Crystal Structures of [Fe]-Hydrogenase from Methanolacinia paynteri Suggest a Path of the FeGP-Cofactor Incorporation Process



Abstract
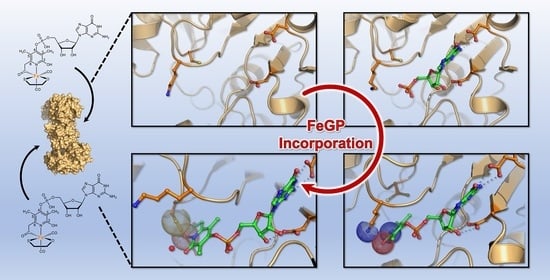
:
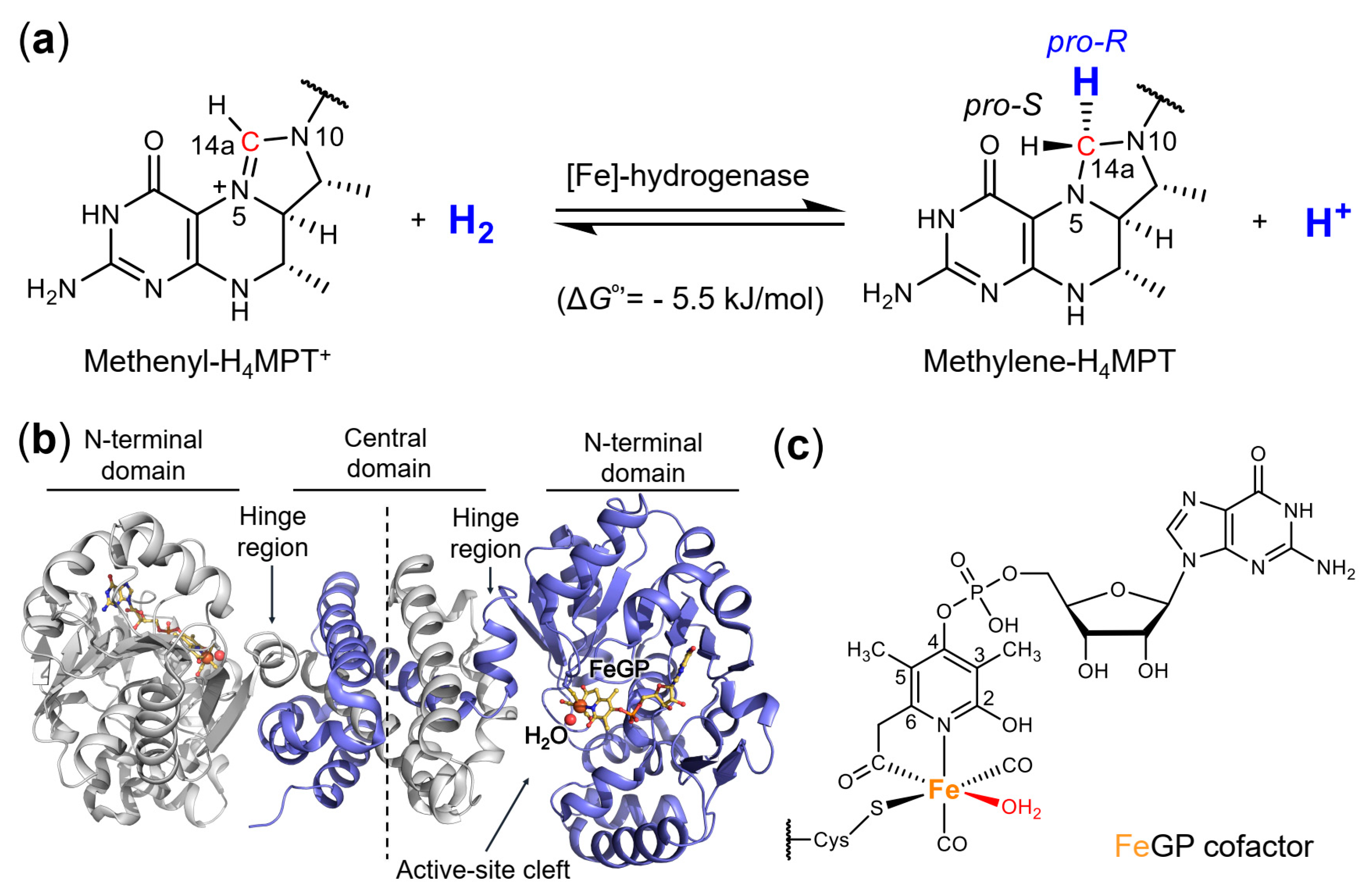
1. Introduction
2. Results and Discussion
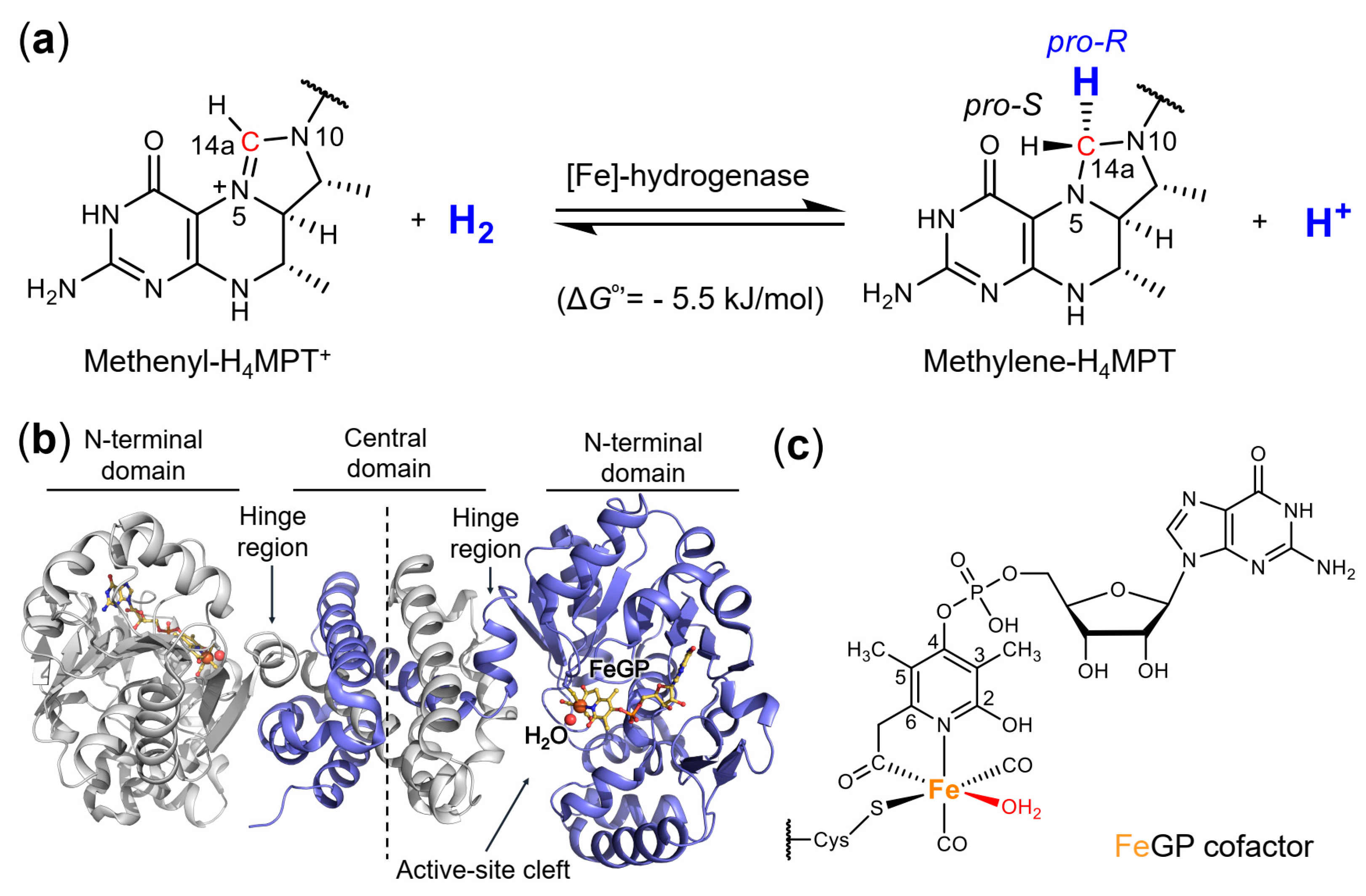
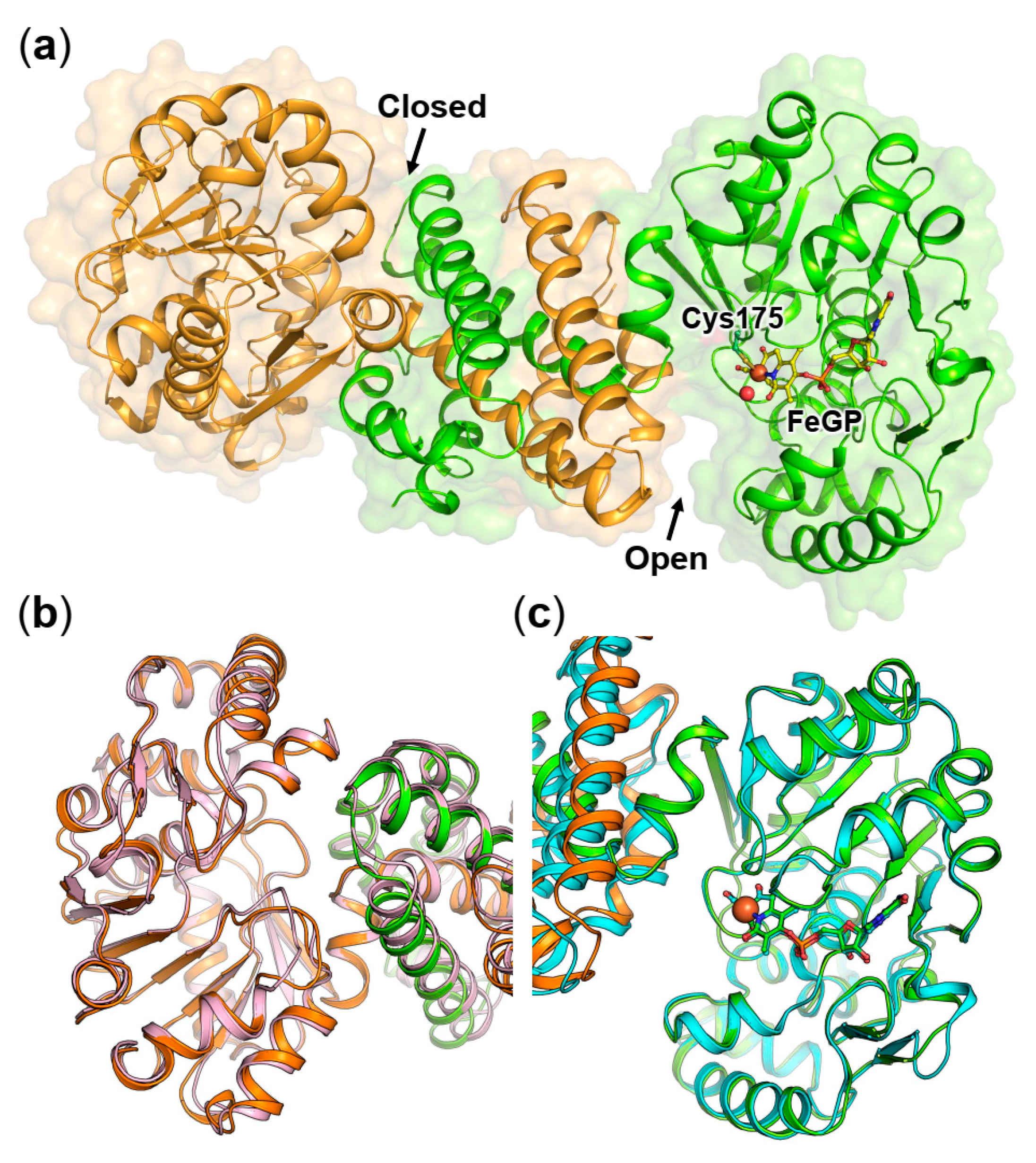
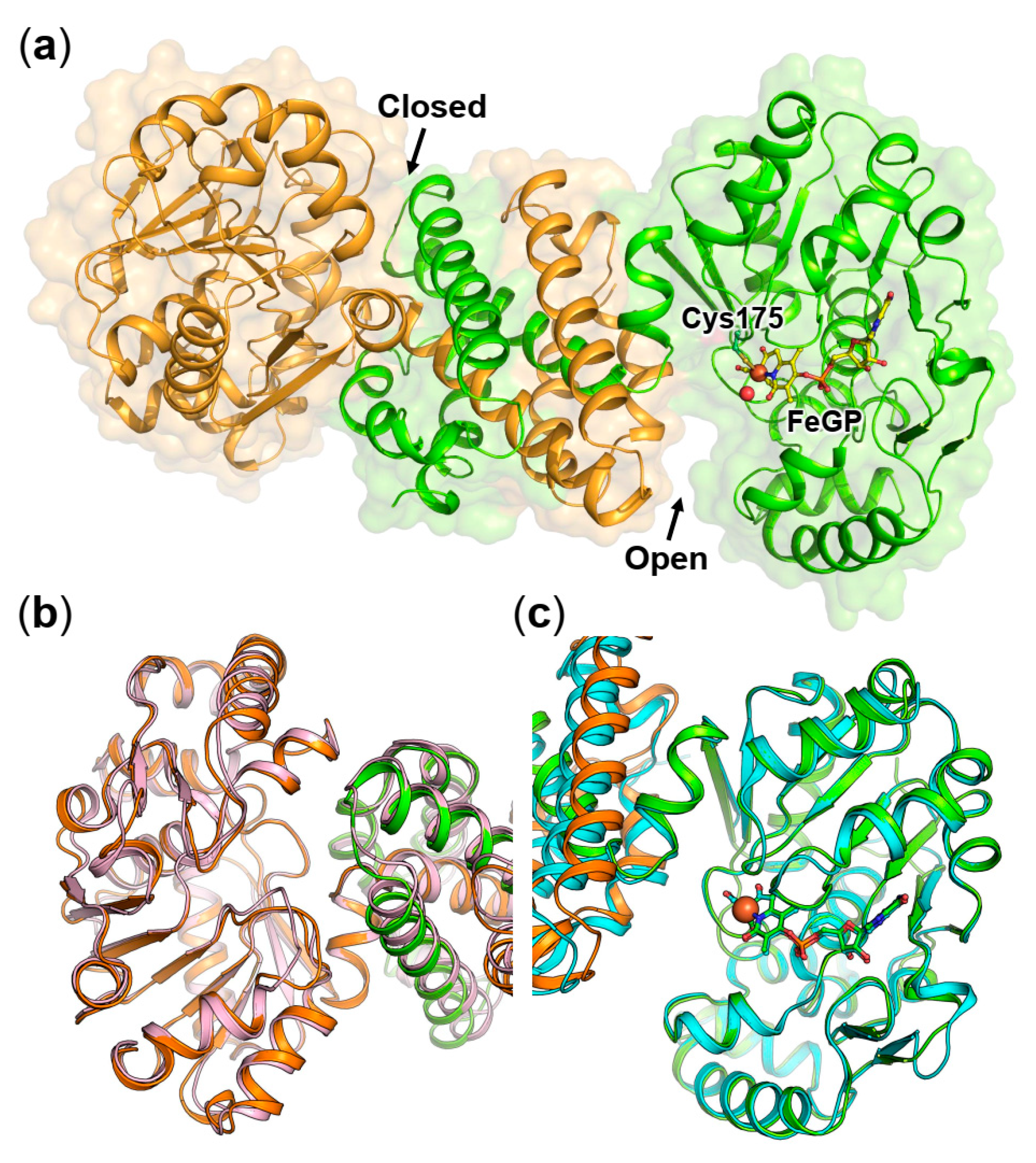
2.1. Crystal Structure of the Asymmetric Homodimer of pHmd
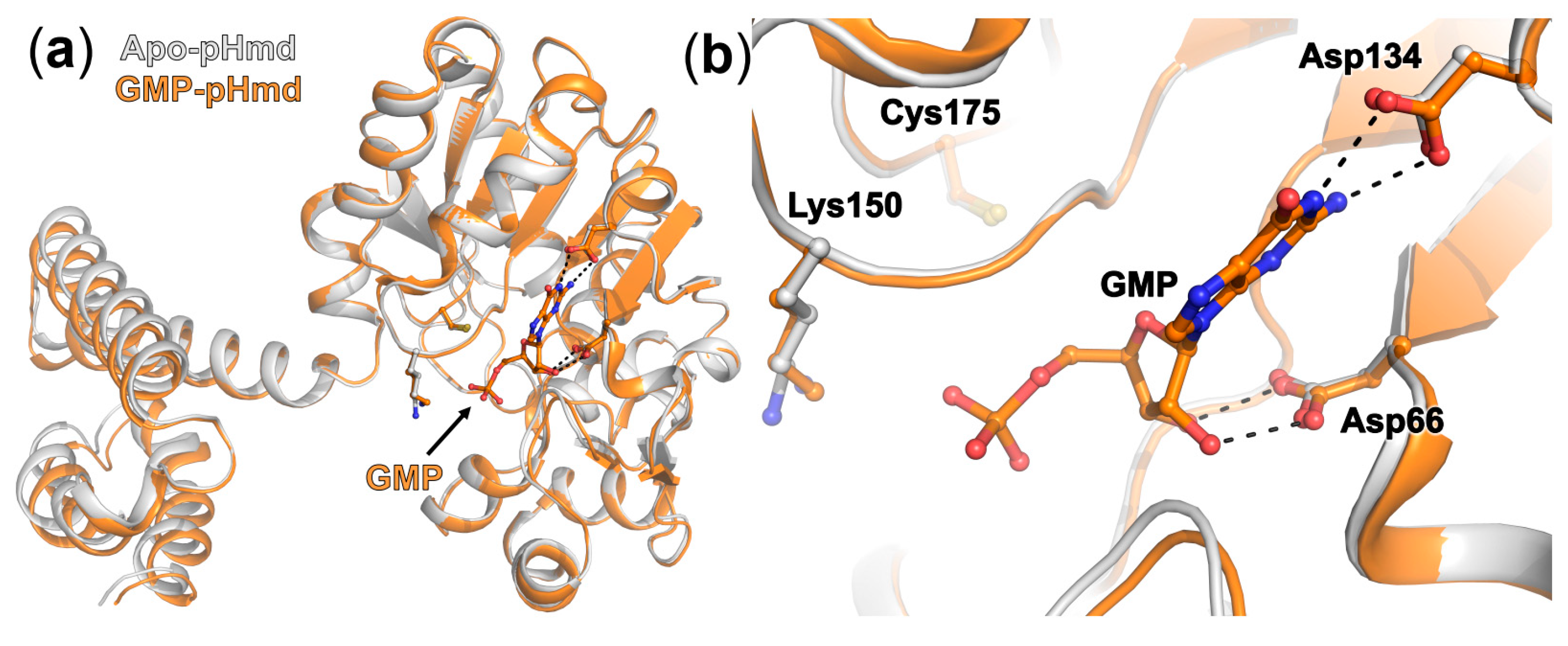
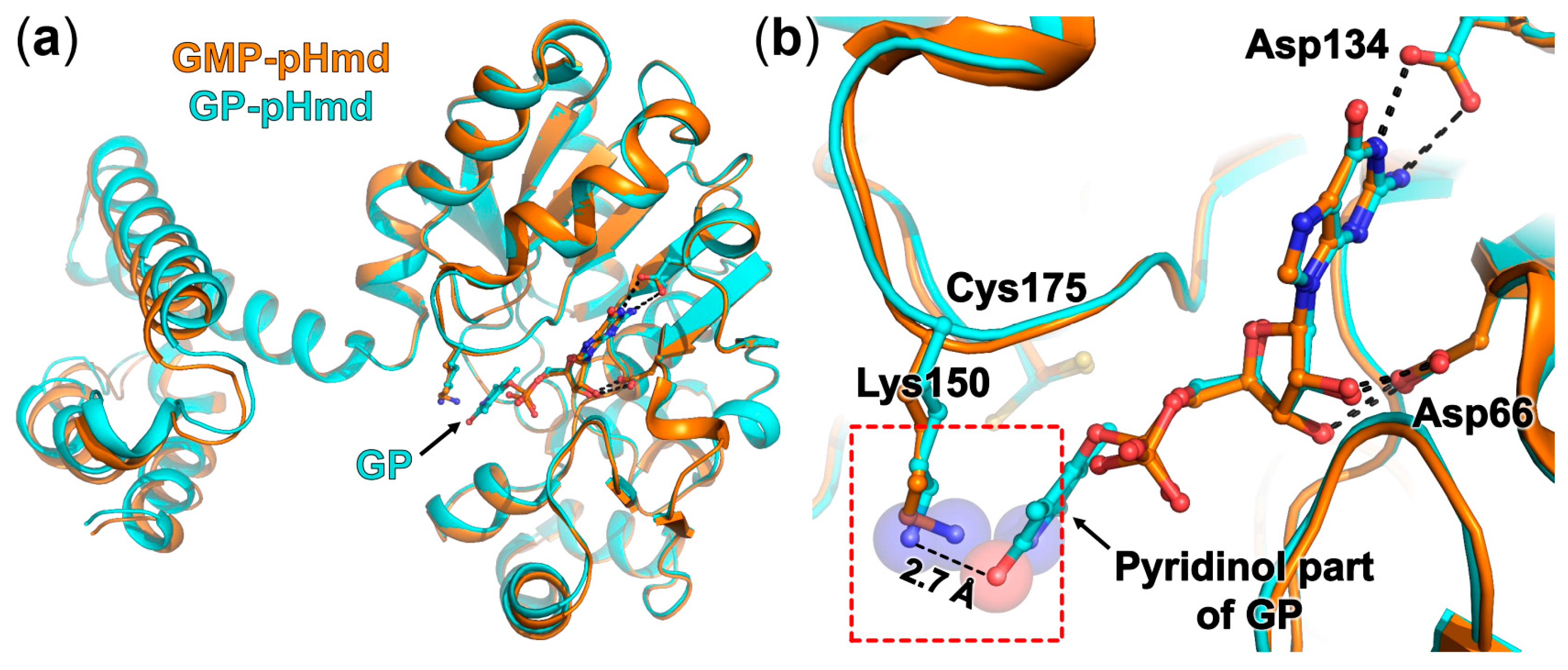
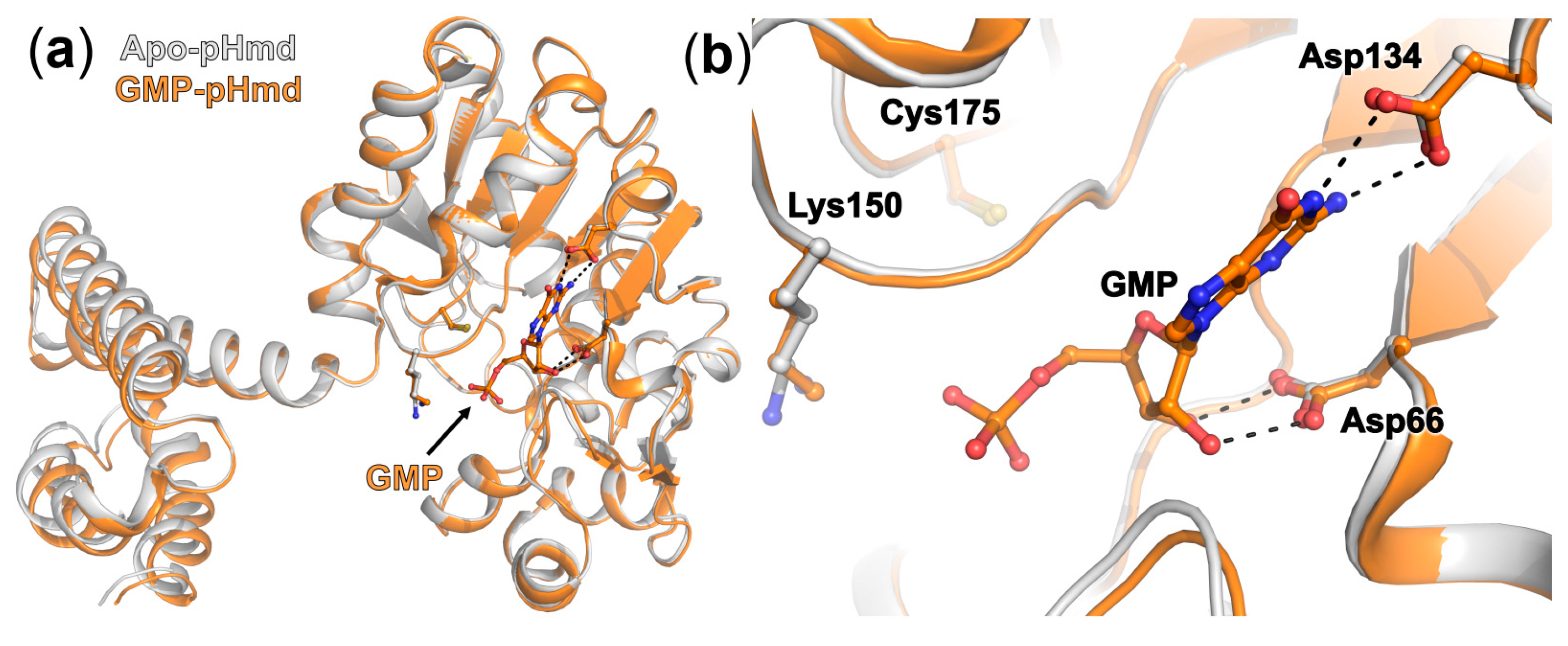
2.2. Crystal Structure of pHmd in Complex with GMP
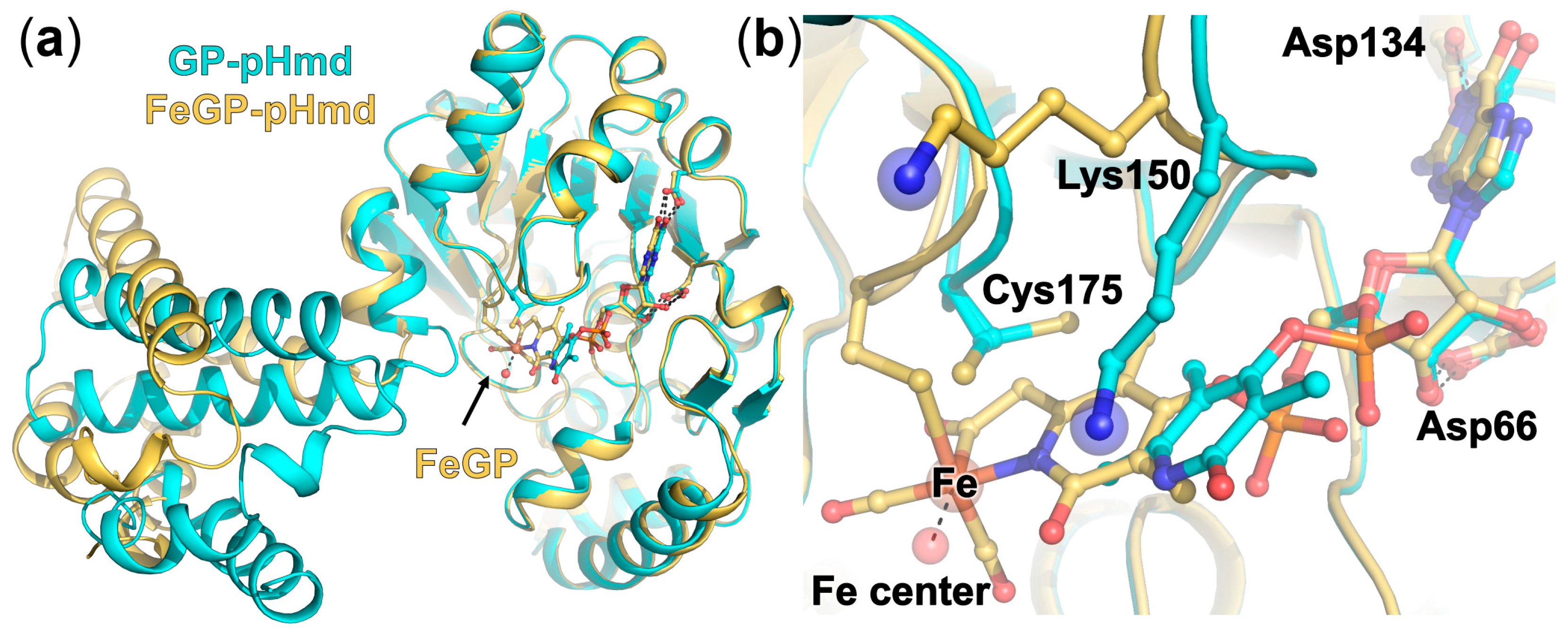
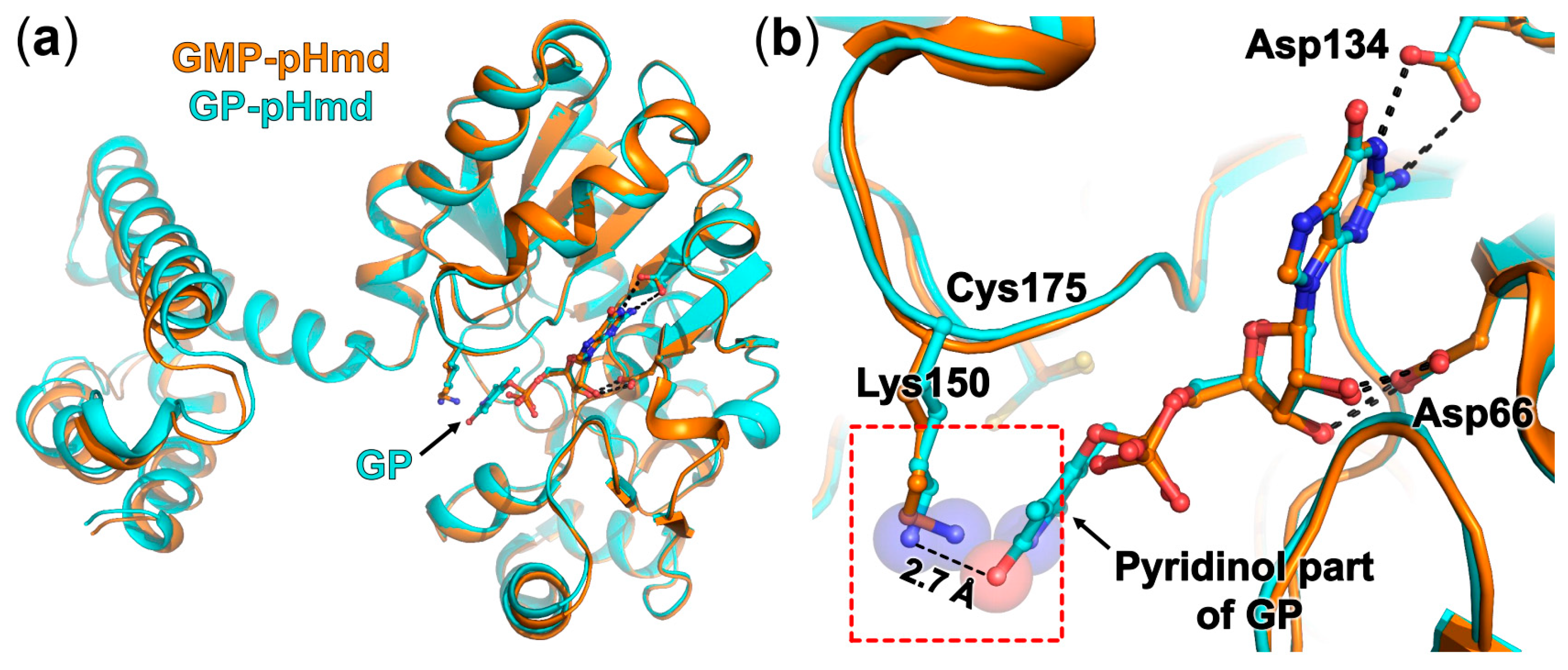
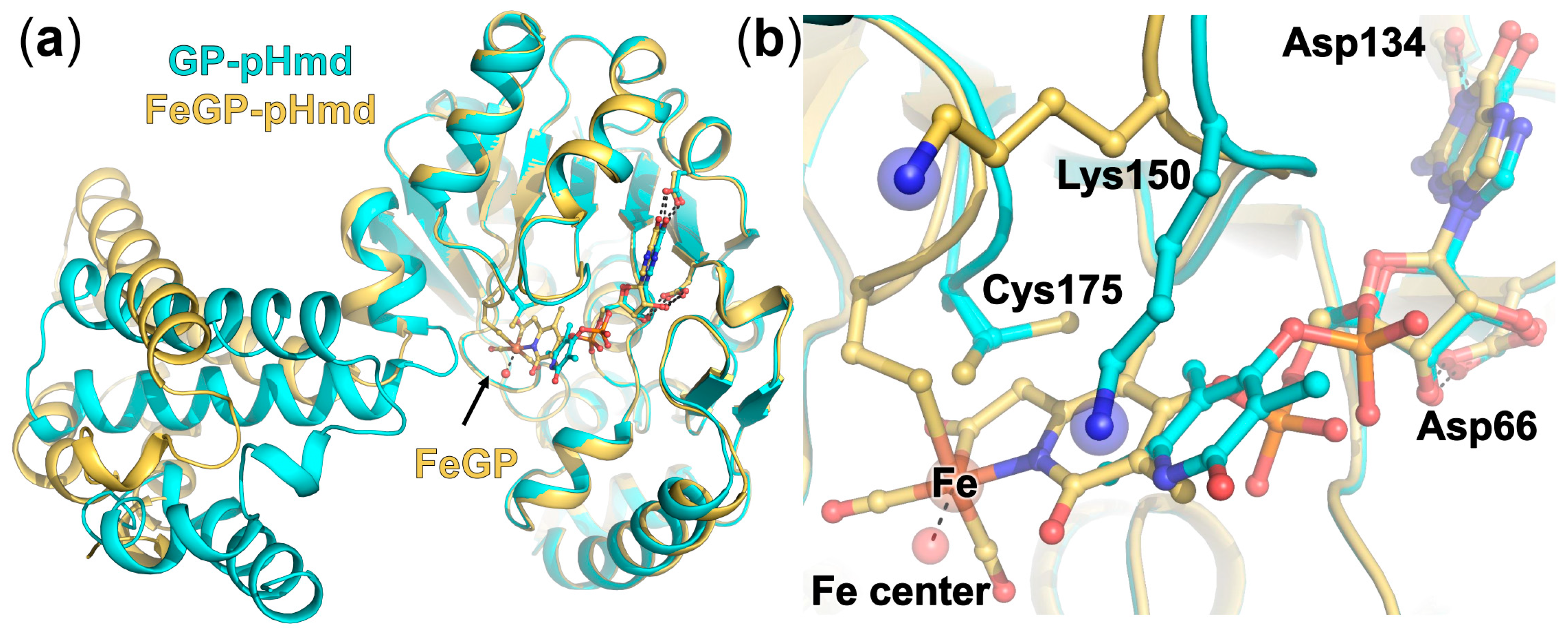
2.3. Crystal Structure of pHmd in Complex with GP
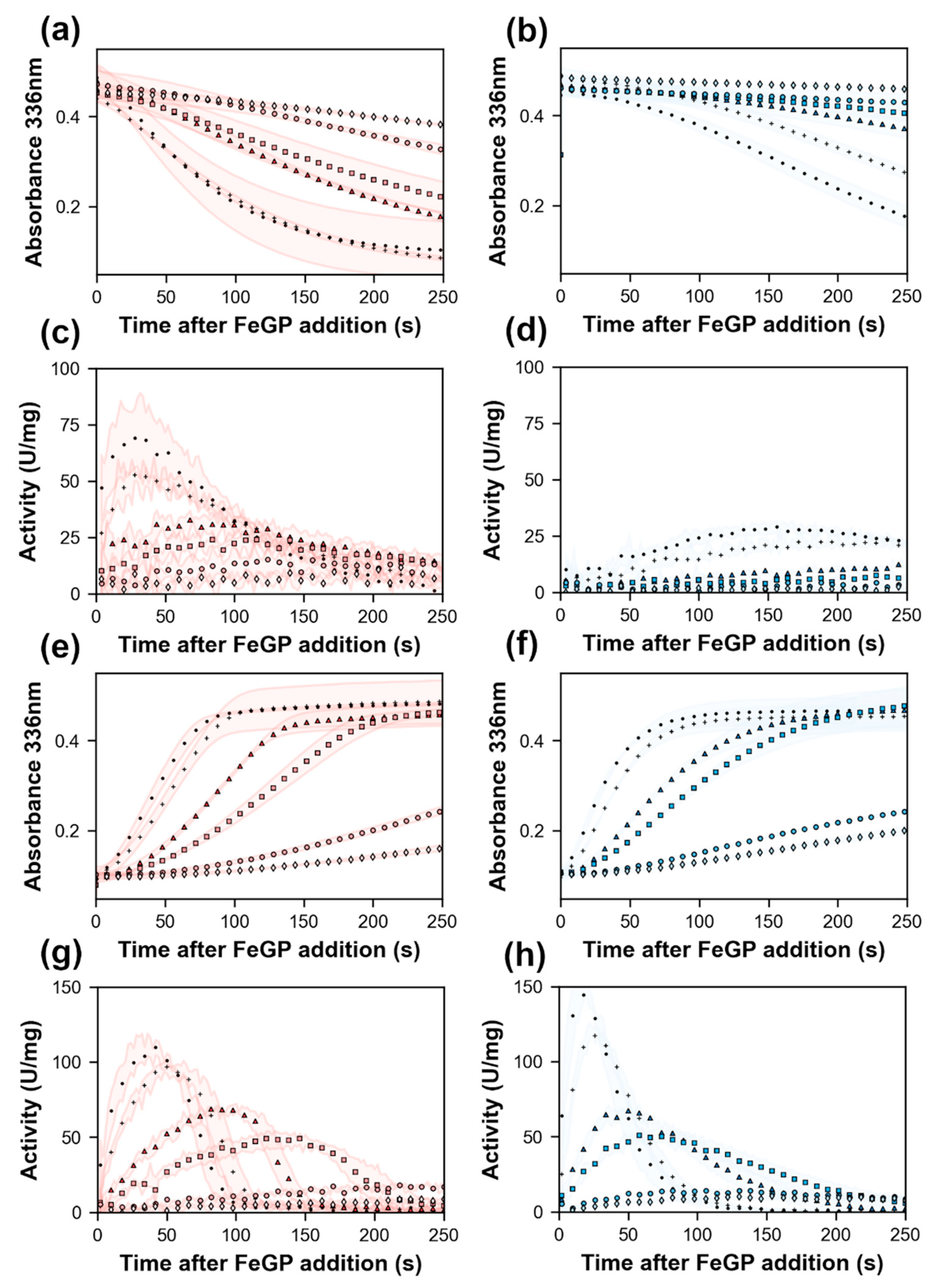
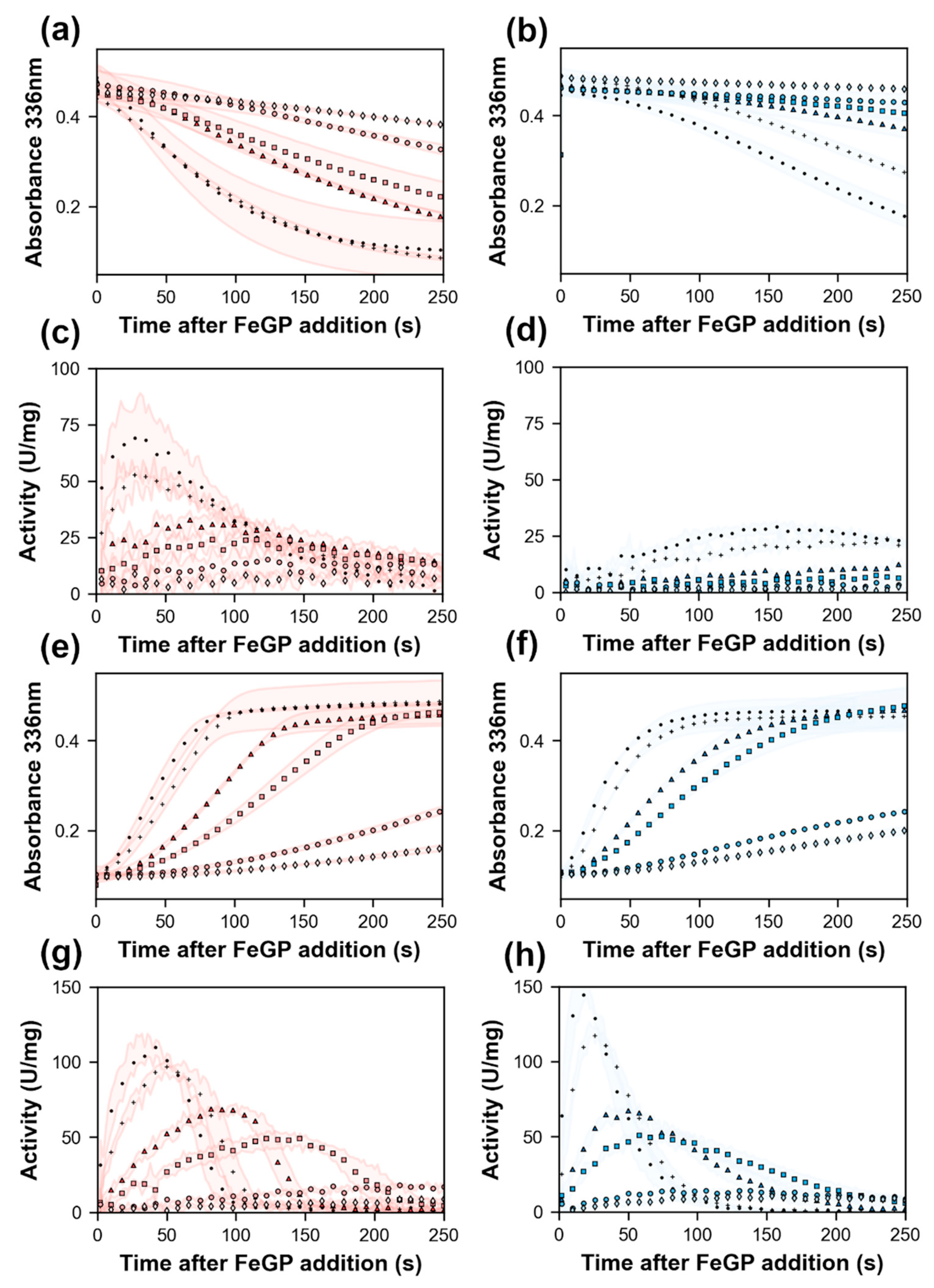
2.4. Effect of Lys150Ala Substitution on the Reconstitution Rate of pHmd
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Gene Synthesis of [Fe]-Hydrogenase from Methanolacinia Paynteri
3.3. Enzyme Production, Purification and Reconstitution
3.4. Enzyme Activity Assay
3.5. Simulation of the Enzyme Kinetics Data
3.6. Crystallization
3.7. Data Collection and Refinement
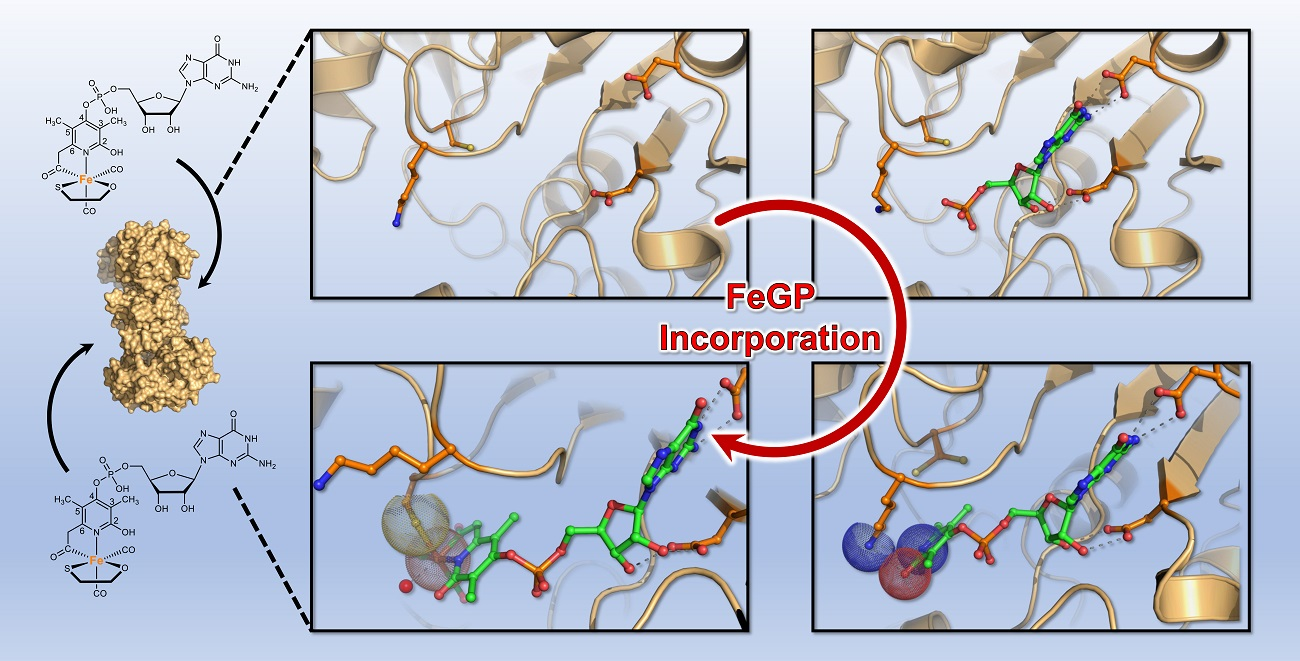
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zirngibl, C.; van Dongen, W.; Schwörer, B.; von Bünau, R.; Richter, M.; Klein, A.; Thauer, R.K. H2-forming methylenetetrahydromethanopterin dehydrogenase, a novel type of hydrogenase without iron-sulfur clusters in methanogenic archaea. Eur. J. Biochem. 1992, 208, 511–520. [Google Scholar]
