Metal–Dithiolene Bonding Contributions to Pyranopterin Molybdenum Enzyme Reactivity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
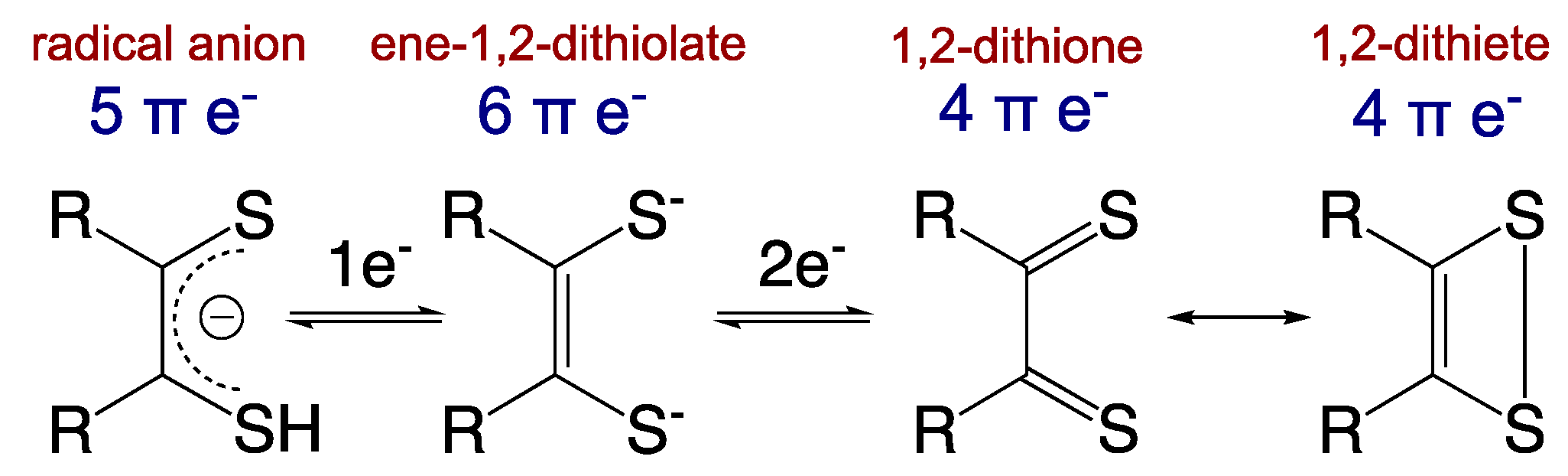
2. Mo–Dithiolene Bonding
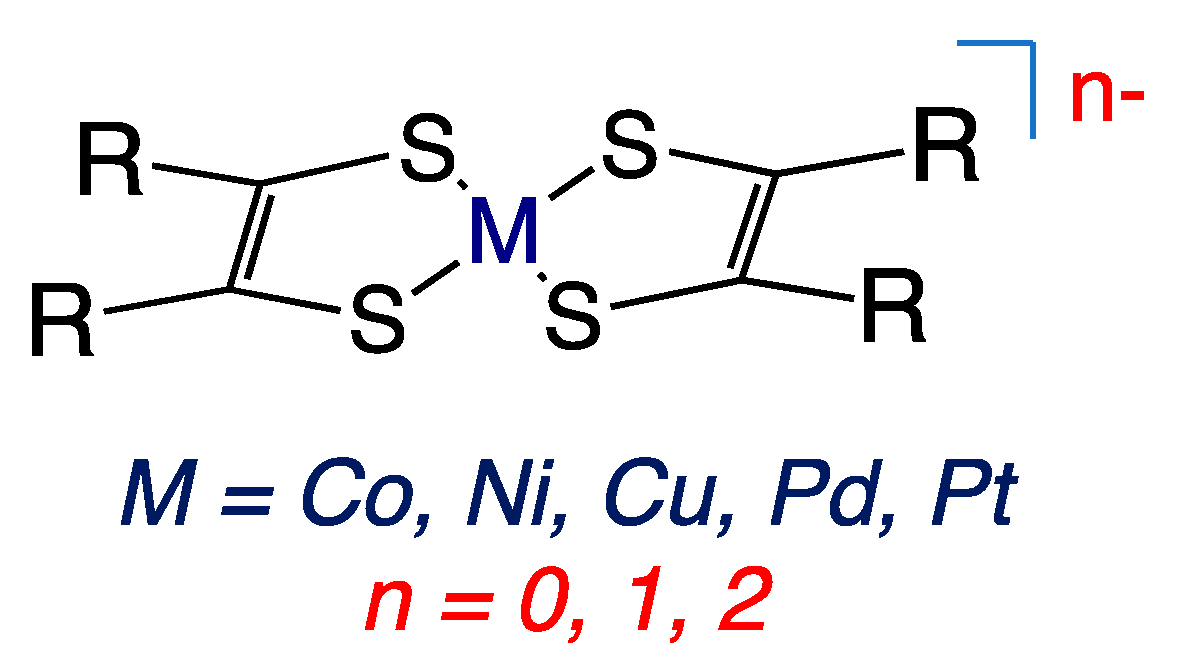
2.1. Early Descriptions of Mo–Dithiolene Bonding
2.2. Spectroscopic Investigations of Mo–Dithiolene Bonding
2.2.1. Electron Paramagnetic Resonance (EPR) Spectroscopy
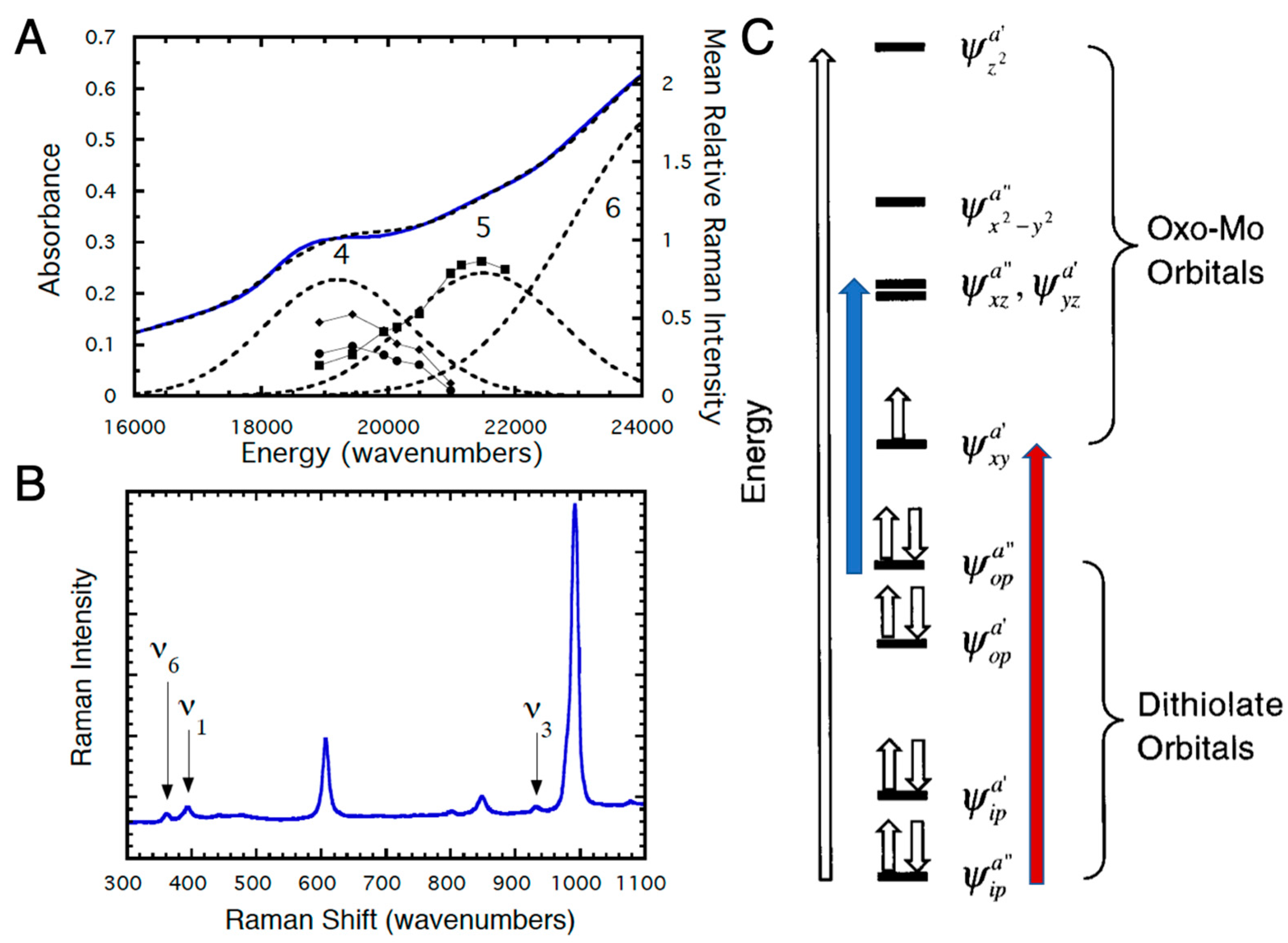
2.2.2. Electronic Absorption and Resonance Raman Spectroscopies
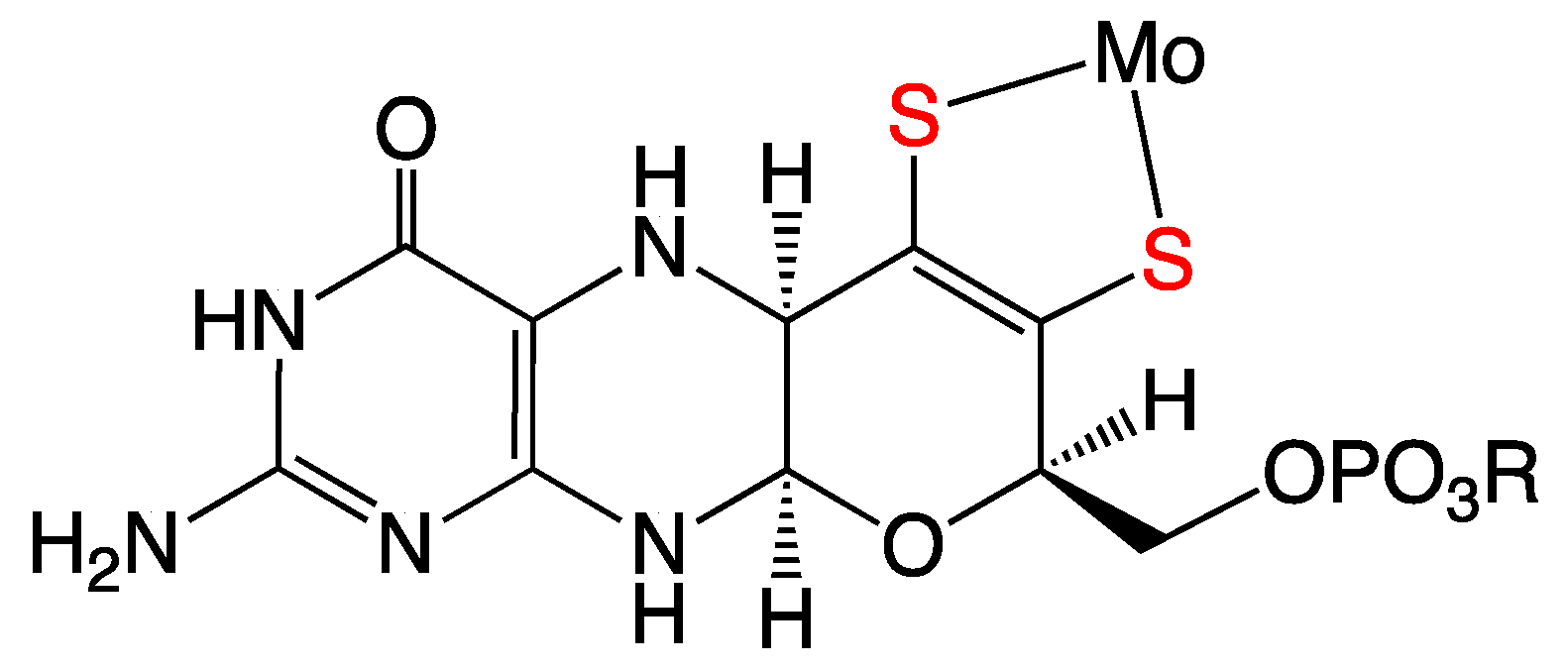
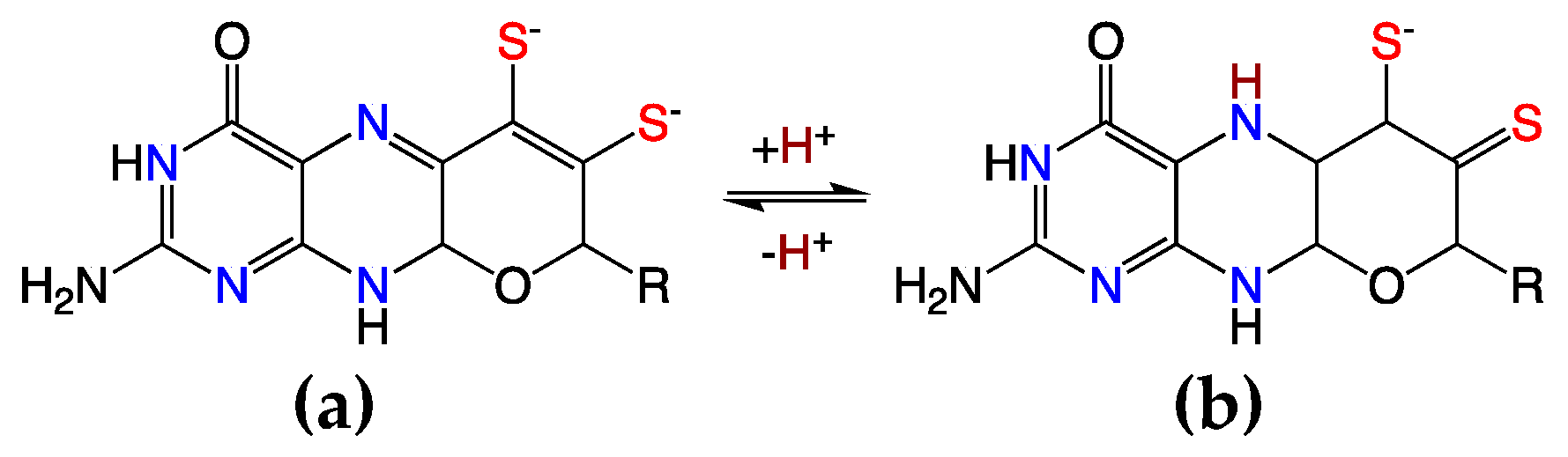
3. Synergistic Interactions between the Dithiolene and Pterin Components of the PDT
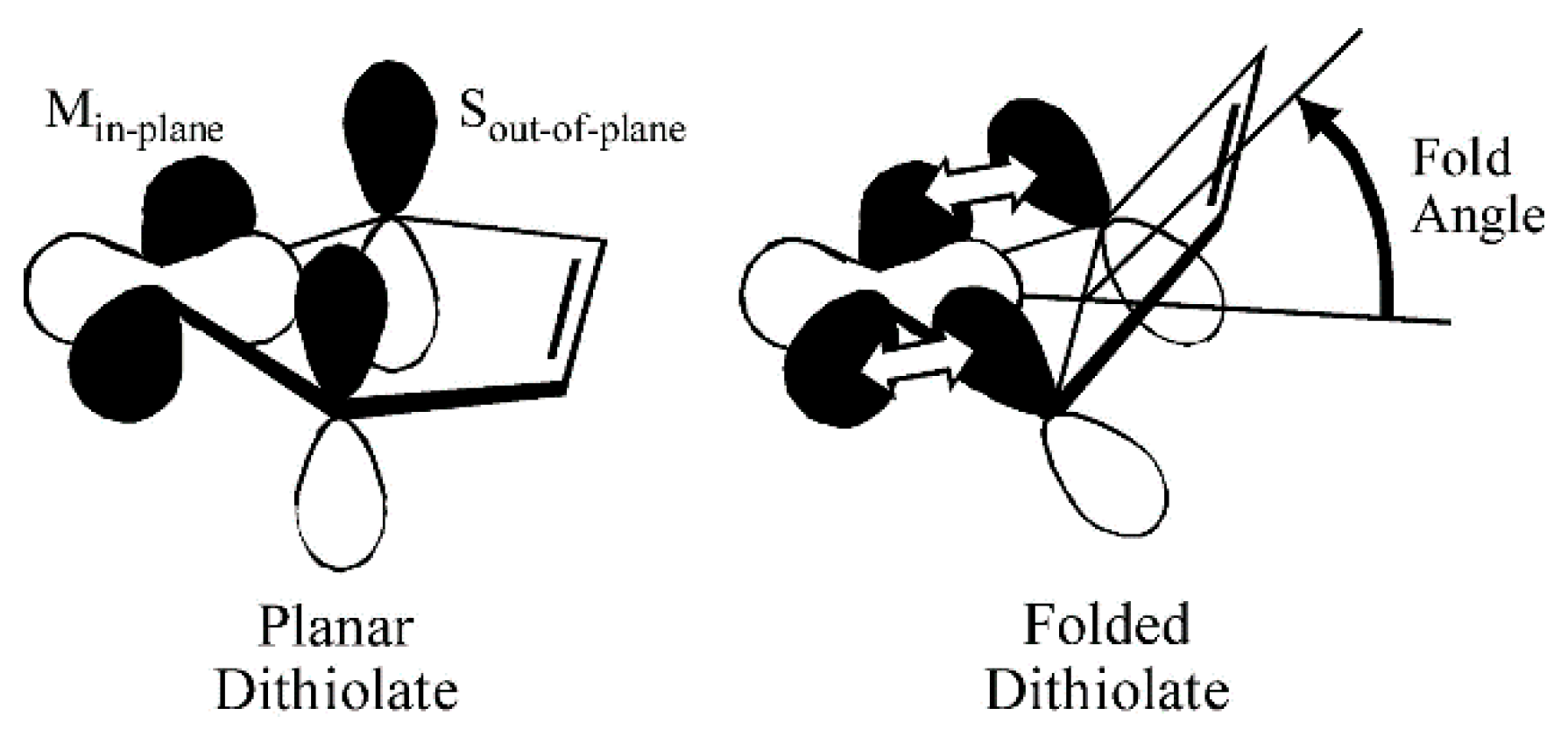
4. The Electronic Buffer Effect and Fold Angle Distortions
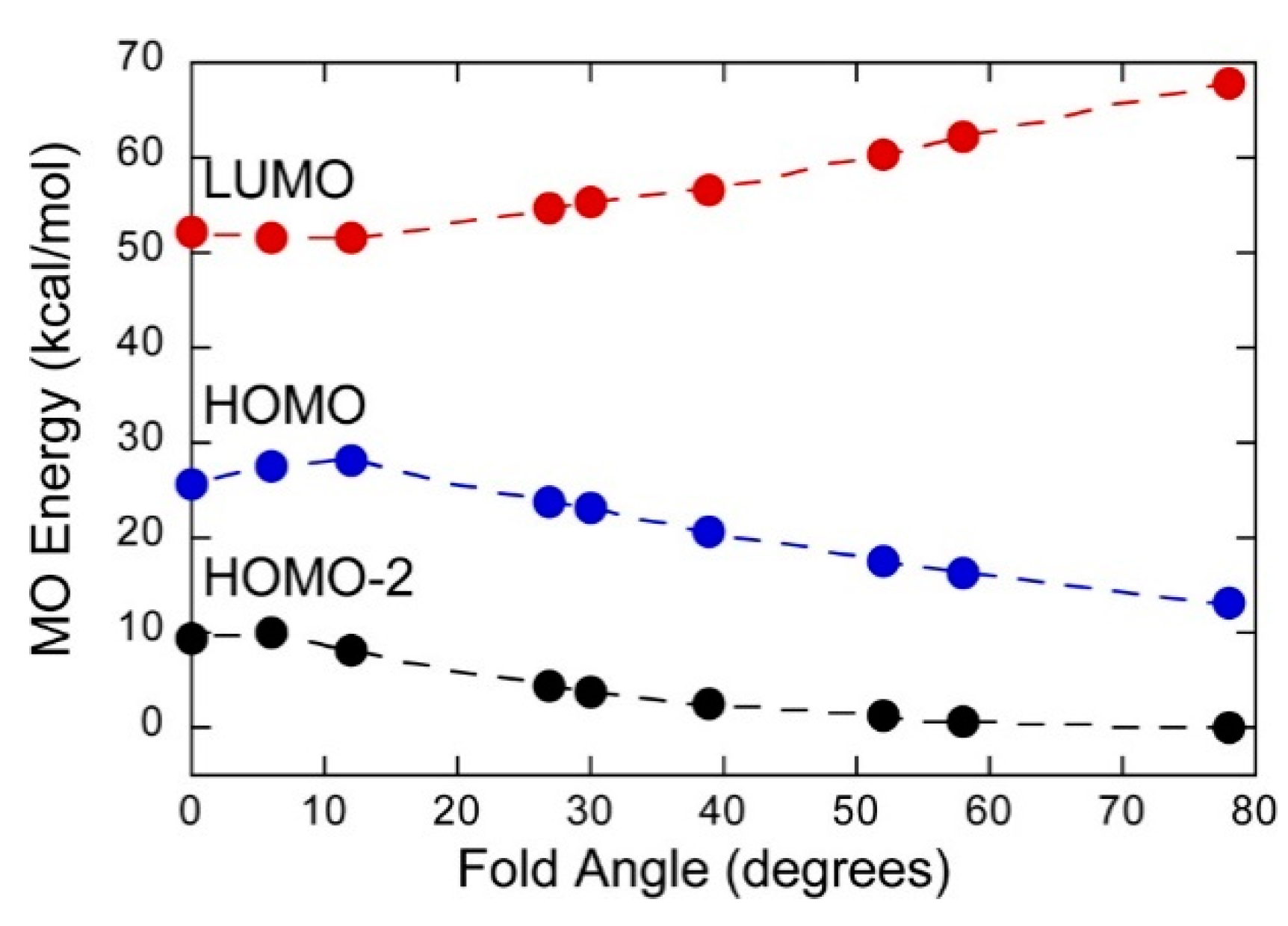
4.1. Photoelectron Spectroscopy (PES) Studies
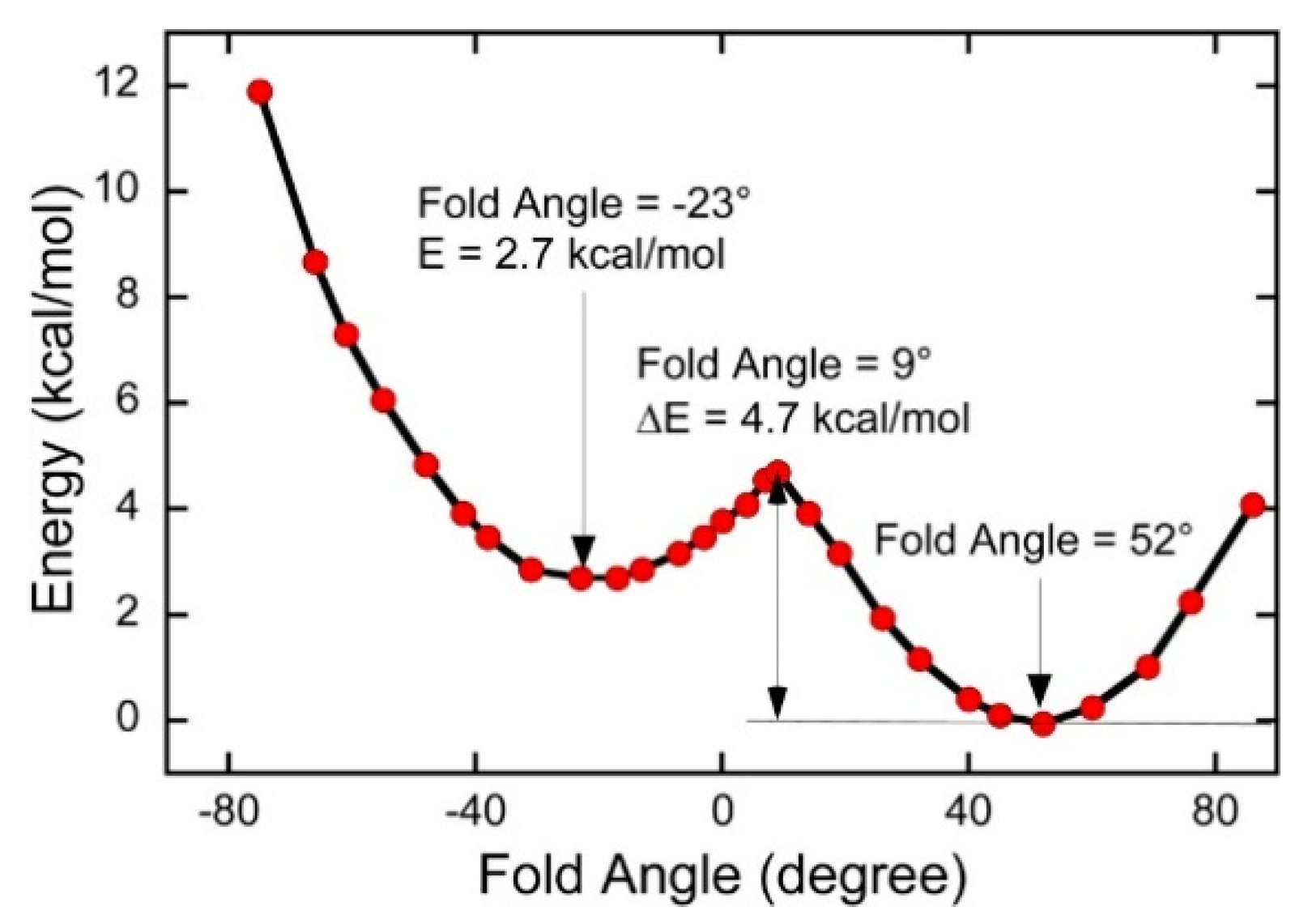
4.2. A Large Fold Angle Distortion in a Mo(IV)–Dithione Complex
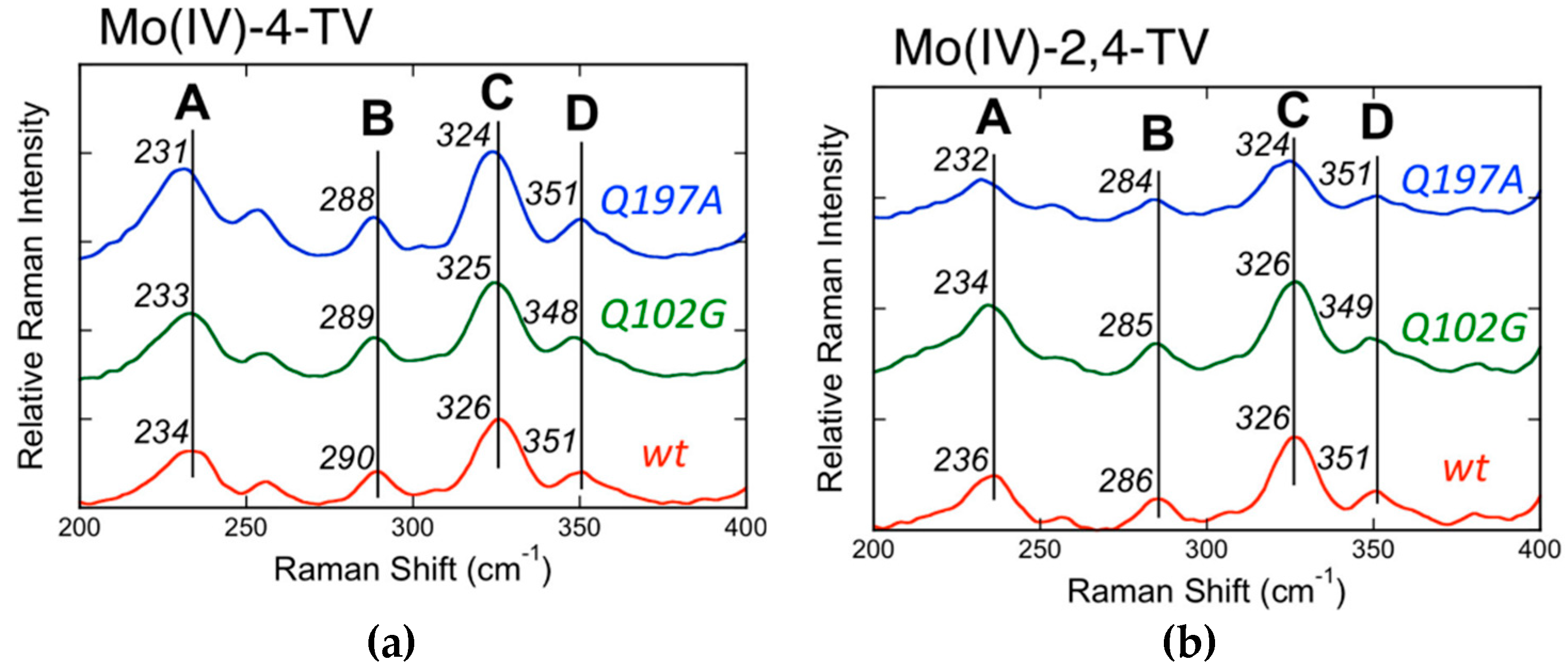
4.3. Low-Frequency Pyranopterin Dithiolene Vibrational Modes in Xanthine Oxidase/Dehydrogenase
5. Vibrational Control of Covalency
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hille, R.; Schulzke, C.; Kirk, M.L. Molybdenum and Tungsten Enzymes; The Royal Society of Chemistry: Cambridge, UK, 2017. [Google Scholar]
- Hille, R.; Hall, J.; Basu, P. The Mononuclear Molybdenum Enzymes. Chem. Rev. 2014, 114, 3963–4038. [Google Scholar] [CrossRef]
- Kirk, M.L.; Stein, B. The Molybdenum Enzymes. In Comprehensive Inorganic Chemistry II, 2nd ed.; Jan, R., Kenneth, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 263–293. [Google Scholar] [CrossRef]
- Nieter Burgmayer, S.J.; Kirk, M.L. The Role of the Pyranopterin Dithiolene Component of Moco in Molybdoenzyme Catalysis. In Metallocofactors that Activate Small Molecules: With Focus on Bioinorganic Chemistry; Ribbe, M.W., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 101–151. [Google Scholar] [CrossRef]
