
Naphthyl-Containing Organophosphonate Derivatives of Keggin-Type Polyoxotungstates


 , and
, and 
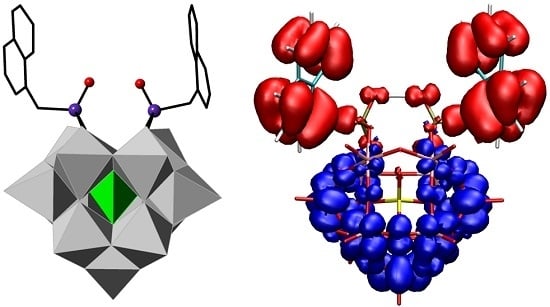
Abstract
:
1. Introduction
2. Results and Discussion
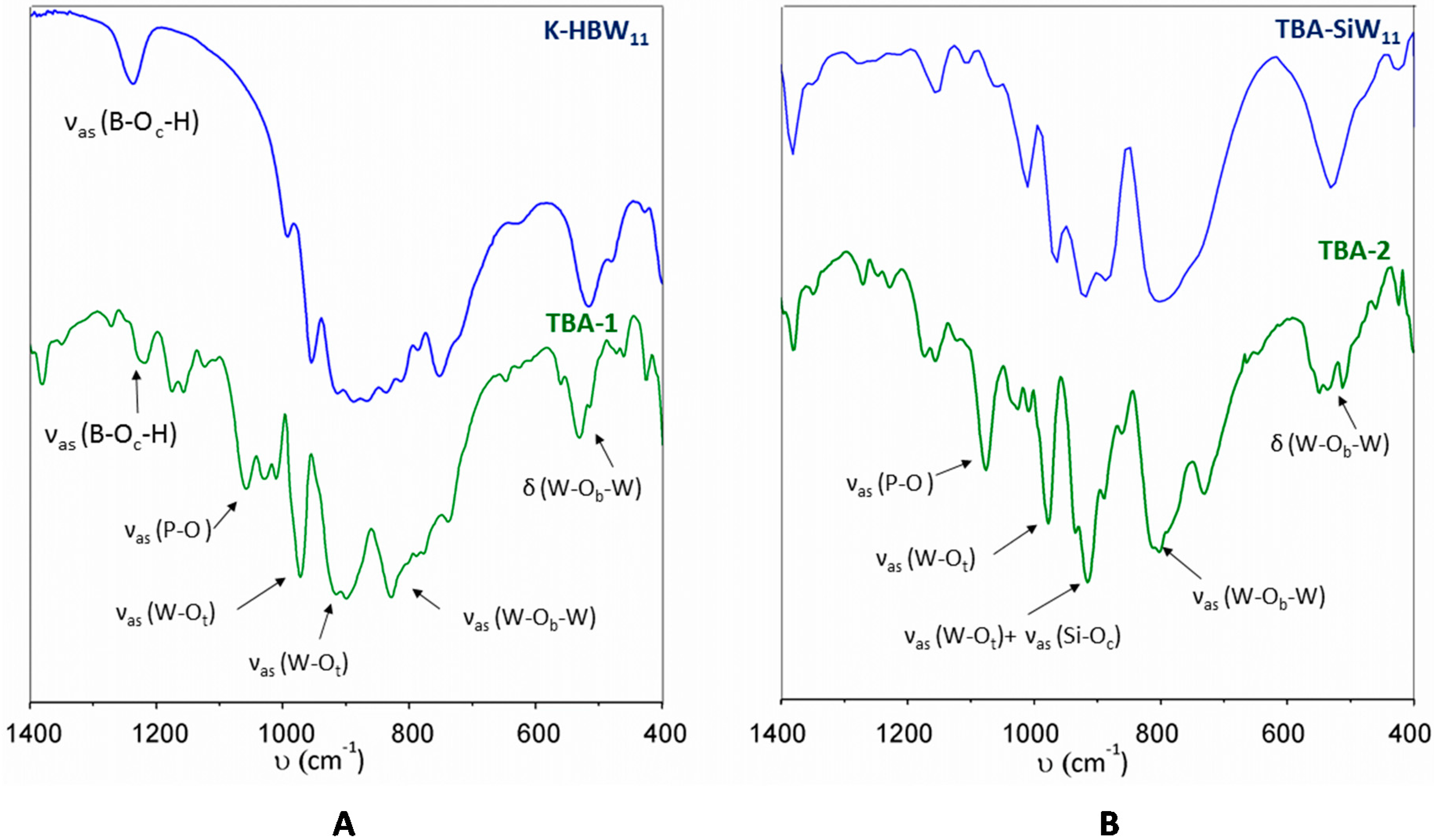
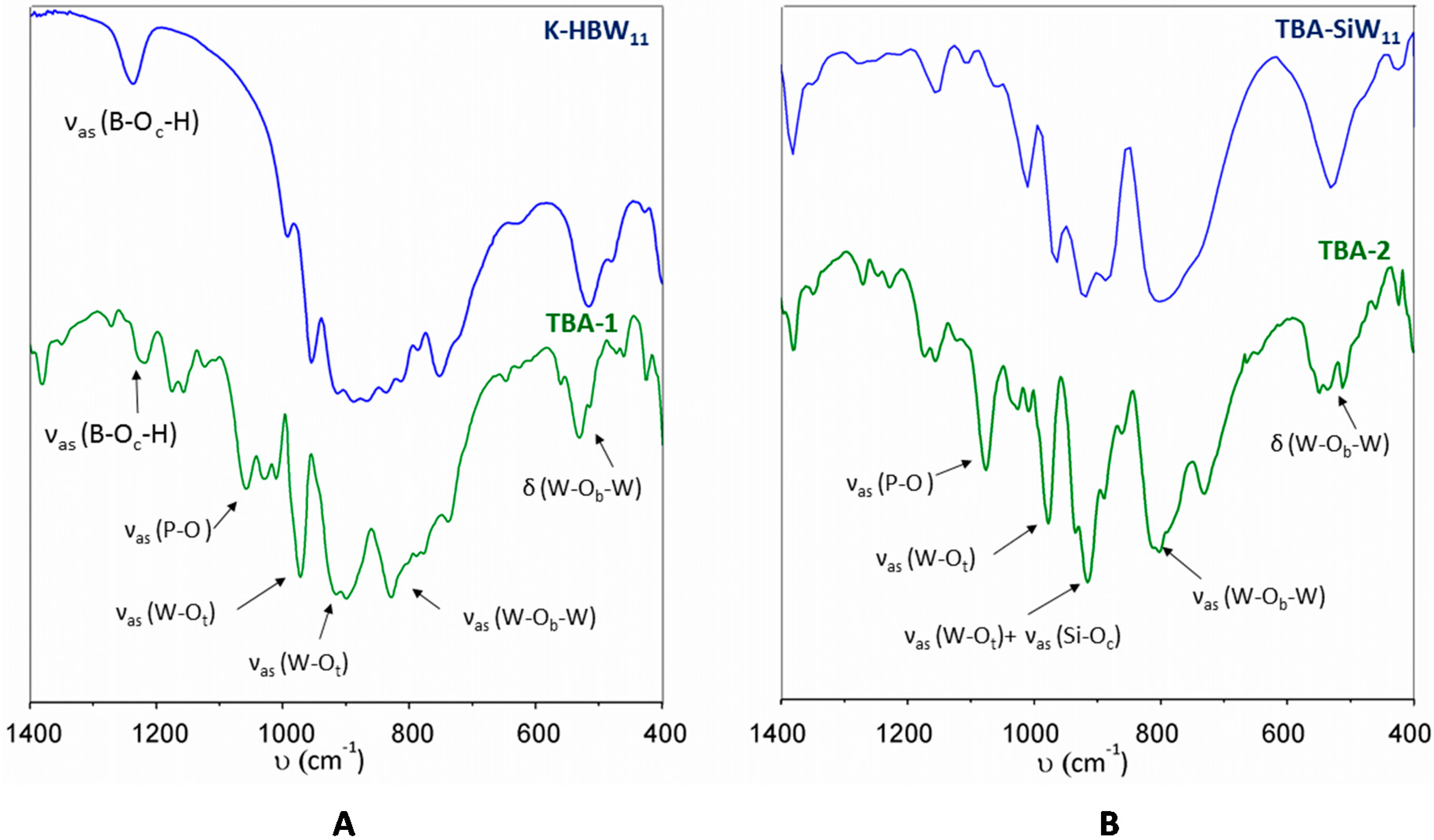
