Redox-Active Manganese Pincers for Electrocatalytic CO2 Reduction


Abstract

1. Introduction
2. Mn-Based Electrocatalysts in CO2 Reduction
3. Pincers in Mn CO2 Reduction Electrocatalysts
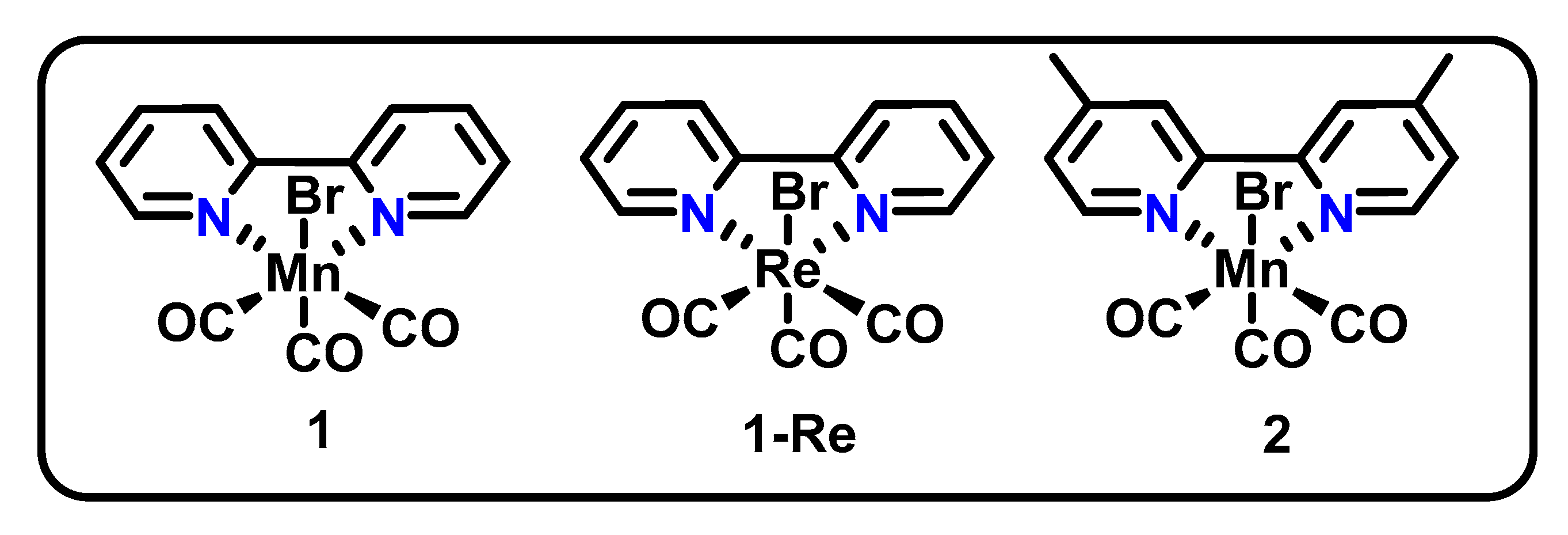
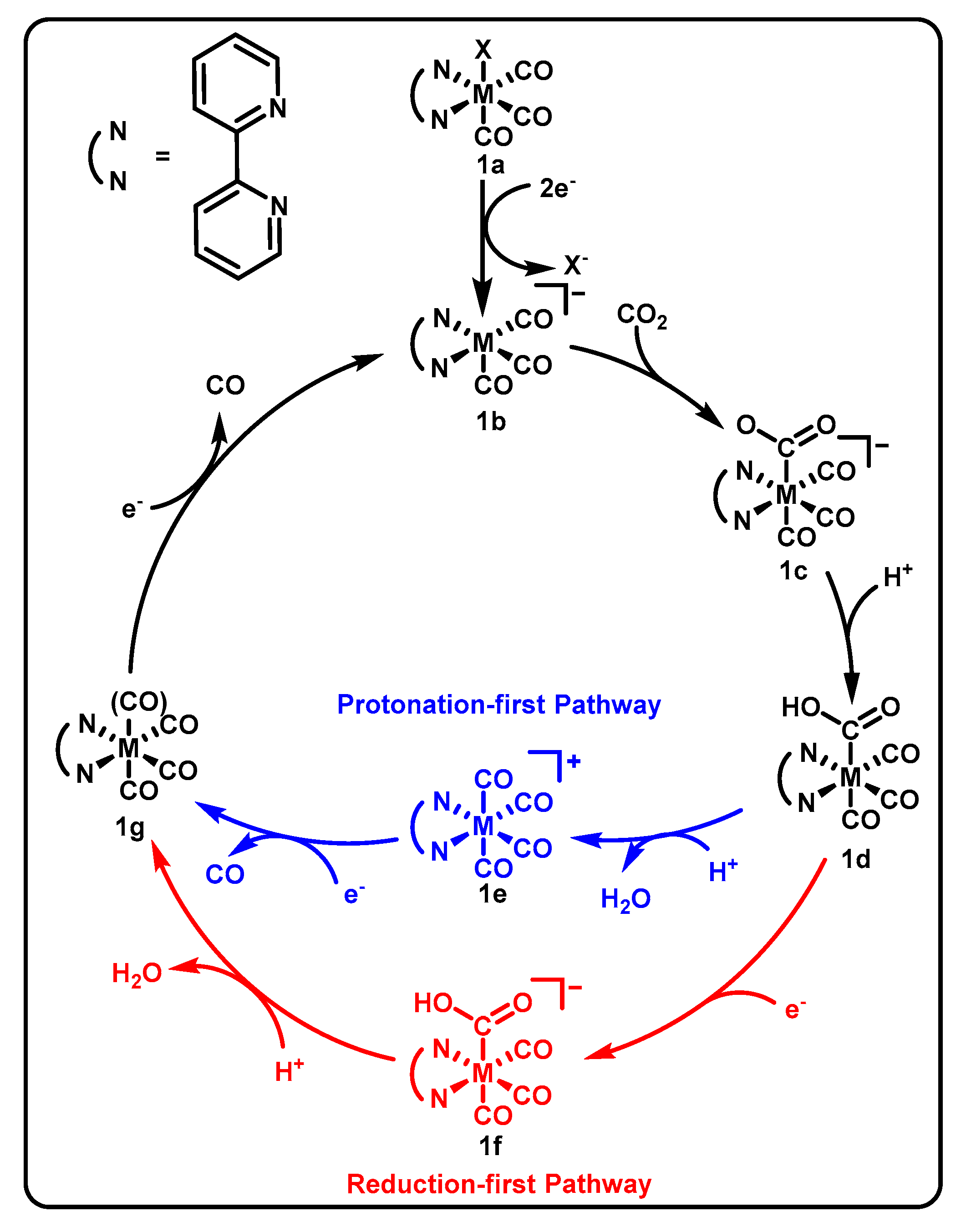
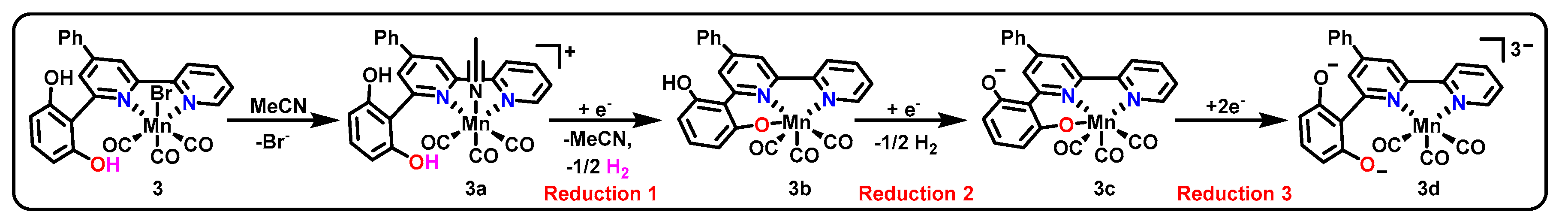
3.1. ONN Pincer Binding in Mn(bpy-R)(CO)3X Catalysts
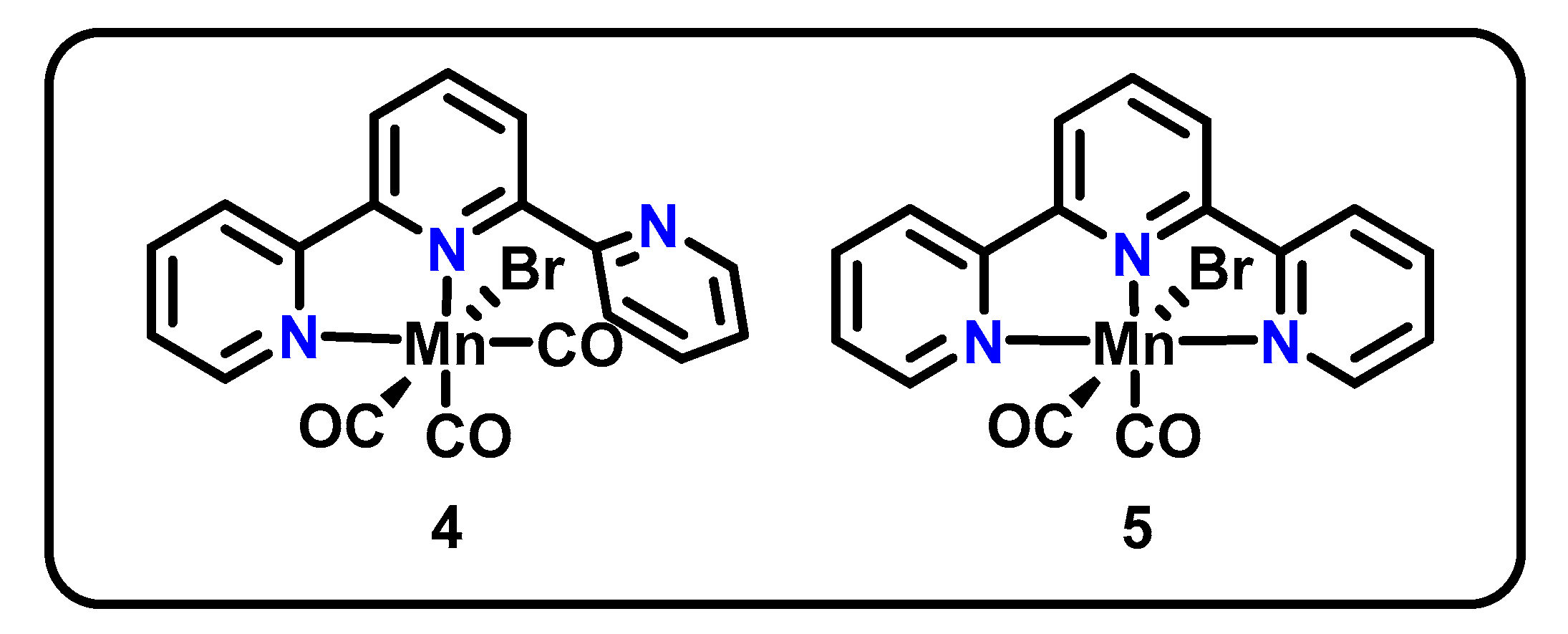
