Synthesis of a Half-Sandwich Hydroxidoiridium(III) Complex Bearing a Nonprotic N-Sulfonyldiamine Ligand and Its Transformations Triggered by the Brønsted Basicity



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
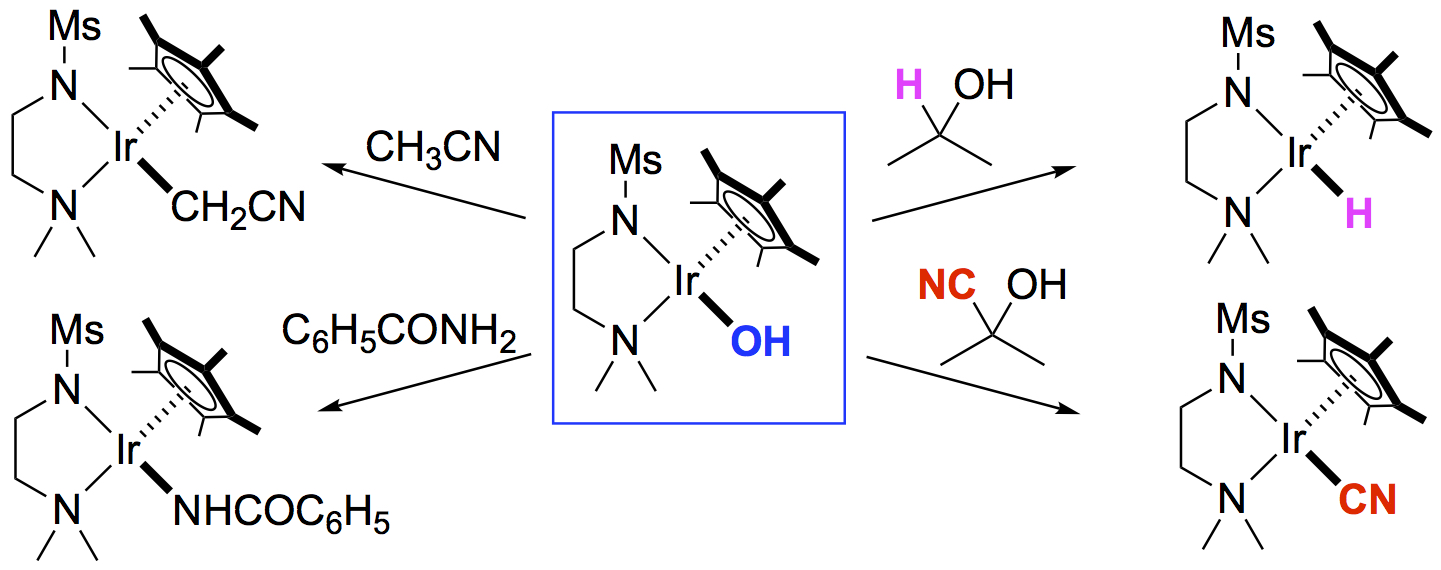
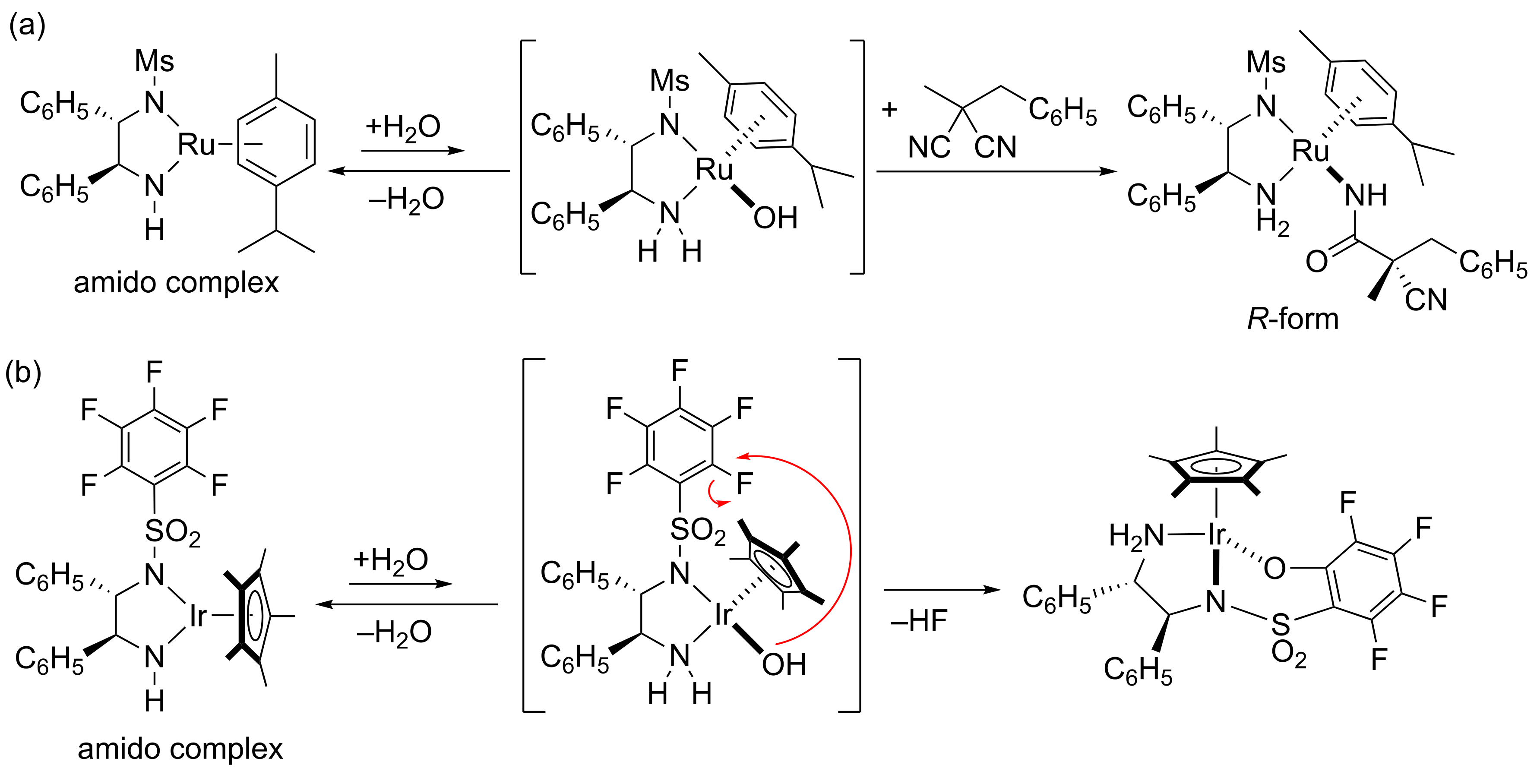
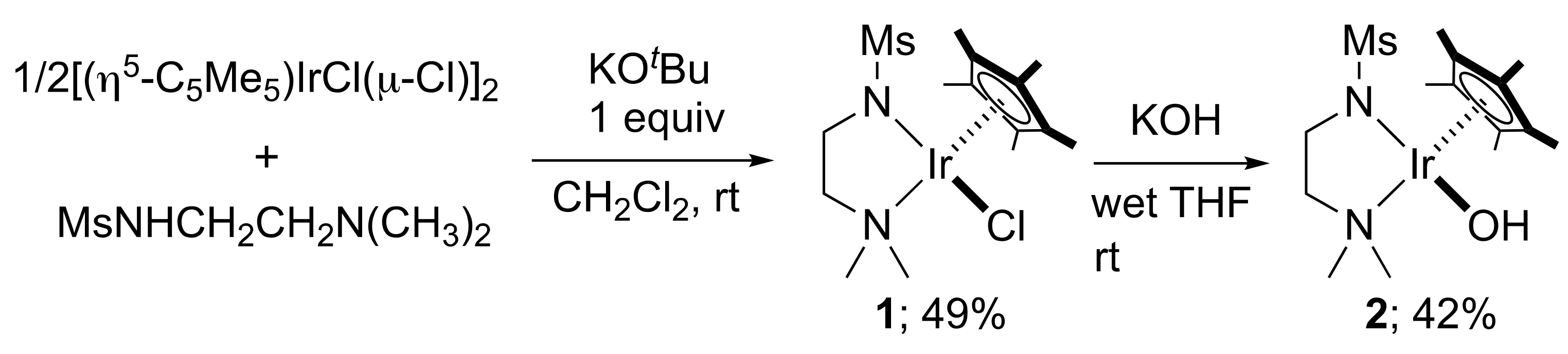
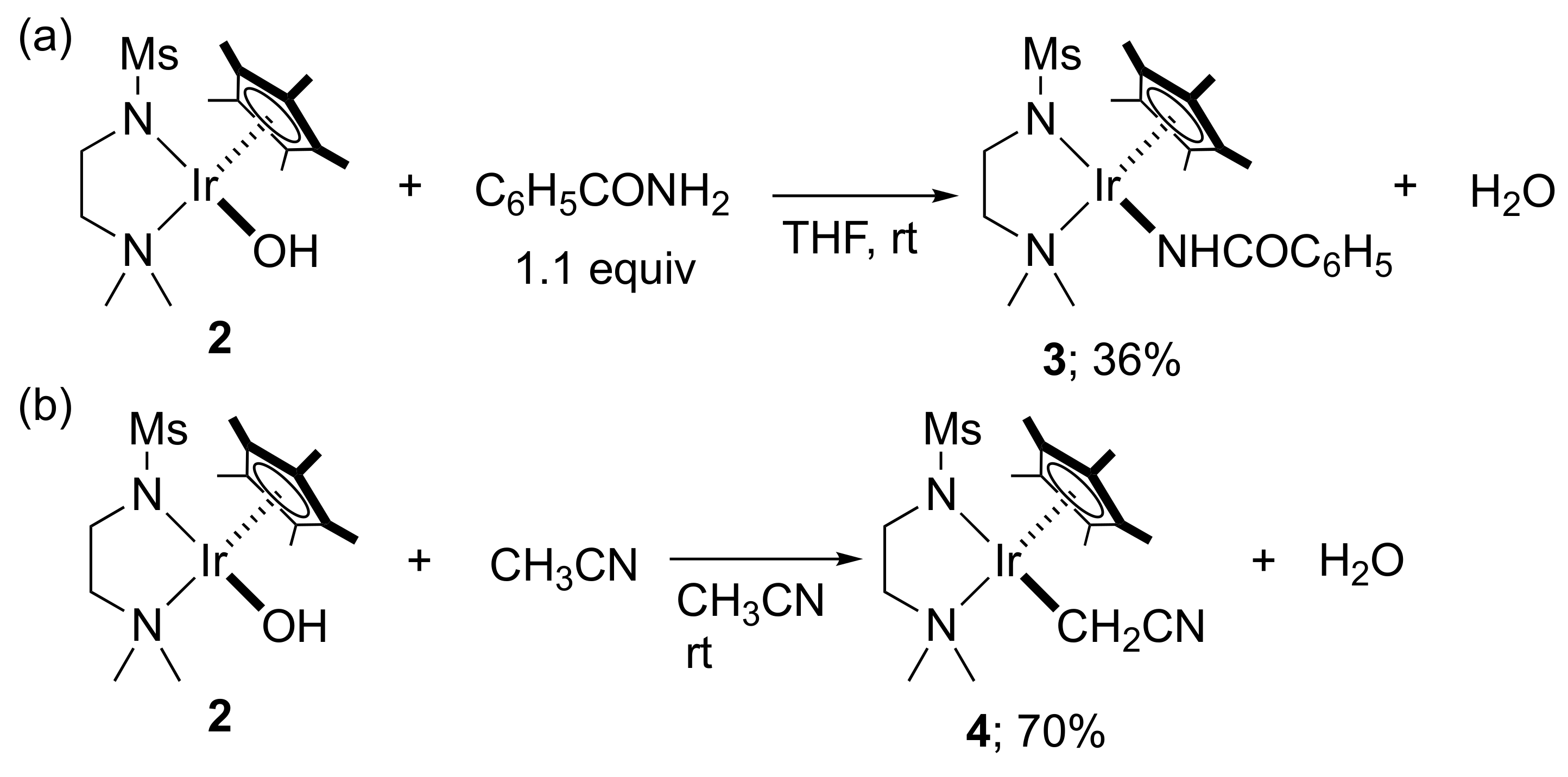
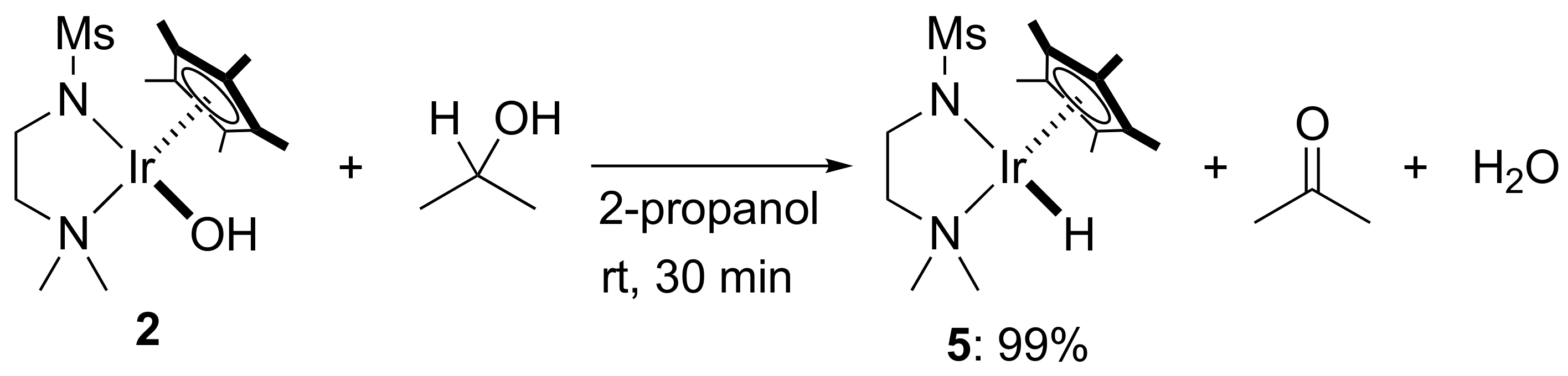
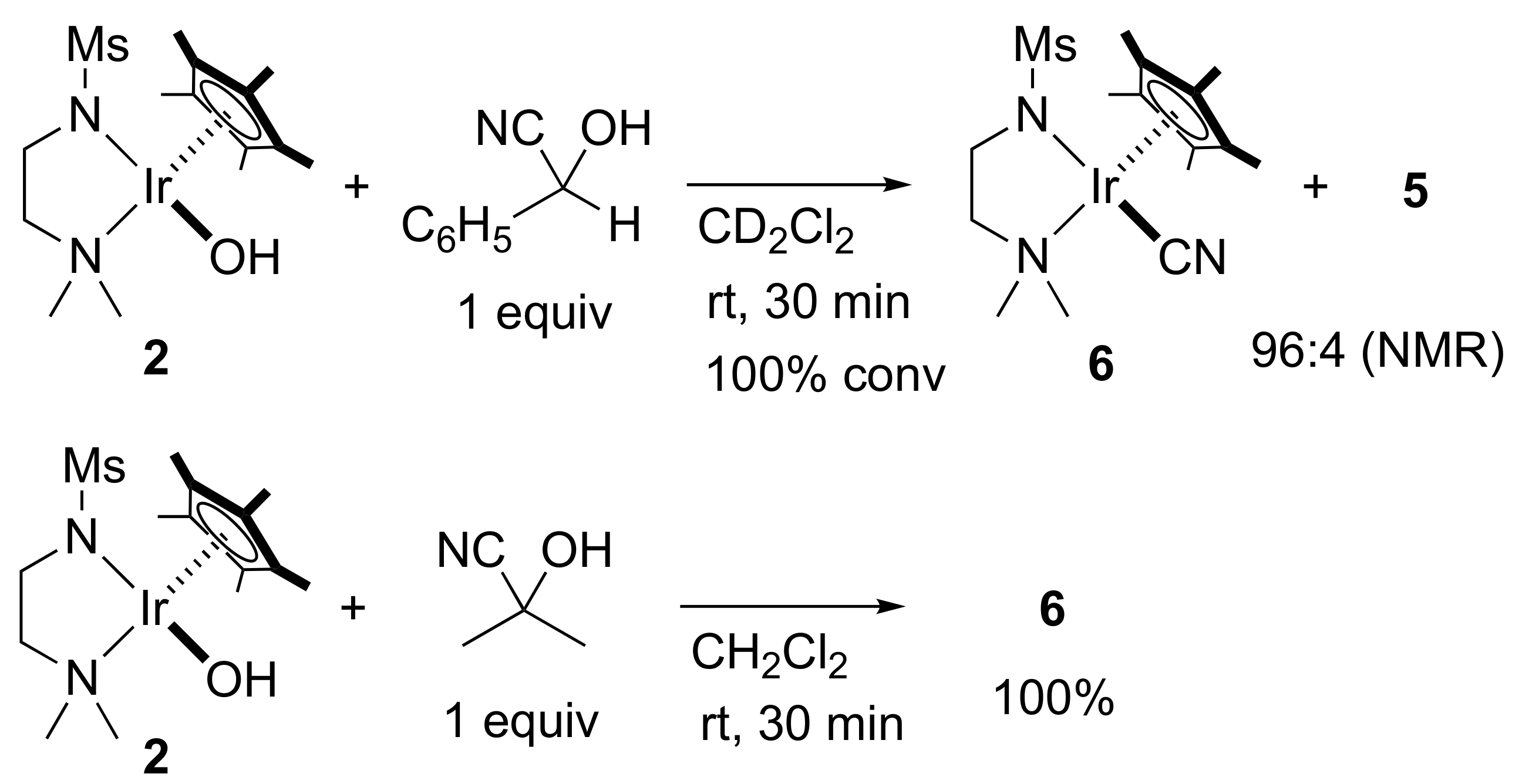
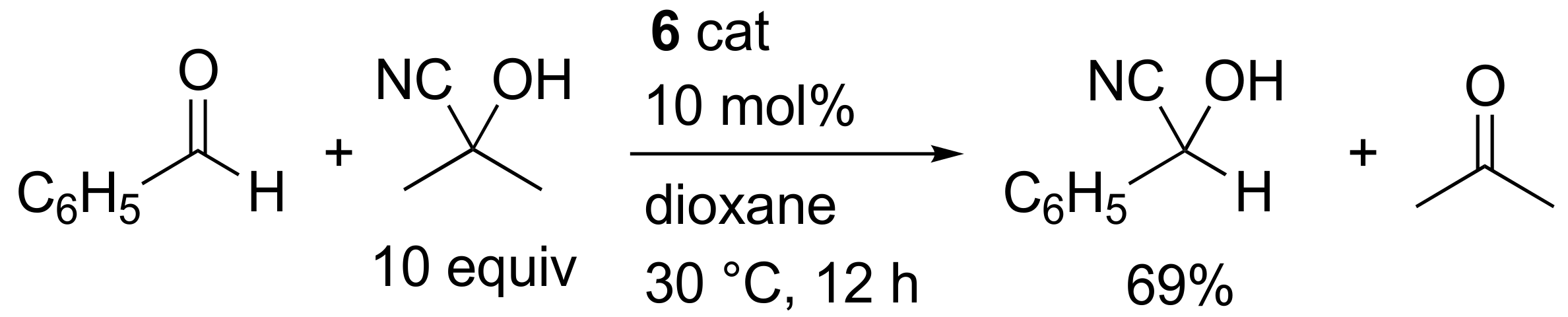
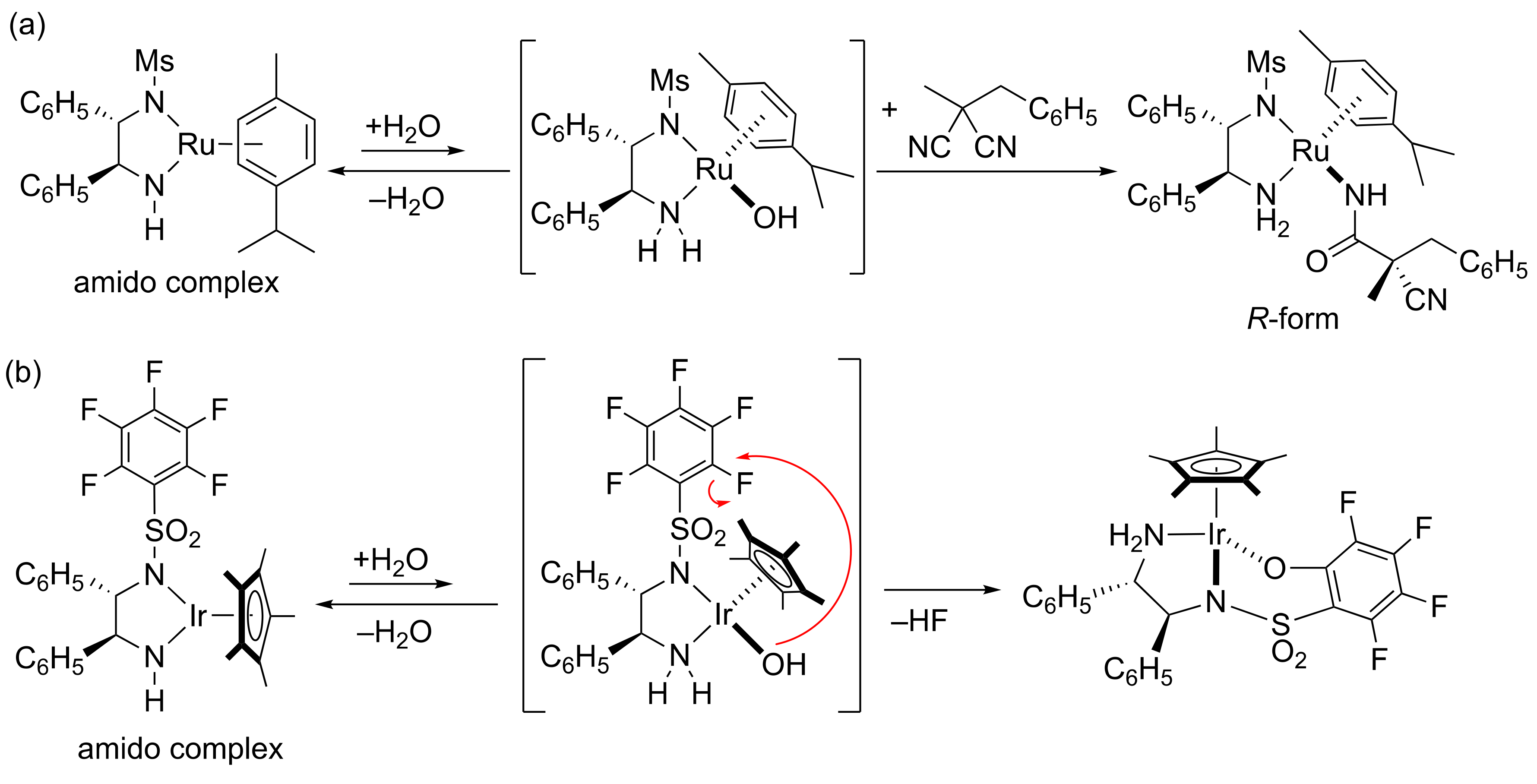
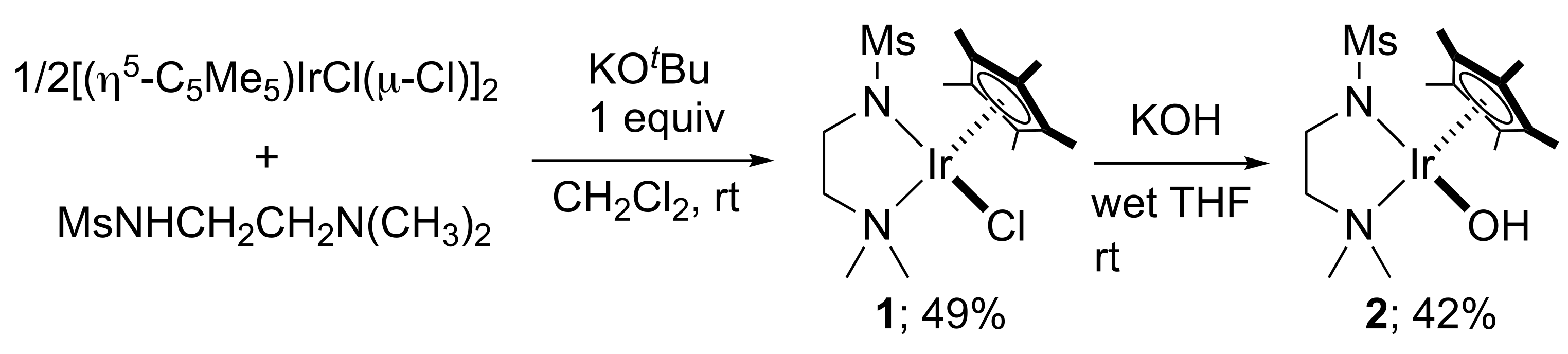
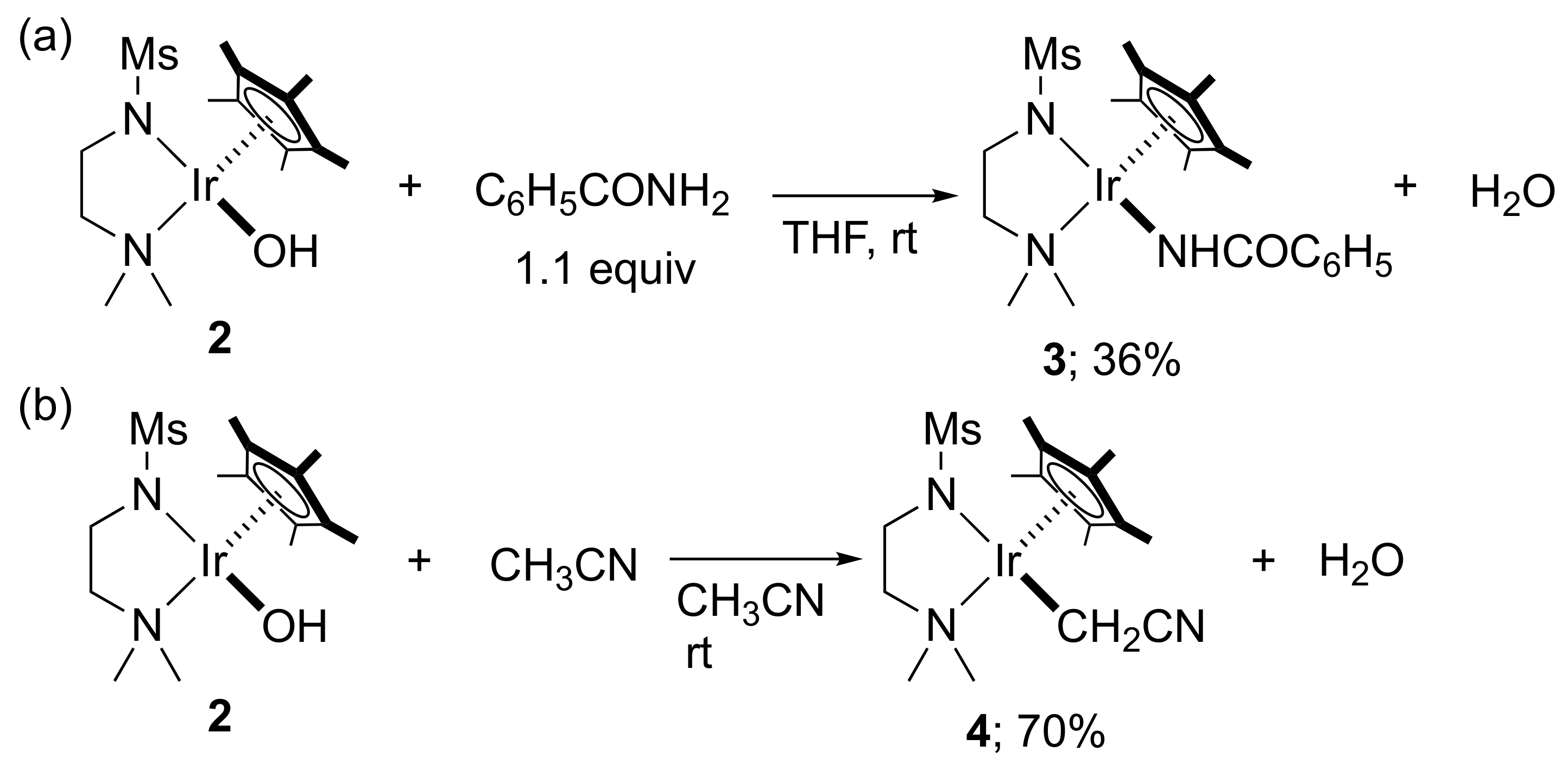
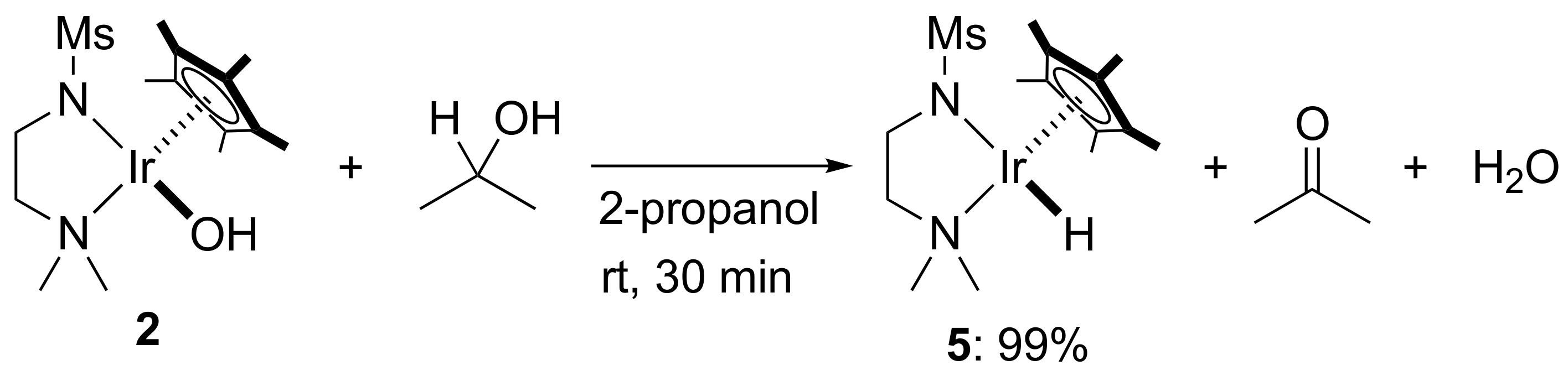
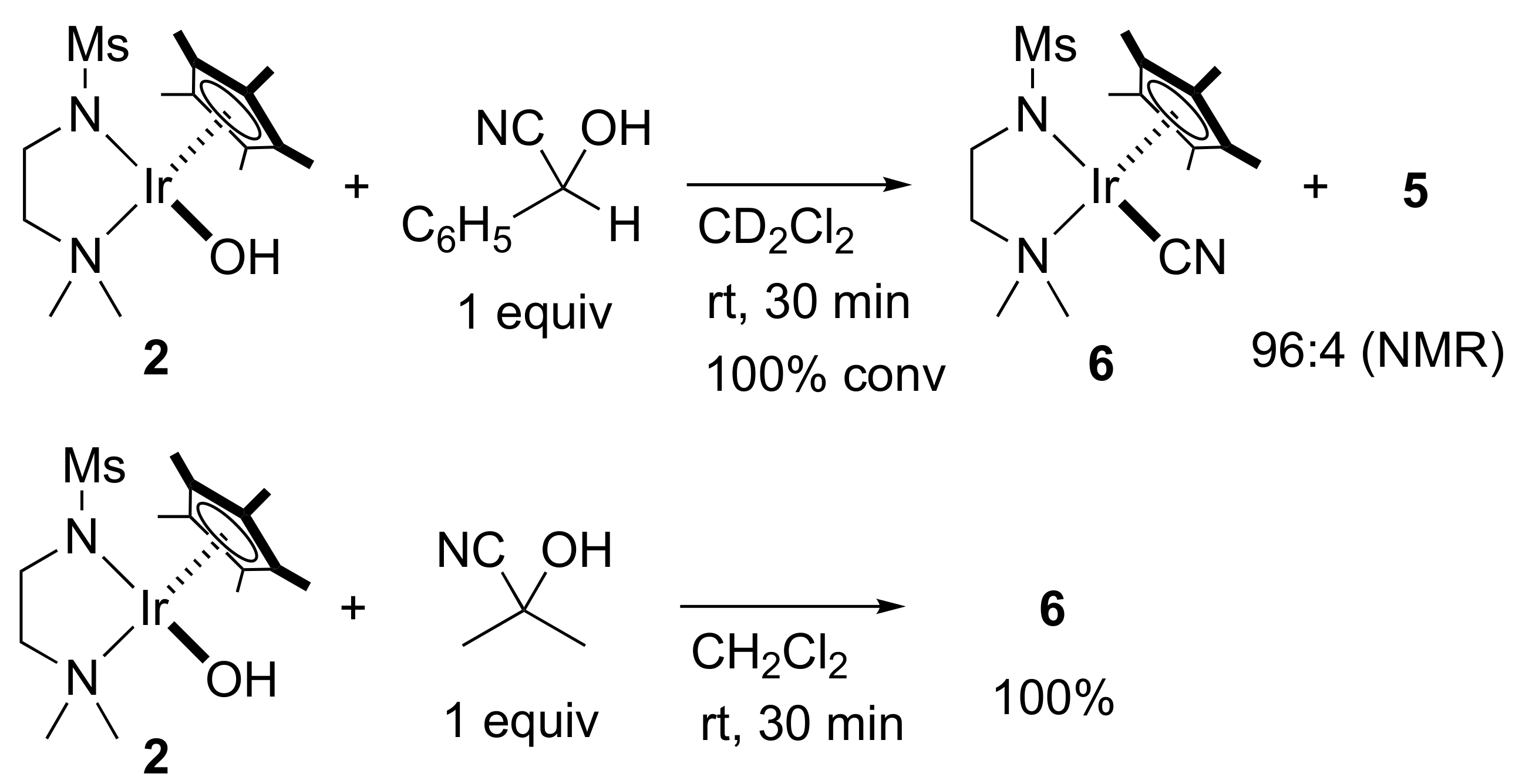
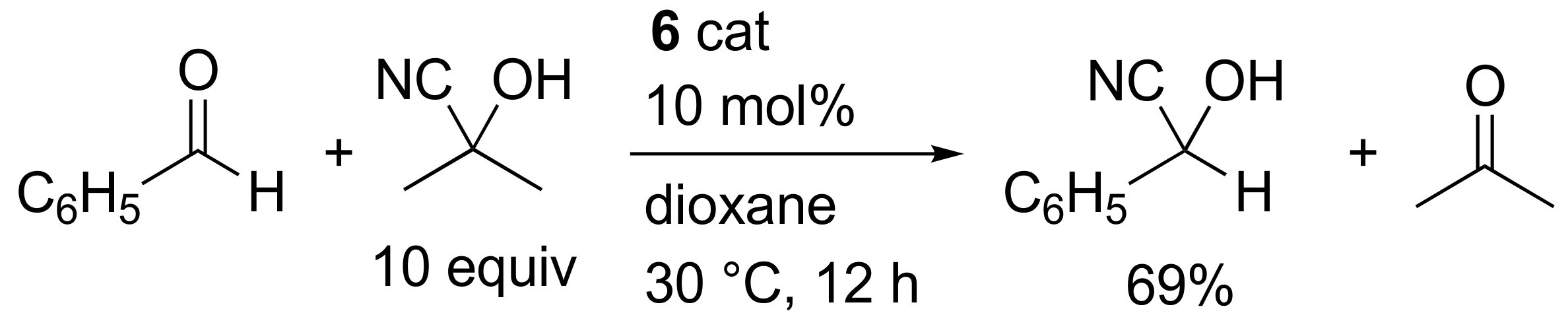
2. Results and Discussion
3. Materials and Methods
3.1. General Information
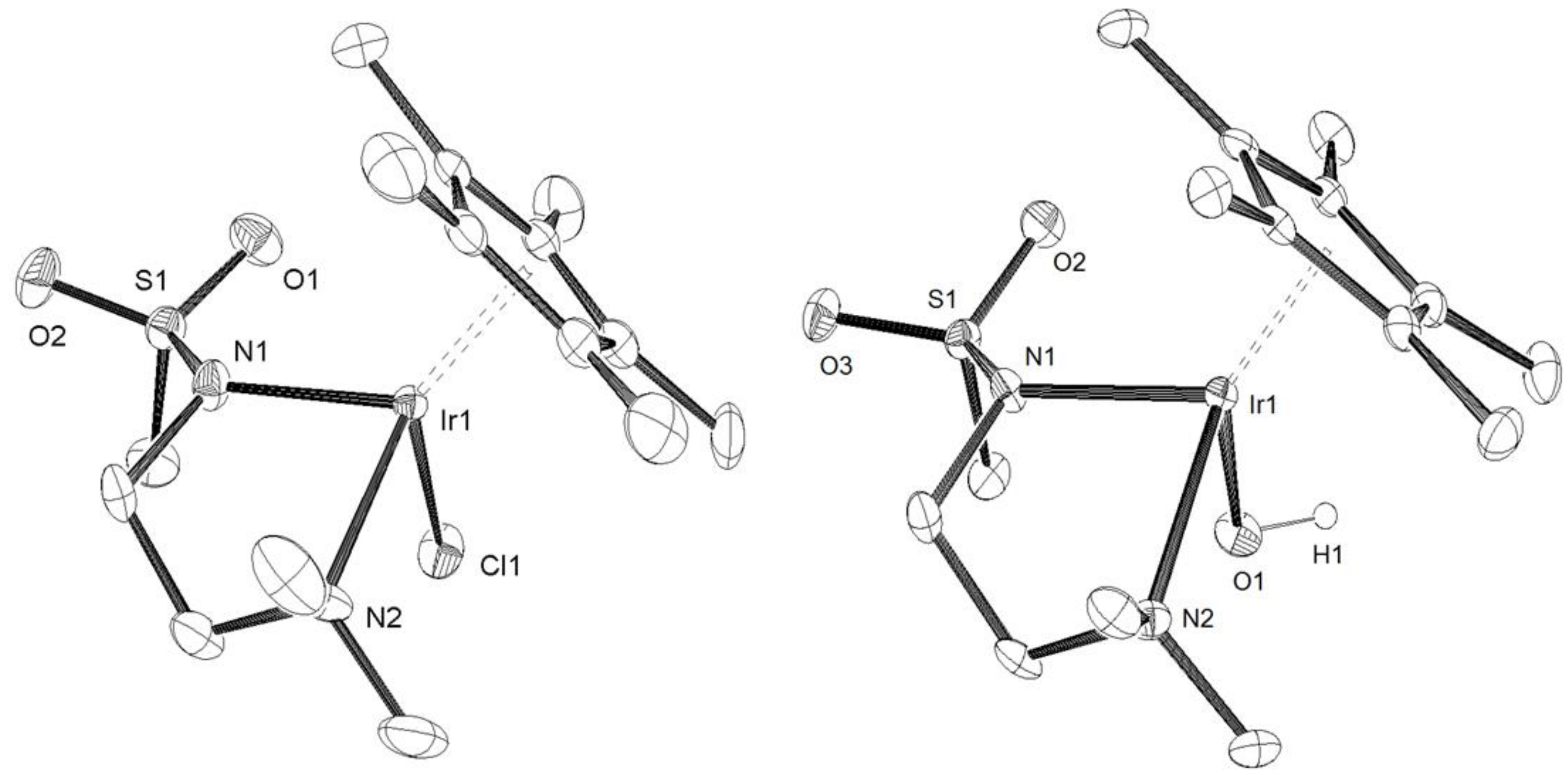
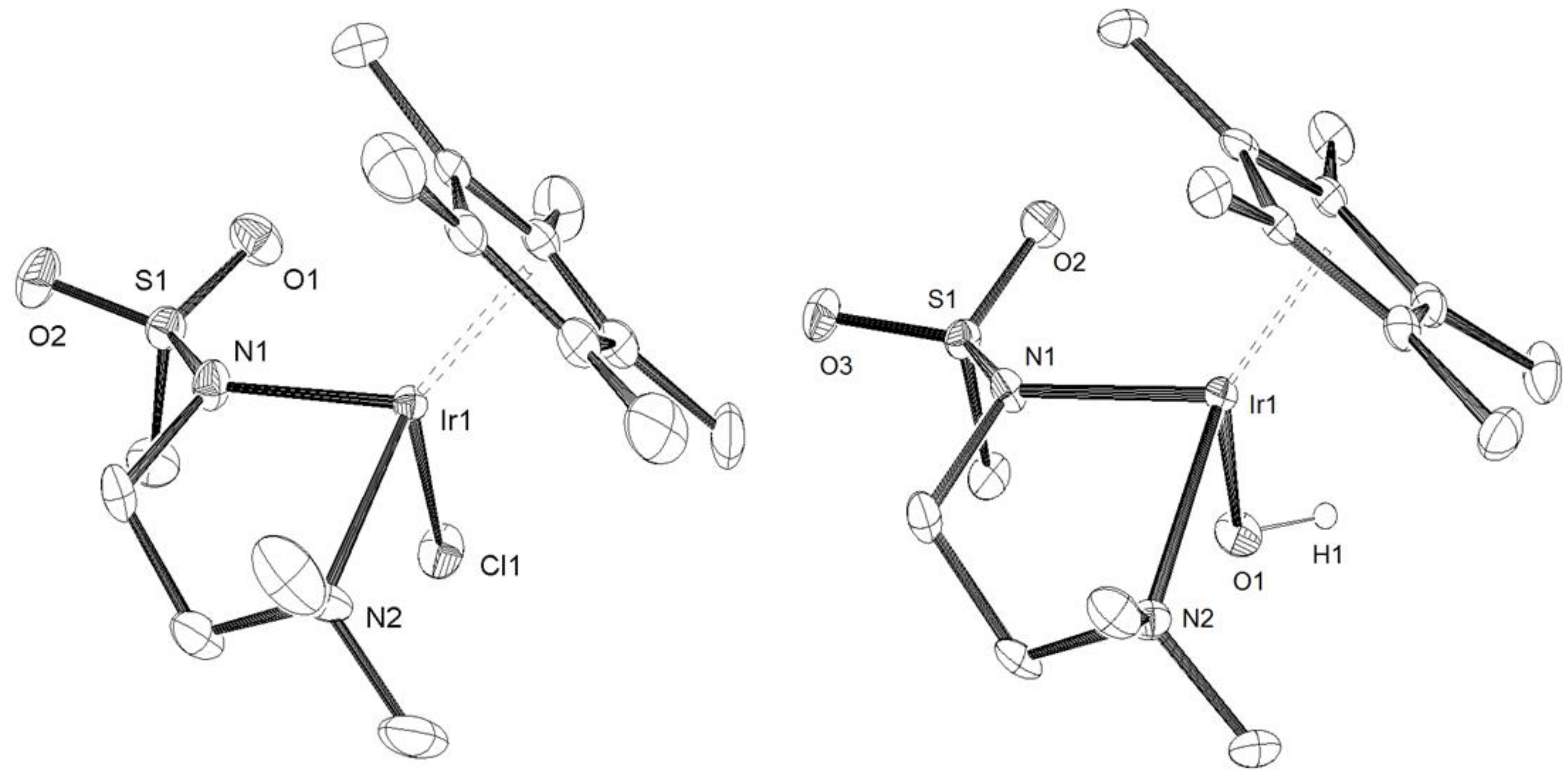
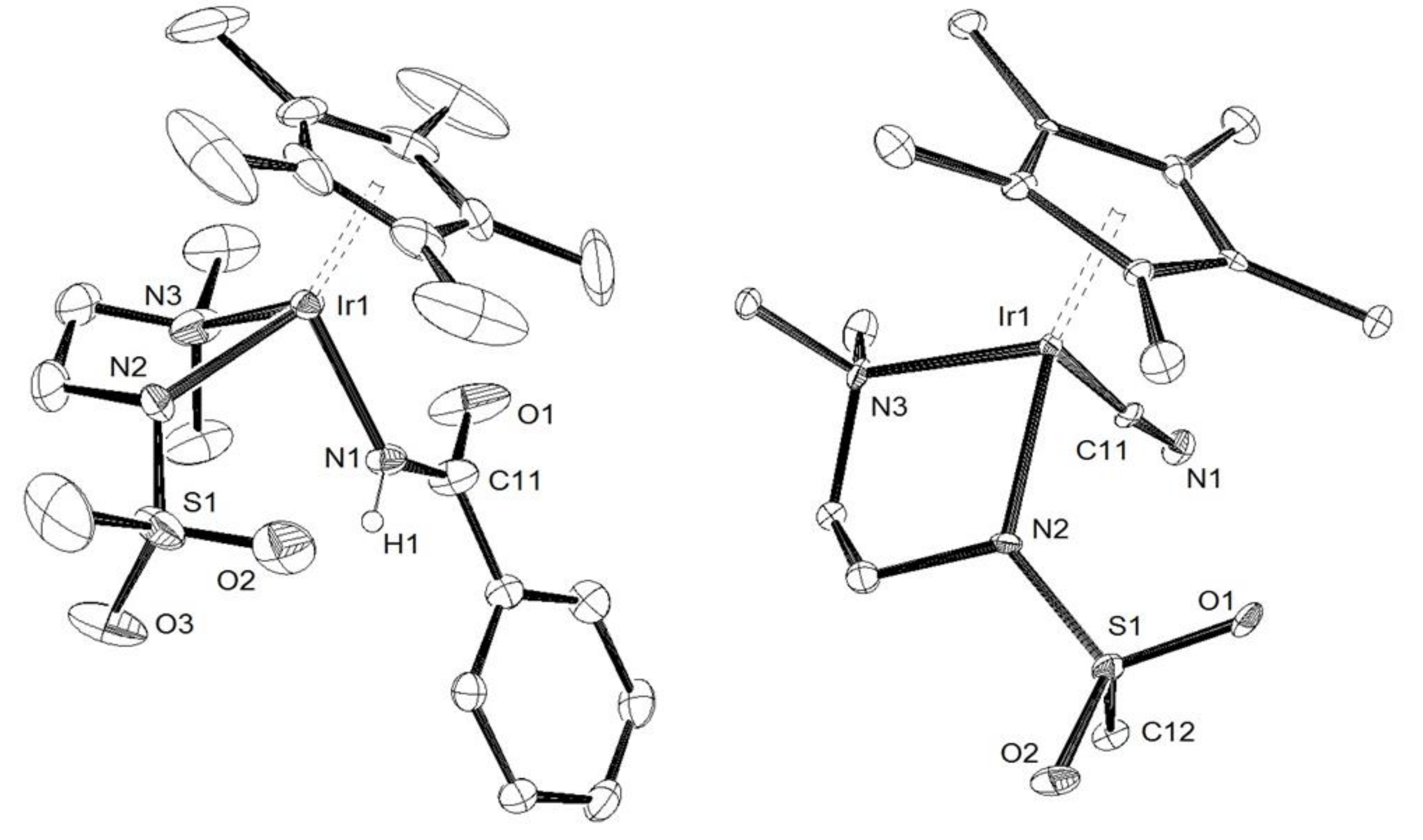
3.2. X-Ray Crystal Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gilje, J.W.; Roesky, H.W. Structurally characterized organometallic hydroxo complexes of the f- and d-block metals. Chem. Rev. 1994, 94, 895–910. [Google Scholar] [CrossRef]