- Huang, G.F.; Wagner, T.; Ermler, U.; Shima, S. Methanogenesis involves direct hydride transfer from H2 to an organic substrate. Nat. Rev. Chem. 2020, 4, 213–221. [Google Scholar]
- Shima, S.; Huang, G.; Wagner, T.; Ermler, U. Structural basis of hydrogenotrophic methanogenesis. Annu. Rev. Microbiol. 2020, 74, 713–733. [Google Scholar]
- Pilak, O.; Mamat, B.; Vogt, S.; Hagemeier, C.H.; Thauer, R.K.; Shima, S.; Vonrhein, C.; Warkentin, E.; Ermler, U. The crystal structure of the apoenzyme of the iron-sulphur cluster-free hydrogenase. J. Mol. Biol. 2006, 358, 798–809. [Google Scholar]
- Shima, S.; Pilak, O.; Vogt, S.; Schick, M.; Stagni, M.S.; Meyer-Klaucke, W.; Warkentin, E.; Thauer, R.K.; Ermler, U. The crystal structure of [Fe]-hydrogenase reveals the geometry of the active site. Science 2008, 321, 572–575. [Google Scholar]
- Shima, S.; Ermler, U. Structure and function of [Fe]-hydrogenase and its iron-guanylylpyridinol (FeGP) cofactor. Eur. J. Inorg. Chem. 2011, 2011, 963–972. [Google Scholar]
- Lyon, E.J.; Shima, S.; Boecher, R.; Thauer, R.K.; Grevels, F.W.; Bill, E.; Roseboom, W.; Albracht, S.P.J. Carbon monoxide as an intrinsic ligand to iron in the active site of the iron-sulfur-cluster-free hydrogenase H2-forming methylenetetrahydromethanopterin dehydrogenase as revealed by infrared spectroscopy. J. Am. Chem. Soc. 2004, 126, 14239–14248. [Google Scholar]
