Abstract
Cyclopentyl substituted silanetriol can be prepared and isolated. Its condensation yields the corresponding disiloxanetetrol as a primary condensation product. Further condensation leads to the hexameric polyhedral silsesquioxane cage T6. The latter has been mentioned in the literature before however, lacking structural data. All compounds have been characterized with multinuclear NMR spectroscopy and, in addition, the molecular structures have been determined in the case of the disiloxanetetrol and the hexasilsesquioxane via single crystal X-ray diffraction.
1. Introduction
Cage silsesquioxanes have attracted much attention in recent years [1,2] owing to their widespread applications for example in catalysis [3], as model systems for silica surfaces [4,5], in the design of superoleophobic surfaces [6], ionic liquids [7], biocompatible materials [8], as well as in polymer chemistry [2]. The synthetic approach towards such octasilsesquioxanes is mainly based on the hydrolytic condensation of trifunctional silanes RSiX3, where R is a stable organic substituent and X a reactive moiety (i.e., X = Cl, OMe etc.) [2,9] and catalysts like tetrabutylammonium fluoride (TBAF) have been shown to improve the yields in the presence of certain organic substituent [10,11,12]. Recently, it has been demonstrated that silanetriols are suitable starting materials for cage silsesquioxanes, giving access to T8 cages in a one-pot synthesis which could not be obtained from the corresponding alkoxysilanes via other routes [13,14]. Here we report our investigation to prepare a cyclopentyl substituted silanetriol and its condensation to the T6 cage via the corresponding tetrahydroxydisiloxane.
2. Results and Discussion
Starting from commercially available cyclopentyltrichlorosilane, the corresponding silanetriol was prepared by careful hydrolysis in ether solution at 0 °C in the presence of three equivalents of aniline in analogy to an established procedure by Takiguchi [15]. Silanetriol 1 has been be isolated from the etheral solution as colorless powder in above 80% yield. The 29Si-NMR resonance of the product was observed at −37.7 ppm in D2O which compares well with the 29Si chemical shifts of related alkylsilanetriols such as tert-butylSi(OH)3 (−36.8 ppm, D2O). While cyclopentyl substituted silanetriol 1 was stable as a solid, it slowly underwent condensation in polar solvents such as THF or DMSO (Scheme 1). The resulting tetrahydroxydisiloxane 2 has been identified as primary condensation product and was characterized by NMR and IR spectroscopy, mass spectrometry and single crystal X-ray diffraction. The 29Si-NMR chemical shift at −51.8 ppm in THF solution is in good agreement with the known chemical shifts of other alkylsubstituted disiloxane tetrols [14,16], resonating at slightly lower field compared with aryl substituted disiloxane tetrols [17,18].
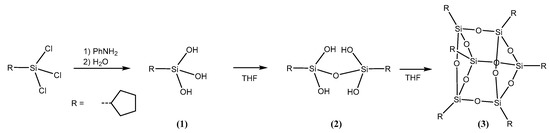
Scheme 1.
Formation of 3 via 1 and 2 starting from cyclopentyl trichlorosilane.
Compound 2 could be obtained as single crystals suitable for X-ray diffraction as two polymorphs, one crystallizing in a monoclinic, the other in an orthorhombic crystal system both confirming the constitution of 1,3-di-cyclopentyl-1,1,3,3-tetrahydroxysiloxane. In the monoclinic crystal of 2, the molecules are lying with O1 on an inversion center resulting in an Si–O–Si angle of 180°. The cyclopentyl rings are disordered over two sites (Figure 1). Each of the four OH groups of the tetrahydroxysiloxanes are involved in one donor and in one acceptor hydrogen bond [O2···O3′ 2.6811(19) Å, O2–H2···O3′ 174.8(9)°; O3···O2″ 2.6718(19) Å, O3–H3···O2″ 177.3(14)°], resp., forming two-dimensional aggregates, in which each molecule is connected to six neighbors showing a two-dimensional closest packing. This planar aggregate is shielded on both sides by the cyclopentyl groups. The molecules show pseudo-mirror planes normal to the c axis; the transformation to orthorhombic symmetry would lead to an angle differing by 0.268(4)° from 90°.