- Roesky, H.W.; Singh, S.; Yusuff, K.K.M.; Maguire, J.A.; Hosmane, N.S. Organometallic hydroxides of the transition elements. Chem. Rev. 2006, 106, 3813–3843. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.J.; Nolan, S.P. Hydroxide complexes of the late transition metal: Organometallic chemistry and catalysis. Coord. Chem. Rev. 2017, 353, 278–294. [Google Scholar] [CrossRef]
- Dorta, R.; Rozenberg, H.; Shimon, L.J.W.; Milstein, D. Oxidative addition of water to novel Ir(I) complexes stabilized by dimethyl sulfoxide ligands. J. Am. Chem. Soc. 2002, 124, 188–189. [Google Scholar] [CrossRef] [PubMed]
- Ozerov, O.V. Oxidative addition of water to transition metal complexes. Chem Soc. Rev. 2009, 38, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Ikariya, T. Chemistry of concerto molecular catalysis based on the metal/NH bifunctionality. Bull. Chem. Soc. Jpn. 2011, 84, 1–16. [Google Scholar] [CrossRef]
- Kamezaki, S.; Akiyama, S.; Kayaki, Y.; Kuwata, S.; Ikariya, T. Asymmetric nitrile-hydration with bifunctional ruthenium catalysts bearing chiral N-sulfonyldiamine ligands. Tetrahedron Asymmetry 2010, 21, 1169–1172. [Google Scholar] [CrossRef]