- Weiss, M.C.; Sousa, F.L.; Mrnjavac, N.; Neukirchen, S.; Roettger, M.; Nelson-Sathi, S.; Martin, W.F. The physiology and habitat of the last universal common ancestor. Nat. Microbiol. 2016, 1, 116. [Google Scholar] [CrossRef]
- Fourmigue, M. Mixed cyclopentadienyl/dithiolene complexes. Coord. Chem. Rev. 1998, 180, 823–864. [Google Scholar] [CrossRef]
- Faulmann, C.; Cassoux, P. Solid-State Properties (Electronic, Magnetic, Optical) of Dithiolene Complex-Based Compounds. Prog. Inorg. Chem. 2004, 52, 399–490. [Google Scholar]
- Deplano, P.; Pilia, L.; Espa, D.; Mercuri, M.L.; Serpe, A. Square-planar d(8) metal mixed-ligand dithiolene complexes as second order nonlinear optical chromophores: Structure/property relationship. Coord. Chem. Rev. 2010, 254, 1434–1447. [Google Scholar] [CrossRef]
- Sproules, S.; Wieghardt, K. o-Dithiolene and o-aminothiolate chemistry of iron: Synthesis, structure and reactivity. Coord. Chem. Rev. 2010, 254, 1358–1382. [Google Scholar] [CrossRef]
- Sproules, S.; Wieghardt, K. Dithiolene radicals: Sulfur K-edge X-ray absorption spectroscopy and Harry’s intuition. Coord. Chem. Rev. 2011, 255, 837–860. [Google Scholar] [CrossRef]
- Eisenberg, R.; Gray, H.B. Noninnocence in Metal Complexes: A Dithiolene Dawn. Inorg. Chem. 2011, 50, 9741–9751. [Google Scholar] [CrossRef]
- Inscore, F.E.; McNaughton, R.; Westcott, B.L.; Helton, M.E.; Jones, R.; Dhawan, I.K.; Enemark, J.H.; Kirk, M.L. Spectroscopic evidence for a unique bonding interaction in oxo-molybdenum dithiolate complexes: Implications for sigma electron transfer pathways in the pyranopterin dithiolate centers of enzymes. Inorg. Chem. 1999, 38, 1401–1410. [Google Scholar] [CrossRef]
- Kirk, M.L.; McNaughton, R.L.; Helton, M.E. The Electronic Structure and Spectroscopy of Metallo-Dithiolene Complexes. In Progress in Inorganic Chemistry: Synthesis, Properties, and Applications; Stiefel, E.I., Karlin, K.D., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 2004; Volume 52, pp. 111–212. [Google Scholar]
