Synthesis, Crystal Structure, Electrochemistry and Electro-Catalytic Properties of the Manganese-Containing Polyoxotungstate, [(Mn(H2O)3)2(H2W12O42)]6−



Abstract
1. Introduction
2. Results and Discussion
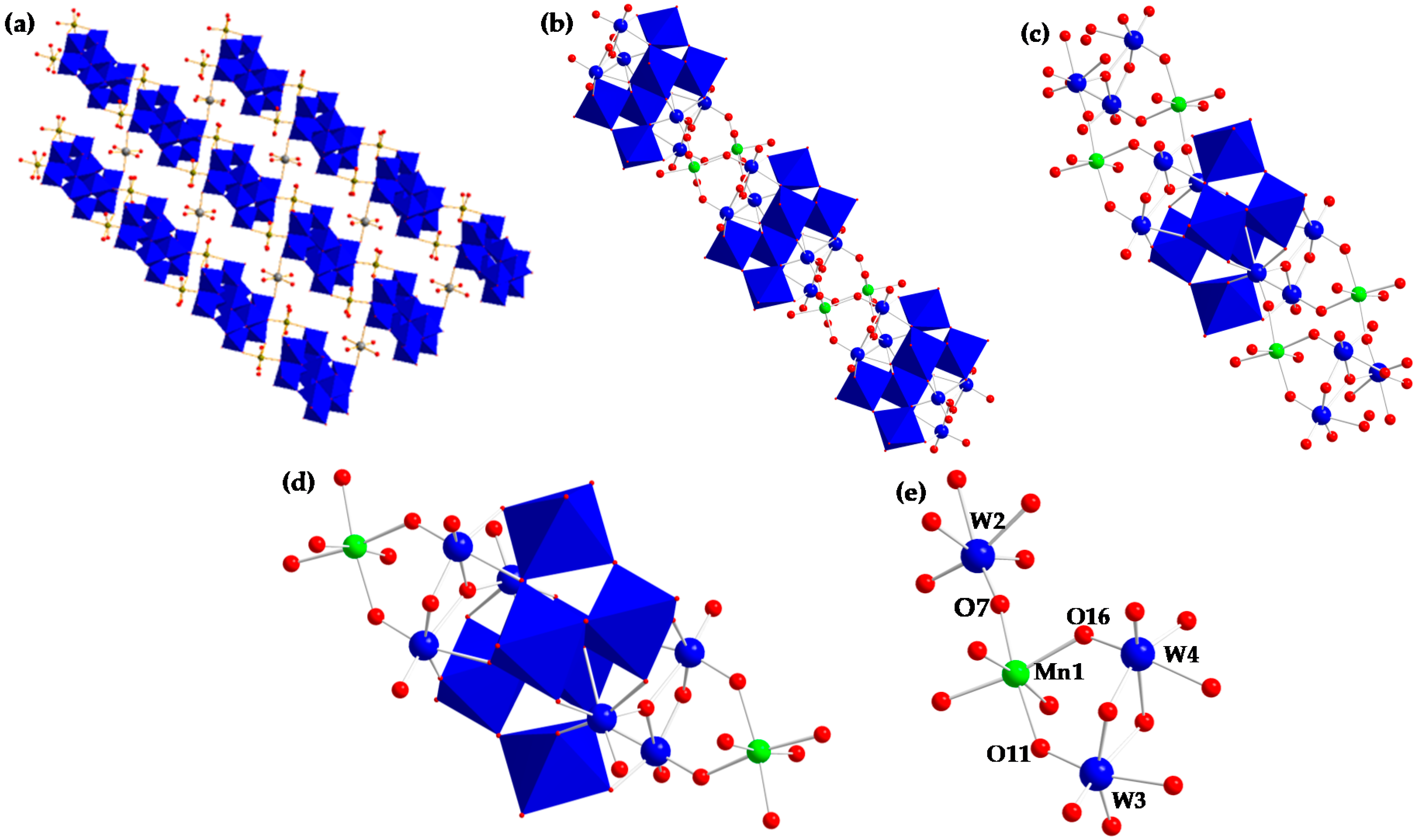
2.1. Synthesis and Structure
2.2. Electrochemistry
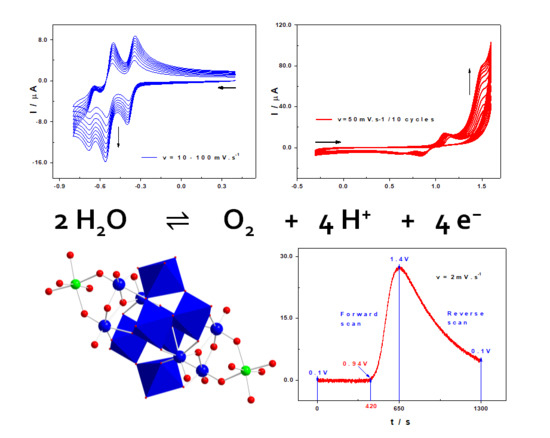
2.2.1. Redox Behaviours and Electro-Catalytic Properties of the Tungstic Framework
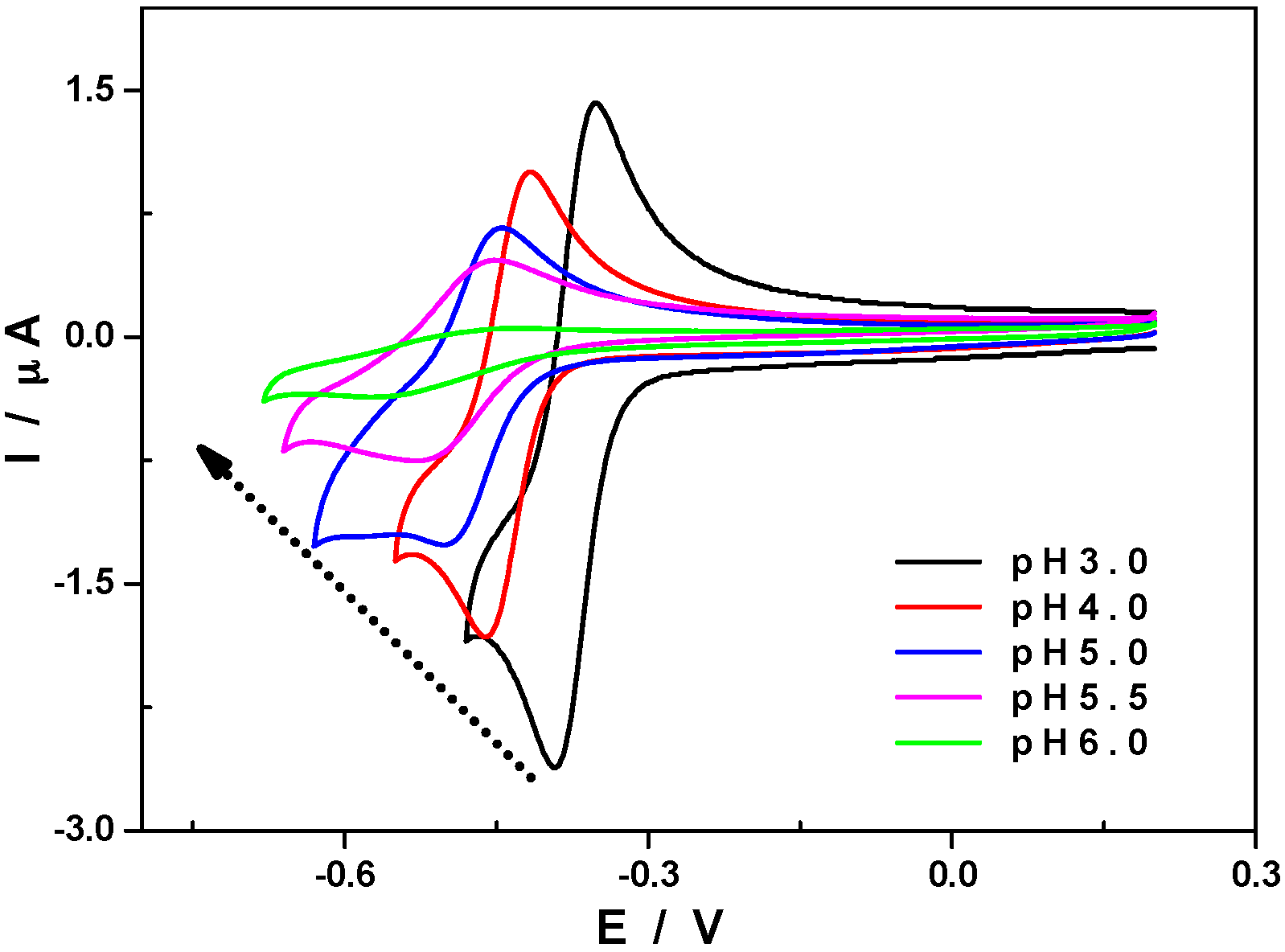
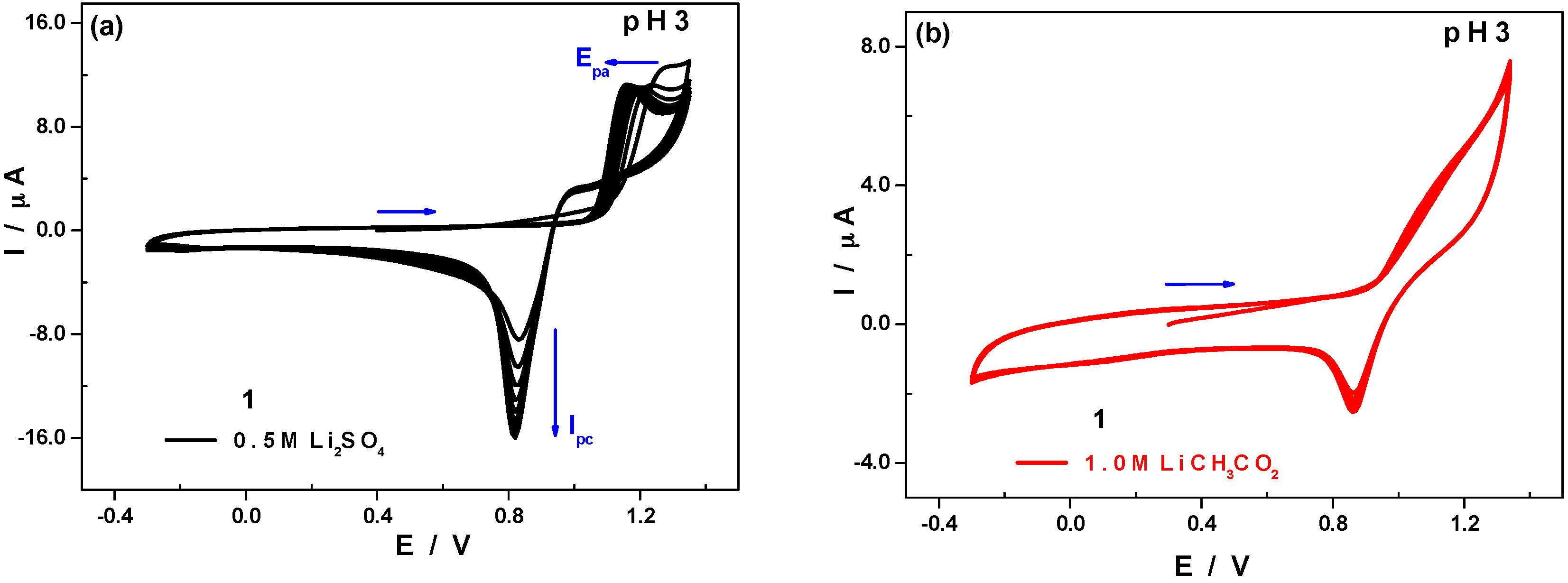
2.2.2. pH Influence
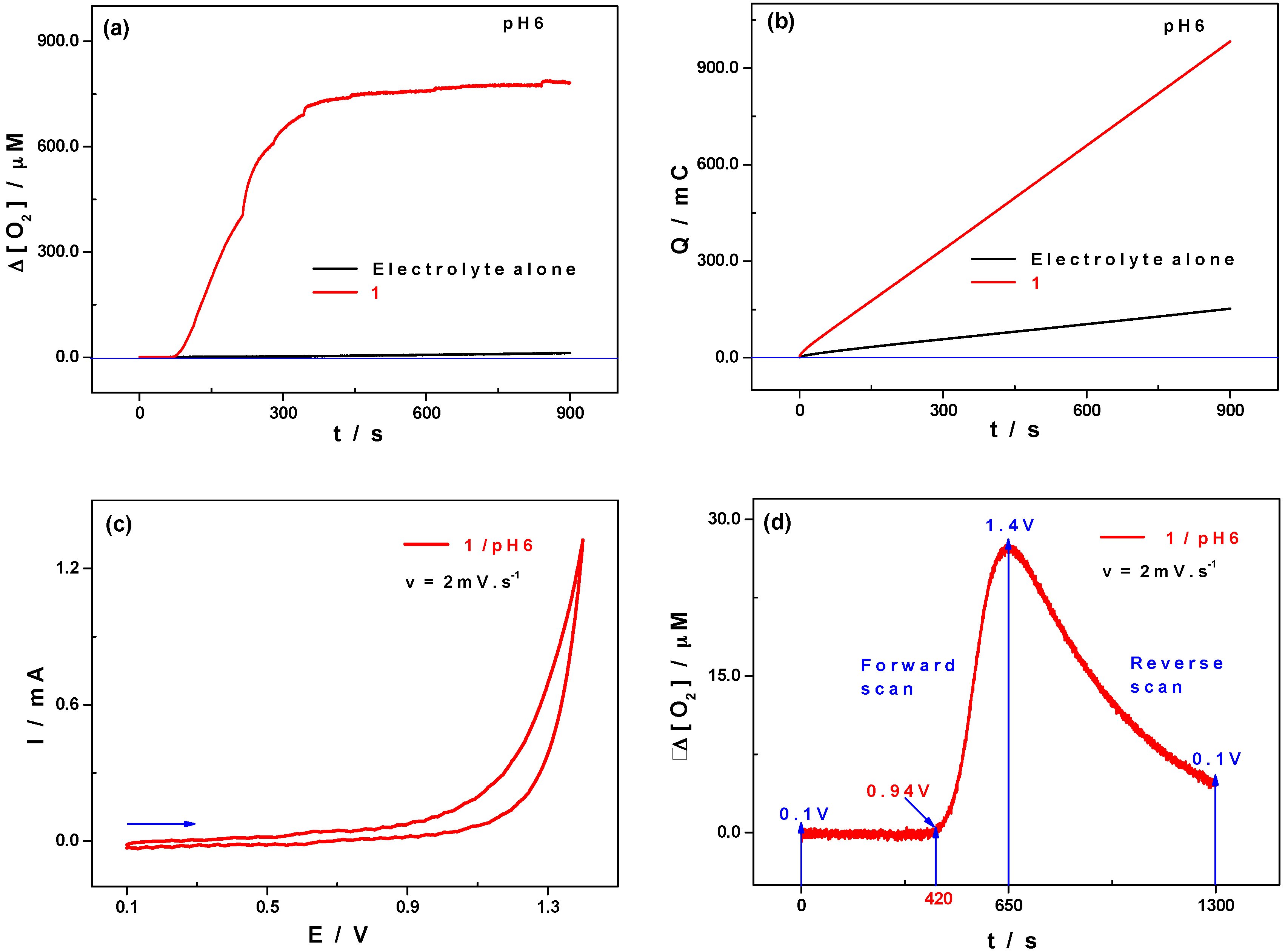
2.3. Electro-Catalytic Reduction Properties
2.4. Redox Behaviour and Influence of the Manganese Centres
2.5. Quartz Crystal Microbalance
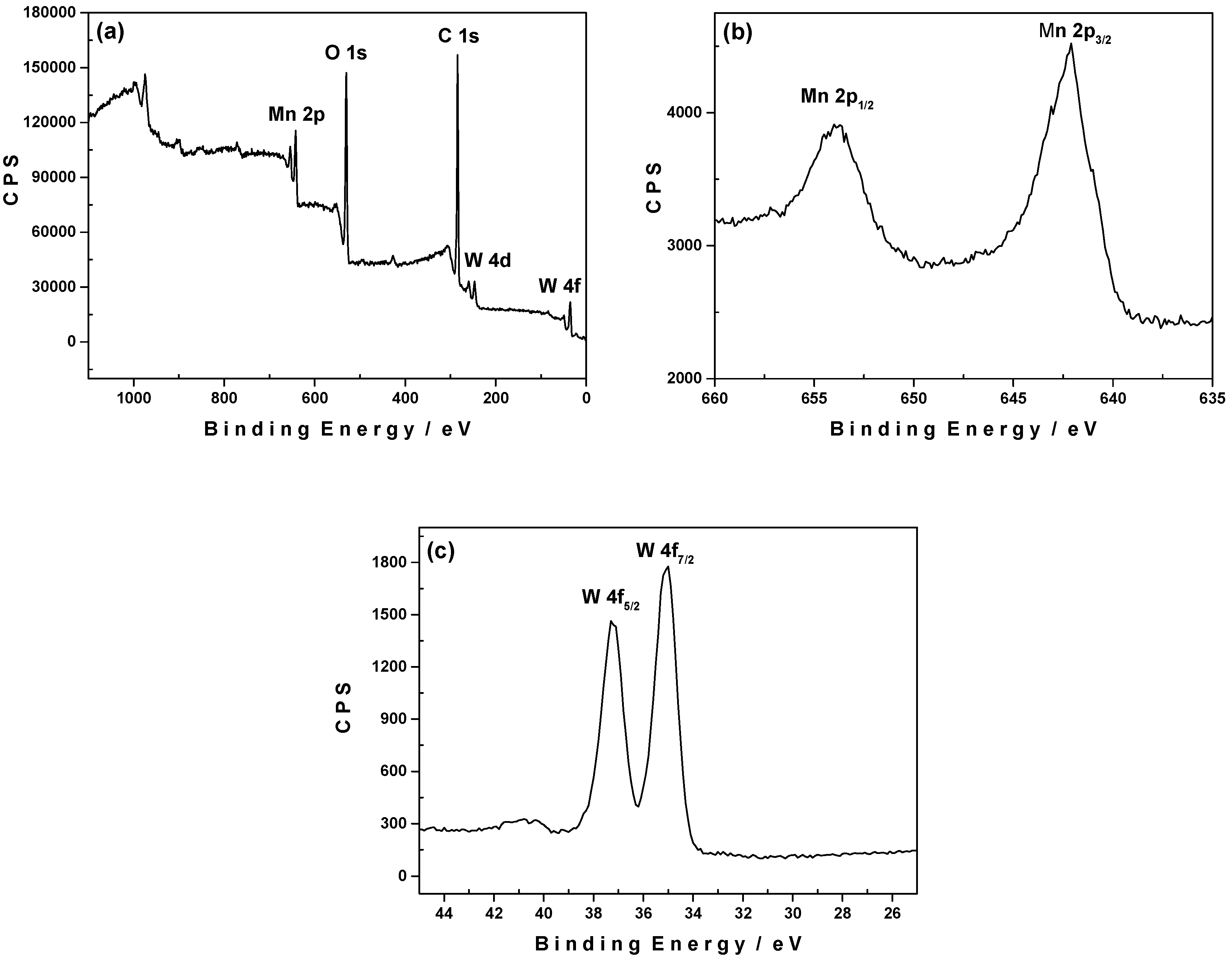
2.6. X-Ray Photoelectron Spectroscopy (XPS)
2.6.1. pH 3.0
2.6.2. pH 6.0
3. Materials and Methods
3.1. Synthesis of [(Mn(H2O)3)2(H2W12O42)]6− (1)
3.2. X-ray Crystallography
3.3. Electrochemical Experiments
3.4. XPS Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gumerova, N.I.; Rompel, A. Synthesis, structures and applications of electron-rich polyoxometalates. Nat. Rev. Chem. 2018, 2, 0112. [Google Scholar] [CrossRef]
- Katsoulis, D.E. A Survey of Applications of Polyoxometalates. Chem. Rev. 1998, 98, 359–388. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.J.; Bond, A.M.; Forster, R.J.; Keyes, T.E. Hybrid polyoxometalate materials for photo(electro-) chemical applications. Coord. Chem. Rev. 2016, 306, 217–234. [Google Scholar] [CrossRef]
- Contant, R.; Herveb, G. The heteropolyoxotungstates: Relationships between routes of formation and structures. Rev. Inorg. Chem. 2002, 22, 63–112. [Google Scholar] [CrossRef]
- Long, D.-L.; Burkholder, E.; Cronin, L. Polyoxometalate clusters, nanostructures and materials: From self assembly to designer materials and devices. Chem. Soc. Rev. 2007, 36, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Long, D.-L.; Tsunashima, R.; Cronin, L. Polyoxometalates: Building Blocks for Functional Nanoscale Systems. Angew. Chem. Int. Ed. 2010, 49, 1736–1758. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Kögerler, P. From simple building blocks to structures with increasing size and complexity. Coord. Chem. Rev. 1999, 182, 3–17. [Google Scholar] [CrossRef]
- Pope, M.T.; Kortz, U. Polyoxometalates: Update Based on the Original Article by Michael T. Pope, Encyclopedia of Inorganic Chemistry © 2005, John Wiley & Sons, Ltd. In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons: New York, NY, USA, 2012. [Google Scholar]
- Pope, M. Heteropoly and Isopoly Oxometalates; Springer: Berlin/Heidelberg, Germany, 1983; p. XIII. 180p. [Google Scholar]
- Keita, B.; Mbomekalle, I.M.; Nadjo, L.; Contant, R. [H4AsW18O62]7−, a novel Dawson heteropolyanion and two of its sandwich-type derivatives [Zn4(H2O)2(H4AsW15O56)2]18−, [Cu4(H2O)2(H4AsW15O56)2]18−: Cyclic voltammetry and electrocatalytic properties towards nitrite and nitrate. Electrochem. Commun. 2001, 3, 267–273. [Google Scholar] [CrossRef]