- Dub, P.A.; Wang, H.; Matsunami, A.; Gridnev, I.D.; Kuwata, S.; Ikariya, T.C.-F. Bond breaking through aromatic nucleophilic substitution with a hydroxy ligand mediated via water bifunctional activation. Bull. Chem. Soc. Jpn. 2013, 86, 557–568. [Google Scholar] [CrossRef]
- Ritter, J.C.M.; Bergman, R.G. The mechanism of addition of an Ir–OH bond to ethylene. Catalytic tandem activation by two [η5-Cp*(Ph)IrPMe3]+ complex fragments. J. Am. Chem. Soc. 1997, 119, 2580–2581. [Google Scholar] [CrossRef]
- Hetterscheid, D.G.H.; Reek, J.N.H. Me2–NHC based robust Ir catalyst for efficient water oxidation. Chem. Commun. 2011, 47, 2712–2714. [Google Scholar] [CrossRef]
- Hintermair, U.; Hashmi, S.M.; Elimelech, M.; Crabtree, R.H. Particle formation during oxidation catalysis with Cp* iridium complexes. J. Am. Chem. Soc. 2012, 134, 9785–9795. [Google Scholar] [CrossRef] [PubMed]
- Hintermair, U.; Sheehan, S.W.; Parent, A.R.; Ess, D.H.; Richens, D.T.; Vaccaro, P.H.; Brudvig, G.W.; Crabtree, R.H. Precursor transformation during molecular oxidation catalysis with organometallic iridium complexes. J. Am. Chem. Soc. 2013, 135, 10837–10851. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Morales, O.; Hersbach, T.J.P.; Hetterscheid, D.G.H.; Reek, J.N.H.; Koper, M.T.M. Electrochemical ans dpectroelectrochemical characterization of an iridium-based molecular catalyst for water splitting: Turnover frequencies, stability, and electrolyte effects. J. Am. Chem. Soc. 2014, 136, 10432–10439. [Google Scholar] [CrossRef] [PubMed]
