Electronic Communication between Dithiolato-Bridged Diiron Carbonyl and S-Bridged Redox-Active Centres

 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
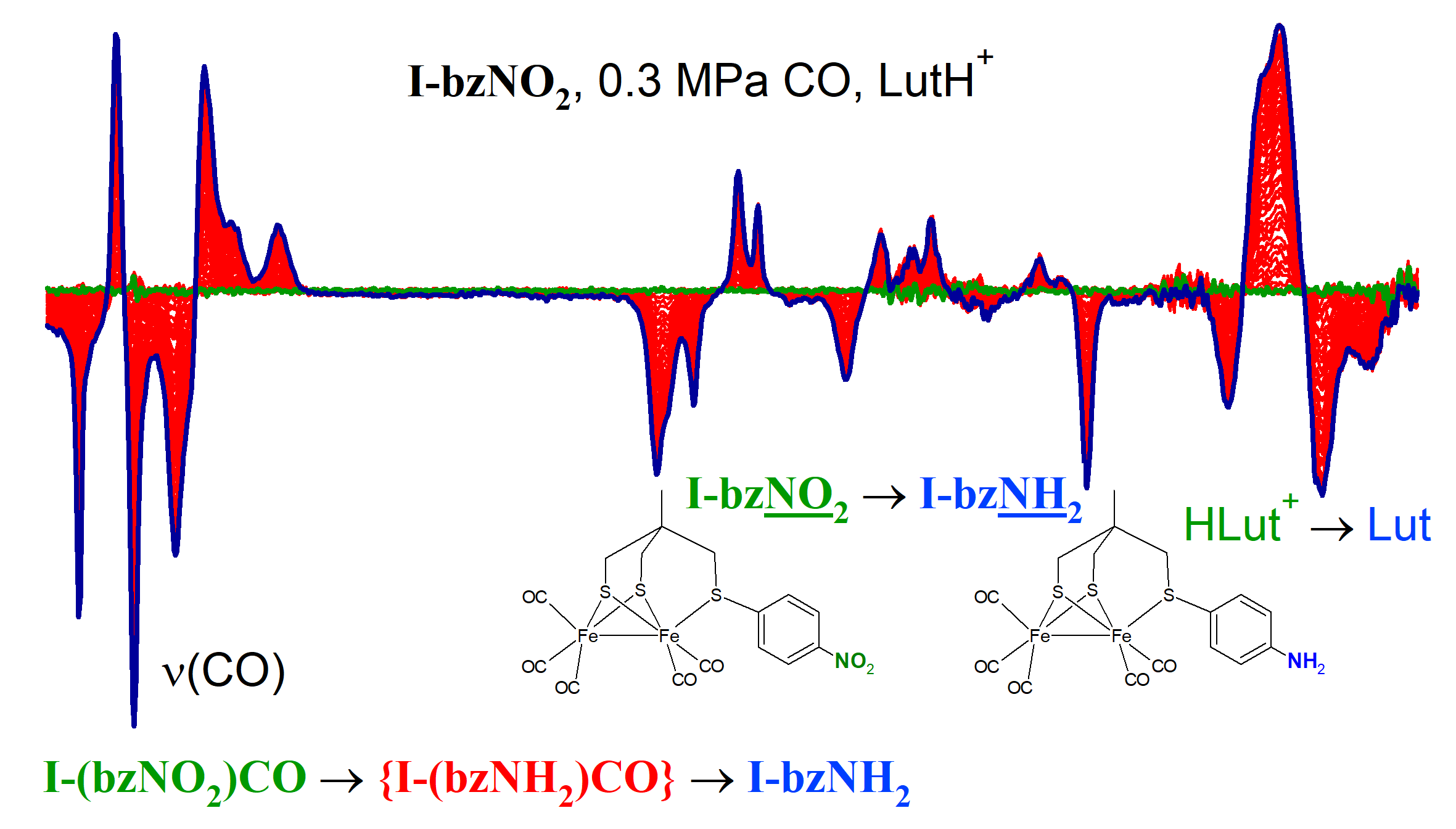
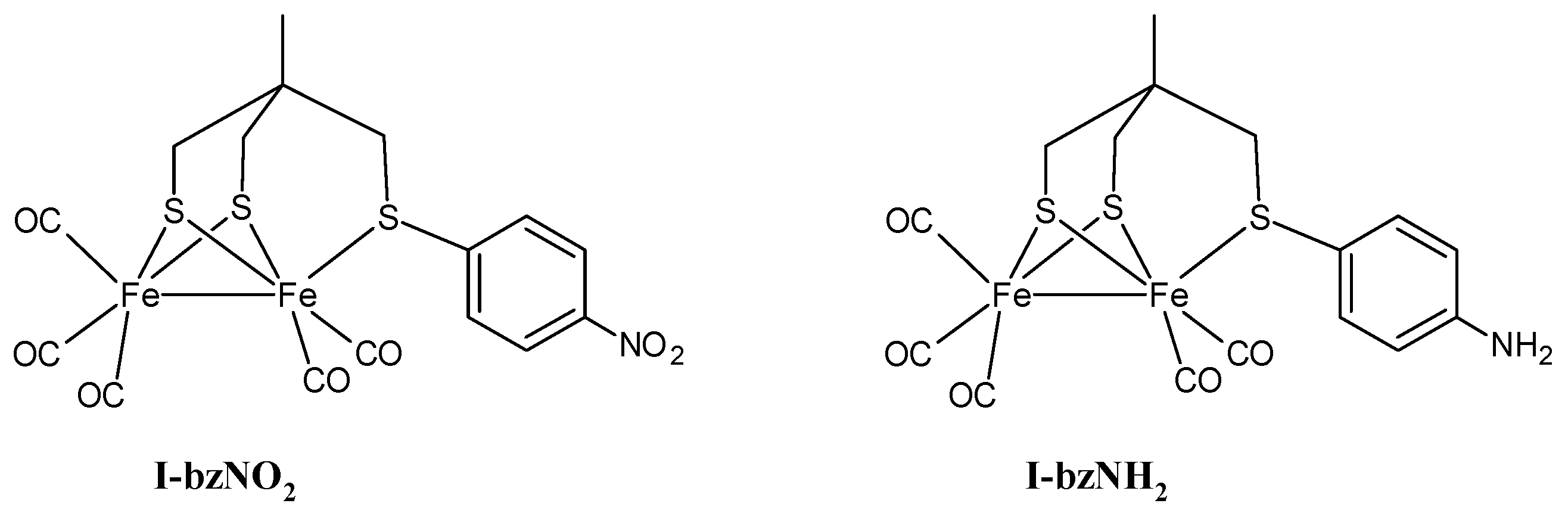
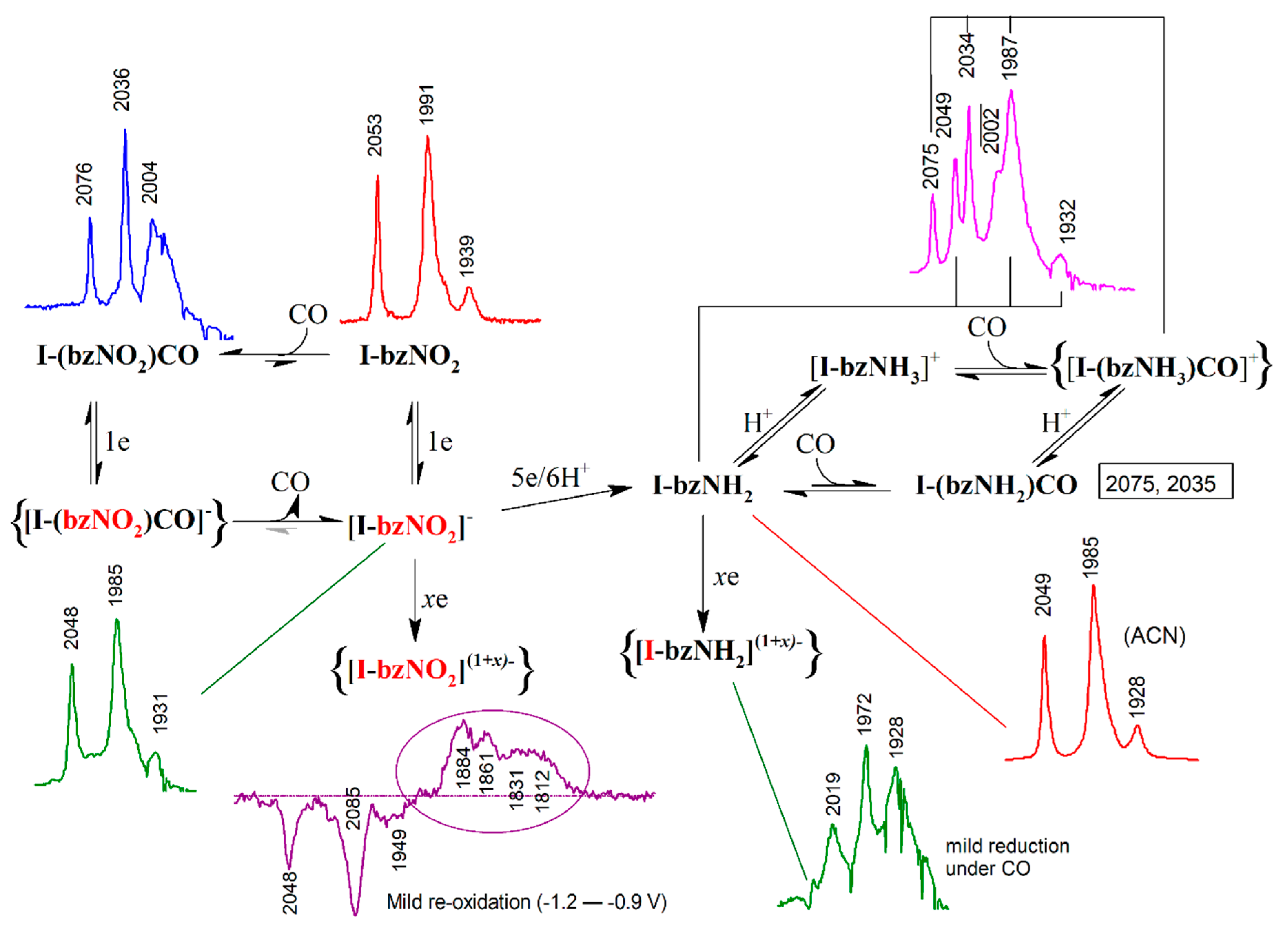
2.1. Electrochemistry of I-bzNO2 and I-bzNH2
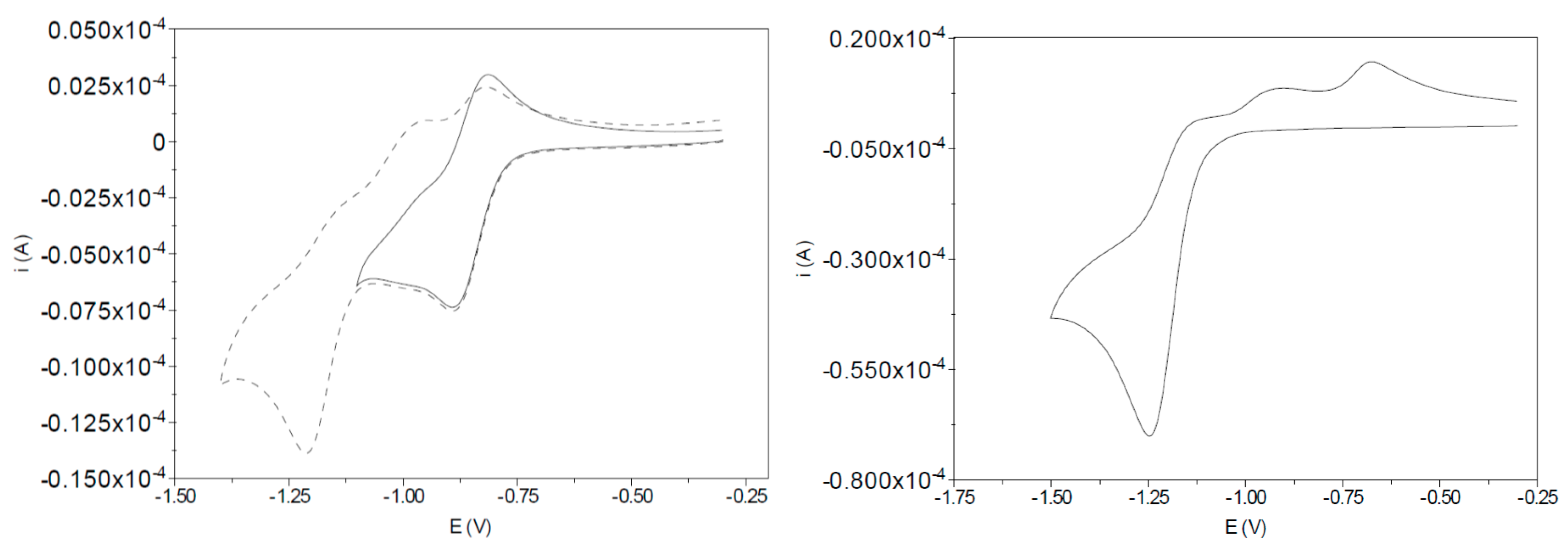
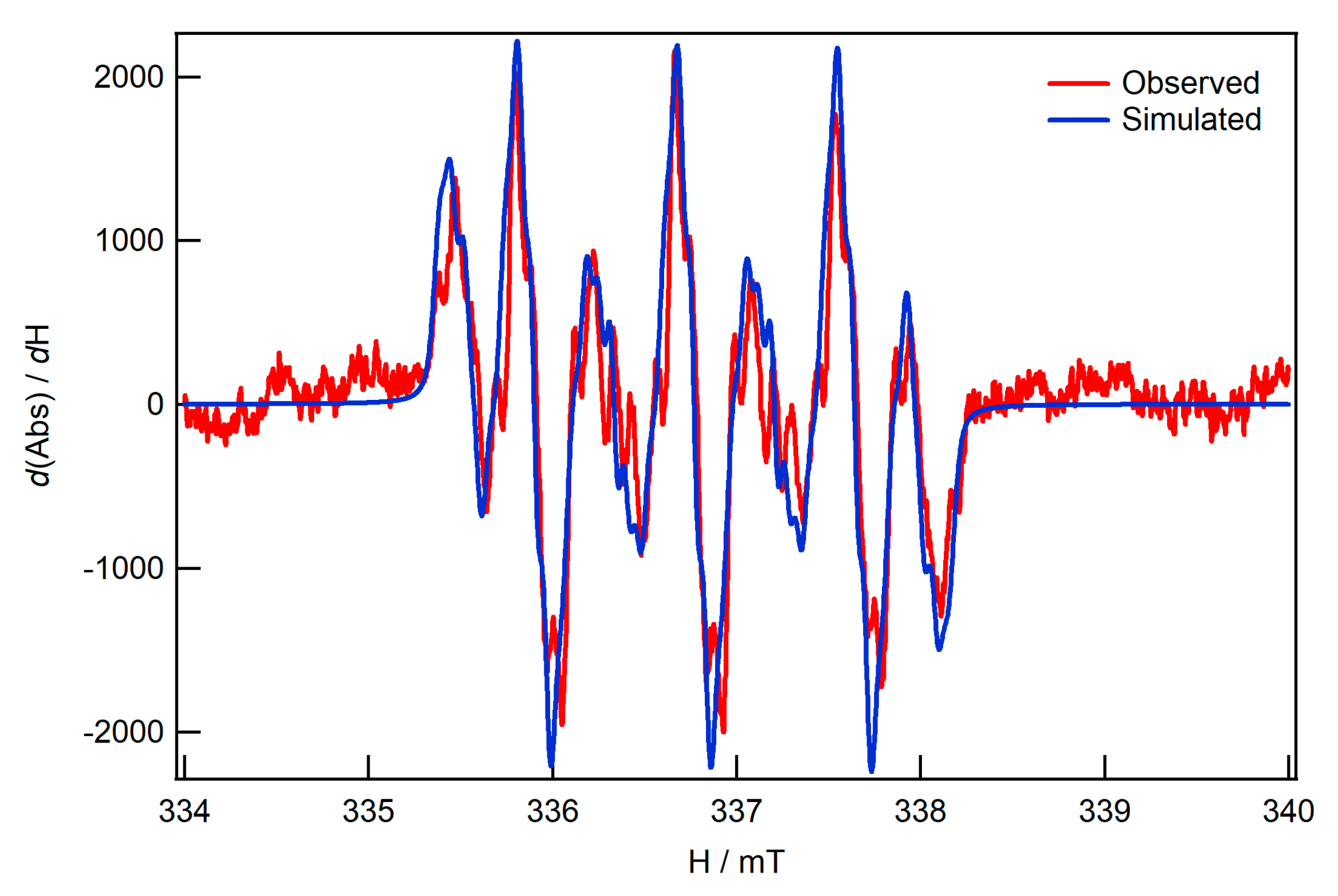
2.2. EPR Specroscopy of the In-Situ Generated Reduction Product of I-bzNO2
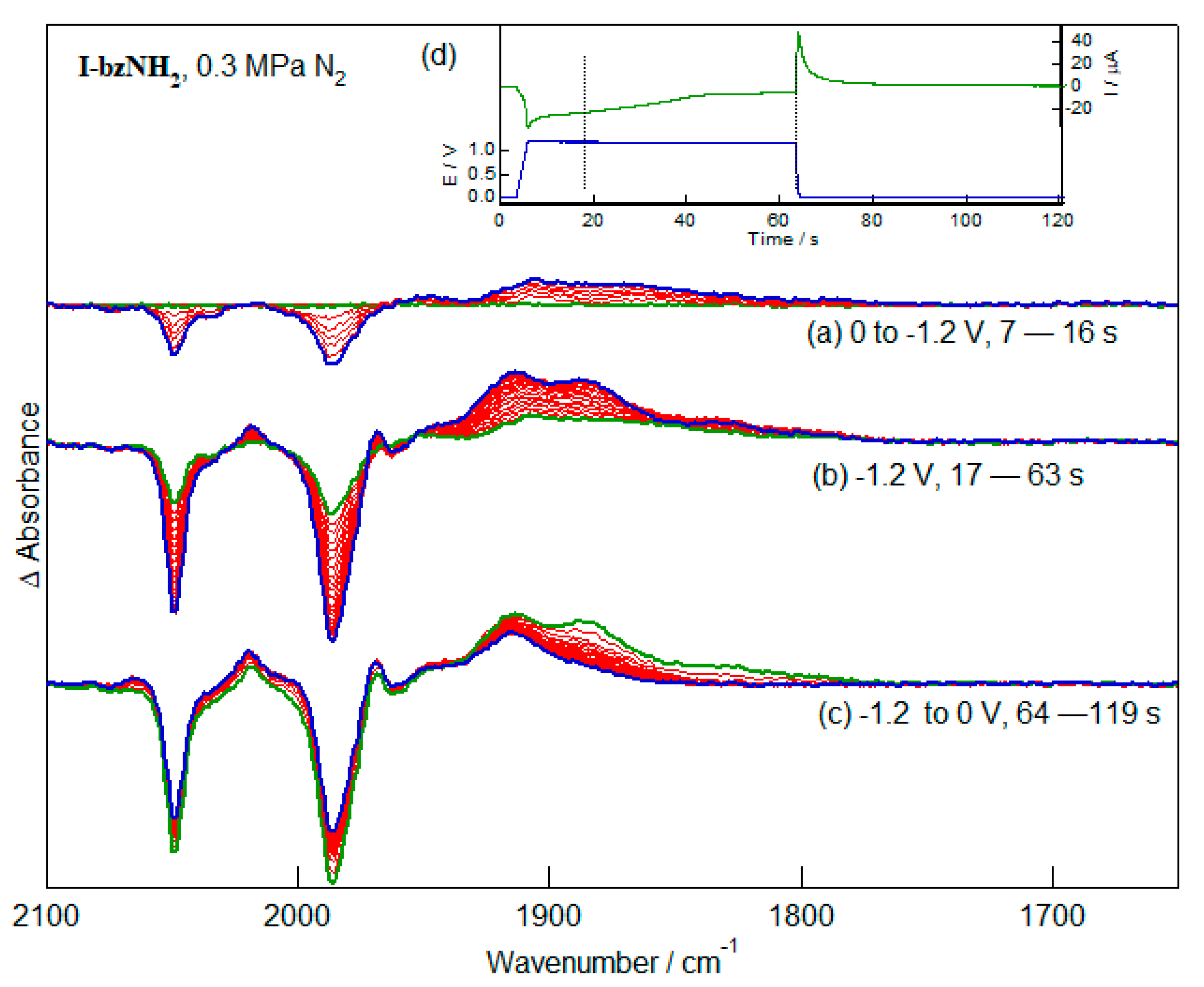
2.3. Infrared Spectroelectrochemistry (IR-SEC) of the Reduction of I-bzNH2
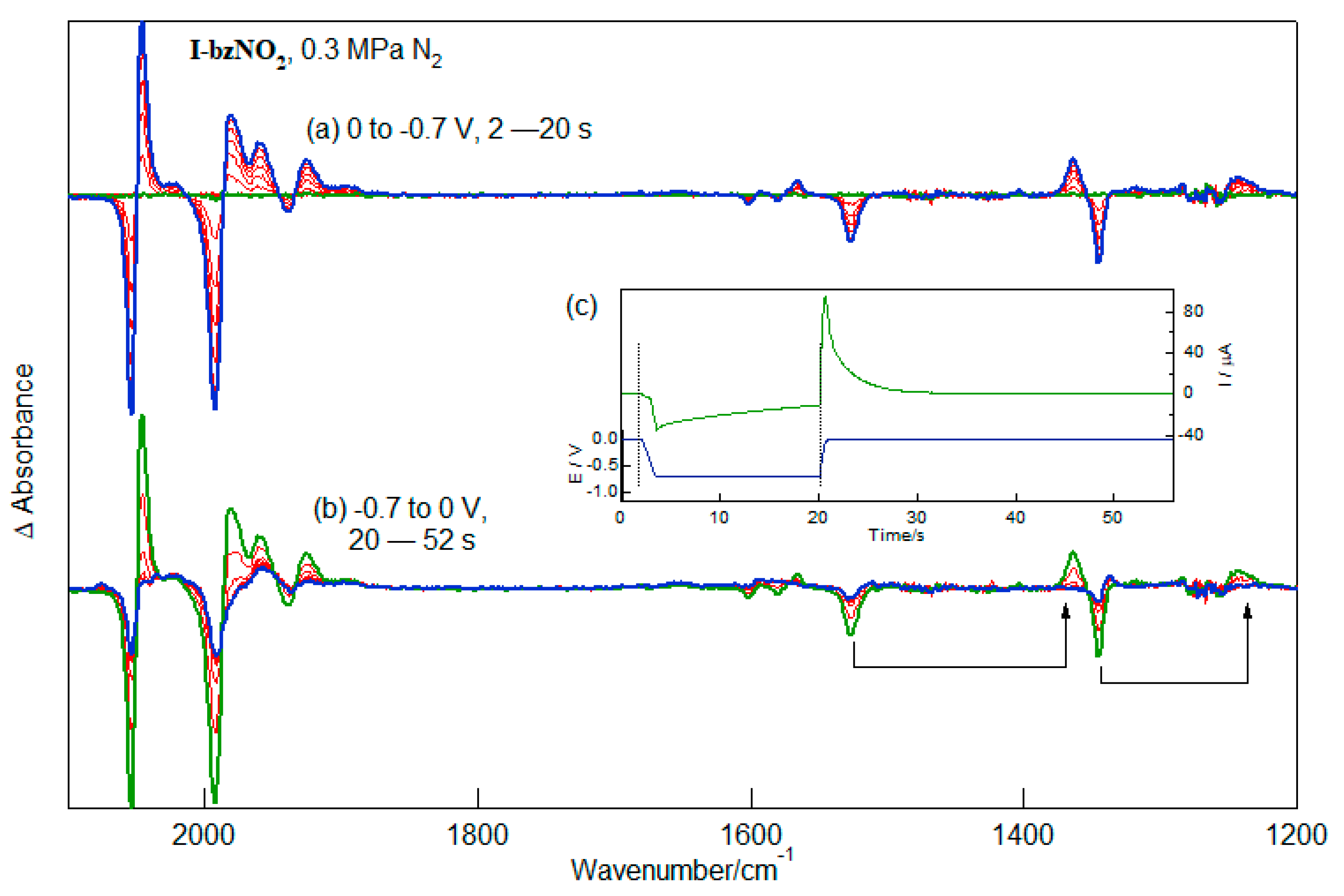
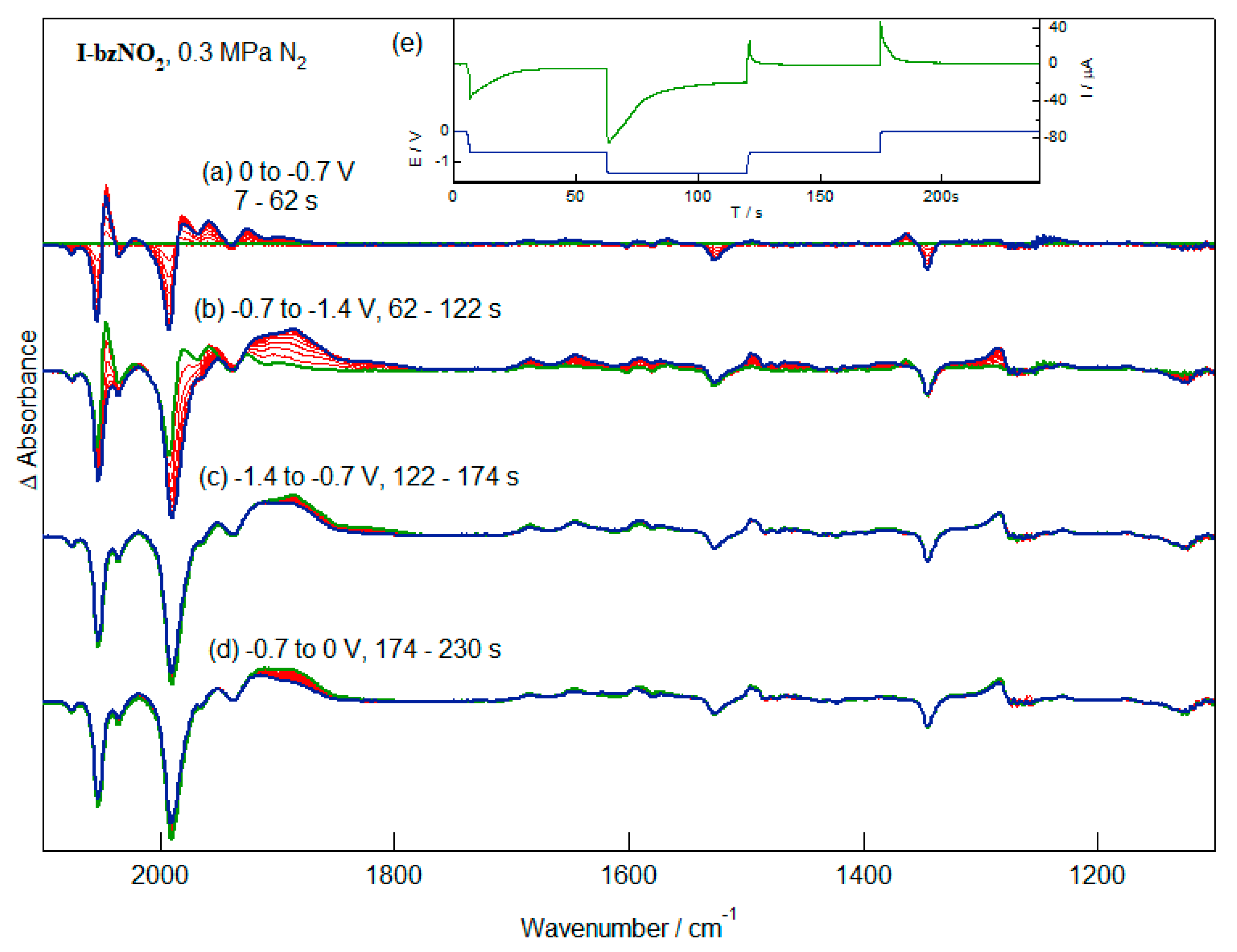
2.4. IR-SEC of the Reduction of I-bzNO2
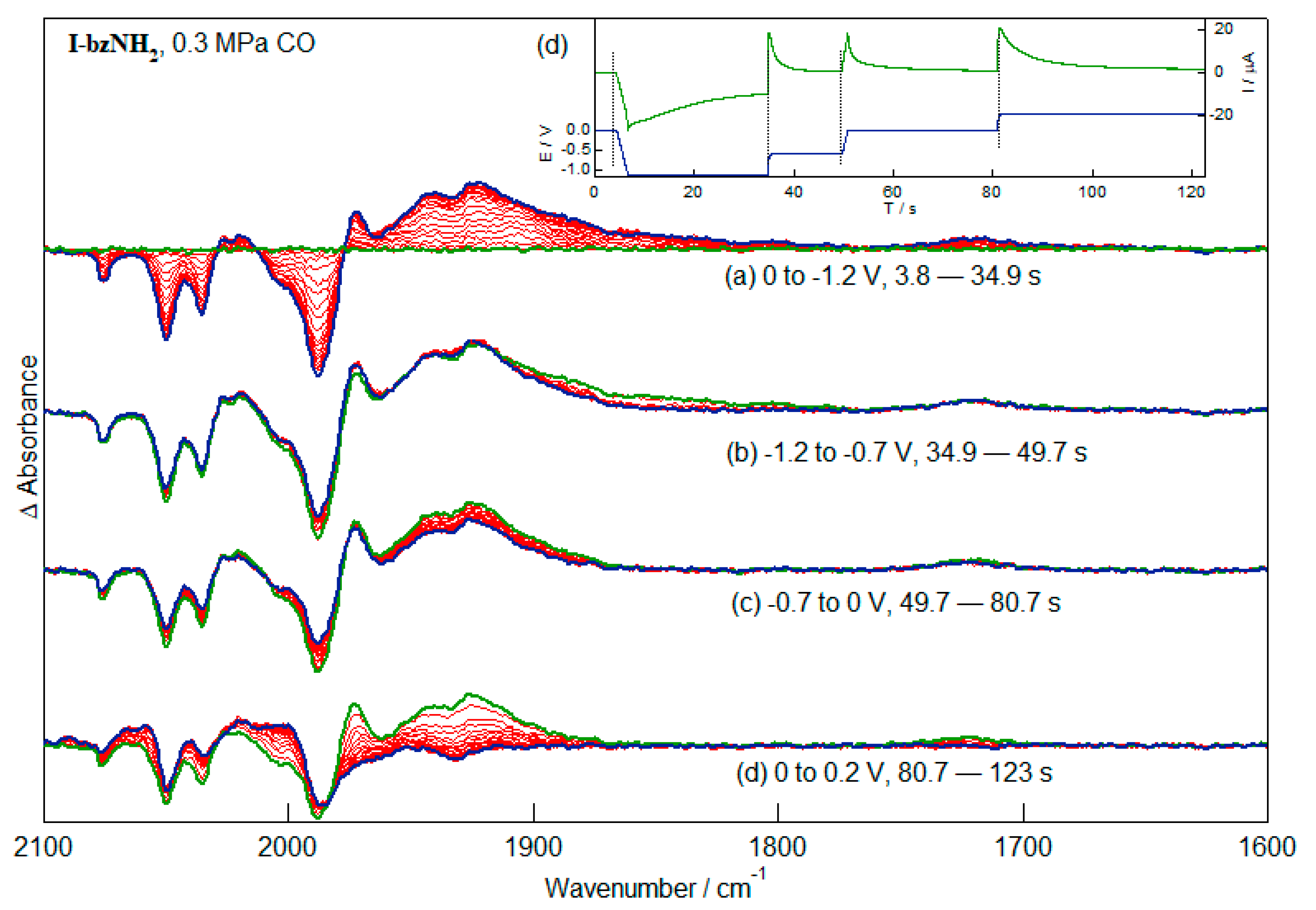
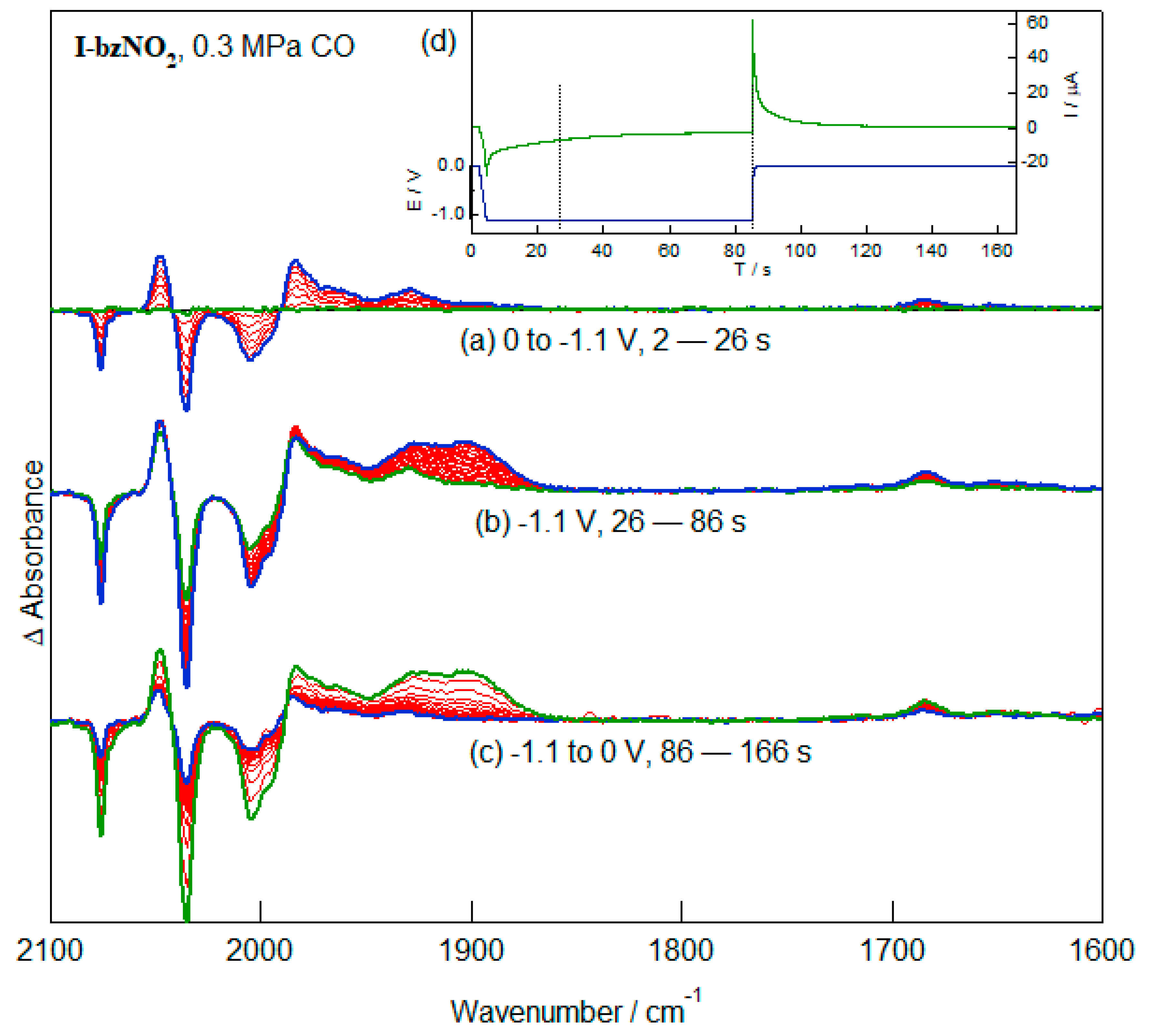
2.5. Redox-Dependent Competitive Binding of CO to I-bzNO2 and I-bzNH2
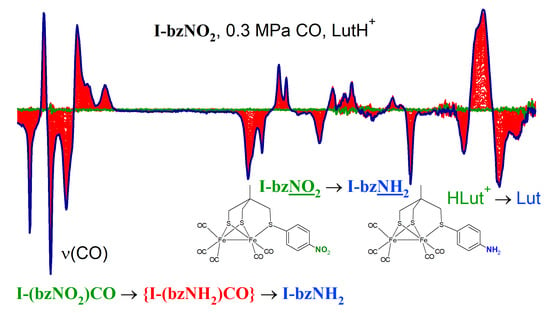
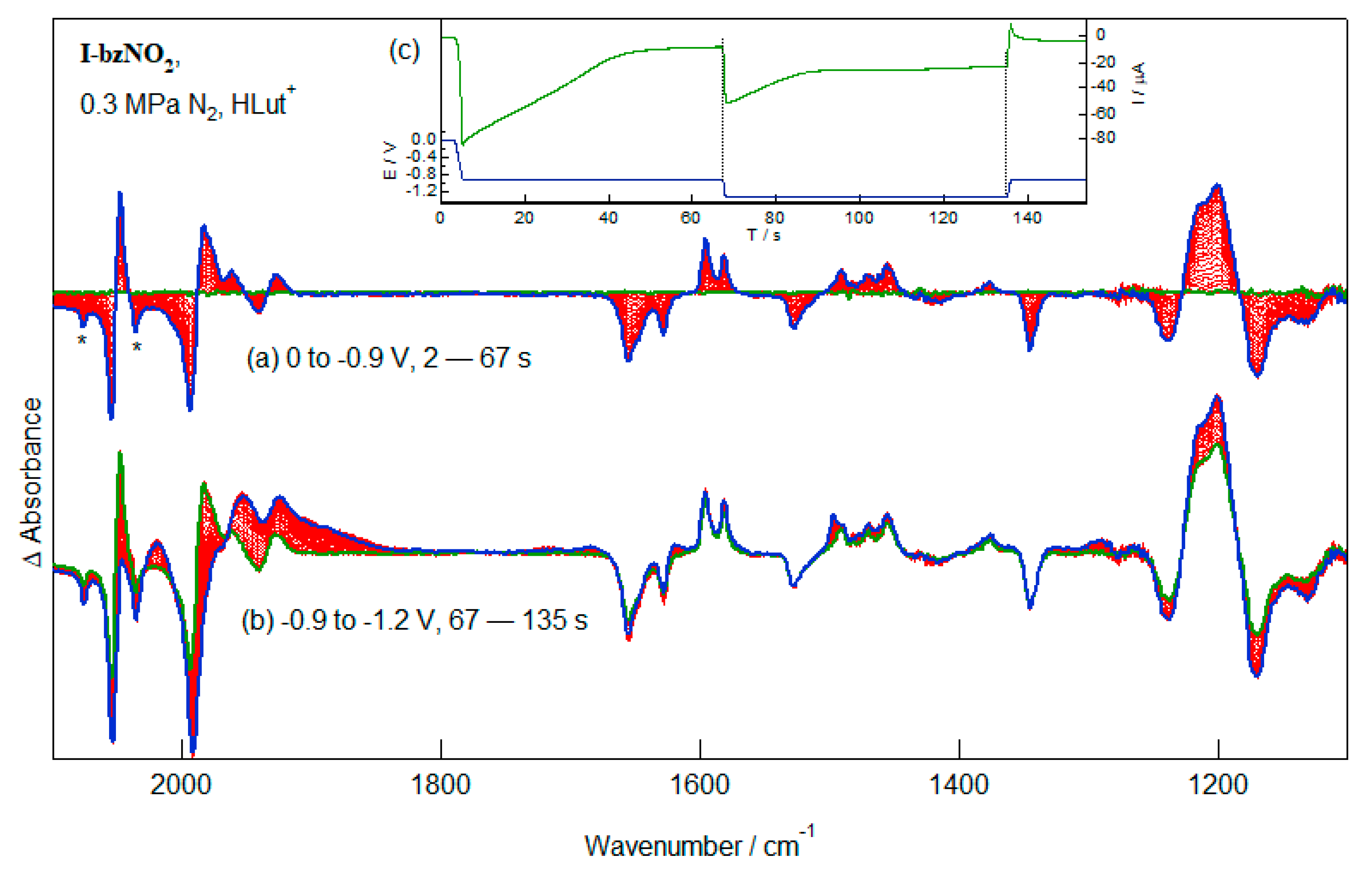
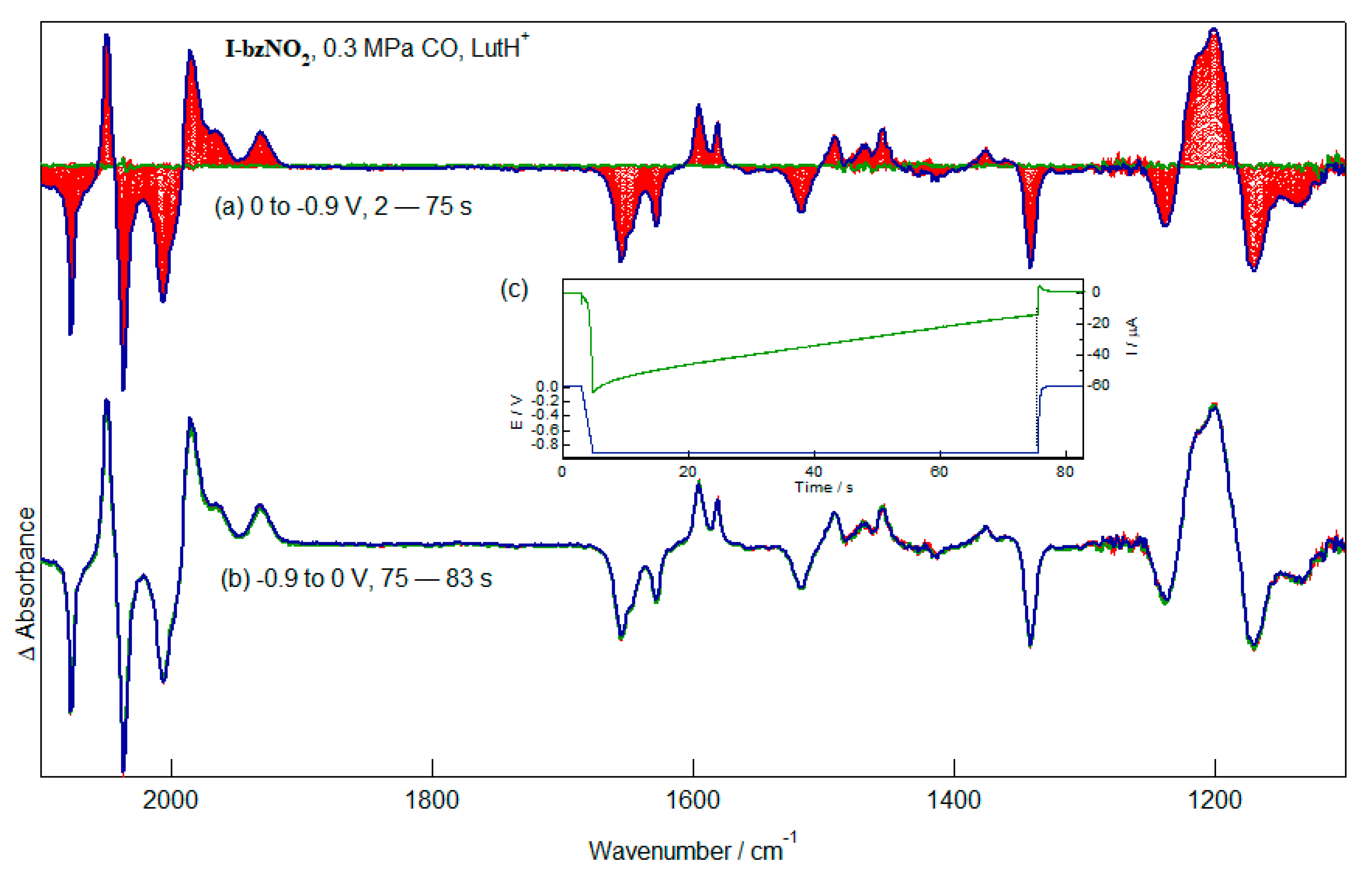
2.6. Reduction of I-bzNO2 in the Presence of Lutidinium, HLut+
3. Discussion
4. Materials and Methods
4.1. General
4.2. Electrochemistry
4.3. Spectroelectrochemistry
4.4. Synthesis
4.5. X-Ray Structure of I-bzNH2
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rauchfuss, T.B. Diiron azadithiolates as models for the [FeFe]-hydrogenase active site and paradigm for the role of the second coordination sphere. Acc. Chem. Res. 2015, 48, 2107–2116. [Google Scholar] [CrossRef] [PubMed]