- Shima, S.; Lyon, E.J.; Sordel-Klippert, M.S.; Kauss, M.; Kahnt, J.; Thauer, R.K.; Steinbach, K.; Xie, X.L.; Verdier, L.; Griesinger, C. The cofactor of the iron-sulfur cluster free hydrogenase Hmd: Structure of the light-inactivation product. Angew. Chem. Int. Ed. 2004, 43, 2547–2551. [Google Scholar]
- Shima, S.; Lyon, E.J.; Thauer, R.K.; Mienert, B.; Bill, E. Mössbauer studies of the iron-sulfur cluster-free hydrogenase: The electronic state of the mononuclear Fe active site. J. Am. Chem. Soc. 2005, 127, 10430–10435. [Google Scholar]
- Hiromoto, T.; Ataka, K.; Pilak, O.; Vogt, S.; Stagni, M.S.; Meyer-Klaucke, W.; Warkentin, E.; Thauer, R.K.; Shima, S.; Ermler, U. The crystal structure of C176A mutated [Fe]-hydrogenase suggests an acyl- iron ligation in the active site iron complex. FEBS Lett. 2009, 583, 585–590. [Google Scholar]
- Hiromoto, T.; Warkentin, E.; Moll, J.; Ermler, U.; Shima, S. The crystal structure of an [Fe]-hydrogenase-substrate complex reveals the framework for H2 activation. Angew. Chem. Int. Ed. 2009, 48, 6457–6460. [Google Scholar]
- Shima, S.; Schick, M.; Kahnt, J.; Ataka, K.; Steinbach, K.; Linne, U. Evidence for acyl–iron ligation in the active site of [Fe]-hydrogenase provided by mass spectrometry and infrared spectroscopy. Dalton Trans. 2012, 41, 767–771. [Google Scholar]
- Korbas, M.; Vogt, S.; Meyer-Klaucke, W.; Bill, E.; Lyon, E.J.; Thauer, R.K.; Shima, S. The iron-sulfur cluster-free hydrogenase (Hmd) is a metalloenzyme with a novel iron binding motif. J. Biol. Chem. 2006, 281, 30804–30813. [Google Scholar]