- Peariso, K.; Helton, M.E.; Duesler, E.N.; Shadle, S.E.; Kirk, M.L. Sulfur K-edge spectroscopic investigation of second coordination sphere effects in oxomolybdenum-thiolates: Relationship to molybdenum-cysteine covalency and electron transfer in sulfite oxidase. Inorg. Chem. 2007, 46, 1259–1267. [Google Scholar] [CrossRef]
- Matz, K.G.; Mtei, R.P.; Leung, B.; Burgmayer, S.J.N.; Kirk, M.L. Noninnocent Dithiolene Ligands: A New Oxomolybdenum Complex Possessing a Donor Acceptor Dithiolene Ligand. J. Am. Chem. Soc. 2010, 132, 7830–7831. [Google Scholar] [CrossRef]
- Mtei, R.P.; Perera, E.; Mogesa, B.; Stein, B.; Basu, P.; Kirk, M.L. A Valence Bond Description of Dizwitterionic Dithiolene Character in an Oxomolybdenum-Bis(dithione) Complex. Eur. J. Inorg. Chem. 2011, 2011, 5467–5470. [Google Scholar] [CrossRef]
- Yang, J.; Mogesa, B.; Basu, P.; Kirk, M.L. Large Ligand Folding Distortion in an Oxomolybdenum Donor Acceptor Complex. Inorg. Chem. 2016, 55, 785–793. [Google Scholar] [CrossRef]
- Yang, J.; Kersi, D.K.; Richers, C.P.; Giles, L.J.; Dangi, R.; Stein, B.W.; Feng, C.; Tichnell, C.R.; Shultz, D.A.; Kirk, M.L. Ground State Nuclear Magnetic Resonance Chemical Shifts Predict Charge-Separated Excited State Lifetimes. Inorg. Chem. 2018, 57, 13470–13476. [Google Scholar] [CrossRef]
- Yang, J.; Kersi, D.K.; Giles, L.J.; Stein, B.W.; Feng, C.J.; Tichnell, C.R.; Shultz, D.A.; Kirk, M.L. Ligand Control of Donor-Acceptor Excited-State Lifetimes. Inorg. Chem. 2014, 53, 4791–4793. [Google Scholar] [CrossRef]
- Matz, K.G.; Mtei, R.P.; Rothstein, R.; Kirk, M.L.; Burgmayer, S.J.N. Study of Molybdenum(4+) Quinoxalyldithiolenes as Models for the Noninnocent Pyranopterin in the Molybdenum Cofactor. Inorg. Chem. 2011, 50, 9804–9815. [Google Scholar] [CrossRef]
- Kirk, M.L. Spectroscopic and Electronic Structure Studies of Mo Model Compounds and Enzymes; The Royal Society of Chemistry: Cambridge, UK, 2016; pp. 13–67. [Google Scholar]
- Schrauzer, G.N.; Mayweg, V. Reaction of Diphenylacetylene with Nickel Sulfides. J. Am. Chem. Soc. 1962, 84, 3221. [Google Scholar] [CrossRef]
- Gray, H.B.; Billig, E.; Williams, R.; Bernal, I. Spin-Free Square Planar Cobaltous Complex. J. Am. Chem. Soc. 1962, 84, 3596. [Google Scholar] [CrossRef]