3.2. NNN and tpy-Based Pincers
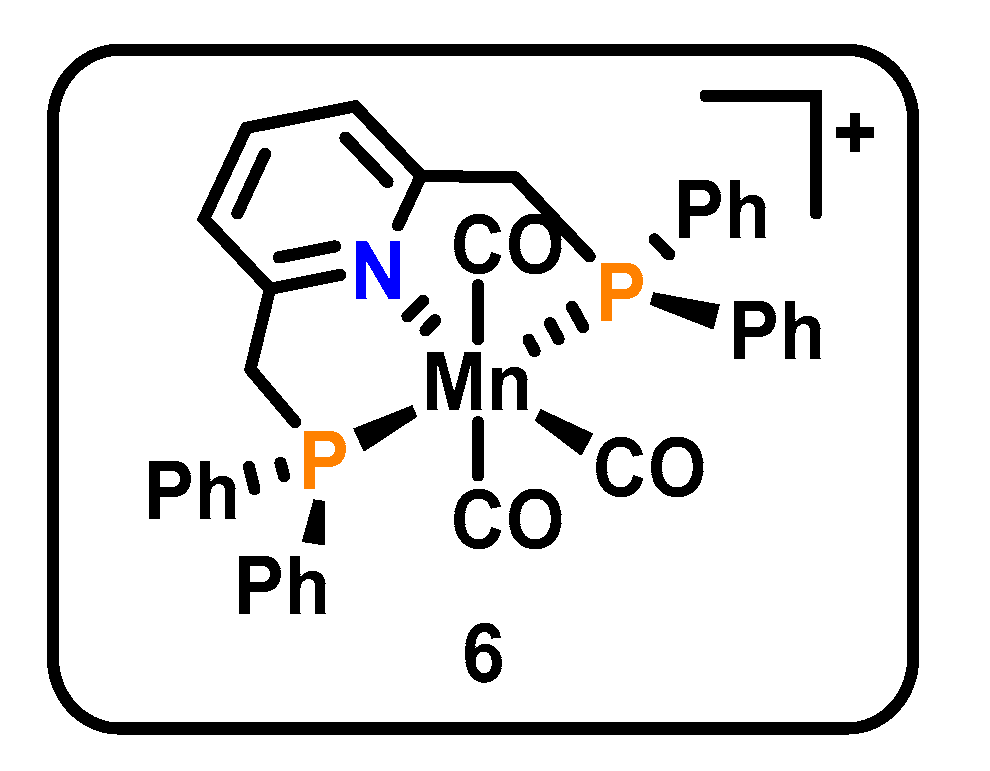
3.3. PNP Pincers
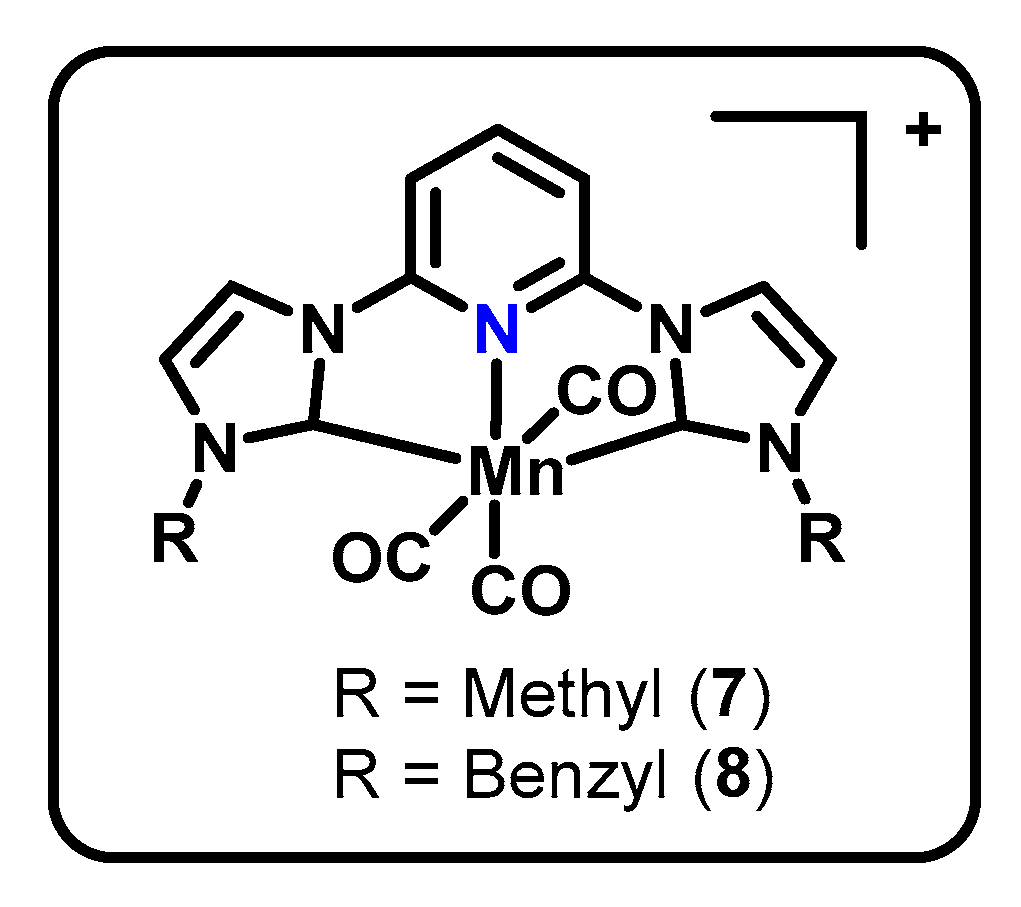
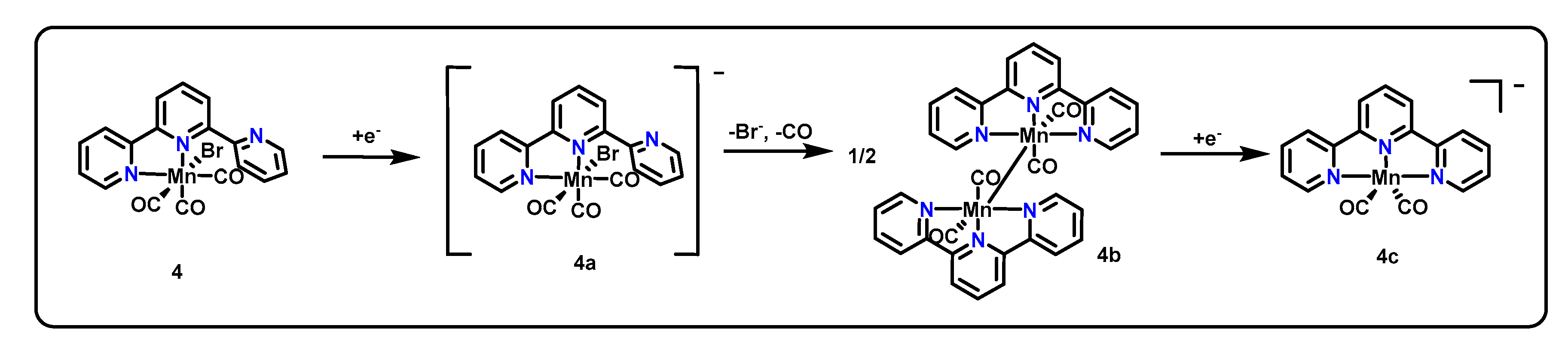
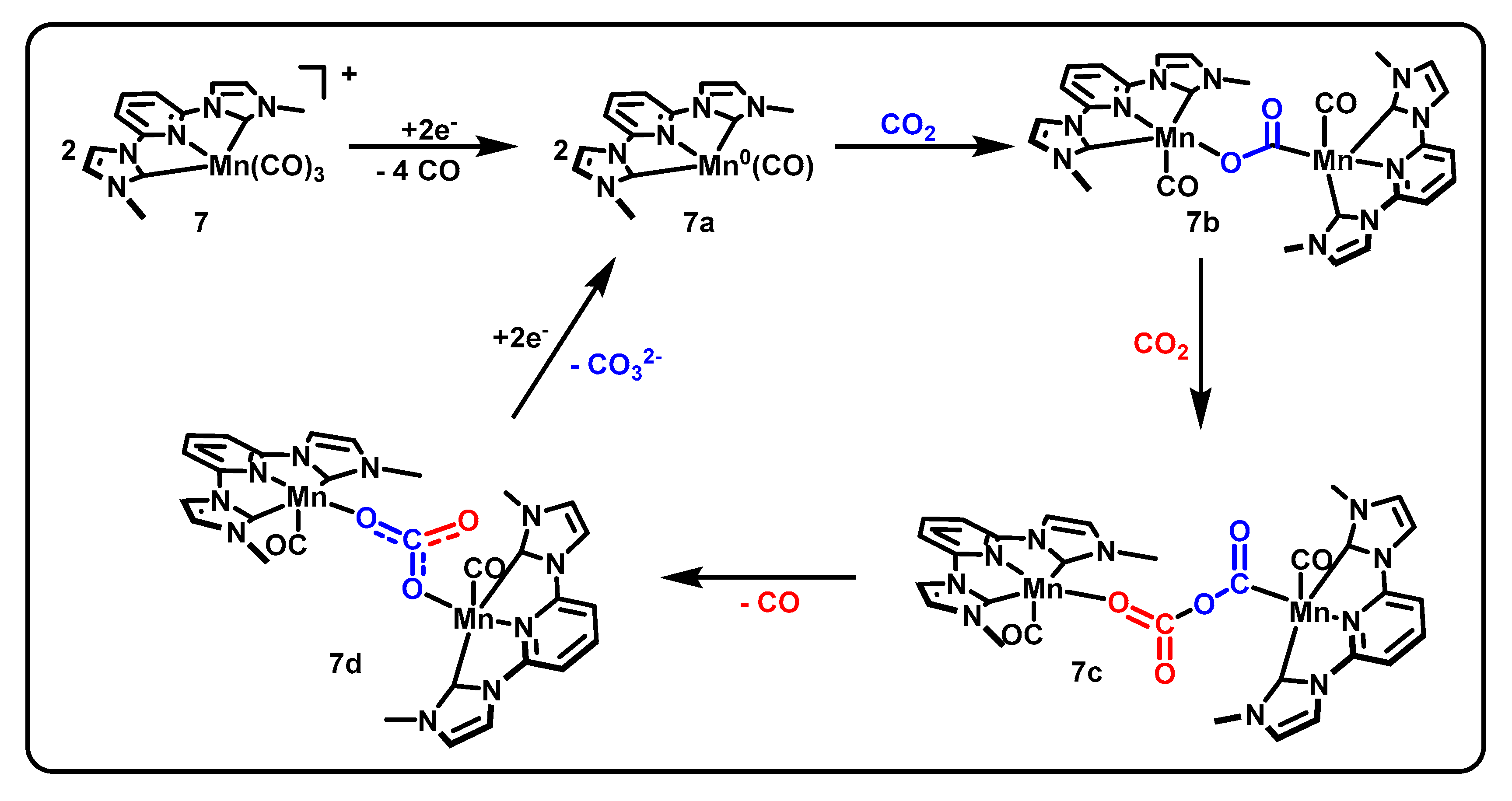
3.4. CNC Pincers
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- IPCC. Climate Change 2014: Synthesis Report; Intergovernmental Panel on Climate Change: Geneva, Switzerland, 2014.
