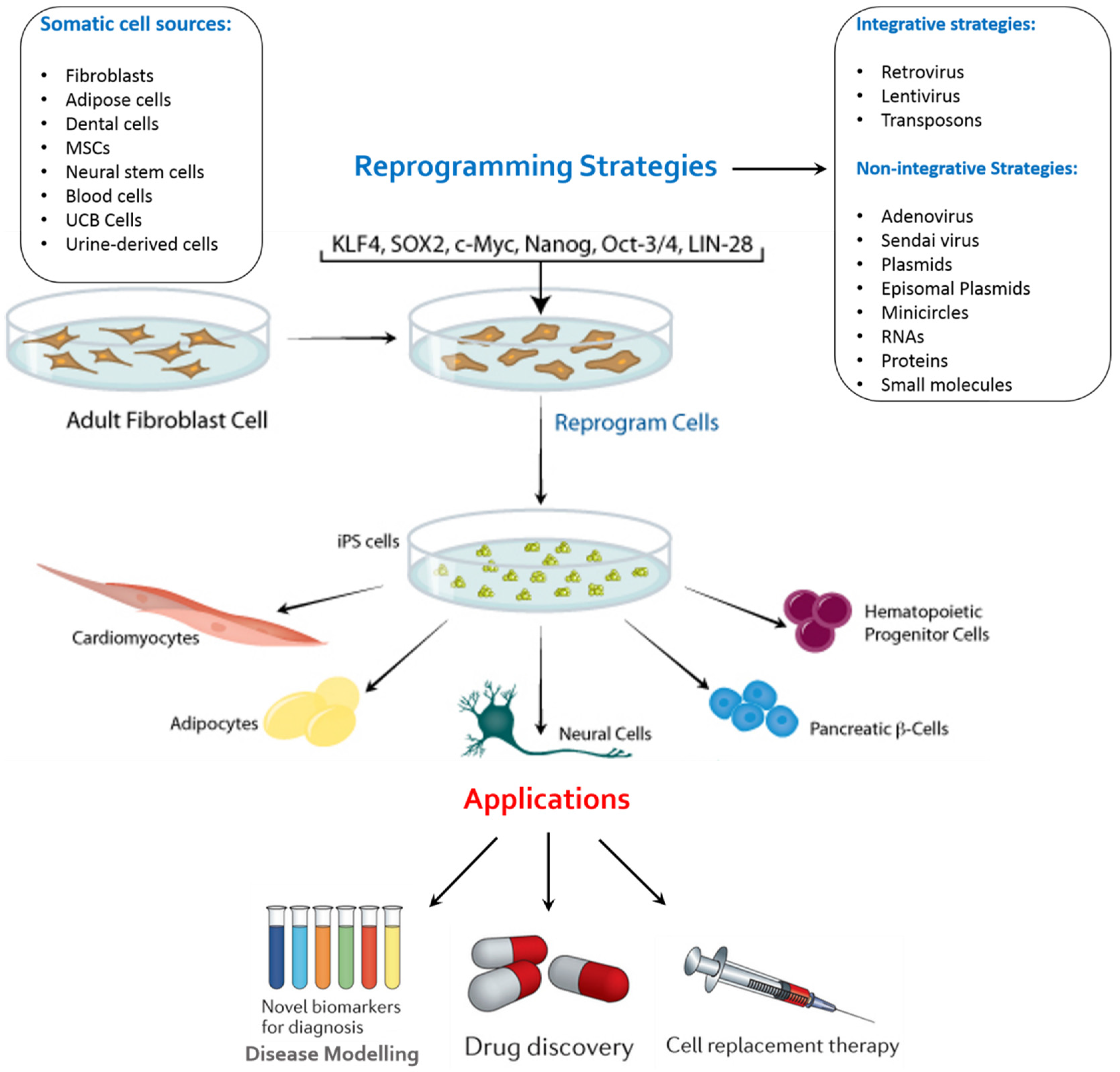
:1. Introduction
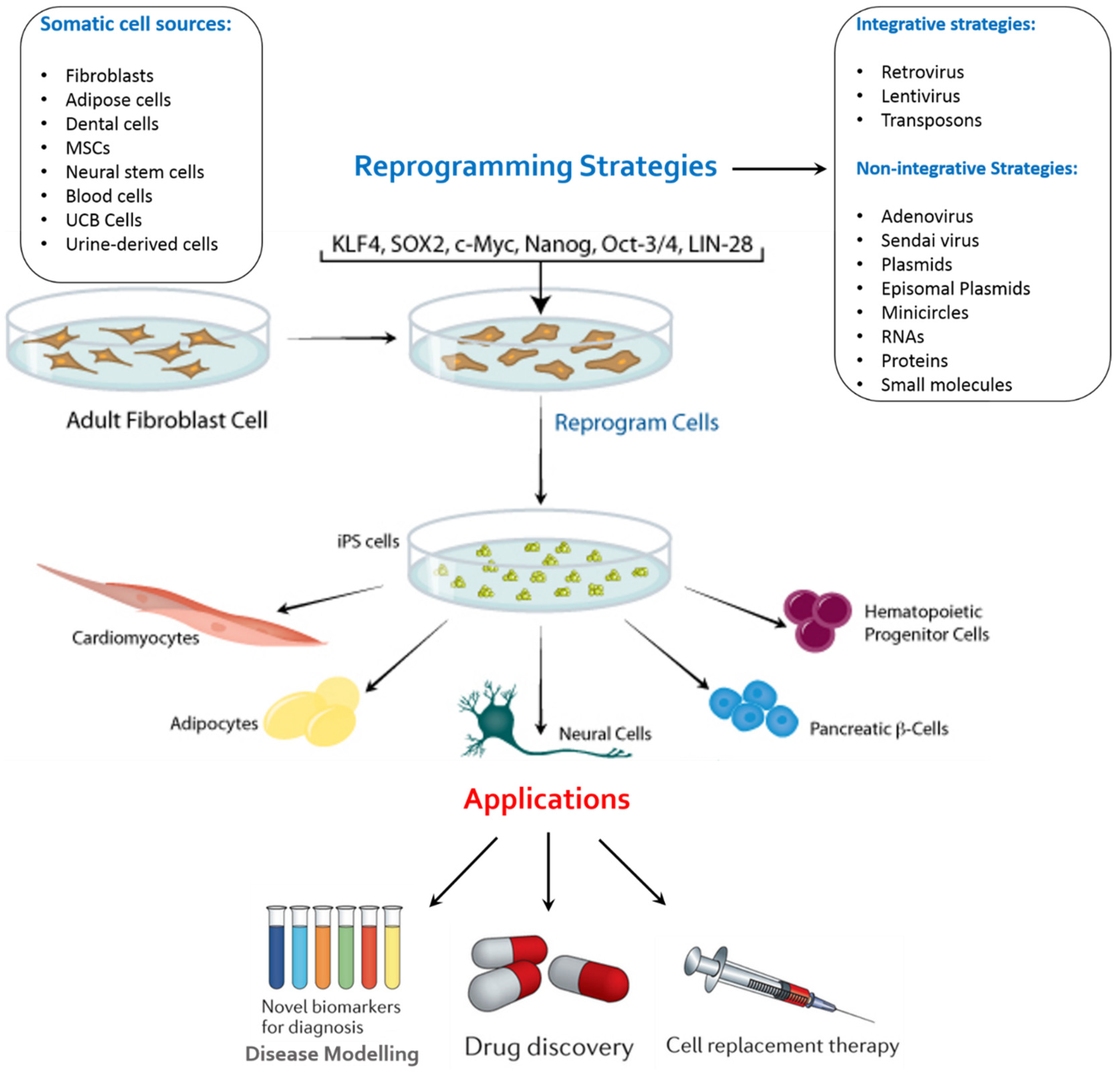
2. Factors of Importance in the Generation of iPSCs
3. A Novel Culture System for iPSCs Derivation
4. Applications of iPSCs
4.1. Disease Remodelling
4.2. Drug Screening
4.3. Regenerative Medicine
4.3.1. First ever Clinical Trial
4.3.2. Ongoing Clinical Trials
4.4. Development of iPSC Library
5. Challenges
5.1. Choosing an Appropriate Somatic Cell Type
5.2. Variability and Heterogeneity
5.3. Validation of Pre-Clinical iPSC Therapies
5.4. Regulatory and Commercial Hurdles
6. Future of iPSCs in Regenerative Dentistry
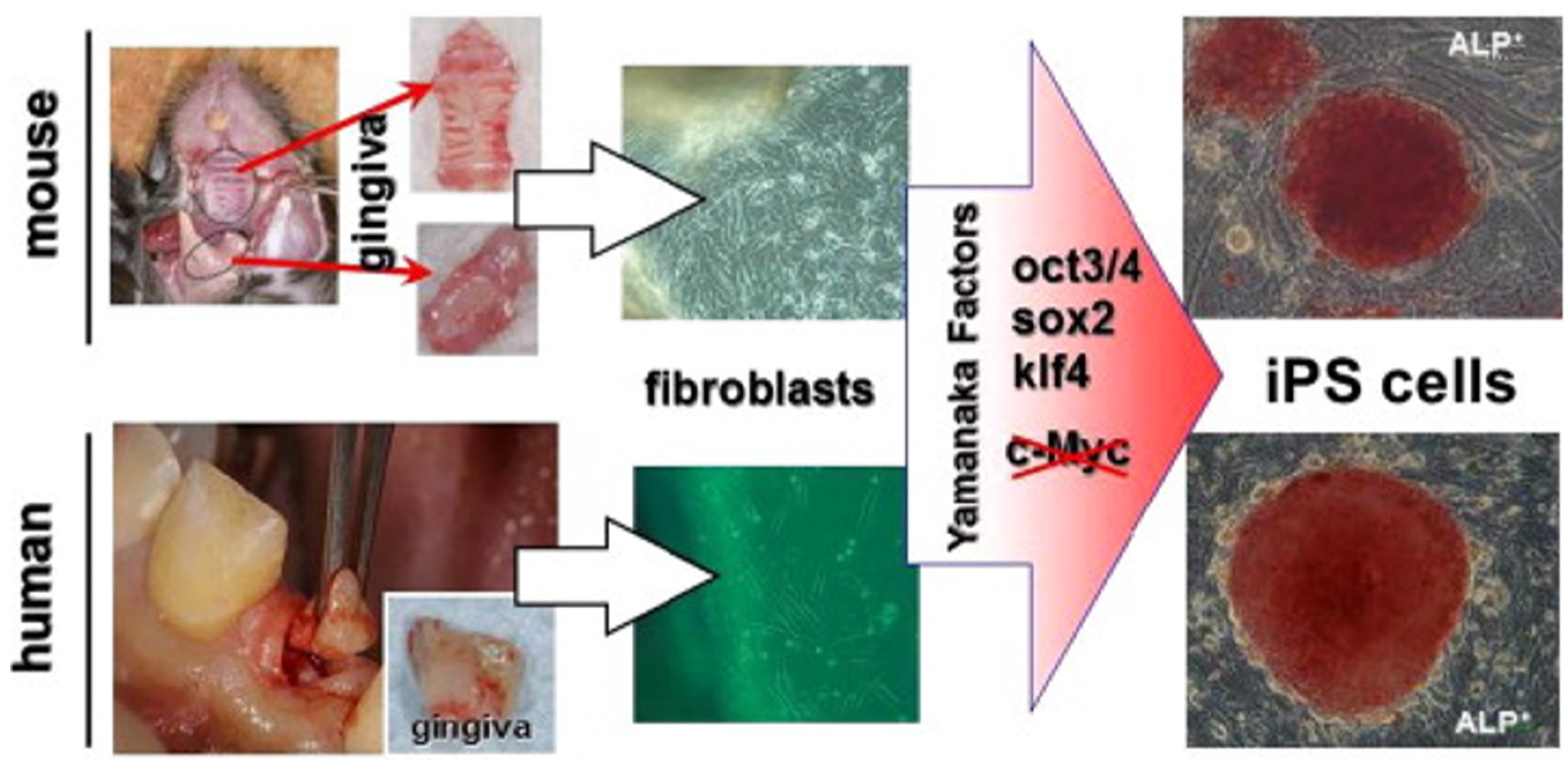
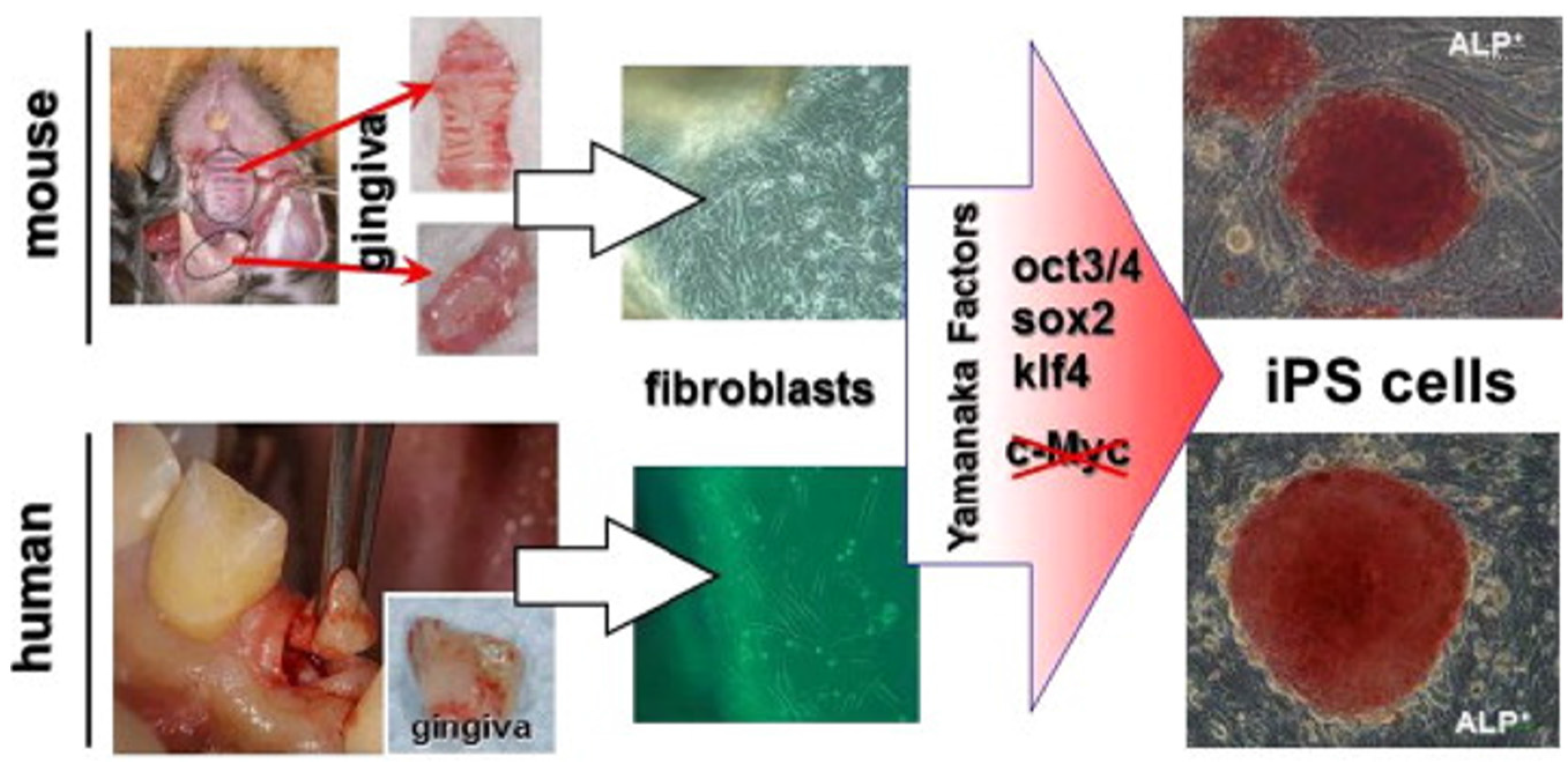
6.1. Gingiva as an iPSC Source
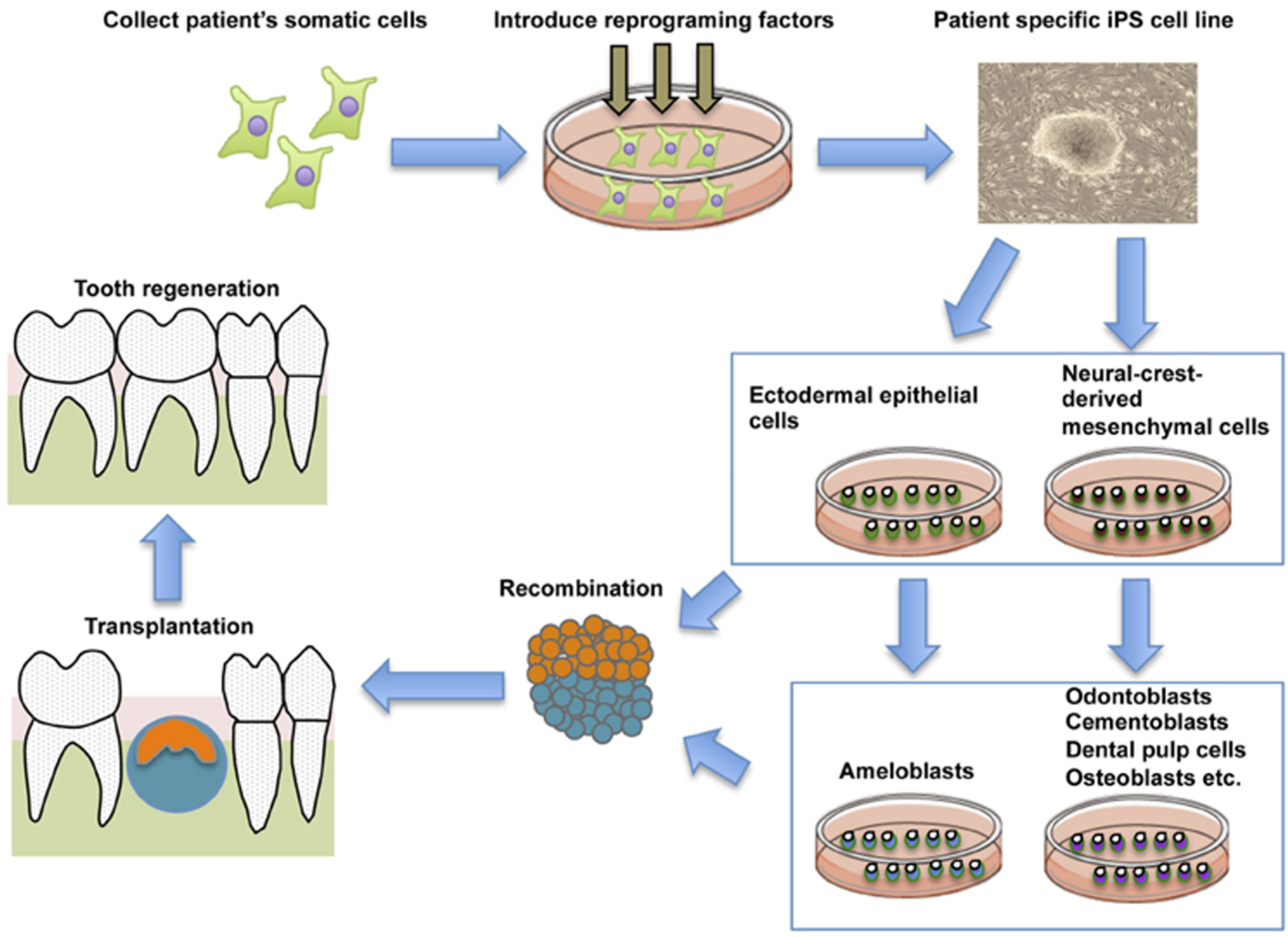
6.2. Tooth Bioengineering
6.3. Periodontal Ligament Regeneration
7. Concluding Remarks and Future Perspectives
Acknowledgments
Conflicts of Interest
Abbreviations
| iPSCs | induced pluripotent stem cells |
| hiPSCs | Human induced pluripotent stem cells |
| OSKM | Oct4, Sox2, Klf4, and c-Myc |
| hESCs | human embryonic stem cells |
| ESC | Embryonic Stem Cells |
| SOX2 | the sex determining region Y-box2 |
| Klf4 | Kruppel-like factor 4 |
| c-Myc | myelocytomatosis oncogene |
| GMP | Good Manufacturing Practice |
| ECM | Extracellular matrix |
| 5hmc | 5-hydroxymethylcytosine |
| RPE | retinal pigment epithelial |
| RBCs | Red Blood Cells |
| DCs | Dedritic cells |
| SSEA | stage-specific embryonic antigen |
| CIRM | California Institute for Regenerative Medicine |
| HLA | human leukocyte antigens |
| CiRA | Center for iPSC Research and Application |
| CiBK | Clinical iPS Cell Bank of Kyoto |
| NC | Neural Crest |
| DF | Dental Follicle |
| PDLSCs | periodontal ligament stem cells |
References
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.; Waknitz, M.; Swiergiel, J.; Marshall, V.; Jones, J. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
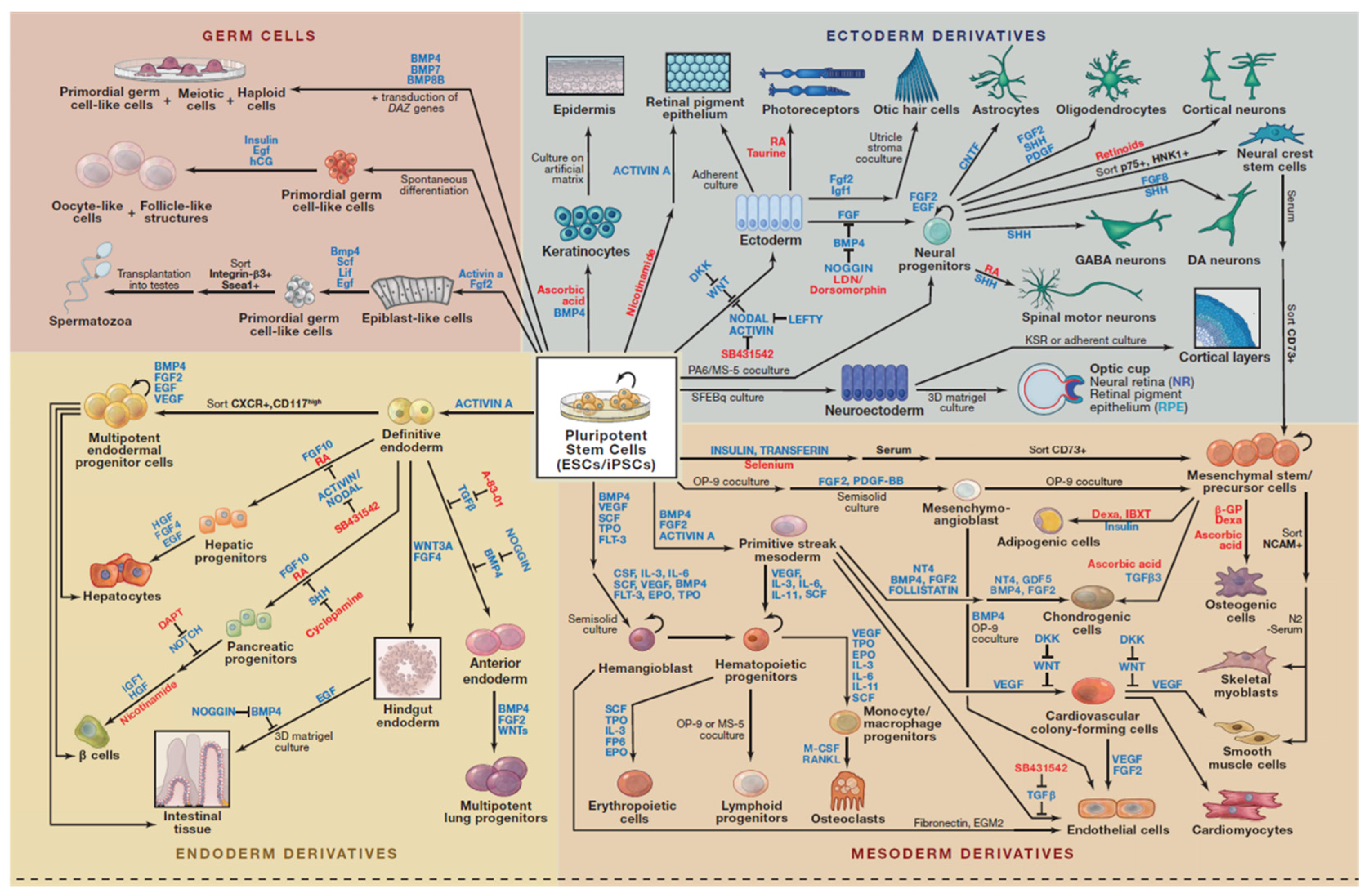
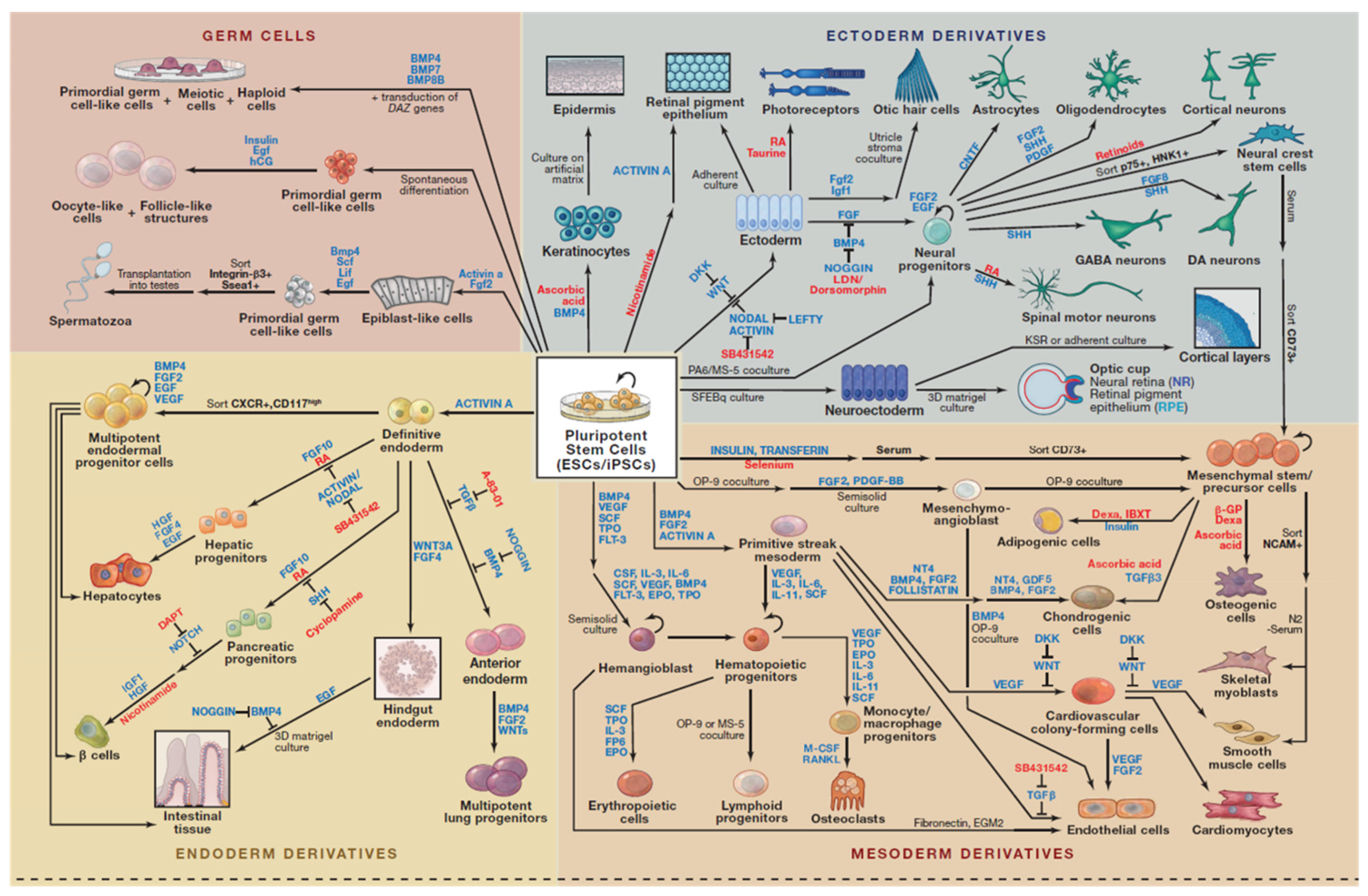
- Williams, L.A.; Davis-Dusenbery, B.N.; Eggan, K.C. SnapShot: Directed Differentiation of Pluripotent Stem Cells. Available online: http://www.sciencedirect.com/science/article/pii/S0092867412005946 (accessed on 29 February 2016).
- Zhao, T.; Zhang, Z.-N.; Rong, Z.; Xu, Y. Immunogenicity of induced pluripotent stem cells. Nature 2011, 474, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced Pluripotent Stem Cells Generated Without Viral Integration. Science 2008, 322, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B 2009, 85, 348–362. [Google Scholar] [CrossRef]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of Mouse Induced Pluripotent Stem Cells without Viral Vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.-H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly Efficient Reprogramming to Pluripotency and Directed Differentiation of Human Cells with Synthetic Modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, C.-H.; Moon, J.-I.; Chung, Y.-G.; Chang, M.-Y.; Han, B.-S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of Human Induced Pluripotent Stem Cells by Direct Delivery of Reprogramming Proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human Induced Pluripotent Stem Cells Free of Vector and Transgene Sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, N.; Gros, E.; Li, H.-R.; Kumar, S.; Deacon, D.C.; Maron, C.; Muotri, A.R.; Chi, N.C.; Fu, X.