- Shirai, S.-Y.; Nara, H.; Kayaki, Y.; Ikariya, T. Remarkable positive effect of silver salts on asymmetric hydrogenation of acyclic imines with Cp*Ir complexes bearing chiral N-sulfonylated diamine ligands. Organometallics 2009, 28, 802–809. [Google Scholar] [CrossRef]
- Bordwell, F.G. Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res. 1988, 21, 456–463. [Google Scholar] [CrossRef]
- Chang, H.-C.; Chang, Y.-F.; Lin, S.-H.; Lin, T.-H.; Lee, W.-Z. Ambient stable cyanomethylcopper(III) complex: A strong Cu–Csp3 bond supported by a PS3-tripodal chelator. Inorg. Chem. 2019, 58, 22–26. [Google Scholar] [CrossRef]
- Maenaka, Y.; Suenobu, T.; Fukuzumi, S. Hydrogen evolution from aliphatic alcohols and 1,4-selective hydrogenation of NAD+ Catalyzed by a [C,N] and a [C,C] cyclometalated organoiridium complex at room temperature in water. J. Am. Chem. Soc. 2012, 134, 9417–9427. [Google Scholar] [CrossRef]
- Letko, C.S.; Heiden, Z.M.; Rauchfuss, T.B.; Wilson, S.R. Coordination chemistry of the soft chiral lewis acid [Cp*Ir(TsDPEN)]+. Inorg. Chem. 2011, 50, 5558–5566. [Google Scholar] [CrossRef]