- Cox, P.M.; Betts, R.A.; Jones, C.D.; Spall, S.A.; Totterdell, I.J. Acceleration of global warming due to carbon-cycle feedbacks in a coupled climate model. Nature 2000, 408, 184–187. [Google Scholar] [CrossRef]
- Solomon, S.; Plattner, G.-K.; Knutti, R.; Friedlingstein, P. Irreversible climate change due to carbon dioxide emissions. Proc. Natl. Acad. Sci. USA 2009, 106, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- IPCC. Global Warming of 1.5 °C; WHO: Geneva, Switzerland, 2018.
- Mikkelsen, M.; Jørgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Sherwood, S.; Webb, M.J.; Annan, J.D.; Armour, K.C.; Forster, P.M.; Hargreaves, J.C.; Hegerl, G.; Klein, S.A.; Marvel, K.D.; Rohling, E.J.; et al. An assessment of Earth’s climate sensitivity using multiple lines of evidence. Rev. Geophys. 2020, 58, e2019RG000678. [Google Scholar] [CrossRef] [PubMed]
- Whittingham, M.S. History, Evolution, and Future Status of Energy Storage. Proc. IEEE 2012, 100, 1518–1534. [Google Scholar] [CrossRef]
- Global CCS Institute. The Global Status of CCS: 2019; Global Carbon Capture and Storage Institute Ltd: Melbourne, Australia, 2019. [Google Scholar]
- Mac Dowell, N.; Fennell, P.S.; Shah, N.; Maitland, G.C. The role of CO2 capture and utilization in mitigating climate change. Nat. Clim. Chang. 2017, 7, 243–249. [Google Scholar] [CrossRef]
- Kar, S.; Goeppert, A.; Kothandaraman, J.; Prakash, G.K.S. Manganese-Catalyzed Sequential Hydrogenation of CO2 to Methanol via Formamide. ACS Catal. 2017, 7, 6347–6351. [Google Scholar] [CrossRef]
- Schneidewind, J.; Adam, R.; Baumann, W.; Jackstell, R.; Beller, M. Low-Temperature Hydrogenation of Carbon Dioxide to Methanol with a Homogeneous Cobalt Catalyst. Angew. Chem. Int. Ed. 2017, 56, 1890–1893. [Google Scholar] [CrossRef]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kuhl, S.; Havecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.L.; et al. The Active Site of Methanol Synthesis over Cu/ZnO/Al2O3 Industrial Catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef]
- Yang, X.; Chen, X. Chapter 5—Mechanistic Insights and Computational Prediction of Base Metal Pincer Complexes for Catalytic Hydrogenation and Dehydrogenation Reactions. In Pincer Compounds; Morales-Morales, D., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 101–110. [Google Scholar]
- Gao, S.; Fan, W.; Liu, Y.; Jiang, D.; Duan, Q. Artificial water-soluble systems inspired by [FeFe]-hydrogenases for electro- and photocatalytic hydrogen production. Int. J. Hydrog. Energy 2020, 45, 4305–4327. [Google Scholar] [CrossRef]
- Peterson, A.A.; Nørskov, J.K. Activity Descriptors for CO2 Electroreduction to Methane on Transition-Metal Catalysts. J. Phys. Chem. Lett. 2012, 3, 251–258. [Google Scholar] [CrossRef]
- Nam, D.-H.; De Luna, P.; Rosas-Hernández, A.; Thevenon, A.; Li, F.; Agapie, T.; Peters, J.C.; Shekhah, O.; Eddaoudi, M.; Sargent, E.H. Molecular enhancement of heterogeneous CO2 reduction. Nat. Mater. 2020, 19, 266–276. [Google Scholar] [PubMed]
- Jiang, C.; Nichols, A.W.; Machan, C.W. A look at periodic trends in d-block molecular electrocatalysts for CO2 reduction. Dalton Trans. 2019, 48, 9454–9468. [Google Scholar] [CrossRef] [PubMed]
- Smieja, J.M.; Kubiak, C.P. Re(bipy-tBu) (CO)3Cl−improved Catalytic Activity for Reduction of Carbon Dioxide: IR-Spectroelectrochemical and Mechanistic Studies. Inorg. Chem. 2010, 49, 9283–9289. [Google Scholar] [CrossRef]
- Sung, S.; Kumar, D.; Gil-Sepulcre, M.; Nippe, M. Electrocatalytic CO2 Reduction by Imidazolium-Functionalized Molecular Catalysts. J. Am. Chem. Soc. 2017, 139, 13993–13996. [Google Scholar] [CrossRef]