-D.; Yu, B.D.; et al. Efficient Generation of Human iPSCs by a Synthetic Self-Replicative RNA. Cell Stem Cell 2013, 13, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, L.; Xie, X. iPSCs and small molecules: A reciprocal effort towards better approaches for drug discovery. Acta Pharmacol. Sin. 2013, 34, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent Stem Cells Induced from Mouse Somatic Cells by Small-Molecule Compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Kiskinis, E.; Eggan, K. Progress toward the clinical application of patient-specific pluripotent stem cells. J. Clin. Investig. 2010, 120, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lengner, C.J.; Kirak, O.; Hanna, J.; Cassady, J.P.; Lodato, M.A.; Wu, S.; Faddah, D.A.; Steine, E.J.; Gao, Q.; et al. Reprogramming of Postnatal Neurons into Induced Pluripotent Stem Cells by Defined Factors. Stem Cells 2011, 29, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.-H.; Agarwal, S.; Park, I.-H.; Urbach, A.; Huo, H.; Heffner, G.C.; Kim, K.; Miller, J.D.; Ng, K.; Daley, G.Q. Generation of induced pluripotent stem cells from human blood. Blood 2009, 113, 5476–5479. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.-H.; Hartung, O.; Li, H.; Guo, C.; Sahalie, J.M.; Manos, P.D.; Urbach, A.; Heffner, G.C.; Grskovic, M.; Vigneault, F.; et al. Reprogramming of T Cells from Human Peripheral Blood. Cell Stem Cell 2010, 7, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Yuasa, S.; Oda, M.; Egashira, T.; Yae, K.; Kusumoto, D.; Nakata, H.; Tohyama, S.; Hashimoto, H.; Kodaira, M.; et al. Generation of Induced Pluripotent Stem Cells from Human Terminally Differentiated Circulating T Cells. Cell Stem Cell 2010, 7, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Staerk, J.; Dawlaty, M.M.; Gao, Q.; Maetzel, D.; Hanna, J.; Sommer, C.A.; Mostoslavsky, G.; Jaenisch, R. Reprogramming of Human Peripheral Blood Cells to Induced Pluripotent Stem Cells. Cell Stem Cell 2010, 7, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.E.; Rondon, E.; Rajesh, D.; Mack, A.; Lewis, R.; Feng, X.; Zitur, L.J.; Learish, R.D.; Nuwaysir, E.F. Derivation of Induced Pluripotent Stem Cells from Human Peripheral Blood T Lymphocytes. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, S. Induced Pluripotent Stem Cells: Past, Present, and Future. Cell Stem Cell 2012, 10, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Gulbranson, D.R.; Hou, Z.; Bolin, J.M.; Ruotti, V.; Probasco, M.D.; Smuga-Otto, K.; Howden, S.E.; Diol, N.R.; Propson, N.E.; et al. Chemically defined conditions for human iPS cell derivation and culture. Nat. Methods 2011, 8, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, T.E.; Levenstein, M.E.; Jones, J.M.; Berggren, W.T.; Mitchen, E.R.; Frane, J.L.; Crandall, L.J.; Daigh, C.A.; Conard, K.R.; Piekarczyk, M.S.; et al. Derivation of human embryonic stem cells in defined conditions. Nat. Biotechnol. 2006, 24, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, T.E.; Bergendahl, V.; Levenstein, M.E.; Yu, J.; Probasco, M.D.; Thomson, J.A. Feeder-independent culture of human embryonic stem cells. Nat. Methods 2006, 3, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Beers, J.; Gulbranson, D.R.; George, N.; Siniscalchi, L.I.; Jones, J.; Thomson, J.A.; Chen, G. Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nat. Protoc. 2012, 7, 2029–2040. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chou, B.-K.; Dowey, S.; He, C.; Gerecht, S.; Cheng, L. Scalable expansion of human induced pluripotent stem cells in the defined xeno-free E8 medium under adherent and suspension culture conditions. Stem Cell Res. 2013, 11, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Taniguchi, Y.; Senda, S.; Takizawa, N.; Ichisaka, T.; Asano, K.; Morizane, A.; Doi, D.; Takahashi, J.; Nishizawa, M.; et al. A novel efficient feeder-free culture system for the derivation of human induced pluripotent stem cells. Sci. Rep. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Baghbaderani, B.A.; Tian, X.; Neo, B.H.; Burkall, A.; Dimezzo, T.; Sierra, G.; Rao, M.S. cGMP-Manufactured Human Induced Pluripotent Stem Cells Are Available for Pre-clinical and Clinical Applications. Stem Cell Rep. 2015, 5, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Lutolf, M.P.; Hubbell, J.A. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat. Biotechnol. 2005, 23, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Loskill, P.; Shao, K.; Huebsch, N.; Hong, S.; Marcus, S.G.; Marks, N.; Mandegar, M.; Conklin, B.R.; Lee, L.P.; et al. Human iPSC-based cardiac microphysiological system for drug screening applications. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Wang, L.; Cai, J.; Yuan, Q.; Yang, X.; Yao, X.; Wong, W.M.; Huang, W.; Li, Z.; Wan, J.B.; et al. Transplanted motoneurons derived from human induced pluripotent stem cells form functional connections with target muscle. Stem Cell Res. 2013, 11, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Chau, M.J.; Deveau, T.C.; Song, M.; Gu, X.; Chen, D.; Wei, L. iPSC Transplantation increases regeneration and functional recovery after ischemic stroke in neonatal rats. Stem Cells 2014, 32, 3075–3087. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cao, J.; Wang, Y.; Lan, T.; Liu, L.; Wang, W.; Jin, N.; Gong, J.; Zhang, C.; Teng, F.; et al. Immunogenicity and functional evaluation of iPSC-derived organs for transplantation. Cell Discov. 2015, 1. [Google Scholar] [CrossRef]
- Korecka, J.A.; Levy, S.; Isacson, O. In vivo modeling of neuronal function, axonal impairment and connectivity in neurodegenerative and neuropsychiatric disorders using induced pluripotent stem cells. Mol. Cell. Neurosci. 2016, 73, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Wang, L.; Xu, L.; Jin, N.; Yan, G.; Xia, J.; Wang, H.; Zhuang, G.; Gao, C.; Meng, L.; et al. Induced Pluripotent Stem Cells Can Effectively Differentiate into Multiple Functional Lymphocyte Lineages in Vivo with Negligible Bias. Stem Cells Dev. 2016, 25, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Cherry, A.B.C.; Daley, G.Q. Reprogramming Cellular Identity for Regenerative Medicine. Cell 2012, 148, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Sterneckert, J.L.; Reinhardt, P.; Schöler, H.R. Investigating human disease using stem cell models. Nat. Rev. Genet. 2014, 15, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef] [PubMed]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Byers, B.; Lee, H.; Reijo Pera, R. Modeling Parkinson’s Disease Using Induced Pluripotent Stem Cells. Curr. Neurol. Neurosci. Rep. 2012, 12, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schöndorf, D.C.; Wagner, L.; Glatza, M.; Höing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Chestkov, I.V.; Vasilieva, E.A.; Illarioshkin, S.N.; Lagarkova, M.A.; Kiselev, S.L. Patient-Specific Induced Pluripotent Stem Cells for SOD1-Associated Amyotrophic Lateral Sclerosis Pathogenesis Studies. Acta Nat. 2014, 6, 54–60. [Google Scholar]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways Disrupted in Human ALS Motor Neurons Identified Through Genetic Correction of Mutant SOD1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.