- Merino, P. CN addition to C=O and C=N bonds. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P., Molander, G.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 1, pp. 697–750. [Google Scholar]
- Ye, F.; Chen, J.; Ritter, T. Rh-catalyzed anti-markovnikov hydrocyanation of terminal alkynes. J. Am. Chem. Soc. 2017, 139, 7184–7187. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Hayashi, H.; Miyaji, K.; Inoue, S. Asymmetric transcyanohydrination. Chem. Lett. 1986, 15, 931–934. [Google Scholar] [CrossRef]
- Mori, A.; Inoue, S. A novel rate enhancement in titanium and zirconium alkoxides mediated cyano group transfers by the addition of a salycylal type schiff base, dl-3-(2-Hydroxy-1-naphthylidene)-imino-ε-caprolactam. A neighboring Amide Effect. Chem. Lett. 1991, 20, 145–148. [Google Scholar] [CrossRef]
- Ohno, H.; Mori, A.; Inoue, S. Lanthanoid(III) alkoxides as novel catalysts for a rapid transhydrocyanation from acetone cyanohydrin to aldehydes and ketones. Chem. Lett. 1993, 22, 375–378. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Fujii, A.; Nakano, Y.; Sakaguchi, S.; Ishii, Y. Acetylcyanation of aldehydes with acetone cyanohydrin and isopropenyl acetate by Cp*2Sm(thf)2. J. Org. Chem. 1999, 64, 4214–4216. [Google Scholar] [CrossRef]