2.1. Synthesis, Infrared Spectroscopy and Thermal Analyses
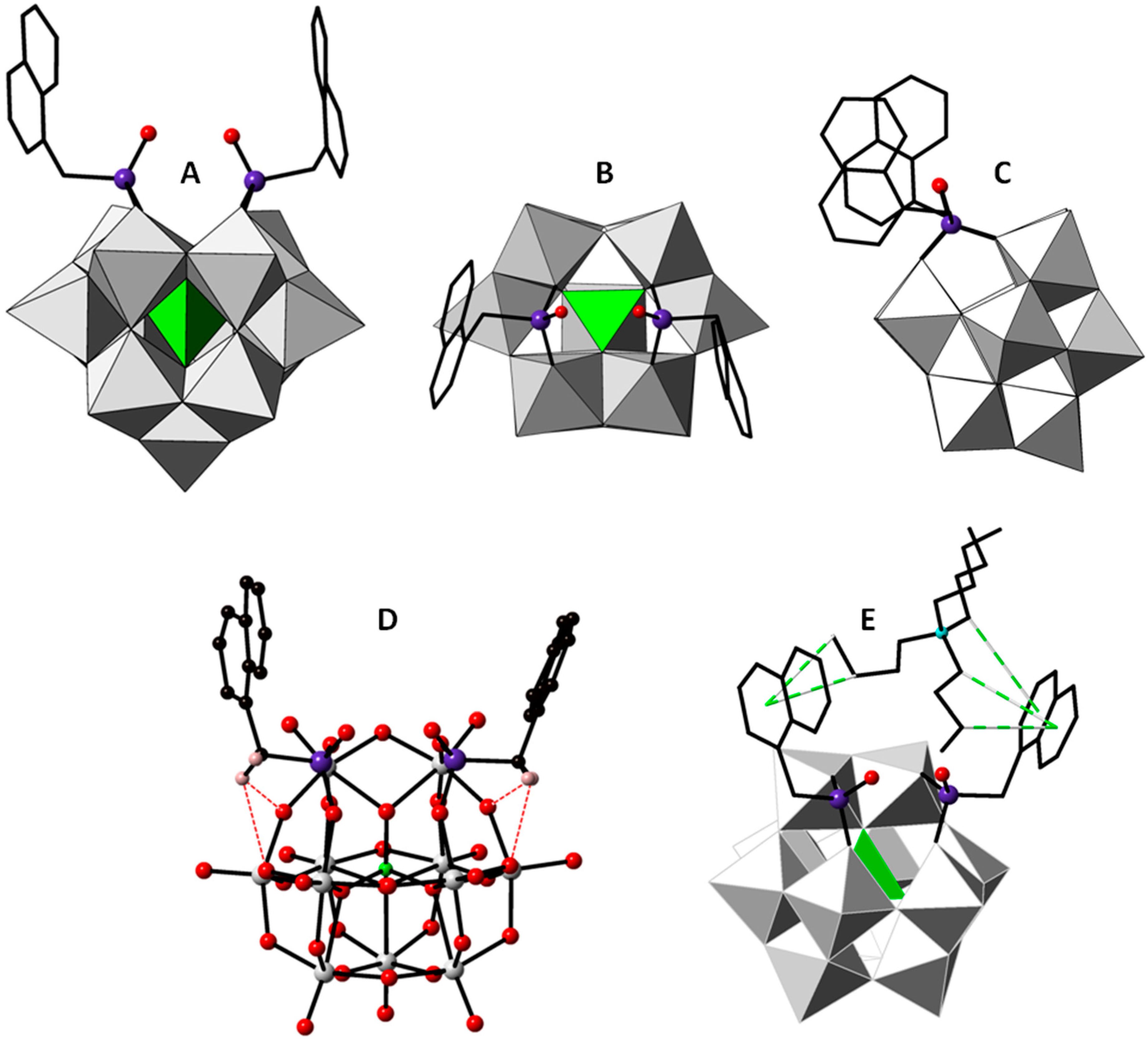
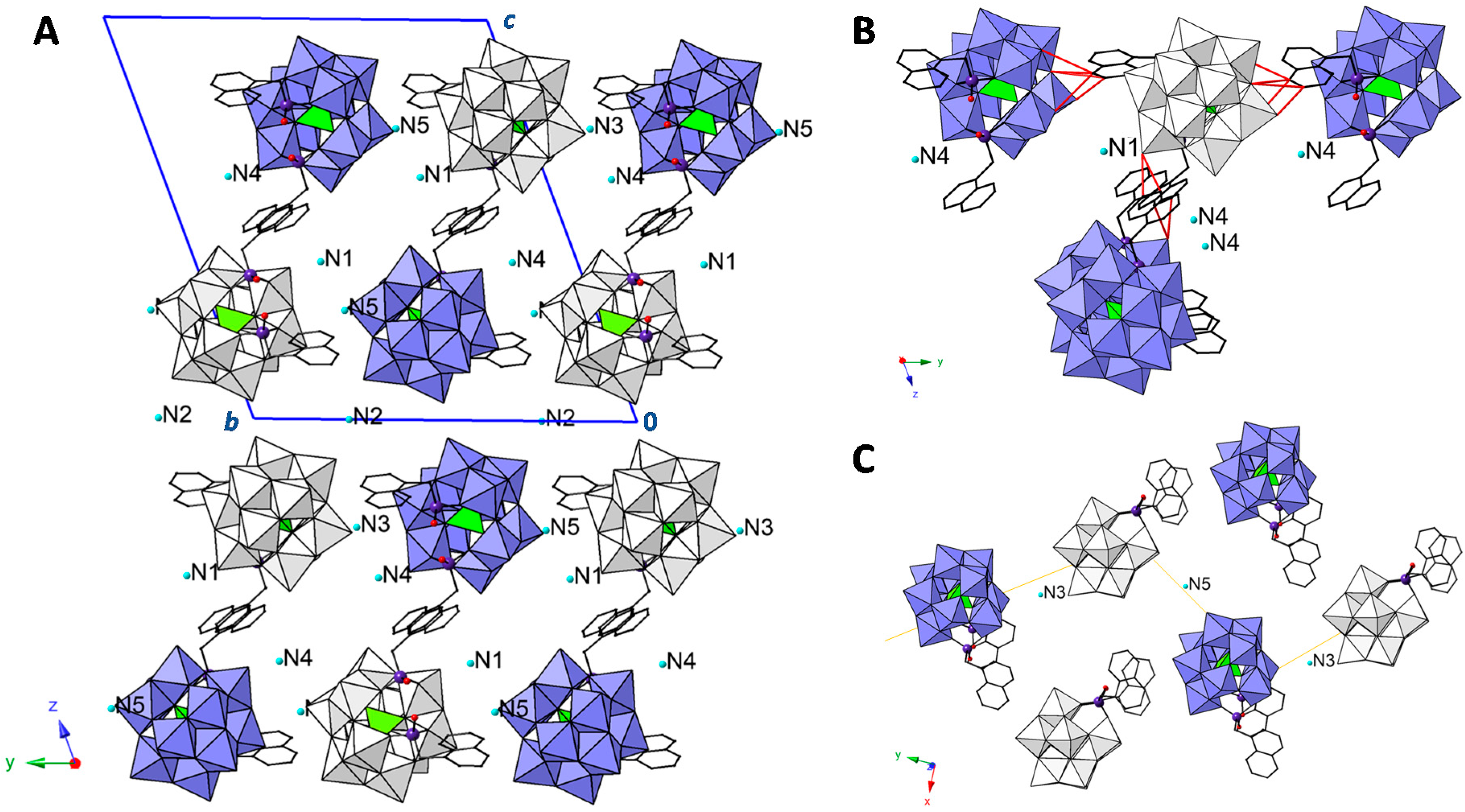
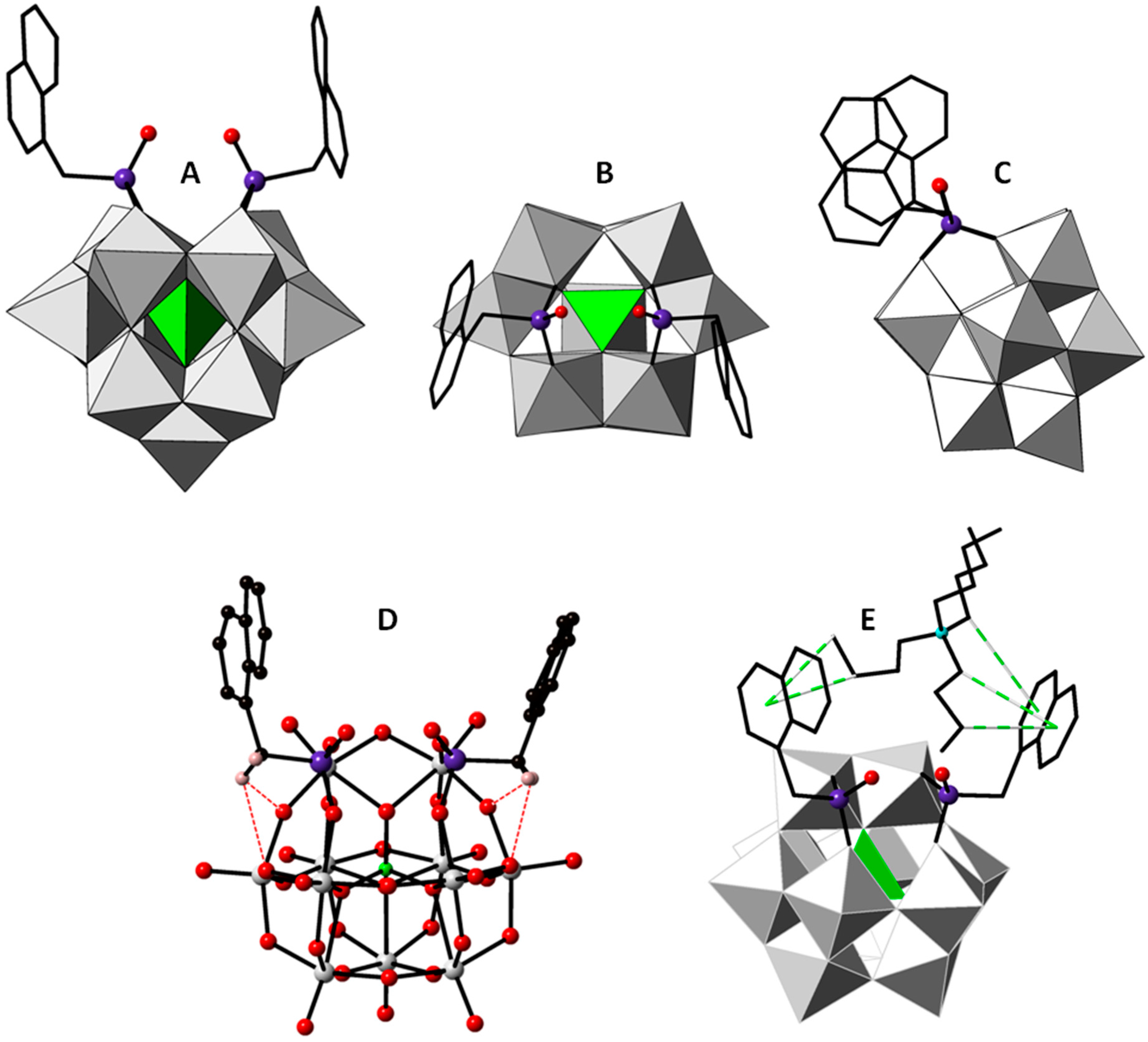
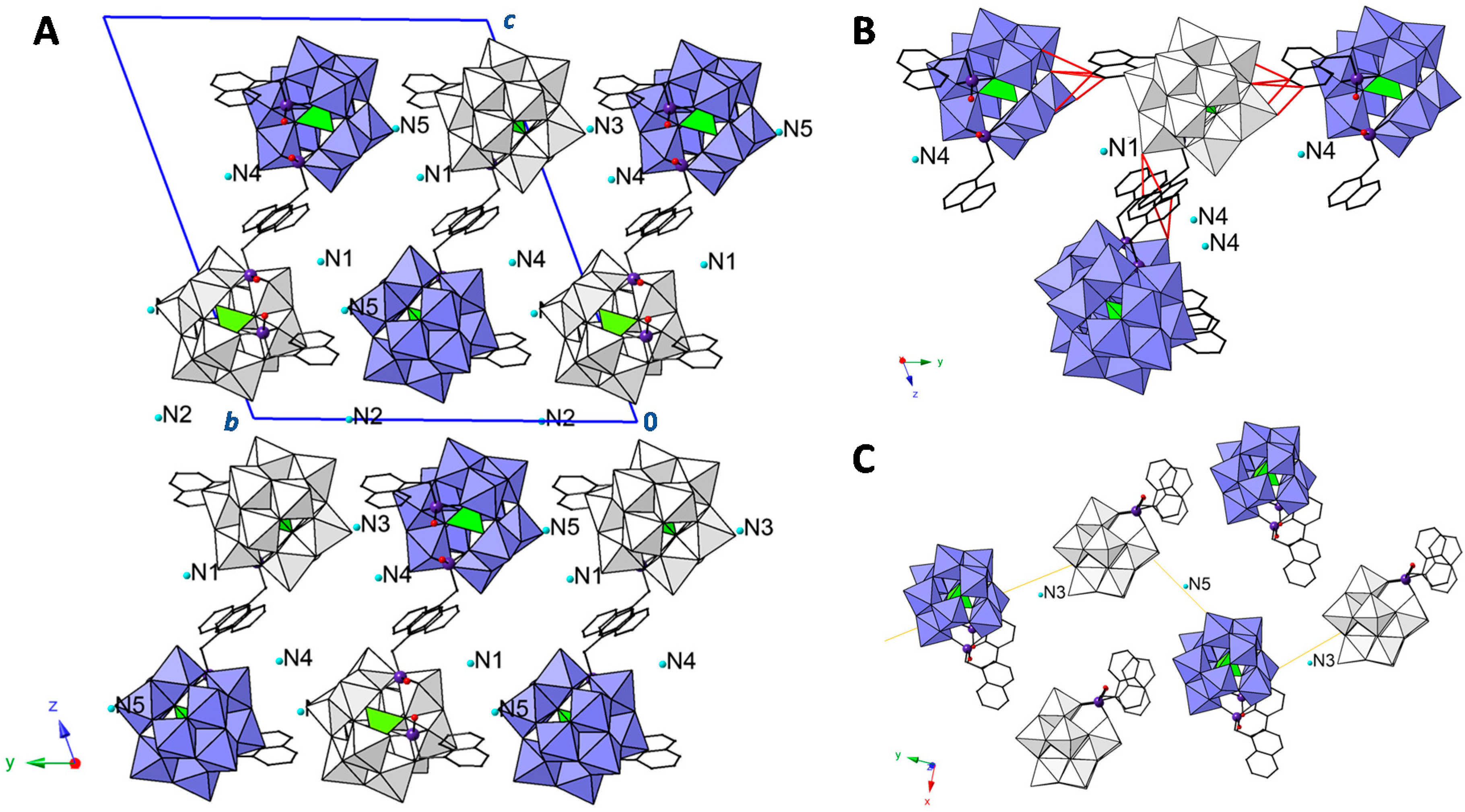
2.2. Crystal Structure
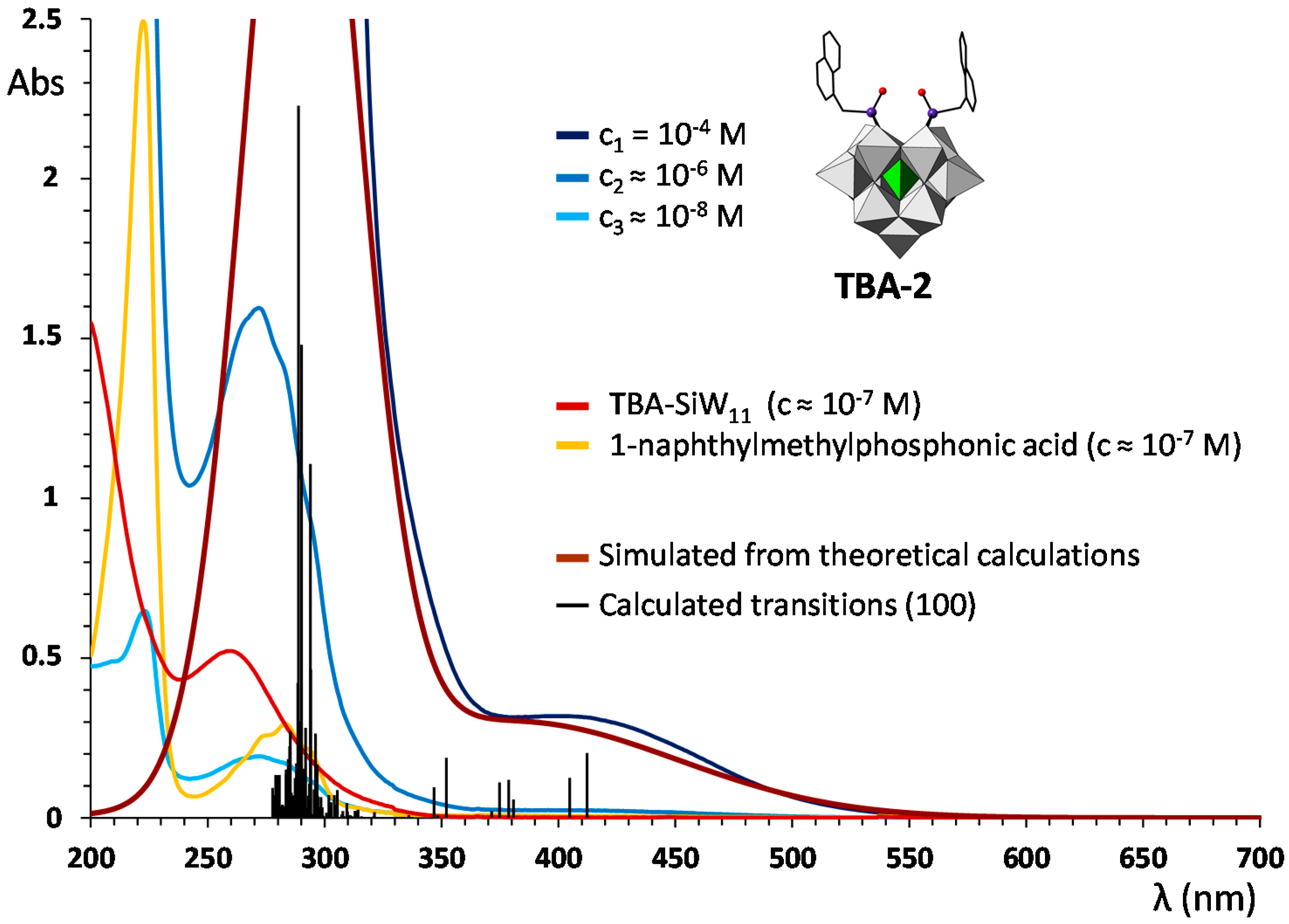
