Abstract
Structural and optical properties as well as chemical bonding of BiI3 at elevated pressures are investigated by means of refinements of X-ray powder diffraction data, measurements of the optical absorption, and calculations of the band structure involving bonding analysis in real space. The data evidence the onset of a phase transition from trigonal (hR24) BiI3 into PuBr3-type (oS16) BiI3 around 4.6 GPa. This high-pressure modification remains stable up to 40 GPa, the highest pressure of this study. The phase exhibits semiconducting properties with constantly decreasing band gap between 5 and 18 GPa. Above this pressure, the absorbance edge broadens significantly. Extrapolation of the determined band gap values implies a semiconductor to metal transition at approximately 35 GPa. The value is in accordance with subtle structural anomalies and the results of band structure calculations. Topological analysis of the computed electron density and the electron-localizability indicator reveal fingerprints for weak covalent Bi-I contributions in addition to dominating ionic interactions for both modifications.
1. Introduction
In the context of current studies of materials with large spin-orbit coupling as topological materials [1,2], compounds of heavy elements attract widespread interest. Bismuth compounds and especially halides are in special focus as their atomic arrangements often exhibit low-symmetry patterns caused by the stereochemical activity of lone pairs [3,4,5,6]. Moreover, their structural and physical properties can be substantially modified by application of hydrostatic pressure [7,8,9,10].
The ambient pressure form of BiI3 [11,12] is one of the most frequent structure types for compounds MX3 (M = metal, X = Cl, Br, I). The motif consists of a slightly distorted hexagonal close packing of halide atoms with the metal atoms occupying 2/3 of the octahedral voids in every second layer in an ordered fashion. Displacement of the metal atoms with respect to the center of the octahedra is attributed to the stereochemical activity of lone pairs located on the bismuth atoms. In AsI3 and SbI3, the difference between short and long bonds is suppressed by applying hydrostatic pressures of 5 and 2 GPa, respectively [13,14,15,16].
Earlier high-pressure investigations on BiI3 [13] revealed a structural transformation at pressures slightly exceeding 4 GPa. The high-pressure structure of BiI3 was assigned to the monoclinic structure type of SbI3, but the diffraction data were measured without using a pressure-transmitting medium and showed clear indications of non-hydrostatic pressure conditions. Thus, we reinvestigated the nature of the high-pressure polymorph of BiI3 by performing high-resolution high-pressure investigations under hydrostatic pressure conditions using helium as the pressure transmitting medium. The pressure-induced changes of the electronic band gap are characterized by the measurements of the optical absorbance in the visible range. In addition, the changes of the electronic structure as well as those of the chemical bonding are studied by means of DFT full-potential electronic band structure calculations.
2. Results and Discussion
Upon hydrostatic compression, the onset of a phase transition is observed at 4.6(2) GPa in agreement with an earlier study [13]. The reflection positions can be indexed using an orthorhombic unit cell, and the crystal structure solution of the high-pressure modification is in accordance with an atomic arrangement of the PuBr3-type (Figure 1 and Supplementary Materials).
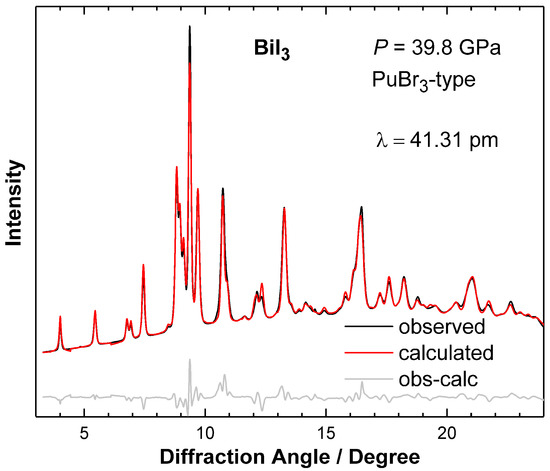
Figure 1.
X-ray diffraction diagram of the PuBr3-type high-pressure modification of BiI3 at the maximum pressure of 39.8 GPa. The diffraction data were measured in situ by means of diamond anvil cells in combination with synchrotron radiation.
The volume of trigonal (hR24) BiI3 shows a continuous decrease up to 4.6(2) GPa (Figure 2) in agreement with earlier data [13]. The predicted transition pressure on basis of the computational results amounts to 2.7 GPa and is, thus, in reasonable agreement with the experimental finding. The experimental volume reduction at 4.6 GPa, which is associated with the phase transition into the PuBr3-type modification, amounts to 6.9%. The bulk modulus of the high-pressure phase (oS16) BiI3 is significantly higher than the corresponding value of the ambient-pressure phase. Concerning the anisotropy of the pressure-induced changes, directions [010] and [001] respond in a very similar way while direction [100] is pronouncedly less compressible (Figure 3a–c). Although the phase transition shows a pronounced hysteresis upon pressure decrease down to 0.7 GPa, the high-pressure phase is not metastable at ambient conditions in accordance with earlier findings [13].