- Shima, S.; Schick, M.; Tamura, H. Preparation of [Fe]-hydrogenase from methanogenic archaea. Methods Enzymol. 2011, 494, 119–137. [Google Scholar]
- Shima, S.; Vogt, S.; Göbels, A.; Bill, E. Iron-chromophore circular dichroism of [Fe]-hydrogenase: The conformational change required for H2 activation. Angew. Chem. Int. Ed. 2010, 49, 9917–9921. [Google Scholar]
- Lyon, E.J.; Shima, S.; Buurman, G.; Chowdhuri, S.; Batschauer, A.; Steinbach, K.; Thauer, R.K. UV-A/blue-light inactivation of the ‘metal-free’ hydrogenase (Hmd) from methanogenic archaea: The enzyme contains functional iron after all. Eur. J. Biochem. 2004, 271, 195–204. [Google Scholar]
- Hidese, R.; Ataka, K.; Bill, E.; Shima, S. CuI and H2O2 Inactivate and FeII Inhibits [Fe]-hydrogenase at very low concentrations. Chembiochem 2015, 16, 1861–1865. [Google Scholar]
- Huang, G.; Wagner, T.; Ermler, U.; Bill, E.; Ataka, K.; Shima, S. Dioxygen sensitivity of [Fe]-hydrogenase in the presence of reducing substrates. Angew. Chem. Int. Ed. 2018, 57, 4917–4920. [Google Scholar]
- Buurman, G.; Shima, S.; Thauer, R.K. The metal-free hydrogenase from methanogenic archaea: Evidence for a bound cofactor. FEBS Lett. 2000, 485, 200–204. [Google Scholar]
- Huang, G.F.; Wagner, T.; Wodrich, M.D.; Ataka, K.; Bill, E.; Ermler, U.; Hu, X.L.; Shima, S. The atomic-resolution crystal structure of activated [Fe]-hydrogenase. Nat. Catal. 2019, 2, 537–543. [Google Scholar]
- Song, L.C.; Zhu, L.; Liu, B.B. A biomimetic model for the active site of [Fe]-H2ase featuring a 2-methoxy-3,5-dimethyl-4-phosphato-6-acylmethylpyridine ligand. Organometallics 2019, 38, 4071–4075. [Google Scholar]