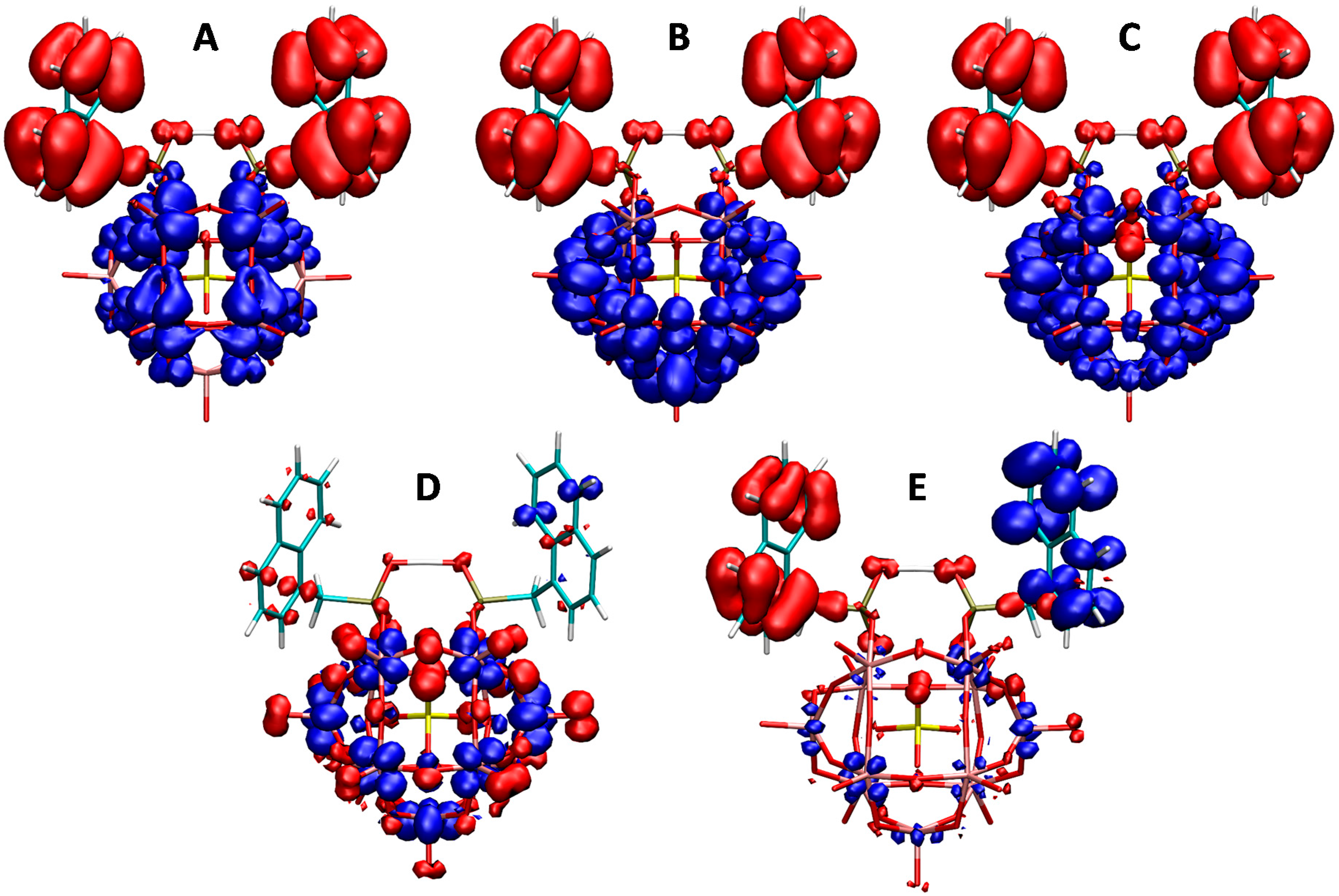
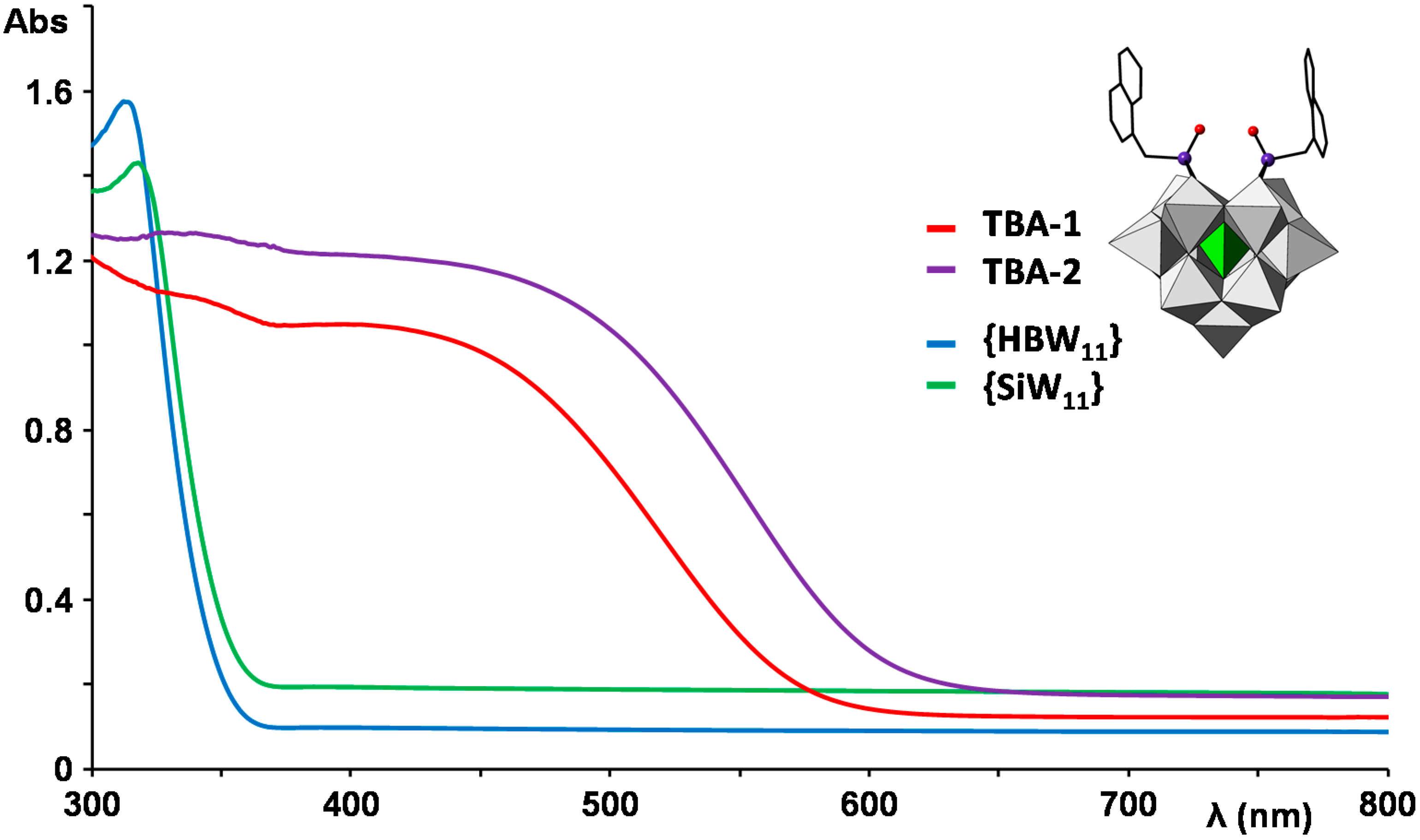
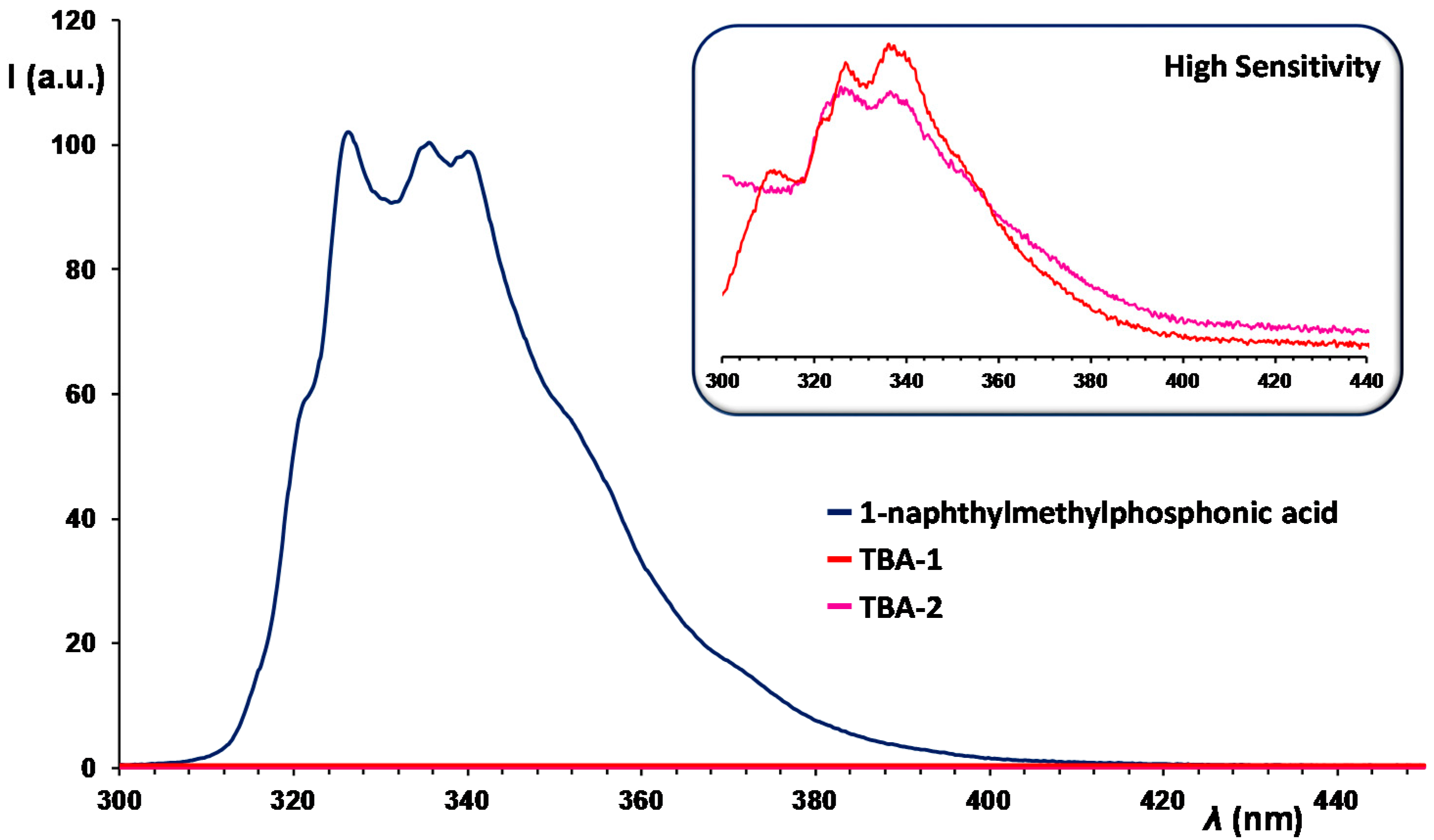
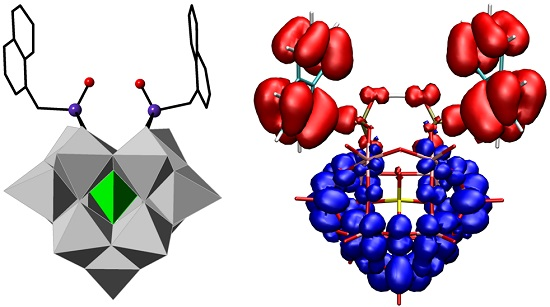
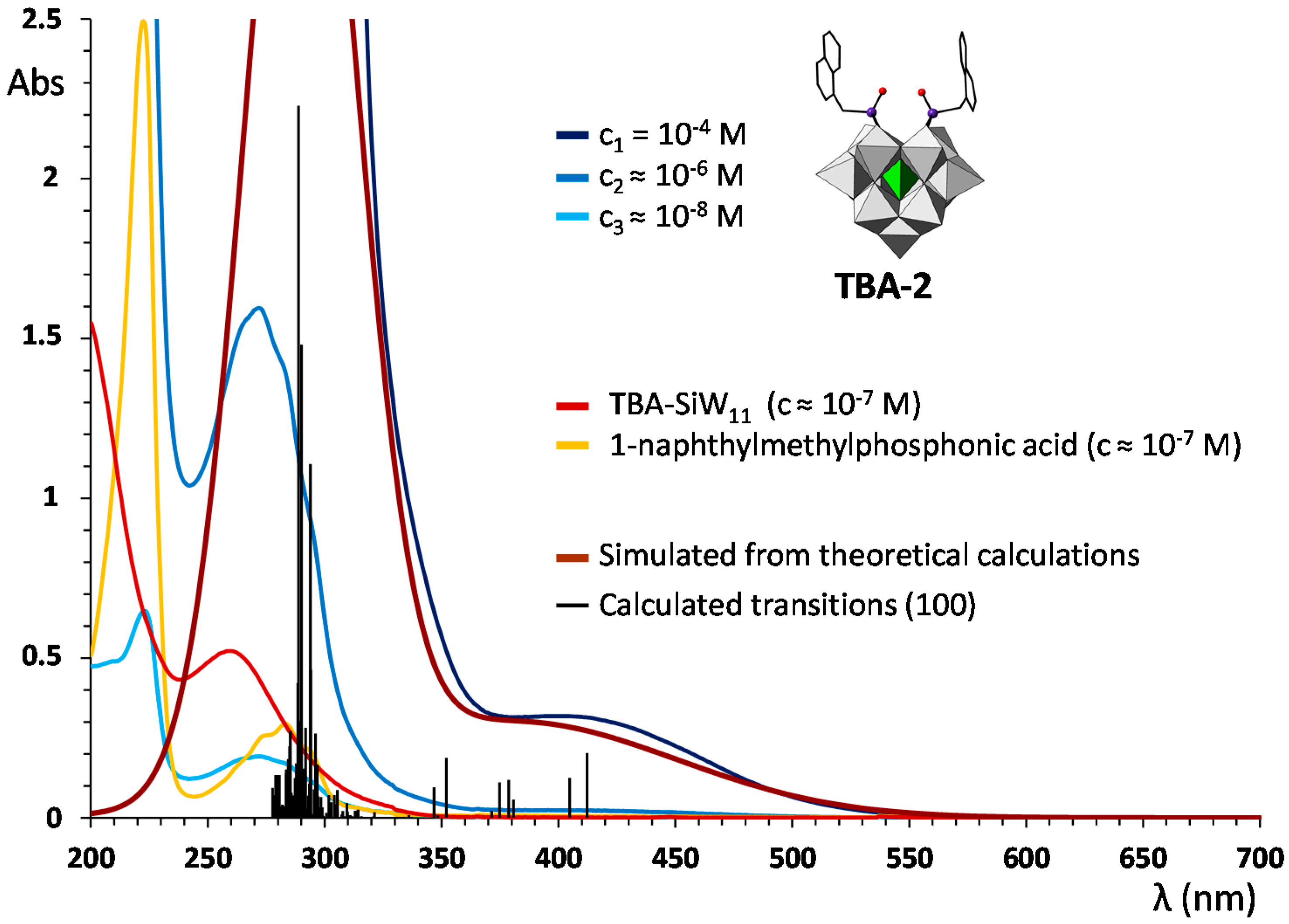
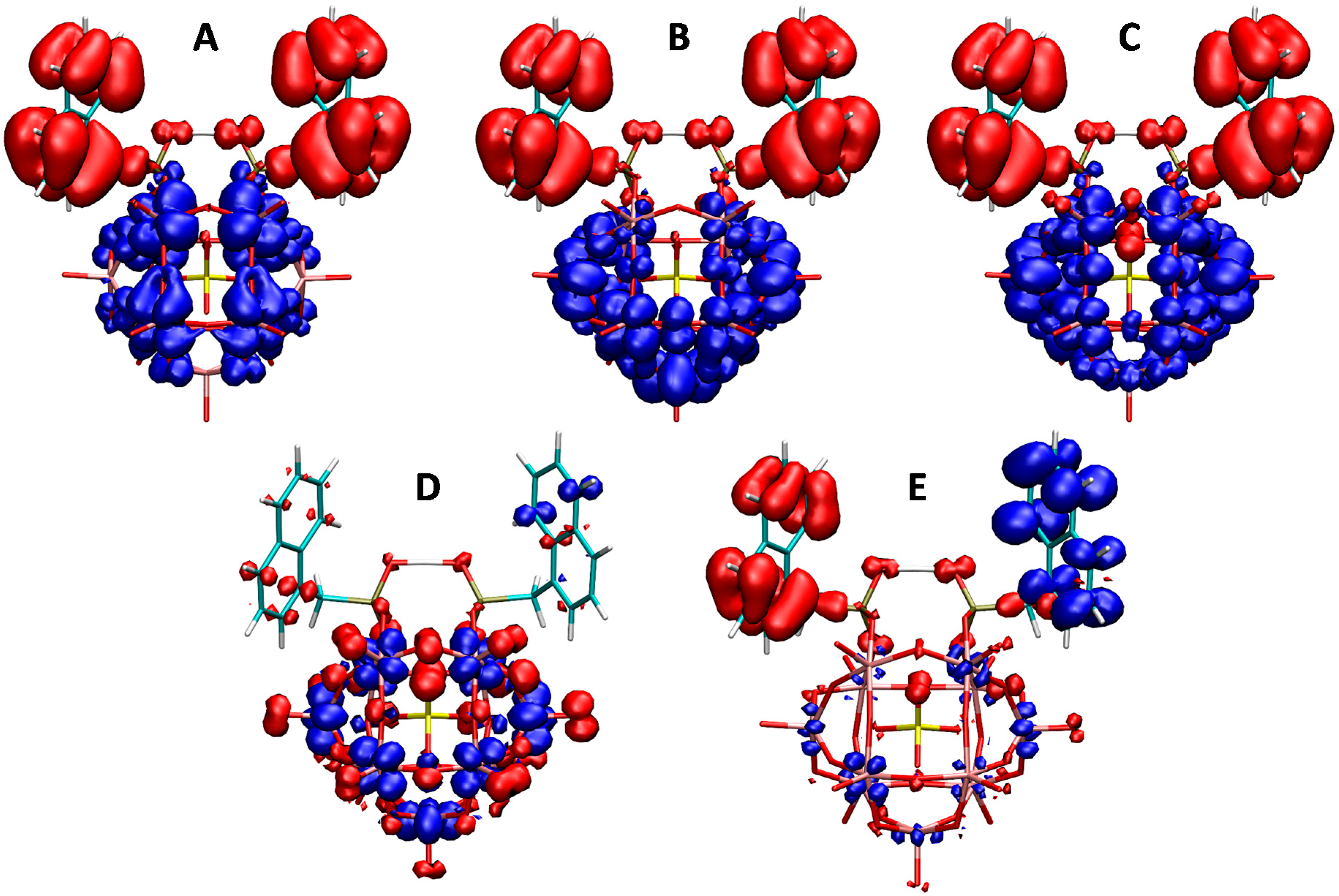
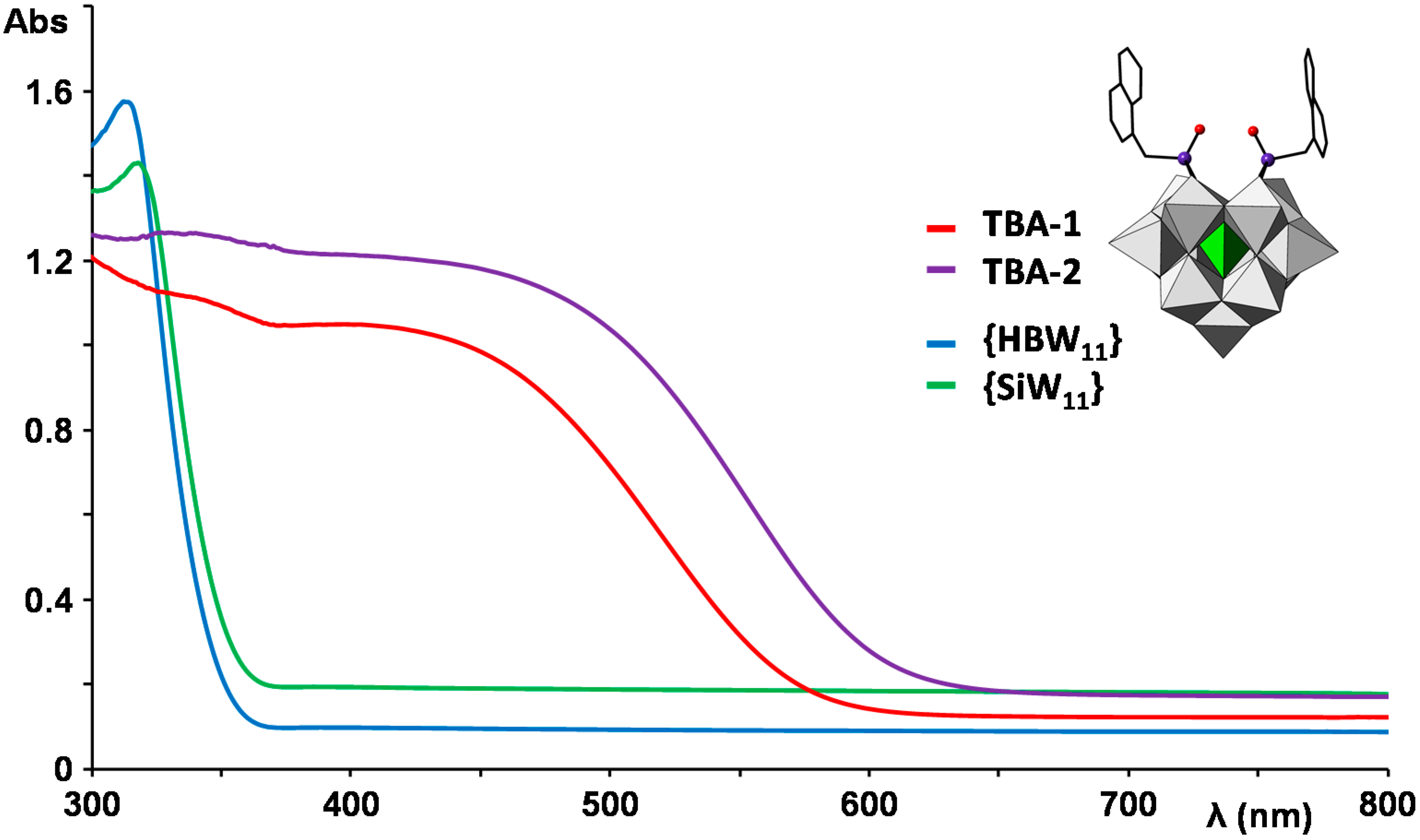