-P.; Maragakis, N.J. Induced pluripotent stem cells from ALS patients for disease modeling. Brain Res. 2015, 1607, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Kaye, J.A.; Finkbeiner, S. Modeling Huntington’s disease with induced pluripotent stem cells. Mol. Cell. Neurosci. 2013, 56, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Huang, J.S.; Han, C.; Zhang, G.X.; Xu, X.Y.; Shen, Y.; Li, J.; Jiang, H.Y.; Lin, Z.C.; Xiong, N.; et al. Induced Pluripotent Stem Cells in Huntington’s Disease: Disease Modeling and the Potential for Cell-Based Therapy. Mol. Neurobiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A.; Mason, E.A.; Ovchinnikov, D.A.; Wells, C.A.; Wolvetang, E.J. Concise Review: New Paradigms for Down Syndrome Research Using Induced Pluripotent Stem Cells: Tackling Complex Human Genetic Disease. Stem Cells Transl. Med. 2013, 2, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Soejitno, A.; Prayudi, A.K.A. The prospect of induced pluripotent stem cells for diabetes mellitus treatment. Ther. Adv. Endocrinol. Metab. 2011, 2, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Mali, P.; Huang, X.; Dowey, S.N.; Cheng, L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood 2011, 118, 4599–4608. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Sweeney, C.L.; Chou, B.K.; Choi, U.; Pan, J.; Wang, H.; Dowey, S.N.; Cheng, L.; Malech, H.L. Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: Functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood 2011, 117, 5561–5572. [Google Scholar] [CrossRef] [PubMed]
- Raya, A.; Rodríguez-Pizà, I.; Guenechea, G.; Vassena, R.; Navarro, S.; Barrero, M.J.; Consiglio, A.; Castellà, M.; Río, P.; Sleep, E.; et al. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature 2009, 460, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Zhan, H.; Mali, P.; Dowey, S.; Williams, D.M.; Jang, Y.Y.; Dang, C.V.; Spivak, J.L.; Moliterno, A.R.; Cheng, L. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood 2009, 114, 5473–5480. [Google Scholar] [CrossRef] [PubMed]
- Sebastiano, V.; Maeder, M.L.; Angstman, J.F.; Haddad, B.; Khayter, C.; Yeo, D.T.; Goodwin, M.J.; Hawkins, J.S.; Ramirez, C.L.; Batista, L.F.; et al. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells 2011, 29, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Fong, H.; Wang, C.; Knoferle, J.; Walker, D.; Balestra, M.E.; Tong, L.M.; Leung, L.; Ring, K.L.; Seeley, W.W.; Karydas, A.; et al. Genetic Correction of Tauopathy Phenotypes in Neurons Derived from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2013, 1, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Wong, J.; Wen, J.; Wang, S.; Wang, C.; Spiering, S.; Kan, N.G.; Forcales, S.; Puri, P.L.; Leone, T.C.; et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 2013, 494, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Kaitin, K. Obstacles and Opportunities in New Drug Development. Clin. Pharmacol. Ther. 2008, 83, 210–212. [Google Scholar] [CrossRef] [PubMed]