- Mbomekalle, I.M.; Keita, B.; Nadjo, L.; Contant, R.; Belai, N.; Pope, M.T. Rationalization and improvement of the syntheses of two octadecatungstoarsenates: The novel α-K7[H4AsW18O62]·18H2O and the well known symmetrical α-K6[As2W18O62]·14H2O. Inorg. Chim. Acta 2003, 342, 219–228. [Google Scholar] [CrossRef]
- Mbomekalle, I.-M.; Lu, Y.W.; Keita, B.; Nadjo, L. Simple, high yield and reagent-saving synthesis of pure α-K6P2W18O62·14H2O. Inorg. Chem. Commun. 2003, 7, 86–90. [Google Scholar] [CrossRef]
- Mbomekalle, I.M.; Lu, Y.W.; Keita, B.; Nadjo, L.; Neiwert, W.A.; Hardcastle, K.I.; Hill, C.L.; Anderson, T.M. Crystallographic studies of a molybdenum-rich diarsenotungstate and reaction of Fe(III) with its isomerically pure α1- and α2-monolacunary derivatives. Eur. J. Inorg. Chem. 2005, 1547–1551. [Google Scholar] [CrossRef]
- Mbomekalle, I.M.; Mialane, P.; Dolbecq, A.; Marrot, J.; Sécheresse, F.; Berthet, P.; Keita, B.; Nadjo, L. Rational Synthesis, Structure, Magnetism and Electrochemistry of Mixed Iron–Nickel-Containing Wells–Dawson-Fragment-Based Sandwich-Type Polyoxometalates. Eur. J. Inorg. Chem. 2009, 2009, 5194–5204. [Google Scholar] [CrossRef]
- Lebrini, M.; Mbomekalle, I.M.; Dolbecq, A.; Marrot, J.; Berthet, P.; Ntienoue, J.; Secheresse, F.; Vigneron, J.; Etcheberry, A. Manganese(III)-containing Wells-Dawson sandwich-type polyoxometalates: Comparison with their manganese(II) counterparts. Inorg. Chem. 2011, 50, 6437–6448. [Google Scholar] [CrossRef] [PubMed]
- Mbomekallé, I.-M.; Bassil, B.S.; Suchopar, A.; Keita, B.; Nadjo, L.; Ammam, M.; Haouas, M.; Taulelle, F.; Kortz, U. Improved Synthesis, Structure and Solution Characterization of the Cyclic 48-Tungsto-8-Arsenate(V), [H4As8W48O184]36−. J. Clust. Sci. 2013, 25, 277–285. [Google Scholar] [CrossRef]
- Parent, L.; de Oliveira, P.; Teillout, A.-L.; Dolbecq, A.; Haouas, M.; Cadot, E.; Mbomekalle, I.M. Synthesis and characterisation of the europium(III) dimolybdo-enneatungsto-silicate dimer, [Eu(α-SiW9Mo2O39)2]13−. Inorganics 2015, 3, 341–354. [Google Scholar] [CrossRef]
- Finke, R.G.; Droege, M.W. Trivacant heteropolytungstate derivatives. 2. Synthesis, characterization and tungsten-183 NMR of P4W30M4(H2O)2O11216− (M = Co, Cu, Zn). Inorg. Chem. 1983, 22, 1006–1008. [Google Scholar] [CrossRef]
- Weakley, T.J.R.; Finke, R.G. Single-crystal x-ray structures of the polyoxotungstate salts K8.3Na1.7[Cu4(H2O)2(PW9O34)2]·24H2O and Na14Cu[Cu4(H2O)2(P2W15O56)2]·53H2O. Inorg. Chem. 1990, 29, 1235–1241. [Google Scholar] [CrossRef]
- Gomez-Garcia, C.J.; Borras-Almenar, J.J.; Coronado, E.; Ouahab, L. Single-Crystal X-ray Structure and Magnetic Properties of the Polyoxotungstate Complexes Na16[M4(H2O)2(P2W15O56)2]·nH2O (M = MnII, n = 53; M = NiII, n = 52): An Antiferromagnetic MnII Tetramer and a Ferromagnetic NiII Tetramer. Inorg. Chem. 1994, 33, 4016–4022. [Google Scholar] [CrossRef]