- Davison, A.; Holm, R.H.; Edelstein, N.; Maki, A.H. Preparation and Characterization of 4-Coordinate Complexes Related by Electron-Transfer Reactions. Inorg. Chem. 1963, 2, 1227. [Google Scholar] [CrossRef]
- McCleverty, J.A. Metal 1,2-Dithiolene and Related Complexes. Prog. Inorg. Chem. 1968, 10, 49–221. [Google Scholar]
- Johnson, J.; Rajagopalan, K. Structural and Metabolic Relationship Between the Molybdenum Cofactor and Urothione. Proc. Natl. Acad. Sci. USA 1982, 79, 6856–6860. [Google Scholar] [CrossRef]
- Chan, M.K.; Mukund, S.; Kletzin, A.; Adams, M.W.W.; Rees, D.C. Structure of a Hyperthermophilic Tungstopterin Enzyme, Aldehyde Ferredoxin Oxidoreductase. Science 1995, 267, 1463–1469. [Google Scholar] [CrossRef]
- Romao, M.J.; Archer, M.; Moura, I.; Moura, J.J.G.; Legall, J.; Engh, R.; Schneider, M.; Hof, P.; Huber, R. Crystal Structure of the Xanthine Oxidase Related Aldehyde Oxidoreductase from D. gigas. Science 1995, 270, 1170–1176. [Google Scholar] [CrossRef]
- Rothery, R.A.; Stein, B.; Solomonson, M.; Kirk, M.L.; Weiner, J.H. Pyranopterin conformation defines the function of molybdenum and tungsten enzymes. Proc. Natl. Acad. Sci. USA 2012, 109, 14773–14778. [Google Scholar] [CrossRef]
- Lauher, J.W.; Hoffmann, R. Structure and Chemistry of Bis(Cyclopentadienyl)-MLn Complexes. J. Am. Chem. Soc. 1976, 98, 1729–1742. [Google Scholar] [CrossRef]
- Joshi, H.K.; Cooney, J.J.A.; Inscore, F.E.; Gruhn, N.E.; Lichtenberger, D.L.; Enemark, J.H. Investigation of metal-dithiolate fold angle effects: Implications for molybdenum and tungsten enzymes. Proc. Natl. Acad. Sci. USA 2003, 100, 3719–3724. [Google Scholar] [CrossRef]
- Inscore, F.E.; Knottenbelt, S.Z.; Rubie, N.D.; Joshi, H.K.; Kirk, M.L.; Enemark, J.H. Understanding the origin of metal-sulfur vibrations in an oxo-molybdenurn dithiolene complex: Relevance to sulfite oxidase. Inorg. Chem. 2006, 45, 967. [Google Scholar] [CrossRef]
- Peariso, K.; Chohan, B.S.; Carrano, C.J.; Kirk, M.L. Synthesis and EPR characterization of new models for the one-electron reduced molybdenum site of sulfite oxidase. Inorg. Chem. 2003, 42, 6194–6203. [Google Scholar] [CrossRef]