- Sollano, J.; Kirsch, J.; Bala, M.; Chambers, M.; Harpole, L. The Economics of Drug Discovery and the Ultimate Valuation of Pharmacotherapies in the Marketplace. Clin. Pharmacol. Ther. 2008, 84, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Gunaseeli, I.; Doss, M.; Antzelevitch, C.; Hescheler, J.; Sachinidis, A. Induced Pluripotent Stem Cells as a Model for Accelerated Patient- and Disease-specific Drug Discovery. Curr. Med. Chem. 2010, 17, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Laustriat, D.; Gide, J.; Peschanski, M. Human pluripotent stem cells in drug discovery and predictive toxicology. Biochem. Soc. Trans. 2010, 38. [Google Scholar] [CrossRef] [PubMed]
- Seiler, A.; Visan, A.; Buesen, R.; Genschow, E.; Spielmann, H. Improvement of an in vitro stem cell assay for developmental toxicity: The use of molecular endpoints in the embryonic stem cell test. Reprod. Toxicol. 2004, 18, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wu, H.; Li, Y.; Szulwach, K.E.; Lin, L.; Li, X.; Chen, I.P.; Goldlust, I.S.; Chamberlain, S.J.; Dodd, A.; et al. Subtelomeric hotspots of aberrant 5-hydroxymethylcytosine-mediated epigenetic modifications during reprogramming to pluripotency. Nat. Cell Biol. 2013, 15, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.S.; Byun, K.; Lee, B. Induced pluripotent stem cells and personalized medicine: Current progress and future perspectives. Anat. Cell Biol. 2011, 44. [Google Scholar] [CrossRef] [PubMed]
- Si-Tayeb, K.; Noto, F.K.; Nagaoka, M.; Li, J.; Battle, M.A.; Duris, C.; North, P.E.; Dalton, S.; Duncan, S.A. Highly Efficient Generation of Human Hepatocyte–like Cells from Induced Pluripotent Stem Cells. Hepatology 2010, 51, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Takebe, T.; Sekine, K.; Koike, H.; Zheng, Y.; Taniguchi, H. Identification of proliferating human hepatic cells from human induced pluripotent stem cells. Transplant. Proc. 2014, 46, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, L. Two Effective Routes for Removing Lineage Restriction Roadblocks: From Somatic Cells to Hepatocytes. Int. J. Mol. Sci. 2015, 16, 20873–20895. [Google Scholar] [CrossRef] [PubMed]
- Yusa, K.; Rad, R.; Takeda, J.; Bradley, A. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBAC transposon. Nat. Methods 2009, 6, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Kaji, K.; Norrby, K.; Paca, A.; Mileikovsky, M.; Mohseni, P.; Woltjen, K. Virus free induction of pluripotency and subsequent excision of reprogramming factors. Nature 2009, 458, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Yamazaki, S.; Yamaguchi, T.; Okabe, M.; Masaki, H.; Takaki, S.; Otsu, M.; Nakauchi, H. Generation of Engraftable Hematopoietic Stem Cells From Induced Pluripotent Stem Cells by Way of Teratoma Formation. Mol. Ther. 2013, 21, 1424–1431. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Kim, Y.; Sharkis, S.; Marchionni, L.; Jang, Y.-Y. In Vivo Liver Regeneration Potential of Human Induced Pluripotent Stem Cells from Diverse Origins. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Nori, S.; Okada, Y.; Yasuda, A.; Tsuji, O.; Takahashi, Y.; Kobayashi, Y.; Fujiyoshi, K.; Koike, M.; Uchiyama, Y.; Ikeda, E.; et al. Grafted human-induced pluripotent stem-cell-derived neurospheres promote motor functional recovery after spinal cord injury in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 16825–16830. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.F.; Inoue-Yokoo, T.; Tan, K.S.; Lai, M.I.; Sugiyama, D. Hematopoietic cell differentiation from embryonic and induced pluripotent stem cells. Stem Cell Res. Ther. 2013, 4. [Google Scholar] [CrossRef]