- Bi, L.-H.; Wang, E.-B.; Peng, J.; Huang, R.-D.; Xu, L.; Hu, C.-W. Crystal Structure and Replacement Reaction of Coordinated Water Molecules of the Heteropoly Compounds of Sandwich-Type Tungstoarsenates. Inorg. Chem. 2000, 39, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Juan, J.M.; Coronado, E.; Gaita-Ariño, A.; Giménez-Saiz, C.; Güdel, H.-U.; Sieber, A.; Bircher, R.; Mutka, H. Magnetic Polyoxometalates: Anisotropic Exchange Interactions in the Co3II Moiety of [(NaOH2)Co3(H2O)(P2W15O56)2]17−. Inorg. Chem. 2005, 44, 3389–3395. [Google Scholar] [CrossRef] [PubMed]
- Ruhlmann, L.; Canny, J.; Contant, R.; Thouvenot, R. Di- and Tricobalt Dawson Sandwich Complexes: Synthesis, Spectroscopic Characterization and Electrochemical Behavior of Na18[(NaOH2)2Co2(P2W15O56)2] and Na17[(NaOH2)Co3(H2O)(P2W15O56)2]. Inorg. Chem. 2002, 41, 3811–3819. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.M.; Fang, X.; Mbomekalle, I.M.; Keita, B.; Nadjo, L.; Hardcastle, K.I.; Farsidjani, A.; Hill, C.L. Structural and Electrochemical Studies of Dicupric Wells–Dawson Sandwich-Type Complexes. J. Clust. Sci. 2006, 17, 183–195. [Google Scholar] [CrossRef]
- Ruhlmann, L.; Costa-Coquelard, C.; Canny, J.; Thouvenot, R. Mixed-Metal Dawson Sandwich Complexes: Synthesis, Spectroscopic Characterization and Electrochemical Behaviour of Na16[MIICo3(H2O)2(P2W15O56)2] (M = Mn, Co, Ni, Zn and Cd). Eur. J. Inorg. Chem. 2007, 2007, 1493–1500. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Q.; Duncan, D.C.; Campana, C.F.; Hill, C.L. Multiiron Polyoxoanions. Syntheses, Characterization, X-ray Crystal Structures and Catalysis of H2O2-Based Hydrocarbon Oxidations by [FeIII4(H2O)2(P2W15O56)2]12−. Inorg. Chem. 1997, 36, 4208–4215. [Google Scholar] [CrossRef]
- Bassil, B.S.; Xiang, Y.; Haider, A.; Hurtado, J.; Novitchi, G.; Powell, A.K.; Bossoh, A.M.; Mbomekallé, I.M.; de Oliveira, P.; Kortz, U. Heptanickel(II) double-cubane core in wells-dawson heteropolytungstate, [Ni7(OH)6(H2O)6(P2W15O56)2]16−. Chem. Commun. 2016, 52, 2601–2604. [Google Scholar] [CrossRef]
- Mbomekalle, I.M.; Keita, B.; Nadjo, L.; Neiwert, W.A.; Zhang, L.; Hardcastle, K.I.; Hill, C.L.; Anderson, T.M. Lacunary Wells-Dawson sandwich complexes—Synthesis, characterization and stability studies of multi-iron species. Eur. J. Inorg. Chem. 2003, 21, 3924–3928. [Google Scholar] [CrossRef]
- Mbomekalle, I.M.; Cao, R.; Hardcastle, K.I.; Hill, C.L.; Ammam, M.; Keita, B.; Nadjo, L.; Anderson, T.M. Synthesis, structural characterization and electrocatalytic studies of αββα-(ZnIIOH2)2(FeIII)2(X2W15O56)214− (X = P or As). C. R. Chim. 2005, 8, 1077–1086. [Google Scholar] [CrossRef]
- Ayingone Mezui, C.S.; de Oliveira, P.; Teillout, A.-L.; Marrot, J.; Berthet, P.; Lebrini, M.; Mbomekallé, I.M. Synthesis, Structure and Magnetic Electrochemical Properties of a Family of Tungstoarsenates Containing Just CoII Centers or Both CoII and FeIII Centers. Inorg. Chem. 2017, 56, 1999–2012. [Google Scholar] [CrossRef]
- Floriant, D.; Charyle, S.A.M.; Pedro de, O.; Anne-Lucie, T.; Israel, M.M. Synthesis, Electrochemistry and Electro-Catalytic Properties of The Mixed Copper–Iron-Containing Sandwich-Type Polyoxometalates [(FeIIIOH)2CuII2(X2W15O56)2]14− and [(CuIIOH2)2FeIII2(X2W15O56)2]14− (with X = AsV and PV). Curr. Inorg. Chem. Discontin. 2017, 7, 28–38. [Google Scholar]