- Best, S.P. Spectroelectrochemistry of hydrogenase enzymes and related compounds. Coord. Chem. Rev. 2005, 249, 1536–1554. [Google Scholar] [CrossRef]
- Lubitz, W.; Ogata, H.; Ruediger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef] [PubMed]
- Capon, J.-F.; Gloaguen, F.; Petillon, F.Y.; Schollhammer, P.; Talarmin, J. On the electrochemistry of diiron dithiolate complexes related to the active site of the [FeFe]H2ase. C. R. Chim. 2008, 11, 842–851. [Google Scholar] [CrossRef]
- Wright, J.A.; Pickett, C.J. Towards a functional model of the [FeFe]-hydrogenase: Dihydrogen oxidation. ChemCatChem 2012, 4, 1723–1724. [Google Scholar] [CrossRef]
- Xu, T.; Chen, D.; Hu, X. Hydrogen-activating models of hydrogenases. Coord. Chem. Rev. 2015, 303, 32–41. [Google Scholar] [CrossRef]
- Tard, C.; Pickett, C.J. Structural and functional analogues of the active sites of the [Fe]-, [NiFe]-, and [FeFe]-hydrogenases. Chem. Rev. 2009, 109, 2245–2274. [Google Scholar] [CrossRef]
- Apfel, U.-P.; Petillon, F.Y.; Schollhammer, P.; Talarmin, J.; Weigand, W. [FeFe] hydrogenase models: An overview. In Bioinspired Catalysis: Metal-Sulfur Complexes; Weigand, W., Schollhammer, P., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; pp. 79–103. [Google Scholar]
- Capon, J.-F.; Gloaguen, F.; Schollhammer, P.; Talarmin, J. Catalysis of the electrochemical H2 evolution by di-iron sub-site models. Coord. Chem. Rev. 2005, 249, 1664–1676. [Google Scholar] [CrossRef]