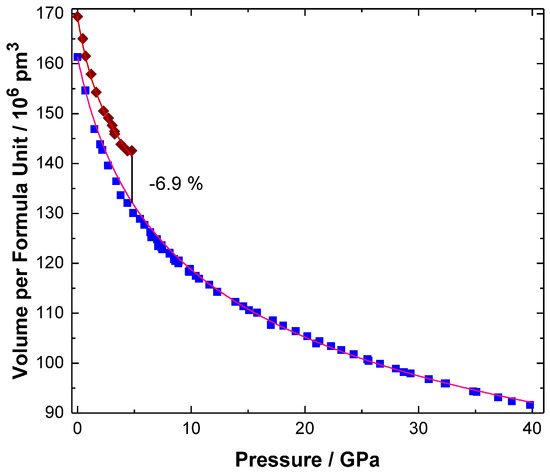
Figure 2.
Pressure-volume relation for BiI3 at pressures up to 40 GPa. Reddish brown symbols indicate data of the low-pressure modification and blue ones those of the high-pressure form. The discontinuous volume change associated with the phase transition is shown by a black line. The fitted equations of state curves are shown in shades of red, the black line visualizes the volume change calculated for P = 4.8 GPa. The compressibility of the low-pressure phase amounts to B0 = 11.7(4), B0′ = 8.1(3) with V0/Z = 169.45 × 106 pm3 and shows only a small anisotropy ([100]: B0 = 39(1) GPa, B0′ = 24 and a0 = 752.49 pm, [001]: B0 = 29.9(9) GPa, B0′ = 24.5(8) and c0 = 2073.3 pm). The bulk modulus of the high-pressure modification (oS16) BiI3 corresponds to B0 = 14.64(1) GPa with B0′ = 4.682(2) and V0/Z = 161.318 × 106 pm3. The anisotropy is shown in Figure 3.
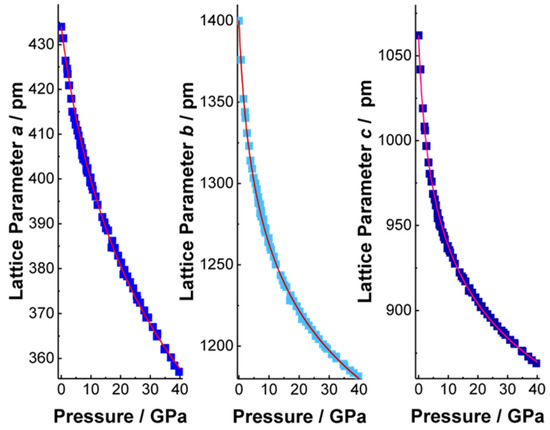
Figure 3.
Lattice parameters a, b and c (from left to right) of the high-pressure phase (oS16) BiI3 at pressures between ambient and 40 GPa. Blue symbols indicate experimental data and the fitted equations of state [17] curves are shown in red ([100]: a0 = 434 pm, B0 = 85.78(5) GPa and B0′ = 8.085(6); [010]: b0 = 1400 pm, B0 = 31.82(2) GPa, B0′ = 18.721(6); [001]: c0 = 1062 pm, B0 = 23.42(2) GPa, B0′ = 17.057(7). Please note the pronounced hardening upon pressure increase in direction [010] and [001], causing the distinct curvature.
In contrast to the earlier study, we find that the high-pressure modification of BiI3 (oS16, Figure 4a) is isopointal to the orthorhombic PuBr3 structure [18] with one bismuth on the 4c position and two independent iodine atoms located on 8f and 4c, respectively. The parent compound PuBr3 is a topological material of type enforced semimetal with degeneracy at the Fermi level [19]. The crystal structures are often described as being built of layers formed by corner-sharing bicapped trigonal prisms with the Bi-I distances d1, d2, and d3 (Figure 4b,c). Distances d4 to d8 in Figure 4a–c correspond to the shortest iodine-iodine contacts within the crystal structure. However, the PuBr3-type is not an intrinsic layer-type structure as the connectivity of the motif is crucially dependent on the specific interatomic distances, here that of bismuth to a third capping iodine atom (distance d9 in Figure 4) [20]. Although this contact is about 20% longer than the Bi-I distances in the first coordination sphere, the direction-dependent compressibility data evidence that the direction perpendicular to the layers is indeed the most compressible one at low pressures (smallest B0), but stiffens upon compression significantly (large B0′). Thus, in accordance with the ambiguous character of PuBr3-type (oS16) atomic arrangements, BiI3 exhibits a compression behavior which is more complex than that of other simple layer-type compounds.
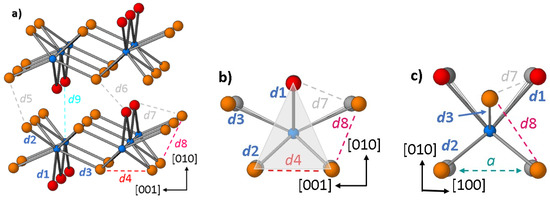
Figure 4.