- Laflamme, M.A.; Chen, K.Y.; Naumova, A.V.; Muskheli, V.; Fugate, J.A.; Dupras, S.K.; Reinecke, H.; Xu, C.; Hassanipour, M.; Police, S.; et al. Cardiomyocytes derived from human embryonic stem cells in pro- survival factors enhance function of infarcted rat hearts. Nat. Biotechnol. 2007, 25, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Wagner, R.A.; Wilson, K.D.; Xie, X.; Fu, J.-D.; Drukker, M.; Lee, A.; Li, R.A.; Gambhir, S.S.; Weissman, I.L.; et al. Transcriptional and Functional Profiling of Human Embryonic Stem Cell-Derived Cardiomyocytes. PLoS ONE 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Levenberg, S.; Ferreira, L.S.; Chen-Konak, L.; Kraehenbuehl, T.P.; Langer, R. Isolation, differentiation and characterization of vascular cells derived from human embryonic stem cells. Nat. Protoc. 2010, 5, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Senju, S.; Haruta, M.; Matsunaga, Y.; Fukushima, S.; Ikeda, T.; Takahashi, K.; Okita, K.; Yamanaka, S.; Nishimura, Y. Characterization of Dendritic Cells and Macrophages Generated by Directed Differentiation from Mouse Induced Pluripotent Stem Cells. Stem Cells 2009, 27, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Kamao, H.; Mandai, M.; Okamoto, S.; Sakai, N.; Suga, A.; Sugita, S.; Kiryu, J.; Takahashi, M. Characterization of Human Induced Pluripotent Stem Cell-Derived Retinal Pigment Epithelium Cell Sheets Aiming for Clinical Application. Stem Cell Rep. 2014, 2, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Kanemura, H.; Go, M.J.; Shikamura, M.; Nishishita, N.; Sakai, N.; Kamao, H.; Mandai, M.; Morinaga, C.; Takahashi, M.; Kawamata, S. Tumorigenicity Studies of Induced Pluripotent Stem Cell (iPSC)-Derived Retinal Pigment Epithelium (RPE) for the Treatment of Age-Related Macular Degeneration. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Stem Cells Cruise to Clinic. Nature News & Comment. Available online: http://www.nature.com/news/stem-cells-cruise-to-clinic-1.12511 (accessed on 29 February 2016).
- Next-Generation Stem Cells Cleared for Human Trial. Nature News & Comment. Available online: http://www.nature.com/news/next-generation-stem-cells-cleared-for-human-trial-1.15897 (accessed on 29 February 2016).
- Japanese Woman is First Recipient of Next-Generation Stem Cells. Nature News & Comment. Available online: http://www.nature.com/news/japanese-woman-is-first-recipient-of-next-generation-stem-cells-1.15915 (accessed on 29 February 2016).
- First iPS Cell Transplant Patient Makes Progress One Year on. Japan Times. Available online: http://www.japantimes.co.jp/news/2015/10/02/national/science-health/first-ips-cell-transplant-patient-makes-progress-oneyear#.VqeIOvmLSUk (accessed on 29 February 2016).
- Zhao, T.; Zhang, Z.N.; Westenskow, P.D.; Todorova, D.; Hu, Z.; Lin, T.; Rong, Z.; Kim, J.; He, J.; Wang, M.; et al. Humanized mice reveal differential immunogenicity of cells derived from autologous induced pluripotent stem cells. Cell Stem Cell 2015, 17, 353–359. [Google Scholar] [CrossRef] [PubMed]
- A Crucial Moment in Time for Stem Cell R&D. Biotechnology Focus. Available online: http://biotechnologyfocus.ca/a-crucial-moment-in-time-forstem-cell-rd/ (accessed on 29 February 2016).