- Schilter, D.; Camara, J.M.; Huynh, M.T.; Hammes-Schiffer, S.; Rauchfuss, T.B. Hydrogenase enzymes and their synthetic models: The role of metal hydrides. Chem. Rev. 2016, 116, 8693–8749. [Google Scholar] [CrossRef]
- Lansing, J.C.; Manor, B.C.; Rauchfuss, T.B. Hydrogenase Models; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2014; pp. 1–21. [Google Scholar]
- Wittkamp, F.; Senger, M.; Stripp, S.T.; Apfel, U.P. [FeFe]-hydrogenases: Recent developments and future perspectives. Chem. Commun. 2018, 54, 5934–5942. [Google Scholar] [CrossRef]
- Razavet, M.; Davies, S.C.; Hughes, D.L.; Barclay, J.E.; Evans, D.J.; Fairhurst, S.A.; Liu, X.; Pickett, C.J. All-iron hydrogenase: Synthesis, structure and properties of {2Fe3S}-assemblies related to the di-iron sub-site of the h-cluster. Dalton Trans. 2003, 586–595. [Google Scholar] [CrossRef]
- Jablonskyte, A.; Wright, J.A.; Fairhurst, S.A.; Webster, L.R.; Pickett, C.J. [FeFe] hydrogenase: Protonation of {2Fe3S} systems and formation of super-reduced hydride states. Angew. Chem. Int. Ed. 2014, 53, 10143–10146. [Google Scholar] [CrossRef]
- Razavet, M.; Borg, S.J.; George, S.J.; Best, S.P.; Fairhurst, S.A.; Pickett, C.J. Transient ftir spectroelectrochemical and stopped-flow detection of a mixed valence {Fe(I)–Fe(II)} bridging carbonyl intermediate with structural elements and spectroscopic characteristics of the di-iron sub-site of all-iron hydrogenase. Chem. Commun. 2002, 700–701. [Google Scholar] [CrossRef]