- Keggin, J.F. Structure of the Molecule of 12-Phosphotungstic Acid. Nature 1933, 131, 908. [Google Scholar] [CrossRef]
- Keita, B.; Mialane, P.; Sécheresse, F.; de Oliveira, P.; Nadjo, L. Electrochemical generation of high-valent manganese catalysts in aqueous solutions from the sandwich-type polyoxoanion [(MnIII(H2O))3(SbW9O33)2]9−. Electrochem. Commun. 2007, 9, 164–172. [Google Scholar] [CrossRef]
- Al-Oweini, R.; Bassil, B.S.; Palden, T.; Keita, B.; Lan, Y.; Powell, A.K.; Kortz, U. The manganese(III)-containing tungstophosphate [MnIII3(H2O)5(A-α-PW9O34)2]9−. Polyhedron 2013, 52, 461–466. [Google Scholar] [CrossRef]
- Friedl, J.; Al-Oweini, R.; Herpich, M.; Keita, B.; Kortz, U.; Stimming, U. Electrochemical studies of tri-manganese substituted Keggin Polyoxoanions. Electrochim. Acta 2014, 141, 357–366. [Google Scholar] [CrossRef]
- Yu, L.; Ding, Y.; Zheng, M. Polyoxometalate-based manganese clusters as catalysts for efficient photocatalytic and electrochemical water oxidation. Appl. Catal. B Environ. 2017, 209, 45–52. [Google Scholar] [CrossRef]
- Han, Q.; Ding, Y. Recent advances in the field of light-driven water oxidation catalyzed by transition-metal substituted polyoxometalates. Dalton Trans. 2018, 47, 8180–8188. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.L.; Brown, R.B. Sustained epoxidation of olefins by oxygen donors catalyzed by transition metal-substituted polyoxometalates, oxidatively resistant inorganic analogs of metalloporphyrins. J. Am. Chem. Soc. 1986, 108, 536–538. [Google Scholar] [CrossRef]
- Lyon, D.K.; Miller, W.K.; Novet, T.; Domaille, P.J.; Evitt, E.; Johnson, D.C.; Finke, R.G. Highly oxidation resistant inorganic-porphyrin analog polyoxometalate oxidation catalysts. 1. The synthesis and characterization of aqueous-soluble potassium salts of α2-P2W17O61(Mn+·OH2)(n−10) and organic solvent soluble tetra-n-butylammonium salts of α2-P2W17O61(Mn+·Br)(n−11) (M = Mn3+,Fe3+,Co2+,Ni2+,Cu2+). J. Am. Chem. Soc. 1991, 113, 7209–7221. [Google Scholar]
- Mansuy, D.; Bartoli, J.F.; Battioni, P.; Lyon, D.K.; Finke, R.G. Highly oxidation resistant inorganic-porphyrin analog polyoxometalate oxidation catalysts. 2. Catalysis of olefin epoxidation and aliphatic and aromatic hydroxylations starting from α2-P2W17O61(Mn+·Br)(n−11) (Mn+ = Mn3+,Fe3+,Co2+,Ni2+,Cu2+), including quantitative comparisons to metalloporphyrin catalysts. J. Am. Chem. Soc. 1991, 113, 7222–7226. [Google Scholar]
- Neumann, R.; Gara, M. Highly Active Manganese-Containing Polyoxometalate as Catalyst for Epoxidation of Alkenes with Hydrogen Peroxide. J. Am. Chem. Soc. 1994, 116, 5509–5510. [Google Scholar] [CrossRef]
- Hill, C.L.; Prosser-McCartha, C.M. Homogeneous catalysis by transition metal oxygen anion clusters. Coord. Chem. Rev. 1995, 143, 407–455. [Google Scholar] [CrossRef]
- Neumann, R.; Gara, M. The Manganese-Containing Polyoxometalate, [WZnMnII2(ZnW9O34)2]12−, as a Remarkably Effective Catalyst for Hydrogen Peroxide Mediated Oxidations. J. Am. Chem. Soc. 1995, 117, 5066–5074. [Google Scholar] [CrossRef]