- Fernandes, S.; Chong, J.J.H.; Paige, S.L.; Iwata, M.; Torok-Storb, B.; Keller, G.; Murry, C.E. Comparison of Human Embryonic Stem Cell-Derived Cardiomyocytes, Cardiovascular Progenitors, and Bone Marrow Mononuclear Cells for Cardiac Repair. Stem Cell Rep. 2015, 5, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.J.H.; Yang, X.; Don, C.W.; Minami, E.; Liu, Y.-W.; Weyers, J.J.; Murry, C.E. Human Embryonic Stem Cell-Derived Cardiomyocytes Regenerate Non-Human Primate Hearts. Nature 2014, 510, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Menasché, P.; Vanneaux, V.; Hagège, A.; Bel, A.; Cholley, B.; Cacciapuoti, I.; Parouchev, A.; Benhamouda, N.; Tachdjian, G.; Tosca, L.; et al. Human embryonic stem cell-derived cardiac progenitors for severe heart failure treatment: First clinical case report. Eur. Heart J. 2015, 36, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Menasché, P.; Vanneaux, V.; Fabreguettes, J.R.; Bel, A.; Tosca, L.; Garcia, S.; Bellamy, V.; Farouz, Y.; Pouly, J.; Damour, O.; et al. Towards a clinical use of human embryonic stem cell-derived cardiac progenitors: A translational experience. Eur. Heart J. 2015, 36, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Spinal Cord Injury and a CIRM-Funded Stem Cell-Based Trial. Available online: http://blog.cirm.ca.gov/2015/10/22/video-spinal-cord-injury-and-a-cirm-funded-stem-cell-based-trial/ (accessed on 29 February 2016).
- Revolutionary Stem Cell Therapy Trial for Parkinson’s Disease to be Held in Australia. ABC News. Available online: http://www.abc.net.au/news/2015-12-15/stem-cell-trial-for-parkinson’s-disease-in-australia/7029722 (accessed on 29 February 2016).
- Turner, M.; Leslie, S.; Martin, N.G.; Peschanski, M.; Rao, M.; Taylor, C.J.; Trounson, A.; Turner, D.; Yamanaka, S.; Wilmut, I. Toward the development of a global induced pluripotent stem cell library. Cell Stem Cell 2013, 13, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Fairchild, P.J. Taming the lion: The challenge of immunity in regenerative medicine. Regen. Med. 2015, 10, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Kiskinis, E.; Verstappen, G.; Gu, H.; Boulting, G.; Smith, Z.D.; Ziller, M.; Croft, G.F.; Amoroso, M.W.; Oakley, D.H.; et al. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 2011, 144, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.J.; Peacock, S.; Chaudhry, A.N.; Bradley, J.A.; Bolton, E.M. Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell 2012, 11, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, F.; Tokunaga, K.; Nakatsuji, N. Human Leukocyte Antigen Matching Estimations in a Hypothetical Bank of Human Embryonic Stem Cell Lines in the Japanese Population for Use in Cell Transplantation Therapy. Stem Cells 2007, 25, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuji, N.; Nakajima, F.; Tokunaga, K. HLA-haplotype banking and iPS cells. Nat. Biotechnol. 2008, 26, 739–740. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.K.; Matsunaga, A.; Takasu, N.; Yamanaka, S. Donor Recruitment and Eligibility Criteria for HLA-Homozygous iPS Cell Bank in Japan. In Stem Cell Banking; Ilic, D., Ed.; Springer: New York, NY, USA, 2014. [Google Scholar]
- Nature. Stem-Cell Pioneer Banks on Future Therapies. Available online: http://www.nature.com/news/stem-cell-pioneer-banks-on-future-therapies-1.11129 (accessed on 29 February 2016).
- Leha, A.; Moens, N.; Meleckyte, R.; Culley, O.J.; Gervasio, M.K.; Kerz, M.; Reimer, A.; Cain, S.; Streeter, I.; Folarin, A.; et al. A high-content platform to characterise human induced pluripotent stem cell lines. Methods 2016, 96, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Moad, M.; Pal, D.; Hepburn, A.C.; Williamson, S.C.; Wilson, L.; Lako, M.; Armstrong, L.; Hayward, S.W.; Franco, O.E.; Cates, J.M.; et al. A Novel Model of Urinary Tract Differentiation, Tissue Regeneration, and Disease: Reprogramming Human Prostate and Bladder Cells into Induced Pluripotent Stem Cells. Eur. Urol. 2013, 64, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Longaker, M.T.; Wu, J.C. Human iPS cell-based therapy: Considerations before clinical applications. Cell Cycle 2010, 9, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilić, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Panetta, N.J.; Gupta, D.M.; Wilson, K.D.; Lee, A.; Jia, F.; Hu, S.; Cherry, A.M.; Robbins, R.C.; Longaker, M.T.; et al. Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 15720–15725. [Google Scholar] [CrossRef] [PubMed]
- Utikal, J.; Maherali, N.; Kulalert, W.; Hochedlinger, K. Sox2 is dispensable for the reprogramming of melanocytes and melanoma cells into induced pluripotent stem cells. J. Cell Sci. 2009, 122, 3502–3510. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, A.; Montserrat, N.; Aasen, T.; Gonzalez, F.; Rodríguez-Pizà, I.; Vassena, R.; Raya, A.; Boué, S.; Barrero, M.J.; Corbella, B.A.; et al. Generation of Induced Pluripotent Stem Cells from Human Cord Blood Using OCT4 and SOX2. Cell Stem Cell 2009, 5, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Haase, A.; Olmer, R.; Schwanke, K.; Wunderlich, S.; Merkert, S.; Hess, C.; Zweigerdt, R.; Gruh, I.; Meyer, J.; Wagner, S.; et al. Generation of Induced Pluripotent Stem Cells from Human Cord Blood. Cell Stem Cell 2009, 5, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Greber, B.; Araúzo-Bravo, M.J.; Meyer, J.; Park, K.I.; Zaehres, H.; Schöler, H.R. Direct reprogramming of human neural stem cells by OCT4. Nature 2009, 461, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, Z.; Wilson, K.D.; Wu, Y.; Hu, S.; Quertermous, T.; Wu, J.C. Persistent Donor Cell Gene Expression among Human Induced Pluripotent Stem Cells Contributes to Differences with Human Embryonic Stem Cells. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhang, Y. Genetic and epigenetic variations in iPSCs: Potential causes and implications for application. Cell Stem Cell 2013, 13, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Zhao, R.; Doi, A.; Ng, K.; Unternaehrer, J.; Cahan, P.; Huo, H.; Loh, Y.H.; Aryee, M.J.; Lensch, M.W.; et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 1117–1119. [Google Scholar] [CrossRef] [PubMed]
- Polo, J.M.; Liu, S.; Figueroa, M.E.; Kulalert, W.; Eminli, S.; Tan, K.Y.; Apostolou, E.; Stadtfeld, M.; Li, Y.; Shioda, T.; et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 2010, 28, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhang, Y. Embryonic stem cell and induced pluripotent stem cell: An epigenetic perspective. Cell Res. 2013, 23, 49–69. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Shoemaker, R.; Xie, B.; Gore, A.; LeProust, E.M.; Antosiewicz-Bourget, J.; Egli, D.; Maherali, N.; Park, I.; Yu, J.; et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat. Biotechnol. 2009, 27, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O’Malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of Aberrant Epigenomic Reprogramming in Human Induced Pluripotent Stem Cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Nishino, K.; Toyoda, M.; Yamazaki-Inoue, M.; Fukawatase, Y.; Chikazawa, E.; Sakaguchi, H.; Akutsu, H.; Umezawa, A. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Diep, D.; Gore, A.; Panopoulos, A.D.; Montserrat, N.; Plongthongkum, N.; Kumar, S.; Fung, H.L.; Giorgetti, A.; Bilic, J.; et al. Identification of a specific reprogramming-associated epigenetic signature in human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 16196–16201. [Google Scholar] [CrossRef] [PubMed]