- George, S.J.; Cui, Z.; Razavet, M.; Pickett, C.J. The di-iron subsite of all-iron hydrogenase: Mechanism of cyanation of a synthetic {2Fe3S}—Carbonyl assembly. Chem. Eur. J. 2002, 8, 4037–4046. [Google Scholar] [CrossRef]
- Tard, C.; Liu, X.; Ibrahim, S.K.; Bruschi, M.; De Gioia, L.; Davies, S.C.; Yang, X.; Wang, L.-S.; Sawers, G.; Pickett, C.J. Synthesis of the H-cluster framework of iron-only hydrogenase. Nature 2005, 433, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Rahaman, A.; Holt, K.B.; Nordlander, E.; Richmond, M.G.; Kabir, S.E.; Hogarth, G. Hydrogenase biomimetics with redox-active ligands: Electrocatalytic proton reduction by [Fe2(CO)4(κ2-diamine)(μ-edt)] (diamine = 2,2′-bipy, 1,10-phen). Polyhedron 2016, 116, 127–135. [Google Scholar] [CrossRef]
- Lansing, J.C.; Camara, J.M.; Gray, D.E.; Rauchfuss, T.B. Hydrogen production catalyzed by bidirectional, biomimetic models of the [FeFe]-hydrogenase active site. Organometallics 2014, 33, 5897–5906. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Groy, T.L.; Jones, A.K. Biomimetic model for [FeFe]-hydrogenase: Asymmetrically disubstituted diiron complex with a redox-active 2,2′-bipyridyl ligand. Dalton Trans. 2013, 42, 3843–3853. [Google Scholar] [CrossRef] [PubMed]