- Neumann, R.; Juwiler, D. Oxidations with hydrogen peroxide catalysed by the [WZnMn(II)2(ZnW9O34)2]12− polyoxometalate. Tetrahedron 1996, 52, 8781–8788. [Google Scholar] [CrossRef]
- Bösing, M.; Nöh, A.; Loose, I.; Krebs, B. Highly Efficient Catalysts in Directed Oxygen-Transfer Processes: Synthesis, Structures of Novel Manganese-Containing Heteropolyanions and Applications in Regioselective Epoxidation of Dienes with Hydrogen Peroxide. J. Am. Chem. Soc. 1998, 120, 7252–7259. [Google Scholar] [CrossRef]
- Neumann, R.; Khenkin, A.M. Alkane oxidation with manganese substituted polyoxometalates in aqueous media with ozone and the intermediacy of manganese ozonide species. Chem. Commun. 1998, 1967–1968. [Google Scholar] [CrossRef]
- Bösing, M.; Krebs, B.; Nestler, B.; Seebach, M.; Reinhardt, G.; Wohlers, M.; Dingerdissen, U. Low-temperature bleaching with manganese-containing heteropolytungstates. Appl. Catal. A Gen. 1999, 184, 273–278. [Google Scholar] [CrossRef]
- Ben-Daniel, R.; Weiner, L.; Neumann, R. Activation of Nitrous Oxide and Selective Epoxidation of Alkenes Catalyzed by the Manganese-Substituted Polyoxometalate, [MnIII2ZnW(Zn2W9O34)2]10−. J. Am. Chem. Soc. 2002, 124, 8788–8789. [Google Scholar] [CrossRef] [PubMed]
- Gamelas, J.A.F.; Gaspar, A.R.; Evtuguin, D.V.; Pascoal Neto, C. Transition metal substituted polyoxotungstates for the oxygen delignification of kraft pulp. Appl. Catal. A Gen. 2005, 295, 134–141. [Google Scholar] [CrossRef]
- Khenkin, A.M.; Kumar, D.; Shaik, S.; Neumann, R. Characterization of Manganese(V)−Oxo Polyoxometalate Intermediates and Their Properties in Oxygen-Transfer Reactions. J. Am. Chem. Soc. 2006, 128, 15451–15460. [Google Scholar] [CrossRef] [PubMed]
- Keita, B.; Nadjo, L. Electrochemistry of Isopoly and Heteropoly Oxometalates. In Encyclopedia of Electrochemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007. [Google Scholar]
- Ruhlmann, L.; Costa-Coquelard, C.; Canny, J.; Thouvenot, R. Electrochemical and electrocatalytical investigations on the trimanganese sandwich complex [NaMn3(H2O)2(P2W15O56)2]17−. J. Electroanal. Chem. 2007, 603, 260–268. [Google Scholar] [CrossRef]
- Ammam, M.; Keita, B.; Nadjo, L.; Mbomekalle, I.-M.; Ritorto, M.D.; Anderson, T.M.; Neiwert, W.A.; Hill, C.L.; Fransaer, J. Cyclic Voltammetry Study of the Mn-Substituted Polyoxoanions [MnII4(H2O)2(H4AsW15O56)2]18− and [((MnIIOH2)MnII2PW9O34)2(PW6O26)]17−: Electrodeposition of Manganese Oxides Electrocatalysts for Dioxygen Reduction. Electroanalysis 2011, 23, 1427–1434. [Google Scholar] [CrossRef]
- Folkman, S.J.; Finke, R.G. Electrochemical Water Oxidation Catalysis Beginning with Co(II) Polyoxometalates: The Case of the Precatalyst Co4V2W18O6810−. ACS Catal. 2017, 7, 7–16. [Google Scholar] [CrossRef]
- Folkman, S.J.; Soriano-Lopez, J.; Galán-Mascarós, J.R.; Finke, R.G. Electrochemically Driven Water-Oxidation Catalysis Beginning with Six Exemplary Cobalt Polyoxometalates: Is It Molecular, Homogeneous Catalysis or Electrode-Bound, Heterogeneous CoOx Catalysis? J. Am. Chem. Soc. 2018, 140, 12040–12055. [Google Scholar] [CrossRef] [PubMed]