- Narsinh, K.H.; Sun, N.; Sanchez-Freire, V.; Lee, A.S.; Almeida, P.; Hu, S.; Jan, T.; Wilson, K.D.; Leong, D.; Rosenberg, J.; et al. Single cell transcriptional profiling reveals heterogeneity of human induced pluripotent stem cells. J. Clin. Investig. 2011, 121, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhu, F.; Yong, J.; Zhang, P.; Hou, P.; Li, H.; Jiang, W.; Cai, J.; Liu, M.; Cui, K.; et al. Generation of Induced Pluripotent Stem Cells from Adult Rhesus Monkey Fibroblasts. Cell Stem Cell 2008, 3, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Esteban, M.A.; Xu, J.; Yang, J.; Peng, M.; Qin, D.; Li, W.; Jiang, Z.; Chen, J.; Deng, K.; Zhong, M.; et al. Generation of Induced Pluripotent Stem Cell Lines from Tibetan Miniature Pig. J. Biol. Chem. 2009, 284, 17634–17640. [Google Scholar] [CrossRef] [PubMed]
- Alexenko, A.P.; Sachdev, S.; Sinha, S.; Roberts, R.M.; Ezashi, T.B.P.; Telugu, V.L. Derivation of induced pluripotent stem cells from pig somatic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 10993–10998. [Google Scholar]
- Wu, Z.; Chen, J.; Ren, J.; Bao, L.; Liao, J.; Cui, C.; Rao, L.; Li, H.; Gu, Y.; Dai, H.; et al. Generation of Pig Induced Pluripotent Stem Cells with a Drug-Inducible System. J. Mol. Cell Biol. 2009, 1, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Swijnenburg, R.-J.; Schrepfer, S.; Govaert, J.A.; Cao, F.; Ransohoff, K.; Sheikh, A.Y.; Haddad, M.; Connolly, A.J.; Davis, M.M.; Robbins, R.C.; et al. Immunosuppressive therapy mitigates immunological rejection of human embryonic stem cell xenografts. Proc. Natl. Acad. Sci. USA 2008, 105, 12991–12996. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.W.; Cavanagro, J.; Deans, R.; Feigel, E.; Horowitz, E.; Keating, A.; Rao, M.; Turner, M.; Wilmut, I.; Yamanaka, S. Harmonizing standards for producing clinical-grade therapies from pluripotent stem cells. Nat. Biotechnol. 2014, 32, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Mehta, J.; Frankfurt, O.; Altman, J.; Evens, A.; Tallman, M.; Gordon, L.; Williams, S.; Winter, J.; Krishnamurthy, J.; Duffey, S.; et al. Optimizing the CD34+cell dose for reduced-intensity allogeneic hematopoietic stem cell transplantation. Leuk. Lymphoma 2009, 50, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, D.E.; Jing, D.; Lock, L.T.; Tzanakakis, E.S. Scalable Stirred-Suspension Bioreactor Culture of Human Pluripotent Stem Cells. Tissue Eng. Part A 2010, 16, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Egusa, H.; Okita, K.; Kayashima, H.; Yu, G.; Fukuyasu, S.; Saeki, M.; Matsumoto, T.; Yamanaka, S.; Yatani, H. Gingival Fibroblasts as a Promising Source of Induced Pluripotent Stem Cells. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Arakaki, M.; Egusa, H.; Otsu, K.; Saitoh, I.; Miura, T.; Harada, H. Frontier dental research on iPS cells. J. Oral Biosci. 2013, 55, 191–199. [Google Scholar] [CrossRef]
- Stephens, P.; Davies, K.J.; Occleston, N.; Pleass, R.D.; Kon, C.; Daniels, J.; Khaw, P.T.; Thomas, D.W. Skin and oral fibroblasts exhibit phenotypic differences in extracellular matrix reorganization and matrix metalloproteinase activity. Bri. J. Dermatol. 2001, 144, 229–237. [Google Scholar] [CrossRef]
- Sciubba, J.J.; Waterhouse, J.P.; Meyer, J. A fine structural comparison of the healing of incisional wounds of mucosa and skin. J. Oral Pathol. Med. 1978, 7, 214–227. [Google Scholar] [CrossRef]
- Walsh, L.J.; L’Estrange, P.R.; Seymour, G.J. High magnification in situ viewing of wound healing in oral mucosa. Aust. Dent. J. 1996, 41, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Egusa, H.; Sonoyama, W.; Nishimura, M.; Atsuta, I.; Akiyama, K. Stem cells in dentistry Part I: Stem cell sources. J. Prosthodont. Res. 2012, 56, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulou, C.; Cimasoni, G. Functional Characteristics of Gingival and Periodontal Ligament Fibroblasts. J. Dent. Res. 1996, 75, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Thesleff, I.; Sharpe, P. Signalling networks regulating dental development. Mech. Dev. 1997, 67, 111–123. [Google Scholar] [CrossRef]
- Jernvall, J.; Thesleff, I. Reiterative signaling and patterning during mammalian tooth morphogenesis. Mech. Dev. 2000, 92, 19–29. [Google Scholar] [CrossRef]
- Ruch, J.; Lesot, H.; Begue-Kirn, C. Odontoblast differentiation. Int. J. Dev. Biol. 1995, 39, 51–68. [Google Scholar] [PubMed]
- Imai, H.; Osumi-Yamashita, N.; Ninomiya, Y.; Eto, K. Contribution of Early-Emigrating Midbrain Crest Cells to the Dental Mesenchyme of Mandibular Molar Teeth in Rat Embryos. Dev. Biol. 1996, 176, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Otsu, K.; Kumakami-Sakano, M.; Fujiwara, N.; Kikuchi, K.; Keller, L.; Lesot, H.; Harada, H. Stem cell sources for tooth regeneration: Current status and future prospects. Front. Physiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Inoue, K.; Nakajima, K.; Tachikawa, T.; Yamazaki, H.; Isobe, T.; Sugawara, A.; Ogawa, M.; Tanaka, C.; Saito, M.; et al. Functional tooth restoration by next-generation bio-hybrid implant as a bio-hybrid artificial organ replacement therapy. Sci. Rep. 2014, 13. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Akamine, A. Quest for the development of tooth root/periodontal ligament complex by tissue engineering. Integr. Mol. Med. 2014, 1. [Google Scholar] [CrossRef]
- Avior, Y.; Sagi, I.; Benvenisty, N. Pluripotent stem cells in disease modelling and drug testing. Nat. Rev. Mol. Cell Biol. 2016, 17, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Trounson, A.; DeWitt, N.D. Pluripotent stem cells progressing to the clinic. Nat. Rev. Mol. Cell Biol. 2016, 17, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Soldner, F.; Beard, C.; Gao, Q.; Mitalipova, M.; DeKelver, R.C.; Jaenisch, R. Highly efficient gene targeting of expressed and silent genes in human ESCs and iPSCs using zinc finger nucleases. Nat. Biotechnol. 2009, 27, 851–857. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Source | Derivation | In vitro Expansion | Reprogramming Efficiency (4 factor) | Reprogramming Speed | Reprogramming Factors | References |
|---|---|---|---|---|---|---|
| Skin Fibroblasts | Skin Biopsy | Yes | ~0.01% (Adult cells) | >21 days | OSKM, OSK, OSNL | [5] |
| Keratinocytes | Skin Biopsy | Yes | ~1% (neonatal and juvenile cells) | >10days | OSKM, OSK | [112] |
| CD34 Blood Cells | Peripheral Blood undergo G-CSF stimulation | No | ~0.01-0.02% (Adult Cells) | >14days | OSKM | [22] |
| Adipose Stem Cells | Lipoaspiration | No | ~0.2% (Adult Cells) | >13–14 days | OSKM | [113] |
| Melanocytes | Skin Biopsy | Yes | ~0.05% (not known) | >10 days | OSKM, OKM | [114] |
| Cord Blood Cells | Collected at birth from cord cells | No | ~0.01% (neonatal cells) | >12–15 days | OSKM, OSNL, OSK, OS | [115,116] |
| Neural Stem Cells | NA | Yes | 0.004% (1 Factor, Fetal cells) | >7–8 Weeks (1Factor) | OK, O | [117] |
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, R. iPS Cells—The Triumphs and Tribulations. Dent. J. 2016, 4, 19. https://doi.org/10.3390/dj4020019
Sharma R. iPS Cells—The Triumphs and Tribulations. Dentistry Journal. 2016; 4(2):19. https://doi.org/10.3390/dj4020019
Chicago/Turabian StyleSharma, Riddhi. 2016. "iPS Cells—The Triumphs and Tribulations" Dentistry Journal 4, no. 2: 19. https://doi.org/10.3390/dj4020019
APA StyleSharma, R. (2016). iPS Cells—The Triumphs and Tribulations. Dentistry Journal, 4(2), 19. https://doi.org/10.3390/dj4020019