- Camara, J.M.; Rauchfuss, T.B. Combining acid-base, redox and substrate binding functionalities to give a complete model for the [FeFe]-hydrogenase. Nat. Chem. 2012, 4, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.B.; Wang, M.; Shi, Z.; Cui, H.G.; Dong, W.B.; Chen, J.S.; Akermark, B.; Sun, L.C. Synthesis, structures and electrochemical properties of nitro- and amino-functionalized diiron azadithiolates as active site models of fe-only hydrogenases. Chem.-Eur. J. 2004, 10, 4474–4479. [Google Scholar] [CrossRef] [PubMed]
- Alberti, A.; Martelli, G.; Pedulli, G.F. Radical anions and nitroxides from alkylthio-, alkylsulfinyl-, and alkylsulfonyl nitrobenzenes. J. Chem. Soc. Perkin Trans. 2 1977, 1252–1255. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. Easyspin, a comprehensive software package for spectral simulation and analysis in epr. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Borg, S.J.; Best, S.P. Spectroelectrochemical cell for the study of interactions between redox-activated species and moderate pressures of gaseous substrates. J. Electroanal. Chem. 2002, 535, 57–64. [Google Scholar] [CrossRef]
- Borg, S.J.; Tye, J.W.; Hall, M.B.; Best, S.P. Assignment of molecular structures to the electrochemical reduction products of diiron compounds related to [Fe–Fe] hydrogenase: A combined experimental and density functional theory study. Inorg. Chem. 2007, 46, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Borg, S.J.; Bondin, M.I.; Best, S.P.; Razavet, M.; Liu, X.; Pickett, C.J. Electrocatalytic proton reduction by dithiolate-bridged diiron carbonyl complexes: A connection to the H-cluster? Biochem. Soc. Trans. 