- Seo, J.; Manes, T.A.; Rose, M.J. Structural and functional synthetic model of mono-iron hydrogenase featuring an anthracene scaffold. Nat. Chem. 2017, 9, 552–557. [Google Scholar]
- Kerns, S.A.; Magtaan, A.C.; Vong, P.R.; Rose, M.J. Functional hydride transfer by a thiolate-containing model of mono-iron hydrogenase featuring an anthracene scaffold. Angew. Chem. Int. Ed. 2018, 57, 2855–2858. [Google Scholar]
- Xu, T.; Yin, C.J.M.; Wodrich, M.D.; Mazza, S.; Schultz, K.M.; Scopelliti, R.; Hu, X. A functional model of [Fe]-hydrogenase. J. Am. Chem. Soc. 2016, 138, 3270–3273. [Google Scholar]
- Pan, H.J.; Huang, G.F.; Wodrich, M.D.; Tirani, F.F.; Ataka, K.; Shima, S.; Hu, X.L. A catalytically active [Mn]-hydrogenase incorporating a non-native metal cofactor. Nat. Chem. 2019, 11, 669–675. [Google Scholar]
- Hu, B.; Chen, D.; Hu, X.L. Synthesis and reactivity of mononuclear iron models of [Fe]-hydrogenase that contain an acylmethylpyridinol ligand. Chem. Eur. J. 2014, 20, 1677–1682. [Google Scholar]
- Chen, D.; Scopelliti, R.; Hu, X. A five-coordinate iron center in the active site of [Fe]-hydrogenase: Hints from a model study. Angew. Chem. Int. Ed. 2011, 50, 5671–5673. [Google Scholar]
- Shima, S.; Chen, D.; Xu, T.; Wodrich, M.D.; Fujishiro, T.; Schultz, K.M.; Kahnt, J.; Ataka, K.; Hu, X.L. Reconstitution of [Fe]-hydrogenase using model complexes. Nat. Chem. 2015, 7, 995–1002. [Google Scholar]