- Clark, M.L.; Cheung, P.L.; Lessio, M.; Carter, E.A.; Kubiak, C.P. Kinetic and Mechanistic Effects of Bipyridine (bpy) Substituent, Labile Ligand, and Brønsted Acid on Electrocatalytic CO2 Reduction by Re(bpy) Complexes. ACS Catal. 2018, 8, 2021–2029. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, C.; Weinberg, D.R.; Kang, P.; Concepcion, J.J.; Harrison, D.P.; Brookhart, M.S.; Meyer, T.J. Electrocatalytic reduction of CO2 to CO by polypyridyl ruthenium complexes. Chem. Commun. 2011, 47, 12607–12609. [Google Scholar] [CrossRef]
- Machan, C.W.; Sampson, M.D.; Kubiak, C.P. A Molecular Ruthenium Electrocatalyst for the Reduction of Carbon Dioxide to CO and Formate. J. Am. Chem. Soc. 2015, 137, 8564–8571. [Google Scholar] [CrossRef]
- Johnson, B.A.; Maji, S.; Agarwala, H.; White, T.A.; Mijangos, E.; Ott, S. Activating a Low Overpotential CO2 Reduction Mechanism by a Strategic Ligand Modification on a Ruthenium Polypyridyl Catalyst. Angew. Chem. Int. Ed. Engl. 2016, 55, 1825–1829. [Google Scholar] [CrossRef]
- Chen, Z.; Kang, P.; Zhang, M.-T.; Meyer, T.J. Making syngas electrocatalytically using a polypyridyl ruthenium catalyst. Chem. Commun. 2014, 50, 335–337. [Google Scholar] [CrossRef]
- Chen, Z.; Concepcion, J.J.; Brennaman, M.K.; Kang, P.; Norris, M.R.; Hoertz, P.G.; Meyer, T.J. Splitting CO2 into CO and O2 by a single catalyst. Proc. Natl. Acad. Sci. USA 2012, 109, 15606–15611. [Google Scholar] [CrossRef] [PubMed]
- Daryanavard, M.; Hadadzadeh, H.; Weil, M.; Farrokhpour, H. Electrocatalytic reduction of CO2 to CO in the presence of a mononuclear polypyridyl ruthenium(II) complex. J. CO2 Util. 2017, 17, 80–89. [Google Scholar] [CrossRef]
- Lilio, A.M. Bi-Functional Molecular Catalysts for CO2 Reduction; UC San Diego: San Diego, CA, USA, 2015. [Google Scholar]
- Hu, G.; Jiang, J.J.; Kelly, H.R.; Matula, A.J.; Wu, Y.; Romano, N.; Mercado, B.Q.; Wang, H.; Batista, V.S.; Crabtree, R.H.; et al. Surprisingly big linker-dependence of activity and selectivity in CO2 reduction by an iridium(I) pincer complex. Chem. Commun. 2020, 56, 9126–9129. [Google Scholar] [CrossRef] [PubMed]
- Kang, P.; Cheng, C.; Chen, Z.; Schauer, C.K.; Meyer, T.J.; Brookhart, M. Selective Electrocatalytic Reduction of CO2 to Formate by Water-Stable Iridium Dihydride Pincer Complexes. J. Am. Chem. Soc. 2012, 134, 5500–5503. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Sun, C.; Sun, N.; Meng, L.; Chen, D. Theoretical mechanism studies on the electrocatalytic reduction of CO2 to formate by water-stable iridium dihydride pincer complex. Dalton Trans. 2013, 42, 5755. [Google Scholar] [CrossRef] [PubMed]
- Feller, M.; Gellrich, U.; Anaby, A.; Diskin-Posner, Y.; Milstein, D. Reductive Cleavage of CO2 by Metal–Ligand-Cooperation Mediated by an Iridium Pincer Complex. J. Am. Chem. Soc. 2016, 138, 6445–6454. [Google Scholar] [CrossRef] [PubMed]
- Weast, R.C. CRC Handbook of Chemistry and Physics; CRC Press: Boca Raton, FL, USA, 1988. [Google Scholar]
- Eberhardt, N.A.; Guan, H. Reduction of CO2 Mediated or Catalyzed by Pincer Complexes; Elsevier: Amsterdam, The Netherlands, 2018; pp. 67–99. [Google Scholar]
- Bourrez, M.; Molton, F.; Chardon-Noblat, S.; Deronzier, A. [Mn(bipyridyl)(CO)3Br]: An Abundant Metal Carbonyl Complex as Efficient Electrocatalyst for CO2 Reduction. Angew. Chem. Int. Ed. 2011, 50, 9903–9906. [Google Scholar] [CrossRef]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Electrocatalytic reduction of carbon dioxide mediated by Re(bipy)(CO)3Cl (bipy = 2,2′-bipyridine). J. Chem. Soc. Chem. Commun. 1984, 328–330. [Google Scholar] [CrossRef]
- Staal, L.H.; Oskam, A.; Vrieze, K. The syntheses and coordination properties of M(CO)3X(DAB) (M = Mn, Re; X = Cl, Br, I.; DAB = 1,4-diazabutadiene). J. Organomet. Chem. 1979, 170, 235–245. [Google Scholar] [CrossRef]