2005, 33, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Pickett, C.J.; Vincent, K.A.; Ibrahim, S.K.; Gormal, C.A.; Smith, B.E.; Fairhurst, S.A.; Best, S.P. Synergic binding of carbon monoxide and cyanide to the femo cofactor of nitrogenase: Relic chemistry of an ancient enzyme? Chem. Eur. J. 2004, 10, 4770–4776. [Google Scholar] [CrossRef] [PubMed]
- Borg, S.J.; Behrsing, T.; Best, S.P.; Razavet, M.; Liu, X.; Pickett, C.J. Electron transfer at a dithiolate-bridged diiron assembly: Electrocatalytic hydrogen evolution. J. Am. Chem. Soc. 2004, 126, 16988–16999. [Google Scholar] [CrossRef]
- Wang, S.; Aster, A.; Mirmohades, M.; Lomoth, R.; Hammarstroem, L. Structural and kinetic studies of intermediates of a biomimetic diiron proton-reduction catalyst. Inorg. Chem. 2018, 57, 768–776. [Google Scholar] [CrossRef]
- Xu, F.; Tard, C.; Wang, X.; Ibrahim, S.K.; Hughes, D.L.; Zhong, W.; Zeng, X.; Luo, Q.; Liu, X.; Pickett, C.J. Controlling carbon monoxide binding at di-iron units related to the iron-only hydrogenase sub-site. Chem. Commun. 2008, 606–608. [Google Scholar] [CrossRef]
- Borg, S.J.; Ibrahim, S.K.; Pickett, C.J.; Best, S.P. Electrocatalysis of hydrogen evolution by synthetic diiron units using weak acids as the proton source: Pathways of doubtful relevance to enzymic catalysis by the diiron subsite of [FeFe] hydrogenase. C. R. Chim. 2008, 11, 852–860. [Google Scholar] [CrossRef]