- Schick, M.; Xie, X.L.; Ataka, K.; Kahnt, J.; Linne, U.; Shima, S. Biosynthesis of the iron-guanylylpyridinol cofactor of [Fe]-hydrogenase in methanogenic archaea as elucidated by stable-isotope labeling. J. Am. Chem. Soc. 2012, 134, 3271–3280. [Google Scholar]
- Schwörer, B.; Thauer, R.K. Activities of formylmethanofuran dehydrogenase, methylenetetrahydromethanopterin dehydrogenase, methylenetetrahydromethanopterin reductase, and heterodisulfide reductase in methanogenic bacteria. Arch. Microbiol. 1991, 155, 459–465. [Google Scholar]
- de Poorter, L.M.I.; Keltjens, J.T. Convenient fluorescence-based methods to measure membrane potential and intracellular pH in the Archaeon Methanobacterium thermoautotrophicum. J. Microbiol. Methods 2001, 47, 233–241. [Google Scholar]
- Shima, S.; Thauer, R.K. Tetrahydromethanopterin-specific enzymes from Methanopyrus kandleri. Methods Enzymol. 2001, 331, 317–353. [Google Scholar]
- Moore, J.T.; Uppal, A.; Maley, F.; Maley, G.F. Overcoming inclusion body formation in a high-level expression system. Protein Expres. Purif. 1993, 4, 160–163. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar]
- Van der Walt, S.; Colbert, S.C.; Varoquaux, G. The NumPy Array: A structure for efficient numerical computation. Comput. Sci. Eng. 2011, 13, 22–30. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar]
- McKinney, W. Data structures for statistical computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28–30 June 2010; pp. 51–56. [Google Scholar]