- Cleland, W.E.; Barnhart, K.M.; Yamanouchi, K.; Collison, D.; Mabbs, F.E.; Ortega, R.B.; Enemark, J.H. Syntheses, Structures, and Spectroscopic Properties of 6-Coordinate Mononuclear Oxo-Molybdenum(V) Complexes Stabilized by the Hydrotris(3,5-Dimethyl-1-Pyrazolyl)Borate Ligand. Inorg. Chem. 1987, 26, 1017–1025. [Google Scholar] [CrossRef]
- Enemark, J.H. Consensus structures of the Mo(V) sites of sulfite-oxidizing enzymes derived from variable frequency pulsed EPR spectroscopy, isotopic labelling and DFT calculations. Dalton Trans. 2017, 46, 13202–13210. [Google Scholar] [CrossRef]
- Klein, E.L.; Belaidi, A.A.; Raitsimring, A.M.; Davis, A.C.; Kramer, T.; Astashkin, A.V.; Neese, F.; Schwarz, G.; Enemark, J.H. Pulsed Electron Paramagnetic Resonance Spectroscopy of S-33-Labeled Molybdenum Cofactor in Catalytically Active Bioengineered Sulfite Oxidase. Inorg. Chem. 2014, 53, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Inscore, F.E.; Hille, R.; Kirk, M.L. Freeze-Quench Magnetic Circular Dichroism Spectroscopic Study of the “Very Rapid” Intermediate in Xanthine Oxidase. Inorg. Chem. 1999, 38, 4963–4970. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Dong, C.; Kirk, M.L. Xanthine oxidase-product complexes probe the importance of substrate/product orientation along the reaction coordinate. Dalton Trans. 2017, 46, 13242–13250. [Google Scholar] [CrossRef]
- Dong, C.; Yang, J.; Reschke, S.; Leimkühler, S.; Kirk, M.L. Vibrational Probes of Molybdenum Cofactor–Protein Interactions in Xanthine Dehydrogenase. Inorg. Chem. 2017, 56, 6830–6837. [Google Scholar] [CrossRef]
- Dong, C.; Yang, J.; Leimkühler, S.; Kirk, M.L. Pyranopterin Dithiolene Distortions Relevant to Electron Transfer in Xanthine Oxidase/Dehydrogenase. Inorg. Chem. 2014, 53, 7077–7079. [Google Scholar] [CrossRef]
- Helton, M.E.; Pacheco, A.; McMaster, J.; Enemark, J.H.; Kirk, M.L. An MCD Spectroscopic Study of the Molybdenum Active Site in Sulfite Oxidase: Insight into the Role of Coordinated Cysteine. J. Inorg. Biochem. 2000, 80, 227–233. [Google Scholar] [CrossRef]