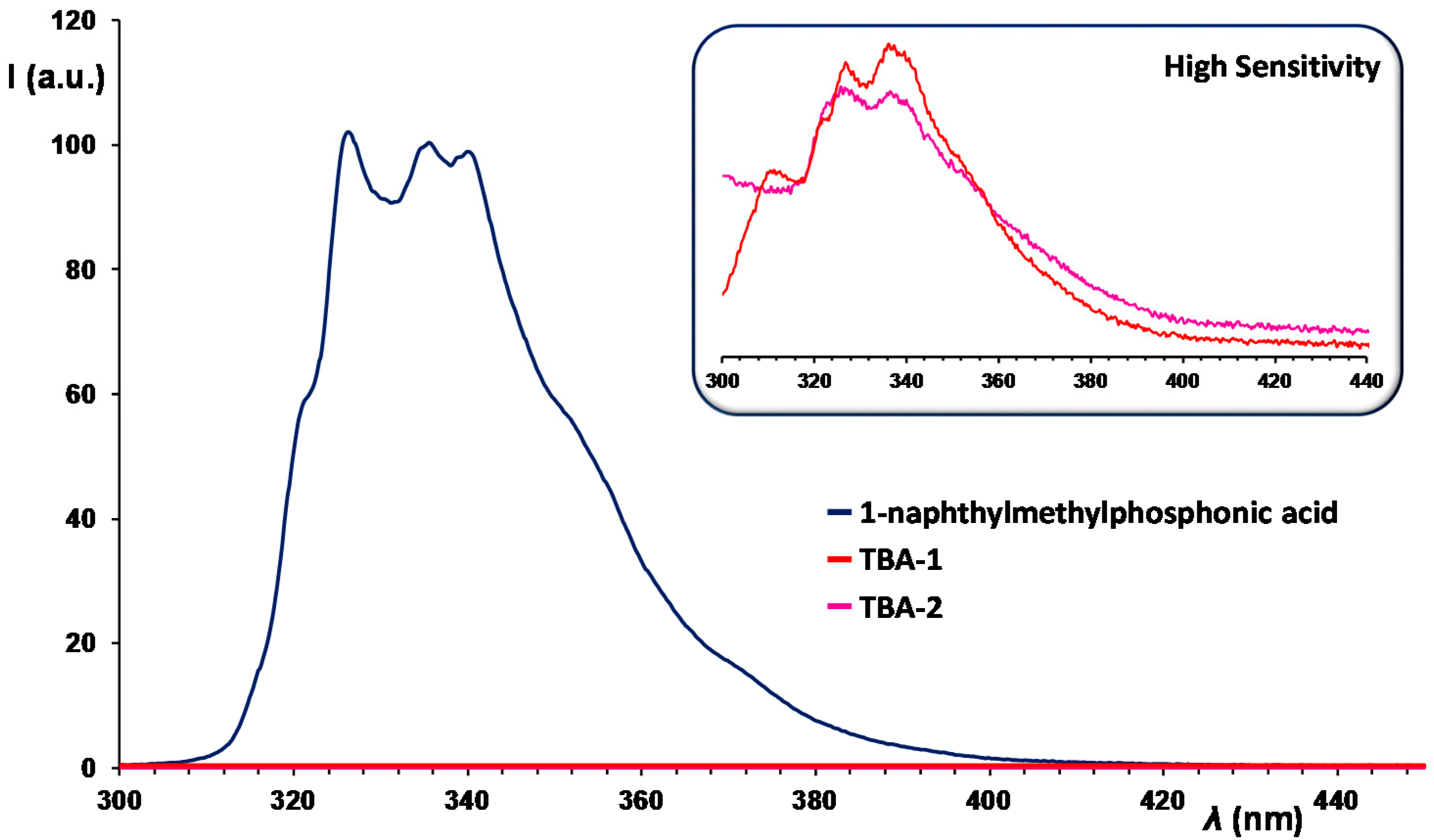
2.3. UV–Vis Spectroscopy and Photoluminescent Properties
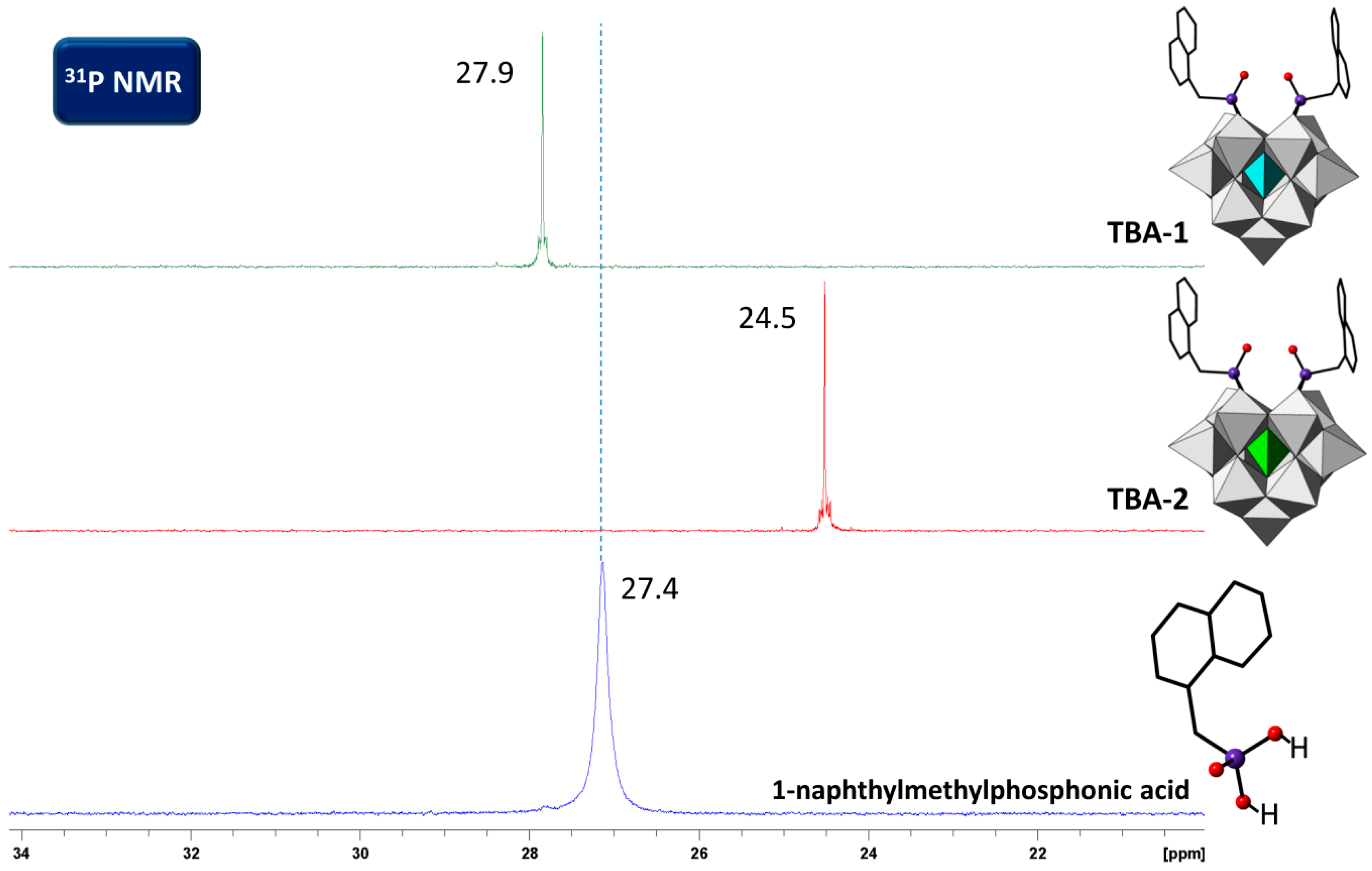
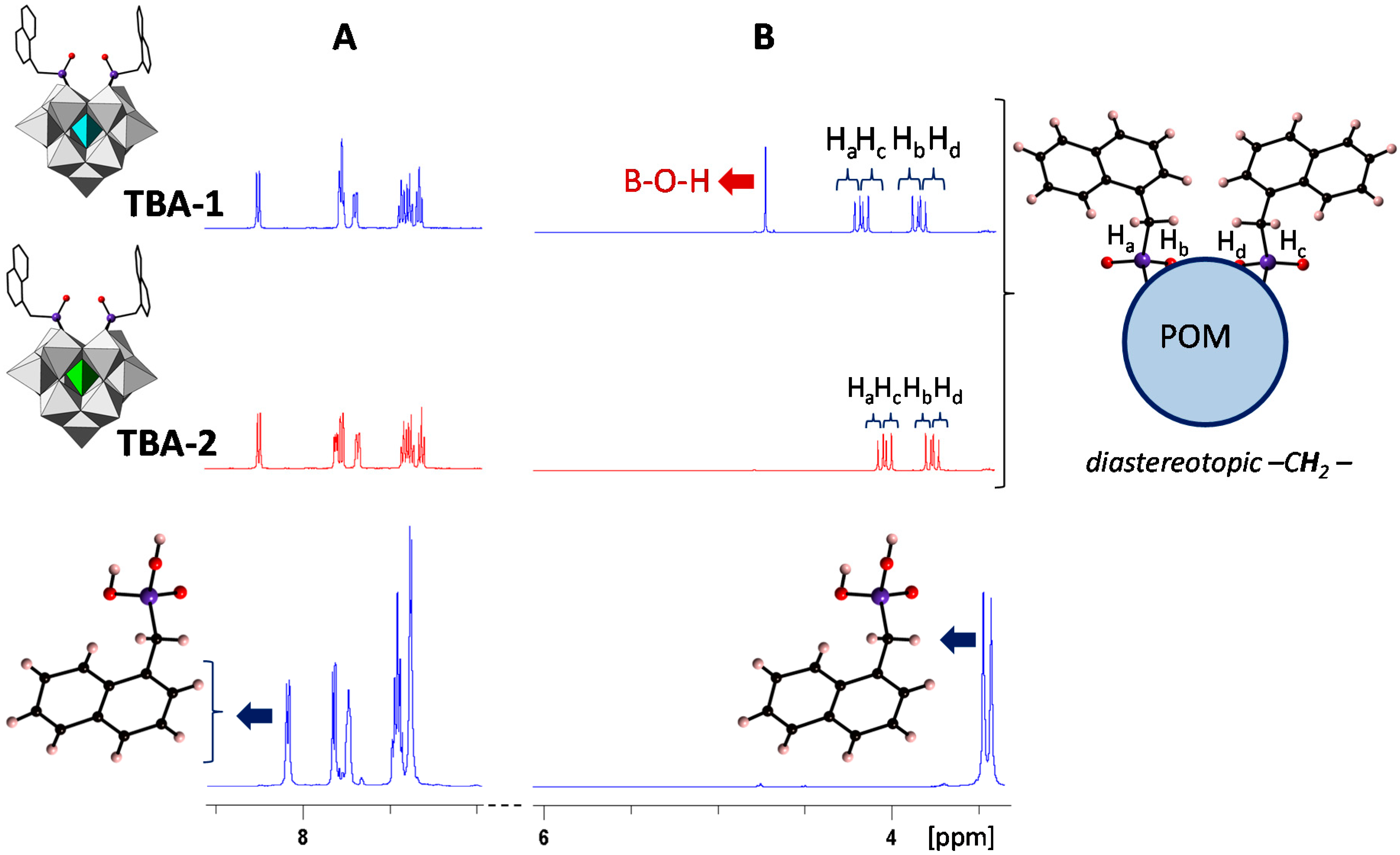
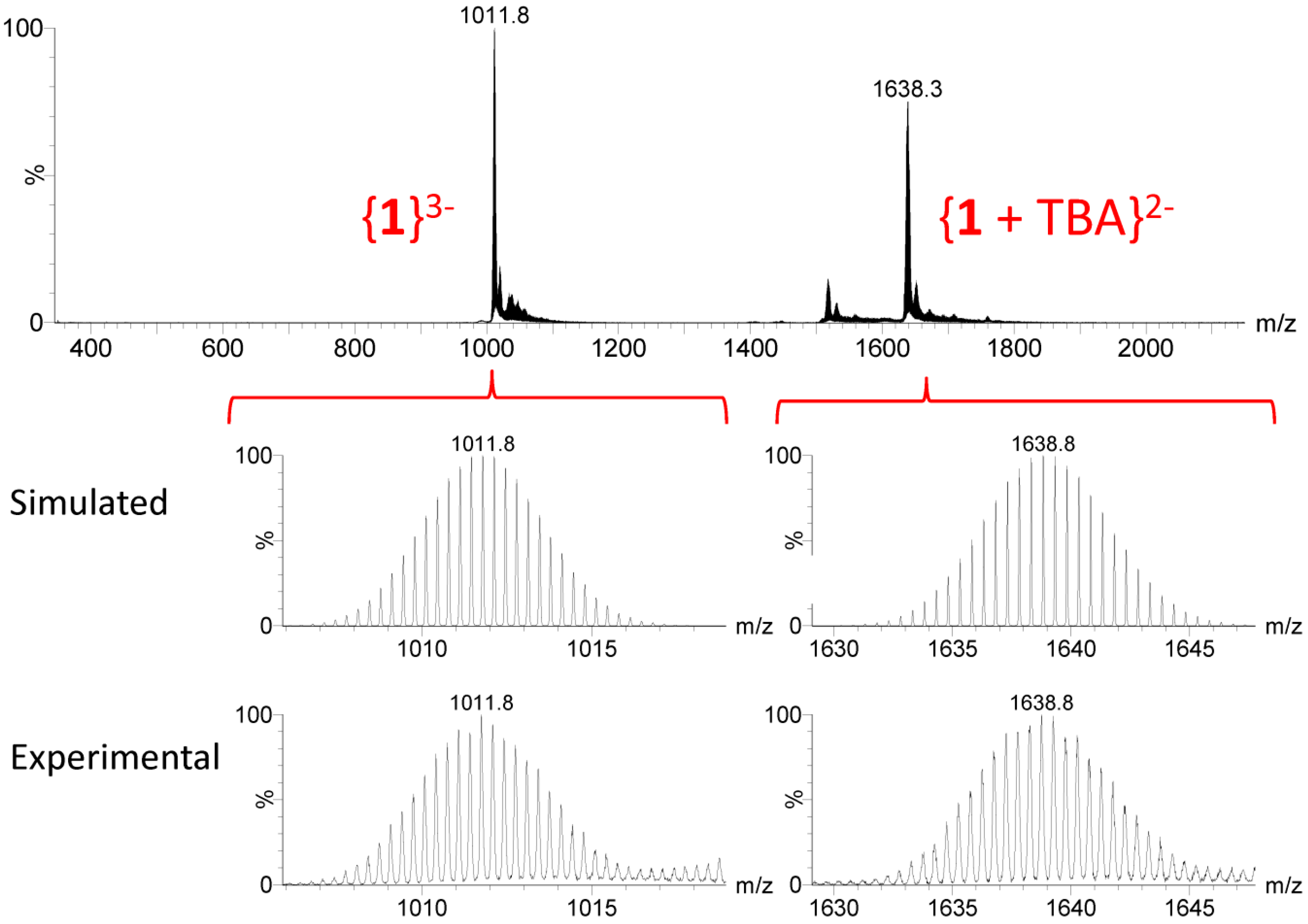
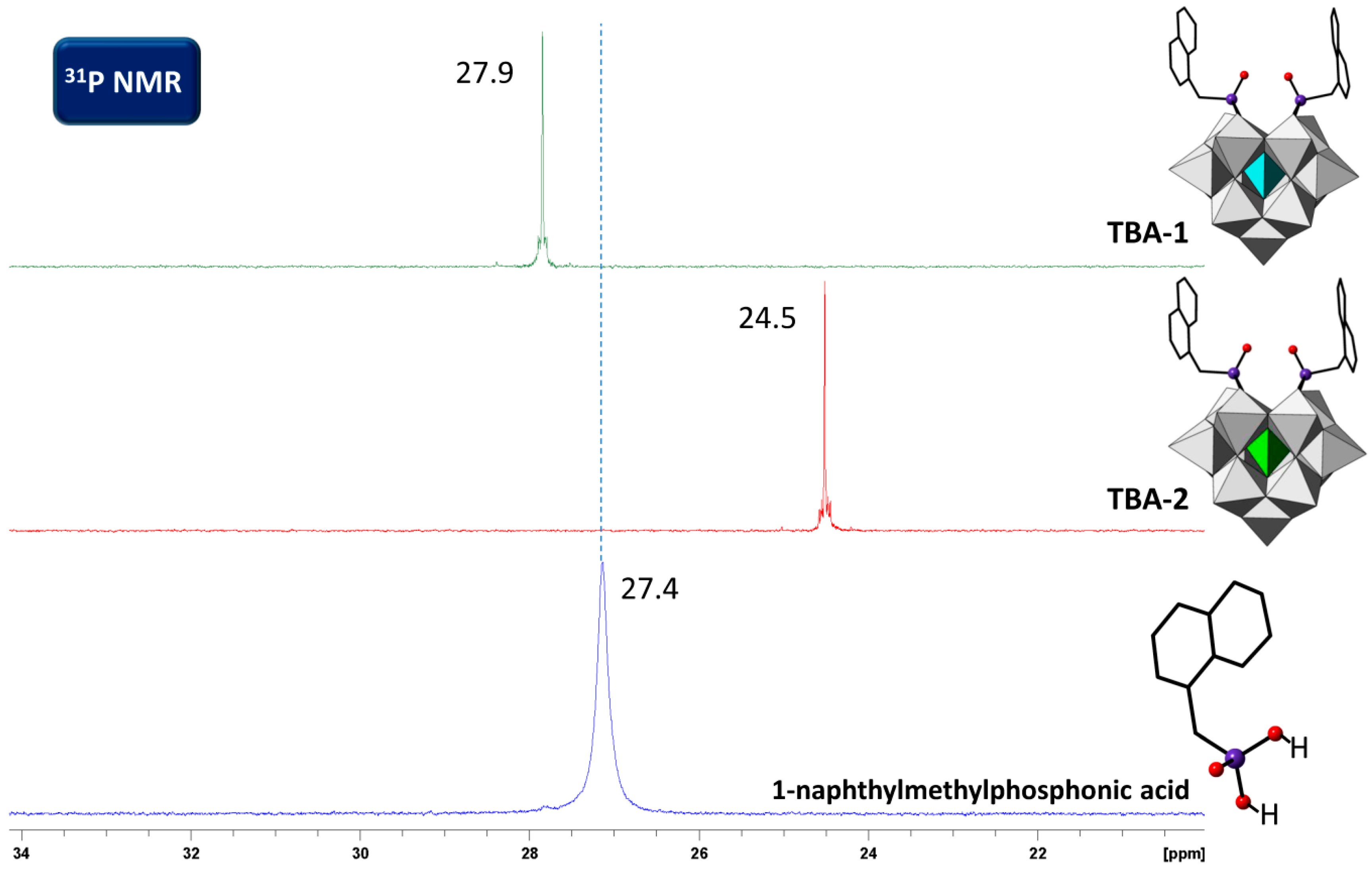
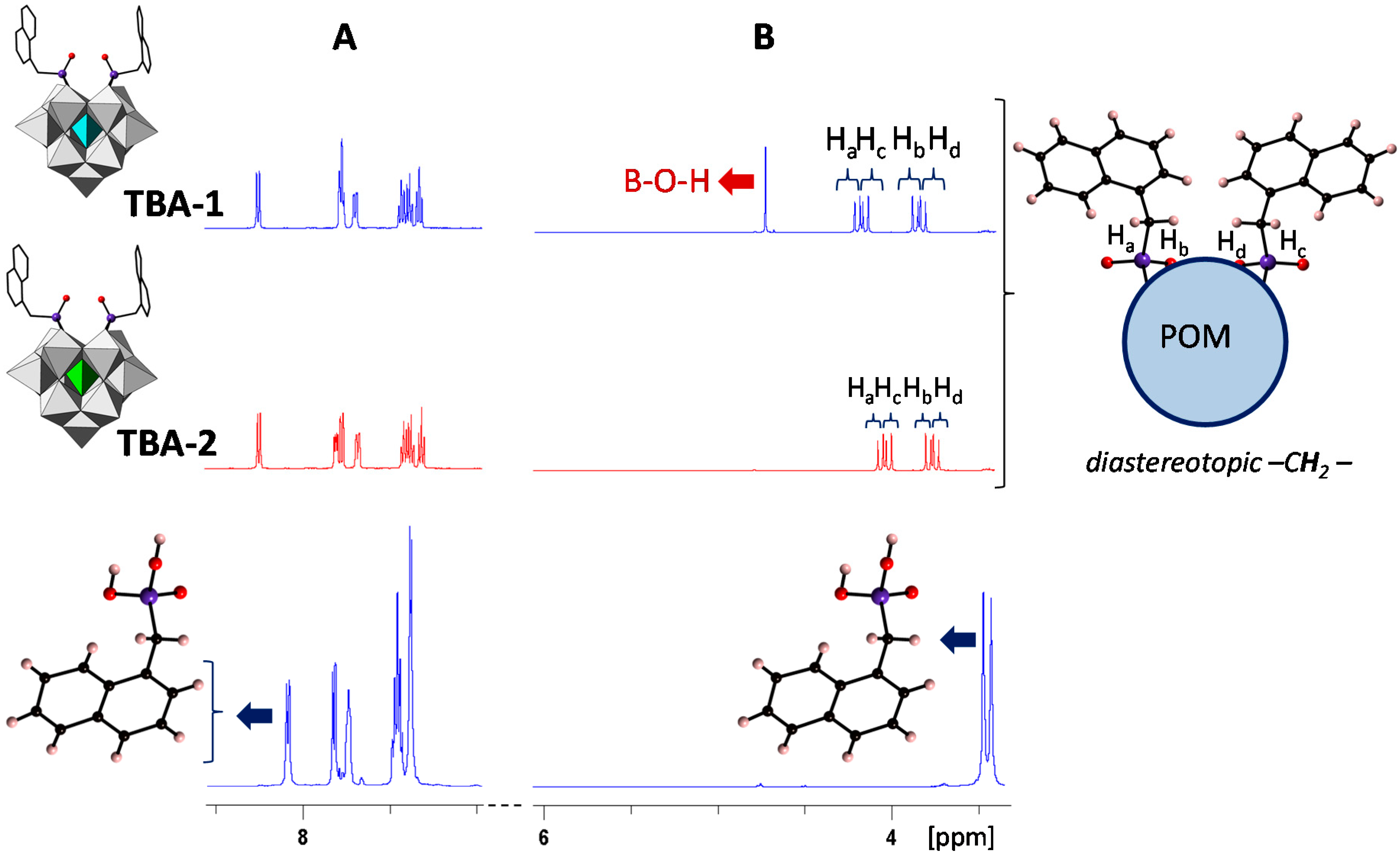