- Zhao, P.-H.; Hu, M.-Y.; Li, J.-R.; Ma, Z.-Y.; Wang, Y.-Z.; He, J.; Li, Y.-L.; Liu, X.-F. Influence of dithiolate bridges on the structures and electrocatalytic performance of small bite-angle PNP-chelated diiron complexes Fe2(μ-xdt)(CO)4{κ2-(Ph2P)2NR} related to [FeFe]-hydrogenases. Organometallics 2019, 38, 385–394. [Google Scholar] [CrossRef]
- Zhao, P.-H.; Ma, Z.-Y.; Hu, M.-Y.; He, J.; Wang, Y.-Z.; Jing, X.-B.; Chen, H.-Y.; Wang, Z.; Li, Y.-L. PNP-chelated and -bridged diiron dithiolate complexes Fe2(μ-pdt)(CO)4{(Ph2P)2NR} together with related monophosphine complexes for the [2Fe]H subsite of [FeFe]-hydrogenases: Preparation, structure, and electrocatalysis. Organometallics 2018, 37, 1280–1290. [Google Scholar] [CrossRef]
- Durgaprasad, G.; Das, S.K. Modeling the active site of [FeFe]-hydrogenase: Electro-catalytic hydrogen evolution from acetic acid catalysed by [Fe2(μ-L)(CO)6] and [Fe2(μ-L(CO)5(PPh3)] (L = pyrazine-2,3-dithiolate, quinoxaline-2,3-dithiolate and pyrido[2,3-b]pyrazine-2,3-dithiolate). J. Chem. Sci. 2015, 127, 295–305. [Google Scholar] [CrossRef]
- Song, L.-C.; Cao, M.; Du, Z.-Q.; Feng, Z.-H.; Ma, Z.; Song, H.-B. CO substitution reactions of diiron complexes [{(μ-SCH2)2x}Fe2(CO)6] and [{(μ-SeCH2)2x}Fe2(CO)6] (x = O, CH2) with Ph2PCl/Me3NO to give Ph2PCl−, Ph2PNMe2−, and Ph2PP(:O)Ph2-substituted complexes related to [FeFe] hydrogenases. Eur. J. Inorg. Chem. 2014, 2014, 1886–1895. [Google Scholar] [CrossRef]