- Stracke, J.J.; Finke, R.G. Electrocatalytic Water Oxidation Beginning with the Cobalt Polyoxometalate [Co4(H2O)2(PW9O34)2]10−: Identification of Heterogeneous CoOx as the Dominant Catalyst. J. Am. Chem. Soc. 2011, 133, 14872–14875. [Google Scholar] [CrossRef] [PubMed]
- Stracke, J.J.; Finke, R.G. Water Oxidation Catalysis Beginning with 2.5 µM [Co4(H2O)2(PW9O34)2]10−: Investigation of the True Electrochemically Driven Catalyst at ≥600 mV Overpotential at a Glassy Carbon Electrode. ACS Catal. 2013, 3, 1209–1219. [Google Scholar] [CrossRef]
- Stracke, J.J.; Finke, R.G. Distinguishing Homogeneous from Heterogeneous Water Oxidation Catalysis when Beginning with Polyoxometalates. ACS Catal. 2014, 4, 909–933. [Google Scholar] [CrossRef]
- Stracke, J.J.; Finke, R.G. Water Oxidation Catalysis Beginning with Co4(H2O)2(PW9O34)210− When Driven by the Chemical Oxidant Ruthenium(III)tris(2,2′-bipyridine): Stoichiometry, Kinetic and Mechanistic Studies en Route to Identifying the True Catalyst. ACS Catal. 2014, 4, 79–89. [Google Scholar] [CrossRef]
- Barats-Damatov, D.; Shimon, L.J.W.; Weiner, L.; Schreiber, R.E.; Jiménez-Lozano, P.; Poblet, J.M.; de Graaf, C.; Neumann, R. Dicobalt-µ-oxo Polyoxometalate Compound, [(α2-P2W17O61Co)2O]14−: A Potent Species for Water Oxidation, C–H Bond Activation and Oxygen Transfer. Inorg. Chem. 2014, 53, 1779–1787. [Google Scholar] [CrossRef]
- Evangelisti, F.; Car, P.-E.; Blacque, O.; Patzke, G.R. Photocatalytic water oxidation with cobalt-containing tungstobismutates: Tuning the metal core. Catal. Sci. Technol. 2013, 3, 3117–3129. [Google Scholar] [CrossRef]
- Lv, H.; Song, J.; Geletii, Y.V.; Vickers, J.W.; Sumliner, J.M.; Musaev, D.G.; Kögerler, P.; Zhuk, P.F.; Bacsa, J.; Zhu, G.; et al. An Exceptionally Fast Homogeneous Carbon-Free Cobalt-Based Water Oxidation Catalyst. J. Am. Chem. Soc. 2014, 136, 9268–9271. [Google Scholar] [CrossRef]
- Vickers, J.W.; Lv, H.; Sumliner, J.M.; Zhu, G.; Luo, Z.; Musaev, D.G.; Geletii, Y.V.; Hill, C.L. Differentiating Homogeneous and Heterogeneous Water Oxidation Catalysis: Confirmation that [Co4(H2O)2(α-PW9O34)2]10− Is a Molecular Water Oxidation Catalyst. J. Am. Chem. Soc. 2013, 135, 14110–14118. [Google Scholar] [CrossRef]
- Zhu, G.; Geletii, Y.V.; Kögerler, P.; Schilder, H.; Song, J.; Lense, S.; Zhao, C.; Hardcastle, K.I.; Musaev, D.G.; Hill, C.L. Water oxidation catalyzed by a new tetracobalt-substituted polyoxometalate complex: [{Co4(μ-OH)(H2O)3}(Si2W19O70)]11−. Dalton Trans. 2012, 41, 2084–2090. [Google Scholar] [CrossRef] [PubMed]
- Soriano-López, J.; Goberna-Ferrón, S.; Vigara, L.; Carbó, J.J.; Poblet, J.M.; Galán-Mascarós, J.R. Cobalt Polyoxometalates as Heterogeneous Water Oxidation Catalysts. Inorg. Chem. 2013, 52, 4753–4755. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.; Zhang, L.; Sang, X.; You, W. Determination of the stability constant of cobalt-substituted mono-lacunary Keggin-type polyoxometalate and its electrocatalytic water oxidation performance. J. Coord. Chem. 2017, 70, 2950–2957. [Google Scholar] [CrossRef]