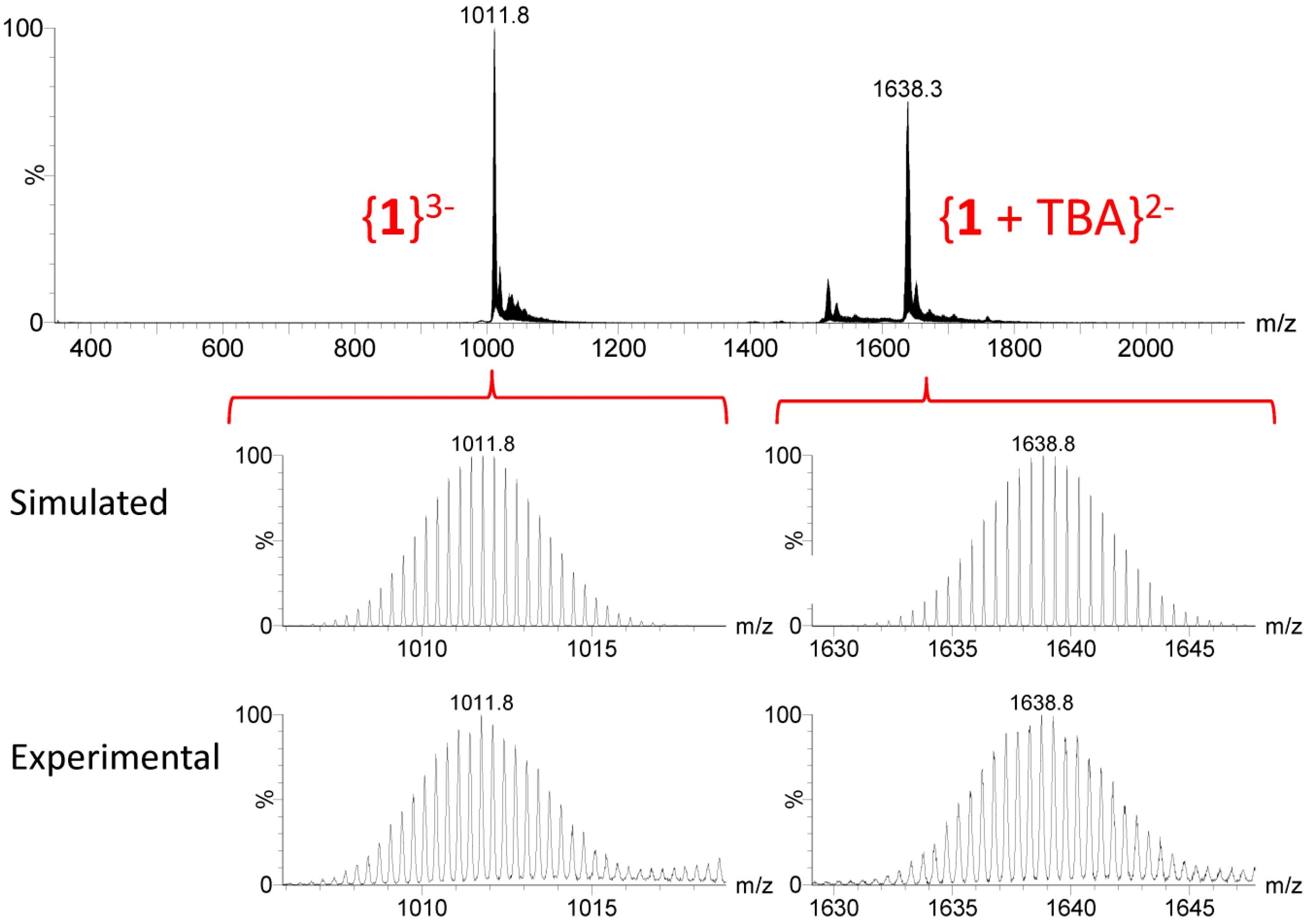
2.4. Solution Behavior
3. Experimental Section
3.1. Materials and Methods
3.2. Synthesis of [N(C4H9)4]3[H(C11H9PO)2(HBW11O39)] (TBA-1)
3.3. Synthesis of [N(C4H9)4]3[H(C11H9PO)2(SiW11O39)] (TBA-2)
3.4. X-ray Crystallography
3.5. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pope, M.T. Heteropoly and Isopoly Oxometalates; Springer: Berlin, Germany, 1983. [Google Scholar]
- Polyoxometalates: From Platonic Solids to Anti-Retroviral Activity; Pope, M.T.; Müller, A. (Eds.) Kluwer: Dordrecht, The Netherlands, 1994.