- Harb, M.K.; Daraosheh, A.; Goerls, H.; Smith, E.R.; Meyer, G.J.; Swenson, M.T.; Sakamoto, T.; Glass, R.S.; Lichtenberger, D.L.; Evans, D.H.; et al. Effects of alkane linker length and chalcogen character in [FeFe]-hydrogenase inspired compounds. Heteroat. Chem. 2014, 25, 592–606. [Google Scholar] [CrossRef]












© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tard, C.; Borg, S.J.; Fairhurst, S.A.; Pickett, C.J.; Best, S.P. Electronic Communication between Dithiolato-Bridged Diiron Carbonyl and S-Bridged Redox-Active Centres. Inorganics 2019, 7, 37. https://doi.org/10.3390/inorganics7030037
Tard C, Borg SJ, Fairhurst SA, Pickett CJ, Best SP. Electronic Communication between Dithiolato-Bridged Diiron Carbonyl and S-Bridged Redox-Active Centres. Inorganics. 2019; 7(3):37. https://doi.org/10.3390/inorganics7030037
Chicago/Turabian StyleTard, Cédric, Stacey J. Borg, Shirley A. Fairhurst, Christopher J. Pickett, and Stephen P. Best. 2019. "Electronic Communication between Dithiolato-Bridged Diiron Carbonyl and S-Bridged Redox-Active Centres" Inorganics 7, no. 3: 37. https://doi.org/10.3390/inorganics7030037
APA StyleTard, C., Borg, S. J., Fairhurst, S. A., Pickett, C. J., & Best, S. P. (2019). Electronic Communication between Dithiolato-Bridged Diiron Carbonyl and S-Bridged Redox-Active Centres. Inorganics, 7(3), 37. https://doi.org/10.3390/inorganics7030037