- Smieja, J.M.; Sampson, M.D.; Grice, K.A.; Benson, E.E.; Froehlich, J.D.; Kubiak, C.P. Manganese as a Substitute for Rhenium in CO2 Reduction Catalysts: The Importance of Acids. Inorg. Chem. 2013, 52, 2484–2491. [Google Scholar] [CrossRef]
- Sampson, M.D.; Kubiak, C.P. Manganese Electrocatalysts with Bulky Bipyridine Ligands: Utilizing Lewis Acids to Promote Carbon Dioxide Reduction at Low Overpotentials. J. Am. Chem. Soc. 2016, 138, 1386–1393. [Google Scholar] [CrossRef]
- Ngo, K.T.; McKinnon, M.; Mahanti, B.; Narayanan, R.; Grills, D.C.; Ertem, M.Z.; Rochford, J. Turning on the Protonation-First Pathway for Electrocatalytic CO2 Reduction by Manganese Bipyridyl Tricarbonyl Complexes. J. Am. Chem. Soc. 2017, 139, 2604–2618. [Google Scholar] [CrossRef]
- Luca, O.R.; Crabtree, R.H. Redox-active ligands in catalysis. Chem. Soc. Rev. 2013, 42, 1440–1459. [Google Scholar] [CrossRef]
- Chirik, P.J.; Wieghardt, K. Radical Ligands Confer Nobility on Base-Metal Catalysts. Science 2010, 327, 794–795. [Google Scholar] [CrossRef] [PubMed]
- Riplinger, C.; Sampson, M.D.; Ritzmann, A.M.; Kubiak, C.P.; Carter, E.A. Mechanistic Contrasts between Manganese and Rhenium Bipyridine Electrocatalysts for the Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2014, 136, 16285–16298. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.A.W.; Green, K.-A.; Nelson, P.N.; Lorraine, S.C. Review: Pincer ligands—Tunable, versatile and applicable. Polyhedron 2018, 143, 11–27. [Google Scholar] [CrossRef]
- Franco, F.; Cometto, C.; Ferrero Vallana, F.; Sordello, F.; Priola, E.; Minero, C.; Nervi, C.; Gobetto, R. A local proton source in a [Mn(bpy-R)(CO)3Br]-type redox catalyst enables CO2 reduction even in the absence of Brønsted acids. Chem. Commun. 2014, 50, 14670–14673. [Google Scholar] [CrossRef] [PubMed]
- Franco, F.; Cometto, C.; Nencini, L.; Barolo, C.; Sordello, F.; Minero, C.; Fiedler, J.; Robert, M.; Gobetto, R.; Nervi, C. Local Proton Source in Electrocatalytic CO2 Reduction with [Mn(bpy-R)(CO)3Br] Complexes. Chem. A Eur. J. 2017, 23, 4782–4793. [Google Scholar] [CrossRef]
- Compain, J.-D.; Bourrez, M.; Haukka, M.; Deronzier, A.; Chardon-Noblat, S. Manganese carbonyl terpyridyl complexes: Their synthesis, characterization and potential application as CO-release molecules. Chem. Commun. 2014, 50, 2539–2542. [Google Scholar] [CrossRef]
- Machan, C.W.; Kubiak, C.P. Electrocatalytic reduction of carbon dioxide with Mn(terpyridine) carbonyl complexes. Dalton Trans. 2016, 45, 17179–17186. [Google Scholar] [CrossRef]
- Rao, G.K.; Pell, W.; Korobkov, I.; Richeson, D. Electrocatalytic reduction of CO2 using Mn complexes with unconventional coordination environments. Chem. Commun. 2016, 52, 8010–8013. [Google Scholar] [CrossRef] [PubMed]
- Myren, T.H.T.; Lilio, A.M.; Huntzinger, C.G.; Horstman, J.W.; Stinson, T.A.; Donadt, T.B.; Moore, C.; Lama, B.; Funke, H.H.; Luca, O.R. Manganese N-Heterocyclic Carbene Pincers for the Electrocatalytic Reduction of Carbon Dioxide. Organometallics 2019, 38, 1248–1253. [Google Scholar] [CrossRef]
- Myren, T.H.T.; Alherz, A.; Thurston, J.R.; Stinson, T.A.; Huntzinger, C.G.; Musgrave, C.B.; Luca, O.R. Mn-Based Molecular Catalysts for the Electrocatalytic Disproportionation of CO2 into CO and CO32–. ACS Catal. 2020, 10, 1961–1968. [Google Scholar] [CrossRef]
- Nagao, H.; Mizukawa, T.; Tanaka, K. Carbon-Carbon Bond Formation in the Electrochemical Reduction of Carbon Dioxide Catalyzed by a Ruthenium Complex. Inorg. Chem. 1994, 33, 3415–3420. [Google Scholar] [CrossRef]
- Narayanan, R.; McKinnon, M.; Reed, B.R.; Ngo, K.T.; Groysman, S.; Rochford, J. Ambiguous electrocatalytic CO2 reduction behaviour of a nickel bis(aldimino)pyridine pincer complex. Dalton Trans. 2016, 45, 15285–15289. [Google Scholar] [CrossRef]