- Ooi, T.; Takaya, K.; Miura, T.; Maruoka, K. Zr(OBut)4 as a new promoter for the meerwein-ponndorf-verley alkynylation and cyanation of aldehydes. Synlett 2000, 2000, 69–70. [Google Scholar]
- Yanagisawa, A.; Matsumoto, T.; Kushihara, N.; Yoshida, K. Dibutyltin dimethoxide-catalyzed cyano transfer to aldehydes and imines. Adv. Synth. Catal. 2010, 352, 2918–2922. [Google Scholar] [CrossRef]
- Klempier, N.; Griengl, H.; Hayn, M. Aliphatic (S)-cyanohydrins by enzyme catalyzed synthesis. Tetrahedron Lett. 1993, 34, 4769–4772. [Google Scholar] [CrossRef]
- Nanda, S.; Kato, Y.; Asano, Y. A new (R)-hydroxynitrile lyase from Prunus mume: Asymmetric synthesis of cyanohydrins. Tetrahedron 2005, 61, 10908–10916. [Google Scholar] [CrossRef]
- Paravidino, M.; Sorgedrager, M.J.; Orru, R.V.A.; Hanefeld, U. Activity and enantioselectivity of the hydroxynitrile lyase MeHNL in dry organic solvents. Chem. Eur. J. 2010, 16, 7596–7604. [Google Scholar] [CrossRef]
- Ooi, T.; Takaya, K.; Miura, T.; Ichikawa, H.; Maruoka, K. Chiral zirconium alkoxides-mediated asymmetric meerwein-ponndorf-verley cyanation of aldehydes. Synlett 2000, 2000, 1133–1134. [Google Scholar] [CrossRef]