Crystal structure segments of the PuBr3-type high-pressure modification (oS16) BiI3 at 7.5 GPa. Bismuth atoms are indicated in blue, I1 and I2 are displayed in red and orange, respectively; (a) illustrates the layer-type character of the atomic arrangement and shows relevant interatomic distances labelled from d1 to d9; (b,c) illustrate the bicapped trigonal-prismatic building units. Gray spheres designate the positions of the iodine atoms at 40 GPa. The trigonal face of the coordination prism is indicated light gray, the height of the trigonal prism is defined by the lattice parameter a shown in turquoise.
The transition from the low-pressure phase (hR24) BiI3 into (oS16) BiI3 is associated with a volume decrease of −6.9%. This experimental finding is in accordance with earlier investigations on selected rare-earth metal tri-halides, which have evidenced a series of pressure-induced transitions from the BiI3-type into PuBr3 type arrangements. These studies demonstrate that the PuBr3 type exhibits a higher packing density [21,22] for a given radius ratio of cations to anions. In addition, the transitions are in accordance with the pressure-distance-paradox and the pressure-coordination rule as it is associated with an increase of the coordination number of bismuth by iodine from 6 to 8.
As a result of the direction dependence of the compressibility, (oS16) BiI3 exhibits pronouncedly anisotropic changes of the lattice parameters (a: −11.7%, b: −7.6%, c: −8.9%, see Figure 3) and interatomic distances (Figure 5) between 7.5 and 40 GPa. The pressure dependence of the Bi-I and I-I contacts reveals overall a largely continuous and smooth decrease with pressure. Despite pronounced anisotropy in the relative decreases of the interatomic distances (d1: −3.9%, d2: −7.5%, d3: −8.8% in comparison to d9: −13%), the relative differences between the short (d1–d3) and the longer (d9) bismuth–iodine distances remain sufficiently large such that the layer-type character of the crystal structure is preserved. The iodine–iodine contacts between the layers (d5: −13.4%, d6: −12.5%) are significantly more compressible than those within the coordination polyhedron of bismuth (d4: −4% and d8: −6.8%), but the strong decrease of d7 (−10.4%) and the change of the lattice parameter a (−11.7%) evidence substantial structure alterations also within the layers. Finally, systematic deviations of several interatomic distances at approximately 32 GPa are in agreement with the insulator-metal transition predicted by band-structure calculations and extrapolation of the optical absorbance data (see below).
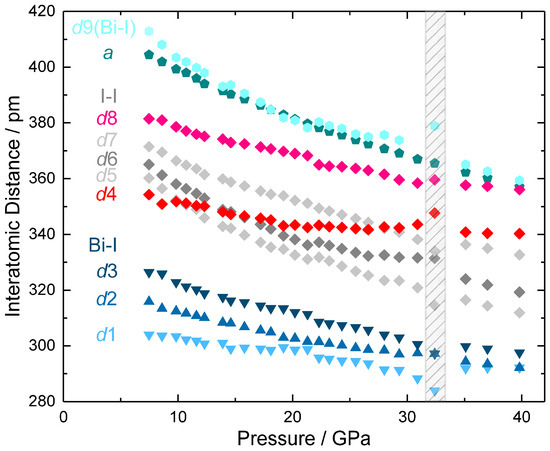
Figure 5.
Interatomic distances d1–d9 (see Figure 4) for the PuBr3-type high-pressure modification (oS16) BiI3 in direction of increasing pressures between 7.5 GPa and 40 GPa. Data for the lattice parameter a also describe the set of intralayer distances d(I1–I1), d(I2–I2), and d(Bi–Bi) between translationally equivalent atoms (see Figure 4c for those between the iodine atoms of adjacent trigonal basal planes of the bicapped coordination prism). The shaded region indicates discontinuities, which occur in the pressure regime of the insulator-metal transition.
The absorbance spectra of BiI3 at selected elevated pressures together with the spectrum of the free-standing crystal (Figure 6) show a well-defined absorption edge of ~1.6 eV at ambient pressure. The sharp decrease and the absorbance spectrum below the steep edge can be described by linear functions, and their crossing points were taken as a measure for the band gap. We apply this method also to the pressure-dependent absorbance spectra, to extract the pressure evolution of the band gap. With increasing pressure, the absorption edge shifts to lower energies, and thus the band gap decreases.
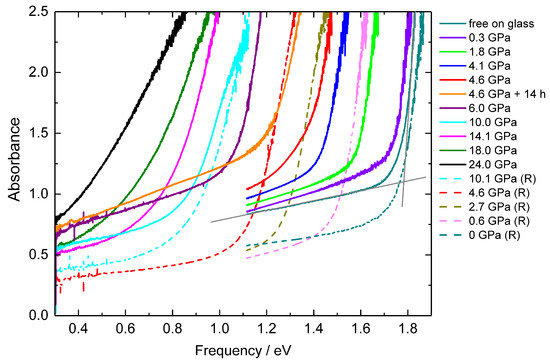
Figure 6.