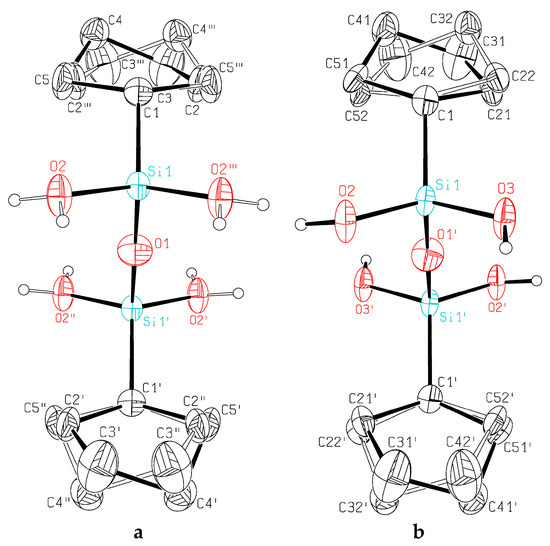
Figure 1.
This ORTEP plot of the molecular structure of 2 from the monoclinic (b) and the orthorhombic (a) polymorph showing the atomic numbering scheme. The probability ellipsoids are drawn at the 50% probability level. The cyclopentyl rings are disordered over two sites. The H atoms bonded to oxygen are drawn with arbitrary radii, the H atoms of the cyclopentyl rings were omitted for the sake of clarity. Red: oxygen; blue: silicon.
In the orthorhombic phase the molecules of 2 adopt C2h (= 2/m) symmetry resulting in a Si–O–Si angle of 180°. Again, the cyclopentyl rings are disordered over two sites (Figure 1). The main difference between the two phases is the fact that the H atoms of the OH groups are ordered in the monoclinic phase but disordered in the orthorhombic phase. Equivalence of the two OH groups bonded to a Si atom and therefore a higher effective symmetry is reached by this disorder in orthorhombic 2. All four OH groups in orthorhombic 2 are equivalent by symmetry and are involved in two hydrogen bonds [O2···O2′ 2.670(2)Å, O2–H3···O2′ 169(2)°; O2···O2″ 2.678(2)Å, O2–H2···O2″ 172.5(19)°] forming two-dimensional aggregates, in which each molecule is connected to six neighbors, showing a two-dimensional closest packing almost identical to monoclinic 2 (Figure 2). In addition, the supramolecular hydrogen bonding the bond distances and angles of the central RSi(OH)2O– units are in the typical range of such disiloxanetetrols [14,16,17,18,19,20].
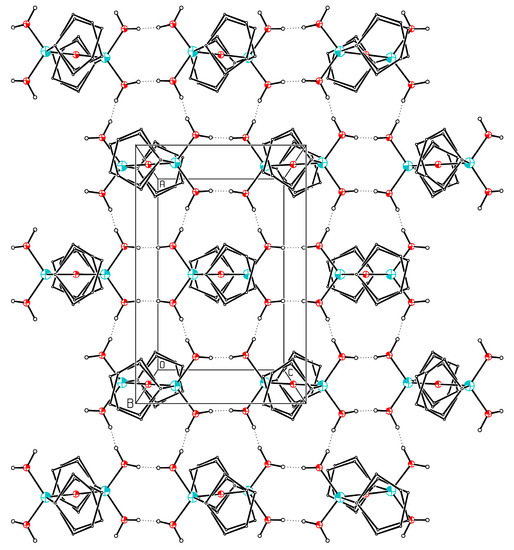
Figure 2.
ORTEP plot of the orthorhombic packing of 2. The atoms are drawn with arbitrary radii, the hydrogen bonds are plotted with dashed lines. Red: oxygen; blue: silicon.
Prolonged condensation of 1 in THF over five months resulted in a mixture containing mainly disiloxane 2 and the corresponding hexasilsesquioxane 3 in a 2:1 ratio. Recrystallization of this mixture in DMSO furnished crystalline 3, which has been identified via spectroscopic methods and single crystal X-ray diffraction. Compound 3 has been already described in the literature together with its spectroscopic data [21]. The 29Si-chemical shift of T6 cage 3 at −56.3 ppm in DMSO solution observed by us is very similar to the previously reported one (−54.4, CDCl3) and fits well in the range observed for alkyl substituted T6 cages [16,21] but resonates at a lower field than the comparable T8-cages [10,12,14,16,22]. Compound 3 could be obtained as single crystals suitable for X-ray diffraction and crystallizes in the orthorhombic space group Ccce. In the crystal structure analysis of 3, the molecules of hexa(cyclopentylsilsesquioxane) are located with one O atom (O5) on a two-fold rotation axis parallel to the crystallographic a-axis (Figure 3).