- Vogt, M.; Nerush, A.; Diskin-Posner, Y.; Ben-David, Y.; Milstein, D. Reversible CO2 binding triggered by metal–ligand cooperation in a rhenium(I) PNP pincer-type complex and the reaction with dihydrogen. Chem. Sci. 2014, 5, 2043–2051. [Google Scholar] [CrossRef]
- Donadt, T.B.; Lilio, A.M.; Stinson, T.A.; Lama, B.; Luca, O.R. DOSY NMR and Normal Pulse Voltammetry for the Expeditious Determination of Number of Electrons Exchanged in Redox Events. Chem. Sel. 2018, 3, 7410–7415. [Google Scholar] [CrossRef]
- Myren, T.H.T.; Alherz, A.; Stinson, T.A.; Huntzinger, C.G.; Lama, B.; Musgrave, C.; Luca, O.R. Metalloradical intermediates in electrocatalytic reduction of CO2 to CO: Mn versus Re bis-N-heterocyclic carbene pincers. Dalton Trans. 2020, 49, 2053–2057. [Google Scholar] [CrossRef]
- Therrien, J.A.; Wolf, M.O. The Influence of para Substituents in Bis(N-Heterocyclic Carbene) Palladium Pincer Complexes for Electrocatalytic CO2 Reduction. Inorg. Chem. 2017, 56, 1161–1172. [Google Scholar] [CrossRef]
- Therrien, J.A.; Wolf, M.O.; Patrick, B.O. Polyannulated Bis(N-heterocyclic carbene)palladium Pincer Complexes for Electrocatalytic CO2 Reduction. Inorg. Chem. 2015, 54, 11721–11732. [Google Scholar] [CrossRef]
- Therrien, J.A.; Wolf, M.O.; Patrick, B.O. Electrocatalytic Reduction of CO2 with Palladium Bis-N-heterocyclic Carbene Pincer Complexes. Inorg. Chem. 2014, 53, 12962–12972. [Google Scholar] [CrossRef] [PubMed]
- Therrien, J.A.; Wolf, M.O.; Patrick, B.O. Synthesis and comparison of nickel, palladium, and platinum bis(N-heterocyclic carbene) pincer complexes for electrocatalytic CO2 reduction. Dalton Trans. 2018, 47, 1827–1840. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (Pre-)Catalyst | Pincer Type | Oxidation States | Products (FE, %) | Bulk Electrolysis Conditions | Proton Source | Ref. |
|---|---|---|---|---|---|---|
| 3a (intermediate, Scheme 2) | ONN | Mn(I), Mn(0) | CO (70%), HCOO− (22%), H2O | −1.8 V vs. SCE, 4 h, MeCN | none | [44] |
| CO (90%), HCOO− (4%) | −1.5 V vs. SCE, 2 h, MeCN, 0.1M TBA PF6 | 2.7 M H2O | [45] | |||
| CO (48%), HCOO− (36%) | −1.5 V vs. SCE, 2 h, MeCN, 0.1 M TBA PF6 | 2.7 M TFE | ||||
| CO (15%), HCOO− (39%) | −1.5 V vs. SCE, 2 h, MeCN, 0.1 M TBA PF6 | 2.7 M phenol | ||||
| Mn(κ2-tpy)(CO)3Br (4, Figure 2) | NNN | Mn(I), Mn(0) | CO (129%) 1, H2O | −2.2 V vs. Fc/Fc+, MeCN, 0.1 M TBA PF6, 4.1 turnovers | 0.5 M phenol | [46,47] |
| Mn(κ3-tpy)(CO)2Br (5, Figure 2) | NNN | Mn(I), Mn(0) | CO (93%), H2O | −2.2 V vs. Fc/Fc+, MeCN, 0.1 M TBA PF6, 4.1 turnovers | 0.5 M phenol | |
| MnPNP(CO)3 (6, Figure 3) | PNP | Mn(I), Mn(0) | CO (96%), CO32− | −2.3 V vs. Fc/Fc+, 125 min, MeCN, 0.1 M TBA PF6 | none | [48] |
| MnCNCMe (7, Figure 4) | CNC | Mn(I), Mn(0) | CO (87%), CO32− | −2.3 V vs. Fc/Fc+, MeCN, 0.1 M TBA PF6 | none | [49] |
| MnCNCBn (8, Figure 4) | CNC | Mn(I), Mn(0) | CO (86%), CO32− (93%) | −2.3 V vs. Fc/Fc+, MeCN, 0.1 M TBA PF6 | none | [49,50] |
| Mn(dmbpy)(CO)3X (2, Figure 1) | - | Mn(I), Mn(0) | CO (100%), H2O | 138 C, −1.70 V vs. Ag/AgNO3, 1mM cat in 95/5 MeCN/H2O, 0.1 M TBAP | 5% H2O | [34] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petersen, H.A.; Myren, T.H.T.; Luca, O.R. Redox-Active Manganese Pincers for Electrocatalytic CO2 Reduction. Inorganics 2020, 8, 62. https://doi.org/10.3390/inorganics8110062
Petersen HA, Myren THT, Luca OR. Redox-Active Manganese Pincers for Electrocatalytic CO2 Reduction. Inorganics. 2020; 8(11):62. https://doi.org/10.3390/inorganics8110062
Chicago/Turabian StylePetersen, Haley A., Tessa H. T. Myren, and Oana R. Luca. 2020. "Redox-Active Manganese Pincers for Electrocatalytic CO2 Reduction" Inorganics 8, no. 11: 62. https://doi.org/10.3390/inorganics8110062
APA StylePetersen, H. A., Myren, T. H. T., & Luca, O. R. (2020). Redox-Active Manganese Pincers for Electrocatalytic CO2 Reduction. Inorganics, 8(11), 62. https://doi.org/10.3390/inorganics8110062