Absorbance spectra for selected pressures. The spectrum below and above the absorption edge is described by linear functions (gray lines), their crossing points are used as measures for the band gaps.
The values of the estimated band gaps as a function of pressure (Figure 7) monotonically decrease with increasing pressures up to a pressure of ~4.6 GPa. At around 4.6 GPa a transition region is entered, in which the absorption edges are not well defined anymore (see Figure 6), and additionally the absorption spectra shift to smaller energies with time. The band gaps abruptly drop at this critical pressure (see Figure 7).
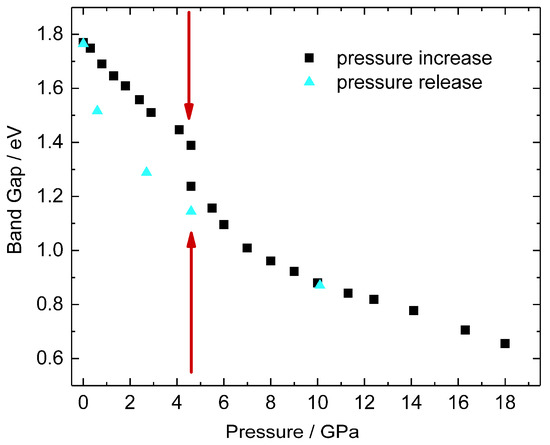
Figure 7.
The evolution of the band gap with pressure increase and release. The structural hysteresis shows in the band-gap differences between pressure increase and decrease in the low-pressure region. Extrapolation of the experimental band gap to 0 eV yields a pressure of 35 GPa for the semiconductor-to metal transition of (oS12)BiI3. The pressure of the phase transition is indicated by dark red arrows.
For pressures between 5 and 18 GPa the absorption edges are well-defined and shift continuously to lower energies with increasing pressures. Accordingly, the band gaps monotonically decrease with increasing pressures in the range 5–18 GPa. Above 18 GPa the absorption edges broaden and are no longer well-defined. Extrapolation of the experimental values in direction of higher pressure yields a predicted closure of the band gap at about 35 GPa in agreement with results of the band structure calculations (see below) and the observed anomalies of the interatomic distances.
During pressure release the absorption edges shift back to higher energies (see Figure 6), and the ambient-pressure band gap value is recovered. We observe a hysteresis in the evolution of the band gap values for pressures below 5 GPa which corresponds to the observed lower transition pressure upon decompression.
The results of calculations in the local density approximation are in agreement with the experimental finding that BiI3 is a semiconductor at ambient pressure. The computed band gaps (Figure 8) amount to 2.12 and 1.11 eV in scalar and fully-relativistic treatments, respectively. The calculated values are larger in the scalar relativistic calculations compared to the fully-relativistic ones, and as a result the semiconductor-to-metal transition occurs at a moderately lower pressure (35.1 GPa in good agreement with the extrapolation of the band gap data) at the fully-relativistic level. For the scalar relativistic level, a higher transition pressure (39.8 GPa) results.
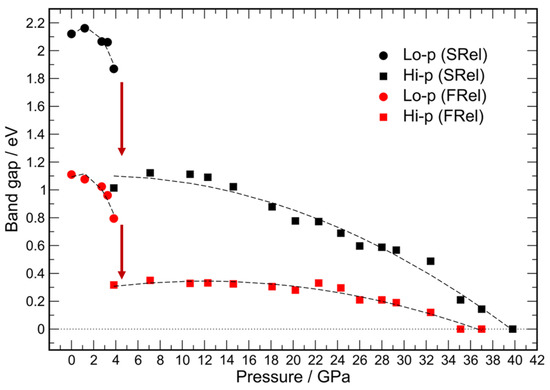
Figure 8.
Band gap values computed at the scalar relativistic level are shown in black, those calculated at the fully-relativistic level are displayed in red; low-pressure modification results are shown as filled circles and high-pressure ones as filled squares. The dashed-lines through the data points are a guide to the eye. The transition pressure is indicated by dark red arrows.
The electronic density of states computed at the fully-relativistic level for the ambient and the high-pressure structures show very similar features (Figure 9). The lowest-lying states are dominated by I 5s contributions (between [−13.0, −11.5] eV for the ambient and [−14.6, −12.1] eV for the high-pressure phase). Bi 6s states dominate the next region, [−10.4, −9.0] eV and [−11.3, −9.7] eV, respectively. I 5p and Bi 6p have similar contributions per atom in the ranges [−4.1, −2.7] eV and [−6, −4] eV, respectively. The densities of states (DOS) above this range up to the top of the valence band (set to 0 eV) is heavily dominated by the I 5p contributions. The main effects of pressure on the occupied electronic states are a shift of the s-dominated bands to lower energies accompanied by some widening and a more pronounced increase of the bandwidth of the p-dominated higher-lying bands. At the semiconductor-to-metal transition pressure (35 GPa), the energy ranges mentioned above become [−17.2, −13.1], [−12.6, −9.9] and [−8.3, 0] eV, respectively. The unoccupied states shown in Figure 9a,b contain 6 electrons per formula unit, formally corresponding to the total number of missing p electrons in one Bi and three I free atoms in comparison to a noble gas configuration.