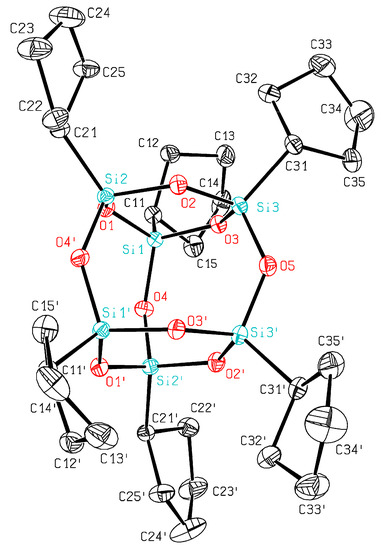
Figure 3.
ORTEP plot of 3 showing the atomic numbering scheme. The probability ellipsoids are drawn at the 50% probability level. The hydrogen atoms were omitted for clarity reasons. Selected bond lengths [Å] and angles [°]: Si1–O4 1.6269(12), Si1–O1 1.6390(11), Si1–O3 1.6430(11), Si1–C11 1.8369(15), Si2–O4′ 1.6274(11), Si2–O1 1.6390(11), Si2–O2 1.6417(12), Si2–C21 1.8385(16), Si3–O5 1.6284(7), Si3–O2 1.6367(11), Si3–O3 1.6400(11), Si3–C31 1.8375(16); Si1–O1–Si2 128.69(7), Si3–O2–Si2 131.35(7), Si3–O3–Si1 131.11(7), Si1–O4–Si2′ 139.84(7), Si3–O5–Si3′ 132.34(10).
The molecules are packed in layers normal to the a-axis, leading to mechanically very soft crystals. The central highly-symmetric Si6O9 tetracycle shows slight but significant deviations from D3h symmetry (e.g., Si1–O4–Si2′ 139.84(7)° vs Si3–O5–Si3′ 132.34(10)°). The variation of the Si–O bond lengths covers a narrow range between 1.627 Å and 1.643 Å which is smaller than in the other structure reports where variations as much as 0.04 to 0.28 Å are reported. Until now, only a few structure determinations of hexa(alkyl/aryl)silsesquioxanes with this Si6O9 cage can be found in the literature including tert-butyl [16], cyclohexyl [23], 1,1,2-trimethylpropyl [24], 2,4,6-triisopropylphenyl [25], trimethoxysilyl [26], and isopropyl [27] substituted hexasilsesquioxanes. For these, the mean value of the Si–O–Si angles in the six-membered rings is 130.2(4)°, the mean value of the other Si–O–Si angles is 140.0(8)° (min. 136.5°). Moreover the topic has been reviewed not long ago and also a structure determination of a T6 cage has been performed in the gas phase [28,29]. The unit cell contains eight equivalent isolated molecules of 3. Relevant geometric parameters of 3 are listed in the caption of Figure 3 and crystallographic details are summarized in Table 1.

Table 1.
Crystal data and structure refinement for 2 and 3.
3. Experimental Details
All manipulations were carried out under inert argon atmosphere using standard Schlenk technique. All solvents were dried and freshly distilled over Na/K-alloy where applicable. Cyclopentyltrichlorosilane has been purchased and used without further purification. 1H– and 13C–NMR-data have been recorded on a Bruker Avance III (Billerica, MA, USA) 300 MHz spectrometer (operating at 300 MHz, 75.4 MHz) or a Varian MR-400 MHz spectrometer (operating at 400 MHz, 100.5 MHz). 29Si-NMR-data have been recorded on a Bruker Avance III 300 MHz spectrometer (operating at 59.6 MHz). All measurements have been performed at room temperature using TMS as external standard. EI-mass spectra have been recorded on an Agilent Technologies 5975C (Santa Clara, CA, USA) inert XL MSD with SIS Direct Insertion Probe. IR-spectra have been recorded using a Perkin-Elmer 1725X FT/IR (Waltham, MA, USA) spectrometer using KBr plates.
3.1. Synthesis of cyclopentylsilanetriol 1
Cyclopentyltrichlorosilane (3.43 g, 16.9 mmol), dissolved in 13 mL diethylether, was added dropwise to a solution of water (0.91 g, 50.6 mmol) and aniline (4.78 g, 51.4 mmol) in 150 mL of diethylether at 0 °C while stirring. A white precipitate is formed in the reaction mixture and stirring is continued for 2 h upon completed addition while slowly warming to room temperature. The precipitate is filtered off with a fritted funnel and discarded. The solvent of the remaining solution is removed in vacuo. Yield 2.02 g (13.7 mmol, 81%). 1H-NMR (300 MHz, D2O): δ(ppm): 1.01, 1.48, 1.59, 1.79; 29Si-NMR (59.6 MHz, D2O): δ(ppm): −37.7.