- Sun, C.-Y.; Liu, S.-X.; Xie, L.-H.; Wang, C.-L.; Gao, B.; Zhang, C.-D.; Su, Z.-M. Synthesis and characterization of one- to three-dimensional compounds composed of paradodecatungstate-B cluster and transition metals as linkers. J. Solid State Chem. 2006, 179, 2093–2100. [Google Scholar] [CrossRef]
- Gimenez-Saiz, C.; Galan-Mascaros, J.R.; Triki, S.; Coronado, E.; Ouahab, L. [(Co(H2O)4)2(H2W12O42)]n6n−: A Novel Chainlike Heteropolyanion Formed by Paradodecatungstate and Cobalt(II) Ions. Inorg. Chem. 1995, 34, 524–526. [Google Scholar] [CrossRef]
- Evans, H.T.; Rollins, O.W. Sodium paradodecatungstate 20-hydrate. Acta Crystallogr. Sect. B 1976, 32, 1565–1567. [Google Scholar] [CrossRef]
- Yuan, L.; Qin, C.; Wang, X.; Wang, E.; Li, Y. A new series of polyoxometalate compounds built up of paradodecatungstate anions and transition metal/alkaline-earth metal cations. Solid State Sci. 2008, 10, 967–975. [Google Scholar] [CrossRef]
- Yang, W.-B.; Lu, C.-Z.; Lin, X.; Zhuang, H.-H. Syntheses, Structures and Magnetic Properties of Three New Extended Iron-containing Heteropolytungstates. Z. Anorg. Allg. Chem. 2003, 629, 2046–2052. [Google Scholar] [CrossRef]
- Xu, X.; Luo, F.; Luo, W.; Chen, J. Synthesis and Crystal Structure of a New 3D Copper B-Paradodecatungstate Compound: [Na2(H2O)8][Na8(H2O)20][Cu(en)2][W12O42]·3H2O. Zeitschrift für Naturforschung B 2009, 64, 269. [Google Scholar] [CrossRef]
- Wu, L.; Yang, W.; Dong, X.; Yu, C.; Liu, B.; Yan, Y.; Hu, H.; Xue, G. A pure inorganic 2-D framework based on paradodecatungstate and Mn2+ ions: Syntheses, structure and properties. J. Coord. Chem. 2015, 68, 2324–2333. [Google Scholar] [CrossRef]
- Wasfi, S.H.; Rheingold, A.L.; Kokoszka, G.F.; Goldstein, A.S. Preparation, structure and magnetic properties of Na10Fe4Cu2W18O70H6·29H2O, containing the double Keggin anion [Fe4Cu2W18O70H6]10−. Inorg. Chem. 1987, 26, 2934–2939. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Laha, S.; Sharma, R.; Bhat, S.V.; Reddy, M.L.P.; Gopalakrishnan, J.; Natarajan, S. Ba3(P1−xMnxO4)2: Blue/green inorganic materials based on tetrahedral Mn(V). Bull. Mater. Sci. 2011, 34, 1257–1262. [Google Scholar] [CrossRef]
- Medina, E.A.; Li, J.; Stalick, J.K.; Subramanian, M.A. Intense turquoise colors of apatite-type compounds with Mn5+ in tetrahedral coordination. Solid State Sci. 2016, 52, 97–105. [Google Scholar] [CrossRef]
- Drewes, D.; Piepenbrink, M.; Krebs, B. The First Structurally Characterized Mn(III) Substituted Sandwich-type Polyoxotungstates. J. Clust. Sci. 2006, 17, 361–374. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS, Program for Scaling and Correction of Area Detector Data; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Blessing, R. An empirical correction for absorption anisotropy. Acta Crystallogr. Sect. A 1995, 51, 33–38. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELX-TL, Software Package for the Crystal Structure Determination, Version 5.03; Siemens Analytical X-ray Instrument Division: Madison, WI, USA, 1994. [Google Scholar]