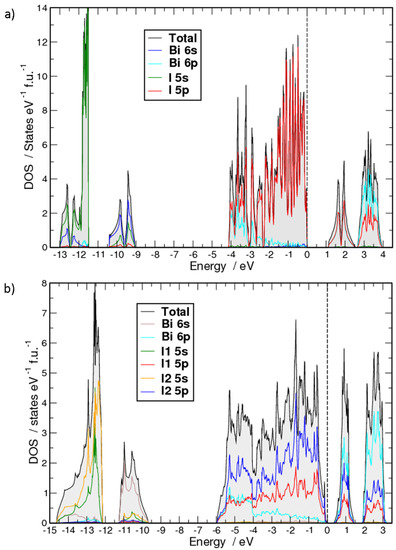
Figure 9.
Computed densities of states (DOS) (fully relativistic) for BiI3: (a) ambient-pressure modification, (b) PuBr3-type (oS16) high-pressure phase of BiI3 at 7.1 GPa. The formula unit contains two I2 atoms, one I1 and on Bi atom.
The chemical bonding in both modifications of BiI3 is investigated by methods operating in real space. The topological analysis yields atomic basins and by integrating the electron density inside these, the total number of electrons belonging to each atom of the crystal structure can be obtained. In accordance with the electronegativity values of the constituting elements, the results show that there is a charge transfer from the bismuth to the iodine atoms. At ambient pressure, Bi atoms donate 0.9 electrons and each iodine atom accepts 0.3.
In the high-pressure modification at 7.1 GPa, Bi is 0.7+ and I2 is slightly more negative than I1, 0.25− versus 0.20−. These effective charges do not change in any significant way with increasing pressures. The ionic nature of the bonding in BiI3 inferred from effective charges turns out to be actually the dominant type of bonding according to the analysis of the electron-localizability indicator (ELI). There are no ELI attractors in the valence region, so a clear sign of covalent interactions is absent (Figure 10). However, an examination of the ELI distribution shows that the shape of the outmost shell (principal quantum number n = 5) of the iodine atoms (Figure 10) deviates from spherical symmetry indicating that the respective electrons participate in the chemical bonding [23]. In the ambient-pressure structure the Bi atoms are coordinated octahedrally with I atoms forming three short (d1 = 3.054 Å) and three longer (d2 = 3.125 Å) contacts, so that each iodine atom has two Bi near neighbors. The basin intersection technique applied to the last shell of the iodine atoms shows that of the 7.83 electrons in the shell’s basin, 0.12 come from the nearest and 0.08 from the next-nearest Bi neighbor. In case of the high-pressure phase, each I1 atom has two Bi contacts at 3.042 Å, and each I2 atom has two nearest Bi neighbors at 3.164 Å as well as a third neighbor at 3.269 Å (Figure 10). Basin intersection analysis finds that (i) the last shell of each I1 atom contains 7.83 electrons to which each Bi atom contributes 0.107 electrons, (ii) the outer shell of each I2 atom contains 7.79 electrons to which the nearest Bi neighbors each contribute 0.057, and the second-nearest Bi 0.038 electrons. These Bi contributions to the fifth shell of the iodine atoms together with the structuring of the shell are the footprints of covalent Bi–I interactions in BiI3 at both ambient and elevated pressures.
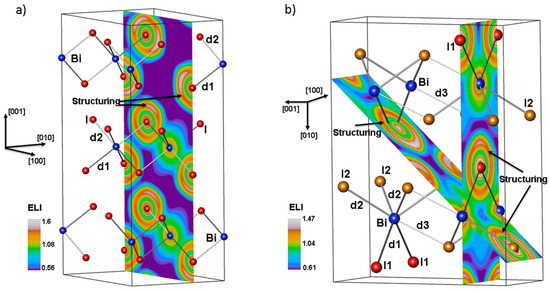
Figure 10.
(a) ELI distribution for the ambient-pressure structure, (hR24) BiI3, in a layer perpendicular to [010]. The layer depicted contains both the short (dark gray) and the long (light gray) Bi–I contacts. The structuring of the fifth shell of I atoms is identified. Shown is the hexagonal setting of the unit cell; (b) ELI distribution for (oS16) BiI3 at7.1 GPa. The labelling of the distances corresponds to that of Figure 4 and Figure 5. The layer perpendicular to [100] highlights Bi–I1, and the oblique layer the shortest Bi–I2 contacts. The structuring of the fifth shell of the iodine atoms is indicated.