- Sugimoto, H.; Sato, M.; Asano, K.; Suzuki, T.; Mieda, K.; Ogura, T.; Matsumoto, T.; Giles, L.J.; Pokhrel, A.; Kirk, M.L.; et al. A Model for the Active-Site Formation Process in DMSO Reductase Family Molybdenum Enzymes Involving Oxido Alcoholato and Oxido Thiolato Molybdenum(VI) Core Structures. Inorg. Chem. 2016, 55, 1542–1550. [Google Scholar] [CrossRef]
- Sugimoto, H.; Sato, M.; Giles, L.J.; Asano, K.; Suzuki, T.; Kirk, M.L.; Itoh, S. Oxo-carboxylato-molybdenum(VI) complexes possessing dithiolene ligands related to the active site of type II DMSOR family molybdoenzymes. Dalton Trans. 2013, 42, 5927–15930. [Google Scholar] [CrossRef][Green Version]
- Sugimoto, H.; Tatemoto, S.; Suyama, K.; Miyake, H.; Mtei, R.P.; Itoh, S.; Kirk, M.L. Monooxomolybdenum(VI) Complexes Possessing Olefinic Dithiolene Ligands: Probing Mo–S Covalency Contributions to Electron Transfer in Dimethyl Sulfoxide Reductase Family Molybdoenzymes. Inorg. Chem. 2010, 49, 5368–5370. [Google Scholar] [CrossRef]
- Sugimoto, H.; Tatemoto, S.; Suyama, K.; Miyake, H.; Itoh, S.; Dong, C.; Yang, J.; Kirk, M.L. Dioxomolybdenum(VI) Complexes with Ene-1,2-dithiolate Ligands: Synthesis, Spectroscopy, and Oxygen Atom Transfer Reactivity. Inorg. Chem. 2009, 48, 10581–10590. [Google Scholar] [CrossRef]
- Burgmayer, S.J.N.; Kim, M.; Petit, R.; Rothkopf, A.; Kim, A.; BelHamdounia, S.; Hou, Y.; Somogyi, A.; Habel-Rodriguez, D.; Williams, A.; et al. Synthesis, characterization, and spectroscopy of model molybdopterin complexes. J. Inorg. Biochem. 2007, 101, 1601–1616. [Google Scholar] [CrossRef]
- Kirk, M.L.; Peariso, K. Ground and excited state spectral comparisons of models for sulfite oxidase. Polyhedron 2004, 23, 499. [Google Scholar] [CrossRef]
- Helton, M.E.; Gebhart, N.L.; Davies, E.S.; McMaster, J.; Garner, C.D.; Kirk, M.L. Thermally Driven Intramolecular Charge Transfer in an Oxo-Molybdenum Dithiolate Complex. J. Am. Chem. Soc. 2001, 123, 10389–10390. [Google Scholar] [CrossRef]