- Vila, N.; Aparicio, P.A.; Secheresse, F.; Poblet, J.M.; Lopez, X.; Mbomekalle, I.M. Electrochemical behavior of α1/α2-[Fe(H2O)P2W17O61](7−) isomers in solution: Experimental and DFT studies. Inorg. Chem. 2012, 51, C-38. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mn1–O22 | 2.163(2) | O7–Mn1–O11 | 168.12(9) | O22–Mn1–O16 | 170.81(9) |
| Mn1–O23 | 2.176(3) | O7–Mn1–O16 | 87.04(10) | O22–Mn1–O23 | 88.48(9) |
| Mn1–O24 | 2.183(3) | O7–Mn1–O22 | 87.13(10) | O22–Mn1–O24 | 86.49(10) |
| Mn1–O7 | 2.163(3) | O7–Mn1–O24 | 96.89(10) | O23–Mn1–O24 | 173.02(10) |
| Mn1–O11 | 2.177(3) | O7–Mn1–O23 | 87.67(10) | O23–Mn1–O11 | 80.56(10) |
| Mn1–O16 | 2.180(2) | O11–Mn1–O16 | 93.04(9) | O23–Mn1–O16 | 98.38(9) |
| W2–O7 | 1.746(3) | O11–Mn1–O24 | 94.97(11) | ||
| W3–O11 | 1.779(2) | O16–Mn1–O24 | 87.16(9) | ||
| W4–O16 | 1.793(2) | O22–Mn1–O11 | 94.12(10) |
| pH | Atomic Concentration % | W/Mn | |||
|---|---|---|---|---|---|
| C 1s | O 1s | W 4f | Mn 2p | ||
| 3.0 | 80.7 | 14.3 | 4.1 | 0.9 | 4.4 |
| 6.0 | 77.3 | 13.3 | 5.1 | 4.3 | 1.2 |
| Empirical formula | H62K2Mn2Na4O76W12 |
| Formula weight, g | 3764.74 |
| Crystal system | Triclinic |
| Space group | P−1 |
| a/Å | 12.132(3) |
| b/Å | 12.290(3) |
| c/Å | 13.130(4) |
| α/deg. | 73.394(10) |
| β/deg. | 67.727(10) |
| α/deg. | 73.205(11) |
| V/Å3 | 1699.7(8) |
| Z | 1 |
| ρcalc/g·cm−3 | 3.678 |
| µ/mm−1 | 20.846 |
| Data/Parameters | 9922/460 |
| Rint | 0.0492 |
| GOF | 1.174 |
| R (>2σ(I)) | R1 a = 0.0340 wR2 b = 0.0914 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teillout, A.-L.; de Oliveira, P.; Marrot, J.; Howell, R.C.; Vilà, N.; Walcarius, A.; Mbomekallé, I.M. Synthesis, Crystal Structure, Electrochemistry and Electro-Catalytic Properties of the Manganese-Containing Polyoxotungstate, [(Mn(H2O)3)2(H2W12O42)]6−. Inorganics 2019, 7, 15. https://doi.org/10.3390/inorganics7020015
Teillout A-L, de Oliveira P, Marrot J, Howell RC, Vilà N, Walcarius A, Mbomekallé IM. Synthesis, Crystal Structure, Electrochemistry and Electro-Catalytic Properties of the Manganese-Containing Polyoxotungstate, [(Mn(H2O)3)2(H2W12O42)]6−. Inorganics. 2019; 7(2):15. https://doi.org/10.3390/inorganics7020015
Chicago/Turabian StyleTeillout, Anne-Lucie, Pedro de Oliveira, Jérôme Marrot, Robertha C. Howell, Neus Vilà, Alain Walcarius, and Israël M. Mbomekallé. 2019. "Synthesis, Crystal Structure, Electrochemistry and Electro-Catalytic Properties of the Manganese-Containing Polyoxotungstate, [(Mn(H2O)3)2(H2W12O42)]6−" Inorganics 7, no. 2: 15. https://doi.org/10.3390/inorganics7020015
APA StyleTeillout, A.-L., de Oliveira, P., Marrot, J., Howell, R. C., Vilà, N., Walcarius, A., & Mbomekallé, I. M. (2019). Synthesis, Crystal Structure, Electrochemistry and Electro-Catalytic Properties of the Manganese-Containing Polyoxotungstate, [(Mn(H2O)3)2(H2W12O42)]6−. Inorganics, 7(2), 15. https://doi.org/10.3390/inorganics7020015