- Polyoxometalate Chemistry: Some Recent Trends; Sécheresse, F. (Ed.) World Scientific: Singapore, 2013.
- Gouzerh, P.; Proust, A. Main-group element, organic, and organometallic derivatives of polyoxometalates. Chem. Rev. 1998, 98, 77–111. [Google Scholar] [CrossRef] [PubMed]
- Long, D.-L.; Tsunashima, R.; Cronin, L. Polyoxometalates: Building blocks for functional nanoscale systems. Angew. Chem. Int. Ed. 2010, 49, 1736–1758. [Google Scholar] [CrossRef] [PubMed]
- Proust, A.; Thouvenot, R.; Gouzerh, P. Functionalization of polyoxometalates: Towards advanced applications in catalysis and materials science. Chem. Commun. 2008, 1837–1852. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.-F.; Tsunashima, R. Recent advances on polyoxometalate-based molecular and composite materials. Chem. Soc. Rev. 2010, 41, 7384–7402. [Google Scholar] [CrossRef] [PubMed]
- Santoni, J.-P.; Hanan, G.S.; Hasenknopf, B. Covalent multi-component systems of polyoxometalates and metal complexes: Toward multi-functional organic–inorganic hybrids in molecular and material sciences. Coord. Chem. Rev. 2014, 281, 64–85. [Google Scholar] [CrossRef]
- Proust, A.; Matt, B.; Villanneau, R.; Guillemot, G.; Gouzerh, P.; Izzet, G. Functionalization and post-functionalization: A step towards polyoxometalate-based materials. Chem. Soc. Rev. 2012, 41, 7605–7622. [Google Scholar] [CrossRef] [PubMed]
- Dolbecq, A.; Dumas, E.; Mayer, C.R.; Mialane, P. Hybrid organic–inorganic polyoxometalate compounds: From structural diversity to applications. Chem. Rev. 2010, 110, 6009–6048. [Google Scholar] [CrossRef] [PubMed]
- Blazevic, A.; Rompel, A. The Anderson–Evans polyoxometalate: From inorganic building blocks via hybrid organic–inorganic structures to tomorrows “Bio-POM”. Coord. Chem. Rev. 2016, 307, 42–64. [Google Scholar] [CrossRef]
- Yin, P.; Wu, P.; Xiao, Z.; Li, D.; Bitterlich, E.; Zhang, J.; Cheng, P.; Vezenov, D.V.; Liu, T.; Wei, Y. A double-tailed fluorescent surfactant with a hexavanadate cluster as the head group. Angew. Chem. Int. Ed. 2011, 50, 2521–2522. [Google Scholar] [CrossRef] [PubMed]
- Pradeep, C.P.; Long, D.-L.; Newton, G.N.; Song, Y.-F.; Cronin, L. Supramolecular metal oxides: Programmed hierarchical assembly of a protein-sized 21 kDa [(C16H36N)19{H2NC(CH2O)3P2V3W15O59}4]5− polyoxometalate assembly. Angew. Chem. Int. Ed. 2008, 47, 4388–4391. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z. Rational synthesis of covalently bonded organic–inorganic hybrids. Angew. Chem. Int. Ed. 2004, 43, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Artetxe, B.; Reinoso, S.; San Felices, L.; Vitoria, P.; Pache, A.; Martín-Caballero, J.; Gutiérrez-Zorrilla, J.M. Functionalization of Krebs-Type polyoxometalates with N,O-chelating ligands: A systematic study. Inorg. Chem. 2015, 54, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, G.; Oms, O.; Dolbecq, A.; Marrot, J.; Mialane, P. Route for the elaboration of functionalized hybrid 3d-substituted trivacant Keggin anions. Inorg. Chem. 2011, 50, 7376–7378. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Anderson, T.M.; Hill, C.L. Enantiomerically pure polytungstates: Chirality transfer through zirconium coordination centers to nanosized inorganic clusters. Angew. Chem. Int. Ed. 2005, 44, 3540–3544. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.-T.; Zhang, J.; Yang, G.-Y. Designed synthesis of POM–organic frameworks from {Ni6PW9} building blocks under hydrothermal conditions. Angew. Chem. Int. Ed. 2008, 47, 3909–3913. [Google Scholar] [CrossRef] [PubMed]
- Zonnevijlle, F.; Pope, M.T. Attachment of organic groups to heteropoly oxometalate anions. J. Am. Chem. Soc. 1979, 101, 2731–2732. [Google Scholar] [CrossRef]
- Kortz, U.; Hussain, F.; Reicke, M. The ball-shaped heteropolytungstates [{Sn(CH3)2(H2O)}24{Sn(CH3)2}12(A-XW9O34)12]36−. Angew. Chem. Int. Ed. 2005, 44, 3773–3777. [Google Scholar] [CrossRef] [PubMed]
- Boglio, C.; Micoine, K.; Derat, E.; Thouvenot, R.; Hasenknopf, B.; Thorimbert, S.; Lacôte, E.; Malacria, M. Regioselective activation of oxo ligands in functionalized Dawson polyoxotungstates. J. Am. Chem. Soc. 2008, 130, 4553–4561. [Google Scholar] [CrossRef] [PubMed]
- Nomiya, K.; Togashi, Y.; Kasahara, Y.; Aoki, S.; Seki, H.; Noguchi, M.; Yoshida, S. Synthesis and structure of Dawson polyoxometalate-based, multifunctional, inorganic–organic hybrid compounds: Organogermyl complexes with one terminal functional group and organosilyl analogues with two terminal functional groups. Inorg. Chem. 2011, 50, 9606–9619. [Google Scholar] [CrossRef] [PubMed]
- Piedra-Garza, L.F.; Dickman, M.H.; Moldovan, O.; Breuning, H.J.; Kortz, U. Organoantimony-containing polyoxometalate: [{PhSbOH}3(A-α-PW9O34)2]9−. Inorg. Chem. 2009, 48, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Knoth, W.H. Derivatives of heteropolyanions. 1. Organic derivatives of W12SiO404−, W12PO403−, and Mo12SiO404−. J. Am. Chem. Soc. 1979, 101, 759–760. [Google Scholar] [CrossRef]
- Judeinstein, P.; Deprunb, C.; Nadjo, L. Synthesis and multispectroscopic characterization of organically modified polyoxometalates. Dalton Trans. 1991, 1991–1997. [Google Scholar] [CrossRef]
- Mayer, C.R.; Roch-Marchal, C.; Lavanant, H.; Thouvenot, R.; Sellier, N.; Blais, J.-C.; Sécheresse, F. New organosilyl derivatives of the Dawson polyoxometalate [α2-P2W17O61(RSi)2O]6−: Synthesis and mass spectrometric investigation. Chem. Eur. J. 2004, 10, 5517–5523. [Google Scholar] [CrossRef] [PubMed]
- Mazeaud, A.; Ammari, N.; Robert, F.; Thouvenot, R. Coordination chemistry of polyoxometalates: Rational synthesis of the mixed organosilyl derivatives of trivacant polyoxotungstates α-A-[PW9O34(tBuSiO)3(RSi)]3− and α-B-[AsW9O33(tBuSiO)3(HSi)]3−. Angew. Chem. Int. Ed. Engl. 1996, 35, 1961–1964. [Google Scholar] [CrossRef]
- Mayer, C.R.; Fournier, I.; Thouvenot, R. Bis- and tetrakis(organosilyl) decatungstosilicate, [γ-SiW10O36(RSi)2O]4− and [γ-SiW10O36(RSiO)4]4−: Synthesis and structural determination by Multinuclear NMR Spectroscopy and Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry. Chem. Eur. J. 2000, 6, 105–110. [Google Scholar] [CrossRef]
- Kim, G.-S.; Hagen, K.S.; Hill, C.L. Synthesis, structure, spectroscopic properties, and hydrolytic chemistry of organophosphonoyl polyoxotungstates of formula [C6H5P(O)]2Xn+WllO39](8−n)− (X = P5+, Si4+)]. Inorg. Chem. 1992, 31, 5316–5324. [Google Scholar] [CrossRef]
- Mayer, C.R.; Thouvenot, R. Organophosphoryl derivatives of trivacant tungstophosphates of general formula α-A-[PW9O34(RPO)2]5−: Synthesis and structure determination by Multinuclear Magnetic Resonance Spectroscopy.(31P, 183W). Dalton Trans. 1998, 7–13. [Google Scholar] [CrossRef]
- Mayer, C.R.; Herson, P.; Thouvenot, R. Organic–inorganic hybrids based on polyoxometalates. Synthesis and structural characterization of bis(organophosphoryl) decatungstosilicates [γ-SiW10O36(RPO)2]4−. Inorg. Chem. 1999, 38, 6152–6158. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.-G.; Liu, Q.; Liu, J.-F. Synthesis and spectroscopic characterization of organophosphoryl polyoxotungstates [C6H11P(O)]2Xn+ W11O39(8−n)− (Xn+ = P5+, Si4+, B3+, Ga3+). Polyhedron 2000, 19, 125–128. [Google Scholar] [CrossRef]
- Boujtita, M.; Boixel, J.; Blart, E.; Mayer, C.R.; Odobel, F. Redox properties of hybrid Dawson type polyoxometalates disubstituted with organo-silyl or organo-phosphoryl moieties. Polyhedron 2008, 27, 688–692. [Google Scholar] [CrossRef]
- Sun, Z.-G.; Zhu, Z.-M.; You, W.-S. Synthesis and characterization of organophosphoryl polyoxotungstates [RP(O)]2Xn+W11O39](8−n)− (Xn+ = P5+, Si4+, Ge4+, Ga3+). Transition Met. Chem. 2003, 28, 849–851. [Google Scholar] [CrossRef]
- Schroden, R.C.; Blanford, C.F.; Melde, B.J.; Johnson, B.J.S.; Stein, A. Direct synthesis of ordered macroporous silica materials functionalized with polyoxometalate clusters. Chem. Mater. 2001, 13, 1074–1081. [Google Scholar] [CrossRef]
- Mayer, C.R.; Cabuil, V.; Lalot, T.; Thouvenot, R. Incorporation of magnetic nanoparticles in new hybrid networks based on heteropolyanions and polyacrylamide. Angew. Chem. Int. Ed. 1999, 38, 3672–3675. [Google Scholar] [CrossRef]
- Mayer, C.R.; Neveu, S.; Cabuil, V. A Nanoscale hybrid system based on gold nanoparticles and heteropolyanions. Angew. Chem. Int. Ed. 2002, 41, 501–503. [Google Scholar] [CrossRef]
- Cannizzo, C.; Mayer, C.R.; Sécheresse, F.; Larpent, C. Covalent hybrid materials based on nanolatex particles and Dawson polyoxometalates. Adv. Mater. 2005, 17, 2888–2892. [Google Scholar] [CrossRef]
- Villanneau, R.; Marzouk, A.; Wang, Y.; Ben Djamaa, A.; Laugel, G.; Proust, A.; Launay, F. Covalent grafting of organic–inorganic polyoxometalates hybrids onto mesoporous SBA-15: A key step for new anchored homogeneous catalysts. Inorg. Chem. 2013, 52, 2958–2965. [Google Scholar] [CrossRef] [PubMed]
- Joo, N.; Renaudineau, S.; Delapierre, G.; Bidan, G.; Chamoreau, L.-S.; Thouvenot, R.; Gouzerh, P.; Proust, A. Organosilyl/-germyl polyoxotungstate hybrids for covalent grafting onto silicon surfaces: Towards molecular memories. Chem. Eur. J. 2010, 16, 5043–5051. [Google Scholar] [CrossRef] [PubMed]
- Mercier, D.; Boujday, S.; Annabi, C.; Villanneau, R.; Pradier, C.-M.; Proust, A. Bifunctional polyoxometalates for planar gold surface nanostructuration and protein immobilization. J. Phys. Chem. C 2012, 116, 13217–13224. [Google Scholar] [CrossRef]
- Duffort, V.; Thouvenot, R.; Afonso, C.; Izzet, G.; Proust, A. Straightforward synthesis of new polyoxometalate-based hybrids exemplified by the covalent bonding of a polypyridyl ligand. Chem. Commun. 2009, 6062–6064. [Google Scholar] [CrossRef] [PubMed]