- Ooi, T.; Miura, T.; Takaya, K.; Ichikawa, H.; Maruoka, K. Zr(OBut)4 as an effective promoter for the meerwein-ponndorf-verley alkynylation and cyanation of aldehydes: Development of new asymmetric cyanohydrin synthesis. Tetrahedron 2001, 57, 867–873. [Google Scholar] [CrossRef]
- Watanabe, A.; Matsumoto, K.; Shimada, Y.; Katsuki, T. Oxovanadium(V)-catalyzed enantioselective meerwein-ponndorf-verley cyanation of aldehydes using acetone cyanohydrin. Tetrahedron Lett. 2004, 45, 6229–6233. [Google Scholar] [CrossRef]
- Takaki, J.; Egami, H.; Matsumoto, K.; Saito, B.; Katsuki, T. Vanadium-catalyzed asymmetric transcyanation of aliphatic aldehydes with acetone cyanohydrin. Chem. Lett. 2008, 37, 502–503. [Google Scholar] [CrossRef]
- Sakai, Y.; Mitote, J.; Matsumoto, K.; Katsuki, T. Room-Temperature synthesis of enantioenriched non-protected cyanohydrins using vanadium(salalen) catalyst. Chem. Commun. 2010, 46, 5787–5789. [Google Scholar] [CrossRef] [PubMed]
- CrystalStructure 4.1: Crystal Structure Analysis Package; Rigaku Coorporation: Tokyo, Japan, 2015.
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.; Polidori, G.; Camalli, M. SIR92—A program for automatic solution of crystal structures by direct methods. J. Appl. Cryst. 1994, 27, 435. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamezaki, S.; Kayaki, Y.; Kuwata, S.; Ikariya, T. Synthesis of a Half-Sandwich Hydroxidoiridium(III) Complex Bearing a Nonprotic N-Sulfonyldiamine Ligand and Its Transformations Triggered by the Brønsted Basicity. Inorganics 2019, 7, 125. https://doi.org/10.3390/inorganics7100125
Kamezaki S, Kayaki Y, Kuwata S, Ikariya T. Synthesis of a Half-Sandwich Hydroxidoiridium(III) Complex Bearing a Nonprotic N-Sulfonyldiamine Ligand and Its Transformations Triggered by the Brønsted Basicity. Inorganics. 2019; 7(10):125. https://doi.org/10.3390/inorganics7100125
Chicago/Turabian StyleKamezaki, Shoko, Yoshihito Kayaki, Shigeki Kuwata, and Takao Ikariya. 2019. "Synthesis of a Half-Sandwich Hydroxidoiridium(III) Complex Bearing a Nonprotic N-Sulfonyldiamine Ligand and Its Transformations Triggered by the Brønsted Basicity" Inorganics 7, no. 10: 125. https://doi.org/10.3390/inorganics7100125
APA StyleKamezaki, S., Kayaki, Y., Kuwata, S., & Ikariya, T. (2019). Synthesis of a Half-Sandwich Hydroxidoiridium(III) Complex Bearing a Nonprotic N-Sulfonyldiamine Ligand and Its Transformations Triggered by the Brønsted Basicity. Inorganics, 7(10), 125. https://doi.org/10.3390/inorganics7100125