3.2. Synthesis of the 1,3-Dicyclopentyldisiloxane-1,1,3,3-tetrol 2
Silanetriol 1 (0.5 g) was dissolved in 20 mL THF at room temperature. Slow evaporation of the volatiles took four weeks. The raw material was extracted with pentane which upon evaporation of the solvent yielded compound 2 as colorless crystalline solid (0.3 g, 1.1 mmol, 64%). 1H-NMR (300 MHz, THF-d8): δ(ppm): 0.83, 1.10, 1.40–1.59, 1.78; 13C-NMR (75.4 MHz, THF-d8): δ(ppm): 25.4, 27.9, 28.7; 29Si-NMR (59.6 MHz, THF-d8): δ(ppm): −51.8; IR: 3251 (OH), 1104 (Si-O-Si); MS/EI (70 eV): m/z (%) = 209 (100) [M-cyc]+, 141 (72) [M-cyc2]+.
3.3. Synthesis of Hexa(cyclopentylsilsesquioxane) 3
2.5 g (16.9 mmol) of silanetriol 1 are dissolved in THF (100 mL) and are stored at room temperature for five months. The solvent is removed and the resulting solid (2.1 g) contains compounds 2 and 3 in a 2:1 ratio. Extraction with DMSO yields 3 as colorless crystalline material (1.1 g, 1.5 mmol, 53%). 1H-NMR (250 MHz, DMSO-d6): δ(ppm): 0.80–1.00 (br), 1.42–1.60 (br), 1.70; 13C-NMR (75.4 MHz, DMSO-d6): δ(ppm): 23.3, 26.38, 27.04; 29Si-NMR (59.6 MHz, DMSO-d6): δ(ppm): −56.3. MS/EI (70 eV): m/z (%) = 726.3 (1) [M]+, 67 (100) [cyc]+.
3.4. X-ray Crystallography
X-ray diffraction measurements were performed on a BRUKER-AXS SMART APEX 2 CCD diffractometer using graphite-monochromatized Mo-Kα radiation. Supplementary crystallographic data for this paper can be obtained free of charge quoting CCDC 1575839–1575841 from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
The structures were solved by direct methods (SHELXS-97)2 and refined by full-matrix least-squares techniques against F2 (SHELXL-97)2. The cyclopentyl groups in 2 are disordered over two orientations and were refined with site occupation factors of 0.5. In 2m the equivalent bonds in these groups were restrained to have the same lengths. In 2o the same anisotropic displacement parameters were used for atoms C2 and C5. The other non-hydrogen atoms were refined with anisotropic displacement parameters. The positions of the H atoms of the OH groups were taken from a difference Fourier map, the O–H distances were fixed to 0.84 Å, and the H atoms were refined with common isotropic displacement parameters without any constraints to the bond angles. The site occupation factors of the disordered H atoms of the OH groups in 2m were fixed to 0.5. The H atoms of the tertiary C–H groups were refined with individual isotropic displacement parameters and all X–C–H angles equal at C–H distances of 1.00 Å. The H atoms of the CH2 groups were refined with common isotropic displacement parameters for the H atoms of the equivalent CH2 groups and idealized geometries with approximately tetrahedral angles and C–H distances of 0.99 Å.
4. Conclusions
In summary, we have shown that cyclopentyl substituted silanetriol can be prepared and isolated. In polar solvents, spontaneous condensation occurs which yields the corresponding disiloxanetetrol as a primary condensation product. Further condensation leads to the hexameric polyhedral silsesquioxane cage T6. The latter has been mentioned in the literature before. However, it lacked structural data. All compounds have been characterized with multinuclear NMR spectroscopy and in addition the molecular structures have been determined in the case of the disiloxanetetrol and the hexasilsesquioxane via single crystal X-ray diffraction. Our results show that silanetriols bearing secondary alkyl substituents may be suitable precursors for the synthesis of POSS cages as well.
Supplementary Materials
The following are available online at www.mdpi.com/2304-6740/5/4/66/s1: Cif and cif-checked files..