- Kabsch, W. Xds. Acta Crystallogr. D 2010, 66, 125–132. [Google Scholar]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D 2011, 67, 235–242. [Google Scholar]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallog. D 2010, 66, 486–501. [Google Scholar]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. D 2019, 75, 861–877. [Google Scholar]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Asymmetric Homodimer-pHmd | GMP Bound | GP Bound | |
|---|---|---|---|
| Data collection | |||
| Wavelength (Å) | 0.97980 | 0.97972 | 0.97970 |
| Space group | P3121 | P21 | C2 |
| Resolution (Å) | 45.13–2.10 (2.21–2.10) | 43.65–1.55 (1.63–1.55) | 46.63–1.70 (1.79–1.70) |
| Cell dimensions | |||
| a, b, c (Å) | 79.48, 79.48, 179.34 | 67.81, 57.06, 95.13 | 160.83, 93.27, 83.64 |
| α, β, γ (°) | 90, 90, 120 | 90, 91.43, 90 | 90, 97.53, 90 |
| Nr. Monomers/asym. Unit | 2 | 2 | 4 |
| open/closed conformation | 1 + 1 | 0/2 | 0/4 |
| Rmerge (%) a | 5.9 (71.1) | 6.8 (35.3) | 10.7 (51.7) |
| Rpim (%) a | 1.9 (22.2) | 4.1 (21.3) | 6.5 (30.8) |
| CC1/2 a | 1.000 (0.755) | 0.997 (0.653) | 0.993 (0.605) |
| I/σI a | 28.8 (3.6) | 12.6 (3.7) | 8.0 (2.5) |
| Completeness (%) a | 100.0 (100.0) | 99.3 (99.7) | 99.1 (99.1) |
| Redundancy a | 10.8 (11.1) | 3.7 (3.7) | 3.7 (3.8) |
| Nr. unique reflections a | 39,181 (5643) | 104,812 (15285) | 133,035 (19398) |
| Refinement | |||
| Resolution (Å) | 39.74–2.10 | 22.69–1.55 | 24.89–1.70 |
| Number of reflections | 39,126 | 104,777 | 132,993 |
| Rwork/Rfree b (%) | 19.5/23.5 | 18.4/20.9 | 18.9/21.6 |
| Number of atoms | |||
| Protein | 5215 | 5256 | 10,490 |