- Elliott, K.J.; Harriman, A.; le Pleux, L.; Pellegrin, Y.; Blart, E.; Mayer, C.R.; Odobel, F.A. Porphyrin–polyoxometallate bio-inspired mimic for artificial photosynthesis. Phys. Chem. Chem. Phys. 2009, 11, 8767–8773. [Google Scholar] [CrossRef] [PubMed]
- Matt, B.; Renaudineau, S.; Chamoreau, L.-M.; Afonso, C.; Izzet, G.; Proust, A. Hybrid polyoxometalates: Keggin and Dawson silyl derivatives as versatile platforms. J. Org. Chem. 2011, 76, 3107–3111. [Google Scholar] [CrossRef] [PubMed]
- Odobel, F.; Severac, M.; Pellegrin, Y.; Blart, E.; Fosse, C.; Cannizzo, C.; Mayer, C.R.; Eliott, K.J.; Harriman, A. Coupled sensitizer–catalyst dyads: Electron-transfer reactions in a perylene–polyoxometalate conjugate. Chem. Eur. J. 2009, 15, 3130–3138. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Hu, L.; Liu, Q.; Du, Z.-L.; Li, F.-B.; Li, G.-H.; Zhu, X.-J.; Wong, W.-Y.; Wang, L.; Li, H. Synthesis and photoelectric properties of new Dawson-type polyoxometalate-based dimeric and oligomeric Pt(II)-acetylide inorganic–organic hybrids. Dalton Trans. 2015, 44, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Villanneau, R.; Ben Djamâa, A.; Chamoreau, L.-M.; Gontard, G.; Proust, A. Bisorganophosphonyl and -organoarsenyl derivatives of heteropolytungstates as hard ligands for early-transition-metal and lanthanide cations. Eur. J. Inorg. Chem. 2013, 1815–1820. [Google Scholar] [CrossRef]
- Villanneau, R.; Racimor, D.; Messner-Henning, E.; Rousselière, H.; Picart, S.; Thouvenot, R.; Proust, A. Insights into the coordination chemistry of phosphonate derivatives of heteropolyoxotungstates. Inorg. Chem. 2011, 50, 1164–1166. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.; Sandei, L.; Sartorel, A.; Scorrano, G.; Bonchio, M. Hybrid polyoxotungstates as second-generation POM-based catalysts for microwave-assisted H2O2 activation. Org. Lett. 2006, 8, 3671–3674. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, G.; Matricardi, E.; Chamoreau, L.-M.; Thouvenot, R.; Proust, A. Oxidovanadium(V) anchored to silanol-functionalized polyoxotungstates: Molecular models for single-site silica-supported vanadium catalysts. ACS Catal. 2015, 5, 7415–7423. [Google Scholar] [CrossRef]
- Tézé, A.; Michelon, M.; Hervé, G. Syntheses and structures of the tungstoborate anions. Inorg. Chem. 1997, 36, 505–509. [Google Scholar] [CrossRef]
- Nsouli, N.H.; Chubarova, E.V.; Al-Oweini, R.; Bassil, B.S.; Sadakane, M.; Kortz, U. Organoruthenium-containing heteropoly-23-tungstate family [{Ru(L)}2(α-XW11O39)2WO2]m− (L = benzene, p-cymene; X = GeIV, SiIV, m = 10; BIII, m = 12). Eur. J. Inorg. Chem. 2013, 1742–1747. [Google Scholar] [CrossRef]
- Reinoso, S.; Dickman, M.H.; Matei, M.F.; Kortz, U. 13-Tungstoborate stabilized by an organostannoxane hexamer. Inorg. Chem. 2007, 46, 4383–4385. [Google Scholar] [CrossRef] [PubMed]
- Reinoso, S.; Vitoria, P.; San Felices, L.; Gutiérrez-Zorrilla, J.M. Diiron(III)-containing 23-tungsto-2-borate: First evidence of 3d metal substitution in the {BW13} framework. Eur. J. Inorg. Chem. 2013, 1644–1648. [Google Scholar] [CrossRef]
- Brown, I.D.; Alternatt, D. Bond-valence parameters obtained from a systematic analysis of the inorganic crystal structure database. Acta Crystallogr. 1985, B41, 244–247. [Google Scholar] [CrossRef]
- Dougherty, D.A. The cation-π interaction. Acc. Chem. Res. 2013, 46, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Umezawa, Y.; Fantini, J.; Weiss, M.S.; Chakrabarti, P. CH−π hydrogen bonds in biological macromolecules. Phys. Chem. Chem. Phys. 2014, 16, 12648–12683. [Google Scholar] [CrossRef] [PubMed]
- Gallivan, J.P.; Dougherty, D.A. A computational study of cation–π interactions vs. salt bridges in aqueous media: Implications for protein engineering. J. Am. Chem. Soc. 2000, 122, 870–874. [Google Scholar] [CrossRef]
- Goto, Y.; Kamata, K.; Yamaguchi, K.; Uehara, K.; Hikichi, S.; Mizuno, N. Synthesis, structural characterization, and catalytic performance of dititanium-substituted γ-Keggin silicotungstate. Inorg. Chem. 2006, 45, 2347–2356. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Maeda, T.; Mizuno, K. Absorption and fluorescence spectroscopic properties of 1- and 1,4-silyl-substituted naphthalene derivatives. Molecules 2012, 17, 5108–5125. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhang, Q.; Zhao, Z.; Wu, X.; Chen, S.; Lu, C. New optical supramolecular compound constructed from a polyoxometalate cluster and an organic substrate. Inorg. Chem. 2008, 47, 8086–8090. [Google Scholar] [CrossRef] [PubMed]
- Matt, B.; Coudret, C.; Viala, C.; Jouvenot, D.; Loiseau, F.; Izzet, G.; Proust, A. Elaboration of covalently linked polyoxometalates with ruthenium and pyrene chromophores and characterization of their photophysical properties. Inorg. Chem. 2011, 50, 7761–7768. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.-G.; You, W.-S.; Li, J.; Liu, J.-F. Synthesis and spectroscopic characterization of organophosphoryl polyoxotungstates α-[{RP(O)}2SiW11O39]4−. Inorg. Chem. Commun. 2003, 6, 238–240. [Google Scholar] [CrossRef]
- Pretsch, E.; Clerc, T.; Seibl, J.; Simon, W. Tables of Spectral Data for Structure Determination of Organic Compounds; Springer-Verlag: Berlin, Germany, 1989. [Google Scholar]
- Hervé, G.; Tézé, A. Study of α and β-enneatungstosilicates and germanates. Inorg. Chem. 1977, 16, 2115–2117. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J. M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR chemical shifts of trace impurities: Common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- CrysAlisPro Software System; Version 171.36.24; Agilent Technologies UK Ltd.: Oxford, UK, 2012.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. 2009, D65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–836. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 73–78. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 2010, 132, 154014. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design an assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Andrae, D.; Haeussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Hirata, S.; Head-Gordon, M. Time-dependent density functional theory within the Tamm-Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘Chain-Of-Spheres’ algorithm for the Hartree–Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Izsak, R.; Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 2011, 135, 144105. [Google Scholar] [CrossRef] [PubMed]
- Petrenko, T.; Kossmann, S.; Neese, F. Efficient time-dependent density functional theory approximations for hybrid density functionals: Analytical gradients and parallelization. J. Chem. Phys. 2011, 134, 054116. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D–H···A | H···A | D···A | <D–H···A> |
|---|---|---|---|
| TBA-1 | |||
| C1–H1B···O21 | 2.85 | 3.47(3) | 121 |
| C1–H1B···O28 | 2.71 | 3.32(3) | 120 |
| C21–H21A···O25 | 2.59 | 3.09(3) | 111 |
| C21–H21A···O31 | 2.63 | 3.29(3) | 124 |
| C201–H20B···O221 | 2.86 | 3.40(3) | 115 |
| C201–H20B···O228 | 2.81 | 3.47(3) | 125 |
| C221–H22A···O225 | 2.61 | 3.27(3) | 123 |
| C221–H22A···O231 | 2.63 | 3.11(3) | 111 |
| TBA-2 | |||
| C1–H1B···O21 | 2.46 | 3.05(5) | 111 |
| C1–H1B···O28 | 2.55 | 3.15(5) | 127 |
| C21–H21A···O25 | 2.77 | 3.46(4) | 127 |
| C21–H21A···O31 | 2.89 | 3.44(4) | 115 |
| C201–H20B···O221 | 2.86 | 3.52(4) | 124 |
| C201–H20B···O228 | 2.94 | 3.47(4) | 115 |
| C221–H22A···O225 | 2.52 | 3.04(5) | 113 |
| C221–H22A···O231 | 2.48 | 3.18(5) | 127 |
| Parameters | TBA-1 | TBA-2 |
|---|---|---|
| Formula | C70H128BN3O41P2W11 | C70H127N3O41P2SiW11 |
| FW (g·mol−1) | 3762.8 | 3779.1 |
| Crystal System | Triclinic | Triclinic |
| Space Group | P−1 | P−1 |
| a (Å) | 17.4020(5) | 16.9676(5) |
| b (Å) | 24.2390(3) | 24.3022(3) |
| c (Å) | 28.6175(5) | 28.5333(7) |
| α (˚) | 70.095(2) | 70.030(2) |
| β (˚) | 72.336(2) | 72.703(2) |
| γ (˚) | 89.976(2) | 89.957(2) |
| V (Å3) | 10743.3(4) | 10491.3(4) |
| Z | 4 | 4 |
| ρcalcd (g·cm−3) | 2.326 | 2.393 |
| μ (mm−1) | 22.022 | 12.124 |
| T (K) | 150(2) | 100(2) |
| λ (Å) | 1.54184 (Cu Kα) | 0.71073 (Mo Kα) |
| Collected Reflections | 80334 | 65644 |
| Unique Reflections (Rint) | 38193 (0.126) | 36457 (0.048) |
| Observed Reflections [I > 2σ(I)] | 23423 | 21696 |
| Parameters/Restraints | 767/250 | 1459/546 |
| R(F)a [I > 2σ(I)] | 0.095 | 0.083 |
| wR(F2)a [all data] | 0.270 | 0.216 |
| GoF | 0.964 | 1.078 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andino, N.; Artetxe, B.; Reinoso, S.; Vitoria, P.; San Felices, L.; Martínez, J.I.; López Arbeloa, F.; Gutiérrez-Zorrilla, J.M. Naphthyl-Containing Organophosphonate Derivatives of Keggin-Type Polyoxotungstates. Inorganics 2016, 4, 14. https://doi.org/10.3390/inorganics4020014
Andino N, Artetxe B, Reinoso S, Vitoria P, San Felices L, Martínez JI, López Arbeloa F, Gutiérrez-Zorrilla JM. Naphthyl-Containing Organophosphonate Derivatives of Keggin-Type Polyoxotungstates. Inorganics. 2016; 4(2):14. https://doi.org/10.3390/inorganics4020014
Chicago/Turabian StyleAndino, Nerea, Beñat Artetxe, Santiago Reinoso, Pablo Vitoria, Leire San Felices, Jose I. Martínez, Fernando López Arbeloa, and Juan M. Gutiérrez-Zorrilla. 2016. "Naphthyl-Containing Organophosphonate Derivatives of Keggin-Type Polyoxotungstates" Inorganics 4, no. 2: 14. https://doi.org/10.3390/inorganics4020014
APA StyleAndino, N., Artetxe, B., Reinoso, S., Vitoria, P., San Felices, L., Martínez, J. I., López Arbeloa, F., & Gutiérrez-Zorrilla, J. M. (2016). Naphthyl-Containing Organophosphonate Derivatives of Keggin-Type Polyoxotungstates. Inorganics, 4(2), 14. https://doi.org/10.3390/inorganics4020014