Acknowledgments
The authors would like to thank the EU-COST network CM1302 “Smart Inorganic Polymers” (SIPs).
Author Contributions
Jürgen Kahr performed the experiments and analyzed the spectroscopic data; Ferdinand Belaj performed the X-ray diffraction and interpretation; Rudolf Pietschnig provided the materials and wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hartmann-Thompson, C. Advances in Silicon Science; Springer: Heidelberg, Germany, 2011; Volume 3. [Google Scholar]
- Cordes, D.B.; Lickiss, P.D.; Rataboul, F. Recent developments in the chemistry of cubic polyhedral oligosilsesquioxanes. Chem. Rev. 2010, 110, 2081–2173. [Google Scholar] [CrossRef] [PubMed]
- Janssen, M.; Wilting, J.; Müller, C.; Vogt, D. Continuous rhodium-catalyzed hydroformylation of 1-octene with polyhedral oligomeric silsesquioxanes (POSS) enlarged triphenylphosphine. Angew. Chem. (Int. Ed.) 2010, 49, 7738–7741. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, U.; Hendan, B.J.; Floerke, U.; Marsmann, H.C. Functionalized octa(propylsilsesquioxanes)(3-xC3H6)8(Si8O12)—Model compounds for surface modified silica gel. J. Organomet. Chem. 1995, 489, 185–194. [Google Scholar] [CrossRef]
- Feher, F.J.; Newman, D.A.; Walzer, J.F. Silsesquioxanes as models for silica surfaces. J. Am. Chem. Soc. 1989, 111, 1741–1748. [Google Scholar] [CrossRef]
- Tuteja, A.; Choi, W.; Ma, M.; Mabry, J.M.; Mazzella, S.A.; Rutledge, G.C.; McKinley, G.H.; Cohen, R.E. Designing superoleophobic surfaces. Science 2007, 318, 1618–1622. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Ishiguro, F.; Chujo, Y. POSS ionic liquid. J. Am. Chem. Soc. 2010, 132, 17649–17651. [Google Scholar] [CrossRef] [PubMed]
- Feher, F.J.; Wyndham, K.D.; Scialdone, M.A. Octafunctionalized polyhedral oligosilsesquioxanes as scaffolds: Synthesis of peptidyl silsesquioxanes. Chem. Commun. 1998, 14, 1469–1470. [Google Scholar] [CrossRef]
- Matisons, J. Applications of Polyhedral Oligomeric Silsesquioxanes; Springer: Dordrecht, The Netherlands; Heidelberg, Germany; London, UK; New York, NY, USA, 2011. [Google Scholar]
- Bassindale, A.R.; Liu, Z.; MacKinnon, I.A.; Taylor, P.G.; Yang, Y.; Light, M.E.; Horton, P.N.; Hursthouse, M.B. A higher yielding route for T8 silsesquioxane cages and X-ray crystal structures of some novel spherosilicates. Dalton Trans. 2003, 14, 2945–2949. [Google Scholar] [CrossRef]
- Bassindale, A.R.; Pourny, M.; Taylor, P.G.; Hursthouse, M.B.; Light, M.E. Fluoride-ion encapsulation within a silsesquioxane cage. Angew. Chem. Int. Ed. 2003, 42, 3488–3490. [Google Scholar] [CrossRef] [PubMed]
- Bassindale, A.R.; Chen, H.; Liu, Z.; MacKinnon, I.A.; Parker, D.J.; Taylor, P.G.; Yang, Y.; Light, M.E.; Horton, P.N.; Hursthouse, M.B. A higher yielding route to octasilsesquioxane cages using tetrabutylammonium fluoride, Part 2: Further synthetic advances, mechanistic investigations and X-ray crystal structure studies into the factors that determine cage geometry in the solid state. J. Organomet. Chem. 2004, 689, 3287–3300. [Google Scholar] [CrossRef]
- Pietschnig, R.; Spirk, S. The chemistry of organo silanetriols. Coord. Chem. Rev. 2016, 323, 87–106. [Google Scholar] [CrossRef]
- Hurkes, N.; Bruhn, C.; Belaj, F.; Pietschnig, R. Silanetriols as powerful starting materials for selective condensation to bulky POSS cages. Organometallics 2014, 33, 7299–7306. [Google Scholar] [CrossRef] [PubMed]