3. Materials and Methods
For the powder X-ray diffraction experiments, commercially available BiI3 (Alfa Aesar, 99.999%; Thermo Fischer GmbH, Kandel, Rhineland-Palatinate, Germany,) was further purified by sublimation. Some of the obtained crystals were ground in the argon atmosphere of a glove box (H2O, O2 < 0.1 ppm) using an agate mortar. The particles smaller than 20 μm as selected by means of sieving were annealed in a sealed quartz ampoule at 500 K for one day in order to reduce the number of stacking faults which were induced by the grinding. Again, particles smaller than 20 μm were separated by sieving and the resulting powder was transferred into diamond anvil cells (Figure 11) with a culet size of 300 and 600 μm, respectively. As gasket materials, stainless steel or rhenium were used. For pressure redistribution, pressurized helium as a liquid medium was used. Pressure was measured using the luminescence shift of a small ruby chip located close to the sample [24]. The collected two dimensional images were integrated [25] and refined by means of full profile analysis [26].
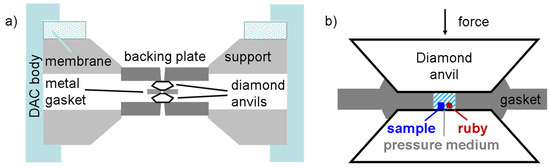
Figure 11.
(a) Schematic drawing of a diamond anvil cell. The diamond pressure anvils are located on seats which transmit the force from the cell body to the diamond tables. (b) Between the diamond anvils there is a metallic gasket positioned with a thickness of typically 30 µm in the pre-indented central part. A hole in the center serves as the high-pressure chamber for sample, pressure sensor (ruby) and medium. The volume not occupied by sample or ruby is filled with helium loaded in a special pressure vessel at approximately 0.2 GPa. When force is applied by pressurizing the outer steel membrane, the diamond tips deform the gasket plastically and hydrostatic pressure is generated.
To determine the compressibilities, second order Murnaghan-type equations of state were fitted by applying least squares routines to the experimental data. The metrical parameters at ambient pressure were taken from X-Ray powder diffraction data on a laboratory source (Huber Guinier G670, Huber Diffractionstechnik, Rimsting, Bavaria, Germany; CuKα1 radiation) with LaB6 as standard using the program WinCSD [26]. The positions at ambient pressure were taken from the most elaborate single crystal refinement [12].
For the optical measurements, ultra-dry BiI3 (Alfa Aesar, 99.998%) was purified by sublimation from 320 to 200 °C. In a second process, crystals of this refined BiI3 were grown by subliming the material from 275 to 200 °C. Suitable specimens of BiI3 were selected and placed in a Syassen-Holzapfel diamond anvil cell (DAC) [27], equipped with type IIa diamonds suitable for infrared measurements. The maximal generated pressure amounted to 24 GPa. Pressure-dependent transmittance measurements were carried out at room temperature in the frequency range 0.3–1.9 eV with a Bruker Vertex 80v Fourier transform infrared spectrometer (Bruker Optik, Ettlingen, Baden-Württemberg, Germany) coupled with a Bruker Hyperion infrared microscope with a 15× Cassegrain objective. For the in situ pressure determination the ruby luminescence technique was applied [24] and CsI powder served as quasi-hydrostatic pressure transmitting medium. For obtaining the transmittance spectra, the intensity transmitted through the sample Is was divided by the intensity transmitted through the pressure transmitting medium Iref, according to T(ω) = Is(ω)/Iref(ω). From the transmittance spectrum T(ω) the absorbance spectrum was calculated according to A(ω) = −log10T(ω).
Electronic structure calculations were performed by using two all electron, atom-centered numerical orbital full-potential methods, namely the Fritz-Haber Institute ab initio molecular simulations (FHI-aims) [28] and the full-potential local orbital (FPLO) [29] methods. The local density approximation (LDA) to the density functional theory as parametrized by Perdew and Wang was employed [30]. The experimentally determined crystal structure data for the ambient and high pressure modifications were used in the calculations. The electronic densities of states (DOS) were computed by the FPLO method within both scalar and fully-relativistic treatments. In these calculations, the first Brillouin zone (BZ) was sampled with meshes of 20 × 20 × 20 and 16 × 16 × 16, respectively. Chemical bonding was analyzed in real space based on the combined topological analysis of electron density (ED) and electron localizability indicator (ELI) [31,32,33]. The ED and ELI were calculated by an own separate code using the output of the FHI-aims method [34]. Topological analysis of the ED and the ELI using the quantum theory of atoms in molecules (QTAIM) [35] was realized by the program DGrid [36]. Determination of which atoms are contributing to a particular bond is done by intersecting the associated ELI bond basin and the atom basins [37].
4. Conclusions
X-ray diffraction data evidence a pressure-induced phase transition of BiI3 into the denser PuBr3-type crystal structure around 4.6 GPa. Optical absorbance measurements show that the structural change is associated with a decrease of the band gap. Within the stability field of the high-pressure form, the decrease proceeds and extrapolation of data measured up to 18 GPa predict a metallization at around 35 GPa, in good agreement with the calculated band gaps determined by fully relativistic density functional calculations. Chemical bonding in both low- and high-pressure form is essentially ionic with small covalent contributions in the Bi–I interactions.