- Davie, S.R.; Rubie, N.D.; Hammes, B.S.; Carrano, C.J.; Kirk, M.L.; Basu, P. Geometric control of reduction potential in oxomolybdenum centers: Implications to the serine coordination in DMSO reductase. Inorg. Chem. 2001, 40, 2632. [Google Scholar] [CrossRef]
- McNaughton, R.L.; Helton, M.E.; Rubie, N.D.; Kirk, M.L. The oxo-gate hypothesis and DMSO reductase: Implications for a psuedo-sigma bonding interaction involved in enzymatic electron transfer. Inorg. Chem. 2000, 39, 4386. [Google Scholar] [CrossRef]
- Helton, M.; Gruhn, N.; McNaughton, R.; Kirk, M. Control of oxo-molybdenum reduction and ionization potentials by dithiolate donors. Inorg. Chem. 2000, 39, 2273–2278. [Google Scholar] [CrossRef]
- Gisewhite, D.R.; Yang, J.; Williams, B.R.; Esmail, A.; Stein, B.; Kirk, M.L.; Burgmayer, S.J.N. Implications of Pyran Cyclization and Pterin Conformation on Oxidized Forms of the Molybdenum Cofactor. J. Am. Chem. Soc. 2018, 140, 12808–12818. [Google Scholar] [CrossRef]
- Paudel, J.; Pokhrel, A.; Kirk, M.L.; Li, F. Remote Charge Effects on the Oxygen-Atom-Transfer Reactivity and Their Relationship to Molybdenum Enzymes. Inorg. Chem. 2019, 58, 2054–2068. [Google Scholar] [CrossRef]
- Westcott, B.L.; Gruhn, N.E.; Enemark, J.H. Evaluation of Molybdenum-Sulfur Interactions in Molybdoenzyme Model Complexes by Gas-Phase Photoelectron Spectroscopy. The ‘‘Electronic Buffer’’ Effect. J. Am. Chem. Soc. 1998, 120, 3382–3386. [Google Scholar] [CrossRef]
- Wiebelhaus, N.J.; Cranswick, M.A.; Klein, E.L.; Lockett, L.T.; Lichtenberger, D.L.; Enemark, J.H. Metal-Sulfur Valence Orbital Interaction Energies in Metal-Dithiolene Complexes: Determination of Charge and Overlap Interaction Energies by Comparison of Core and Valence Ionization Energy Shifts. Inorg. Chem. 2011, 50, 11021–11031. [Google Scholar] [CrossRef][Green Version]
- Davis, M.; Olson, J.; Palmer, G. The Reaction of Xanthine Oxidase with Lumazine: Characterization of the Reductive Half-reaction. J. Biol. Chem. 1984, 259, 3526–3533. [Google Scholar] [PubMed]