- Takiguchi, T. Preparation of some organosilanediols and phenylsilanetriol by direct hydrolysis using aniline as hydrogen chloride acceptor. J. Am. Chem. Soc. 1959, 81, 2359–2361. [Google Scholar] [CrossRef]
- Spirk, S.; Nieger, M.; Belaj, F.; Pietschnig, R. Formation and hydrogen bonding of a novel POSS-trisilanol. Dalton Trans. 2009, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Čas, D.; Hurkes, N.; Spirk, S.; Belaj, F.; Bruhn, C.; Rechberger, G.N.; Pietschnig, R. Dimer formation upon deprotonation: Synthesis and structure of a m-terphenyl substituted (R,S)-dilithium disiloxanolate disilanol. Dalton Trans. 2015, 44, 12818–12823. [Google Scholar] [CrossRef] [PubMed]
- Hurkes, N.; Spirk, S.; Belaj, F.; Pietschnig, R. At the edge of stability—Preparation of methyl-substituted arylsilanetriols and investigation of their condensation behavior. Z. Anorg. Allg. Chem. 2013, 639, 2631–2636. [Google Scholar] [CrossRef]
- Suyama, K.-I.; Nakatsuka, T.; Gunji, T.; Abe, Y. Synthesis and crystal structure of disiloxane-1,3-diols and disiloxane-1,1,3,3-tetraol. J. Organomet. Chem. 2007, 692, 2028–2035. [Google Scholar] [CrossRef]
- Lickiss, P.D.; Litster, S.A.; Redhouse, A.D.; Wisener, C.J. Isolation of a tetrahydroxydisiloxane formed during hydrolysis of an alkyltrichlorosilane: Crystal and molecular structure of [But(OH)2Si]2O. J. Chem. Soc. Chem. Commun. 1991, 3, 173–174. [Google Scholar] [CrossRef]
- Bassindale, A.R.; MacKinnon, I.A.; Maesano, M.G.; Taylor, P.G. The preparation of hexasilsesquioxane (T6) cages by “non aqueous” hydrolysis of trichlorosilanes. Chem. Commun. 2003, 12, 1382–1383. [Google Scholar] [CrossRef]
- Unno, M.; Alias, S.B.; Arai, M.; Takada, K.; Tanaka, R.; Matsumoto, H. Synthesis and characterization of cage and bicyclic silsesquioxanes via dehydration of silanols. Appl. Organometal. Chem. 1999, 13, 303–310. [Google Scholar] [CrossRef]
- Behbehani, T.; Brisdon, H.B.J.; Mahon, M.F.; Molloy, K.C. The structure of hexa(cyclohexylsesquisiloxane), (C6H11)6Si6O9. J. Organomet. Chem. 1994, 469, 19–24. [Google Scholar] [CrossRef]
- Unno, M.; Alias, S.B.; Saito, H.; Matsumoto, H. Synthesis of hexasilsesquioxanes bearing bulky substituents: Hexakis((1,1,2-trimethylpropyl)silsesquioxane) and hexakis(tert-butylsilsesquioxane). Organometallics 1996, 15, 2413–2414. [Google Scholar] [CrossRef]
- Unno, M.; Imai, Y.; Matsumoto, H. Hexakis(2,4,6-triisopropylphenylsilsesquioxane). Silicon Chem. 2003, 2, 175–178. [Google Scholar] [CrossRef]
- Hoebbel, D.; Engelhardt, G.; Samoson, A.; Újszászy, K.; Smolin, Y.I. Preparation and constitution of the crystalline silicic acid trimethylsilyl ester [(CH3)3Si]6Si6O15. Z. Anorg. Allg. Chem. 1987, 552, 236–240. [Google Scholar] [CrossRef]
- Unno, M.; Suto, A.; Takada, K.; Matsumoto, H. Synthesis of Ladder and Cage Silsesquioxanes from 1,2,3,4-Tetrahydroxycyclotetrasiloxane. Bull. Chem. Soc. Jpn. 2000, 73, 215–220. [Google Scholar] [CrossRef]
- Lickiss, P.D.; Rataboul, F. Fully condensed polyhedral oligosilsesquioxanes (POSS): From synthesis to application. In Advances in Organometallic Chemistry; Academic Press: Oxford, UK, 2008; Volume 57, pp. 1–116. [Google Scholar]
- Wann, D.A.; Reilly, A.M.; Rataboul, F.; Lickiss, P.D.; Rankin, D.W.H. The gas-phase structure of the hexasilsesquioxane Si6O9(OSiMe3)6. Z. Naturforsch. B 2009, 64, 1269–1275. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).