Supplementary Materials
The following are available online at https://www.mdpi.com/2304-6740/7/12/143/s1, CIF, and the checkCIF output files. CCDC 1969518–1969543 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Author Contributions
Conceptualization by A.W., C.K., and U.S.; methodology, M.H., A.O., and C.K.; software, L.A., A.O., and U.S.; validation, U.S., L.A., and C.K.; formal analysis, A.W., U.S., L.A., A.O., and C.K.; investigation, A.W., M.S., M.H., and V.H.; data curation, U.S. and C.K.; writing—original draft preparation, U.S., A.O., and C.K.; writing—review and editing, U.S.; visualization, U.S., A.O., and C.K.; supervision, U.S. and C.K.; project administration, U.S.; funding acquisition, A.W.
Funding
This research was supported by a beam time grants of the European Synchrotron Radiation Facility (ESRF, Grenoble) under proposal numbers hs2739 and hs3112.
Acknowledgments
The authors thank Stefano Leoni and S. E. Boulfelfel for discussions on chemical bonding. Support of Carola J. Müller and Miriam Schmidt for the synchrotron experiments is gratefully acknowledged. Thorough discussions with Michael Ruck and Luis Elcoro are appreciated.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Manna, K.; Sun, Y.; Muechler, L.; Kübler, J.; Felser, C. Heusler, weyl and berry. Nat. Rev. Mater. 2018, 3, 244–256. [Google Scholar] [CrossRef]
- Schoop, L.M.; Pielnhofer, F.; Lotsch, B.V. Chemical principles of topological semimetals. Chem. Mater. 2018, 30, 3155–3176. [Google Scholar] [CrossRef]
- Von Schnering, H.G.; von Benda, H.; Kalveram, C. Wismutmonojodid bij, eine verbindung mit Bi(0) und Bi(II). ZAAC 1978, 438, 37–52. [Google Scholar] [CrossRef]
- Noguchi, R.; Takahashi, T.; Kuroda, K.; Ochi, M.; Shirasawa, T.; Sakano, M.; Bareille, C.; Nakayama, M.; Watson, M.D.; Yaji, K.; et al. A weak topological insulator state in quasi-one-dimensional bismuth iodide. Nature 2019, 566, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Autes, G.; Isaeva, A.; Moreschini, L.; Johannsen, J.C.; Pisoni, A.; Mori, R.; Zhang, W.; Filatova, T.G.; Kuznetsov, A.N.; Forró, L.; et al. A novel quasi-one-dimensional topological insulator in bismuth iodide β-Bi4I4. Nat. Mater. 2016, 15, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Zhou, J.-J.; Yao, Y.; Zhang, F. Weak topological insulators and composite weyl semimetals: β-Bi4X4 (X=Br, I). Phys. Rev. Lett. 2016, 116, 066801. [Google Scholar] [CrossRef] [PubMed]
- Lia, X.; Chena, D.; Jinc, M.; Maa, D.; Ged, Y.; Sune, J.; Guof, W.; Suna, H.; Hana, J.; Xiaoa, W.; et al. Pressure-induced phase transitions and superconductivity in a quasi–1-dimensional topological crystalline insulator α-Bi4Br4. Proc. Natl. Acad. Sci. USA 2019, 116, 17696–17700. [Google Scholar] [CrossRef]
- Qi, Y.; Shi, W.; Werner, P.; Naumov, P.G.; Schnelle, W.; Wang, L.; Rana, K.G.; Parkin, S.; Medvedev, S.A.; Yan, B.; et al. Pressure-induced superconductivity and topological quantum phase transitions in a quasi-one-dimensional topological insulator: Bi4I4. npj Quantum Mater. 2018, 3, 4. [Google Scholar] [CrossRef]
- Pisoni, A.; Gaál, R.; Zeugner, A.; Falkowski, V.; Isaeva, A.; Huppertz, H.; Autès, G.; Yazyev, O.V.; Forró, L. Pressure effect and superconductivity in the β-Bi4I4 topological insulator. Phys. Rev. B 2017, 95, 235149. [Google Scholar] [CrossRef]
- Wang, X.; Wu, J.; Wang, J.; Chen, T.; Gao, H.; Lu, P.; Chen, Q.; Ding, C.; Wen, I.; Sun, J. Pressure-induced structural and electronic transitions in bismuth iodide. Phys. Rev. B 2018, 98, 174112. [Google Scholar] [CrossRef]
- Trotter, J.; Zobel, T. The crystal structure of SbI3 and BiI3. Zeitschrift für Kristallographie 1966, 123, 67–72. [Google Scholar] [CrossRef]
- Ruck, M. Darstellung und kristallstruktur von fehlordnungsfreiem bismuthtriiodid. Zeitschrift für Kristallographie 1995, 210, 650–655. [Google Scholar]
- Hsueh, H.C.; Chen, R.K.; Vass, H.; Clark, S.J.; Ackland, G.J.; Poon, W.C.K.; Crain, J. Compression mechanisms in quasimolecular XI3 (X = As,Sb,Bi) solids. Phys. Rev. B. 1998, 22, 14812. [Google Scholar] [CrossRef]