| Ligands/ions | 103 | 78 | 114 |
| Solvent | 224 | 640 | 1068 |
| Mean B-value (Å2) | 61.9 | 22.2 | 23.1 |
| Molprobity clash score, all atoms | 1.49 | 1.39 | 0.79 |
| Ramachandran plot | |||
| Favored regions (%) | 97.05 | 96.31 | 96.31 |
| Outlier regions (%) | 0.15 | 0 | 0 |
| Rmsd c bond lengths (Å) | 0.008 | 0.010 | 0.010 |
| Rmsd c bond angles (°) | 1.01 | 1.10 | 1.08 |
| PDB code | 6YKA | 6YKB | 6YK9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, G.; Arriaza-Gallardo, F.J.; Wagner, T.; Shima, S. Crystal Structures of [Fe]-Hydrogenase from Methanolacinia paynteri Suggest a Path of the FeGP-Cofactor Incorporation Process. Inorganics 2020, 8, 50. https://doi.org/10.3390/inorganics8090050
Huang G, Arriaza-Gallardo FJ, Wagner T, Shima S. Crystal Structures of [Fe]-Hydrogenase from Methanolacinia paynteri Suggest a Path of the FeGP-Cofactor Incorporation Process. Inorganics. 2020; 8(9):50. https://doi.org/10.3390/inorganics8090050
Chicago/Turabian StyleHuang, Gangfeng, Francisco Javier Arriaza-Gallardo, Tristan Wagner, and Seigo Shima. 2020. "Crystal Structures of [Fe]-Hydrogenase from Methanolacinia paynteri Suggest a Path of the FeGP-Cofactor Incorporation Process" Inorganics 8, no. 9: 50. https://doi.org/10.3390/inorganics8090050
APA StyleHuang, G., Arriaza-Gallardo, F. J., Wagner, T., & Shima, S. (2020). Crystal Structures of [Fe]-Hydrogenase from Methanolacinia paynteri Suggest a Path of the FeGP-Cofactor Incorporation Process. Inorganics, 8(9), 50. https://doi.org/10.3390/inorganics8090050