- Pauff, J.M.; Cao, H.; Hille, R. Substrate Orientation and Catalysis at the Molybdenum Site in Xanthine Oxidase Crystal structures in complex with xanthine and lumazine. J. Biol. Chem. 2009, 284, 8751–8758. [Google Scholar] [CrossRef] [PubMed]
- Hemann, C.; Ilich, P.; Stockert, A.L.; Choi, E.Y.; Hille, R. Resonance Raman studies of xanthine oxidase: The reduced enzyme–Product complex with violapterin. J. Phys. Chem. B 2005, 109, 3023–3031. [Google Scholar] [CrossRef] [PubMed]
- Hemann, C.; Ilich, P.; Hille, R. Vibrational spectra of lumazine in water at pH 2–13: Ab initio calculation and FTIR/Raman spectra. J. Phys. Chem. B 2003, 107, 2139–2155. [Google Scholar] [CrossRef]
- Stein, B.W.; Yang, J.; Mtei, R.; Wiebelhaus, N.J.; Kersi, D.K.; LePluart, J.; Lichtenberger, D.L.; Enemark, J.H.; Kirk, M.L. Vibrational Control of Covalency Effects Related to the Active Sites of Molybdenum Enzymes. J. Am. Chem. Soc. 2018, 140, 14777–14788. [Google Scholar] [CrossRef]
- Bersuker, I.B. The Jahn-Teller Effect; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Bersuker, I.B. Pseudo-Jahn-Teller Effect-A Two-State Paradigm in Formation, Deformation, and Transformation of Molecular Systems and Solids. Chem. Rev. 2013, 113, 1351–1390. [Google Scholar] [CrossRef]












© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Enemark, J.H.; Kirk, M.L. Metal–Dithiolene Bonding Contributions to Pyranopterin Molybdenum Enzyme Reactivity. Inorganics 2020, 8, 19. https://doi.org/10.3390/inorganics8030019
Yang J, Enemark JH, Kirk ML. Metal–Dithiolene Bonding Contributions to Pyranopterin Molybdenum Enzyme Reactivity. Inorganics. 2020; 8(3):19. https://doi.org/10.3390/inorganics8030019
Chicago/Turabian StyleYang, Jing, John H. Enemark, and Martin L. Kirk. 2020. "Metal–Dithiolene Bonding Contributions to Pyranopterin Molybdenum Enzyme Reactivity" Inorganics 8, no. 3: 19. https://doi.org/10.3390/inorganics8030019
APA StyleYang, J., Enemark, J. H., & Kirk, M. L. (2020). Metal–Dithiolene Bonding Contributions to Pyranopterin Molybdenum Enzyme Reactivity. Inorganics, 8(3), 19. https://doi.org/10.3390/inorganics8030019