- Saitoh, A.; Komatsu, T.; Karasawa, T.; Ohtake, H.; Suemoto, T. Raman scattering under hydrostatic pressures in layered BiI3 and SbI3 crystals. Phys. Stat. Sol. 2001, 2, 357–367. [Google Scholar] [CrossRef]
- Saitoh, A. Raman scattering, luminescence, and absorption edge under hydrostatic pressures of layered BiI3 and SbI3. J. Raman. Spectrosc. 2007, 38, 537–542. [Google Scholar] [CrossRef]
- Hsueh, H.C.; Poon, W.C.K.; Vass, H.; Crain, J. Pressure-induced electron transfer in quasi-molecular solids. EPL 1996, 35, 689–694. [Google Scholar] [CrossRef]
- Murnaghan, F.D. The compressibility of media under extreme pressures. Proc. Natl. Acad. Sci. USA 1944, 30, 244–247. [Google Scholar] [CrossRef]
- Zachariasen, W.H. Crystal chemical studies of the 5f-series of elements. I. New structure types. Acta Crystallographica 1948, 1, 265–268. [Google Scholar] [CrossRef]
- Vergniory, M.G.; Elcoro, L.; Felser, C.; Regnault, N.; Bernevig, B.A.; Wang, Z. A complete catalogue of high-quality topological materials. Nature 2019, 566, 480–485. [Google Scholar] [CrossRef]
- Hulliger, F. Structural Chemistry of Layer-Type Phases; Lévy, F.A., Ed.; Physics and Chemistry of Materials with Layered Structures Volume 5; D. Reidel Publishing Company: Dordrecht, The Netherlands, 1976. [Google Scholar]
- Beck, H.K.; Gladow, E. Zur hochdruckpolymorphie der seltenerd-trihalogenide. ZAAC 1979, 453, 79–92. [Google Scholar] [CrossRef]
- Beck, H.K.; Gladow, E. Der PuBr3-typ als hochdruckmodifikation bei seltenerdtrihalogeniden LnX3 (X = Cl, Br, I). ZAAC 1983, 502, 178–184. [Google Scholar]
- Kohout, M.; Wagner, F.R.; Grin, Y. Electron localization function for transition-metal compounds. Theor. Chem. Acc. 2002, 108, 150–156. [Google Scholar] [CrossRef]
- Piermarini, G.J.; Block, S.; Barnett, J.D.; Forman, R.A. Calibration of the pressure dependence of the R1 ruby fluorescence line to 195 kbar. J. Appl. Phys. 1975, 46, 2774–2780. [Google Scholar] [CrossRef]
- Hammersley, A.P.; Svensson, S.O.; Hanfland, M.; Fitch, A.N.; Häussermann, D. Two-dimensional detector software: From real detector to idealised image or two-theta scan. High Press. Res. 1996, 14, 235–248. [Google Scholar] [CrossRef]
- Akselrud, L.; Grin, Y. WinCSD: Software package for crystallographic calculations (Version 4). J. Appl. Crystallogr. 2014, 47, 803–805. [Google Scholar] [CrossRef]
- Huber, G.; Syassen, K.; Holzapfel, W.B. Pressure dependence of 4f levels in europium pentaphosphate up to 400 kbar. Phys. Rev. B 1977, 15, 5123–5128. [Google Scholar] [CrossRef]
- Blum, V.; Gehrke, R.; Hanke, F.; Havu, P.; Havu, V.; Ren, X.; Reuter, K.; Scheffler, M. Ab initio molecular simulations with numeric atom-centered orbitals. Comput. Phys. Commun. 2009, 180, 2175–2196. [Google Scholar] [CrossRef]
- Koepernik, K.; Eschrig, H. Full-potential nonorthogonal local-orbital minimum-basis band-structure scheme. Phys. Rev. B 1999, 59, 1743–1757. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Kohout, M. A measure of electron localizability. Int. J. Quantum Chem. 2004, 97, 651–658. [Google Scholar] [CrossRef]
- Kohout, M.; Wagner, F.R.; Grin, Y. Atomic shells from the electron localizability in momentum space. Int. J. Quantum Chem. 2006, 106, 1499–1507. [Google Scholar] [CrossRef]
- Kohout, M. Bonding indicators from electron pair density functionals. Faraday Discuss. 2007, 135, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Villaseca, S.A.; Ormeci, A.; Levchenko, S.V.; Schlögl, R.; Grin, Y.; Armbrüster, M. CO adsorption on GaPd—Unraveling the chemical bonding in real space. Chem. Phys. Chem. 2017, 18, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1995. [Google Scholar]
- Kohout, M. Program DGrid, Version 4.6; Radebeul, Germany, 2011–2017.
- Raub, S.; Jansen, G. A quantitative measure of bond polarity from the electron localization function and the theory of atoms in molecules. Theor. Chem. Acc. 2001, 106, 223–232. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).