
Synthesis, Characterization and Applications in Catalysis of Polyoxometalate/Zeolite Composites


Abstract
:1. Introduction
2. Synthesis of Molybdic Keggin Species or Their Salts in/on Zeolites
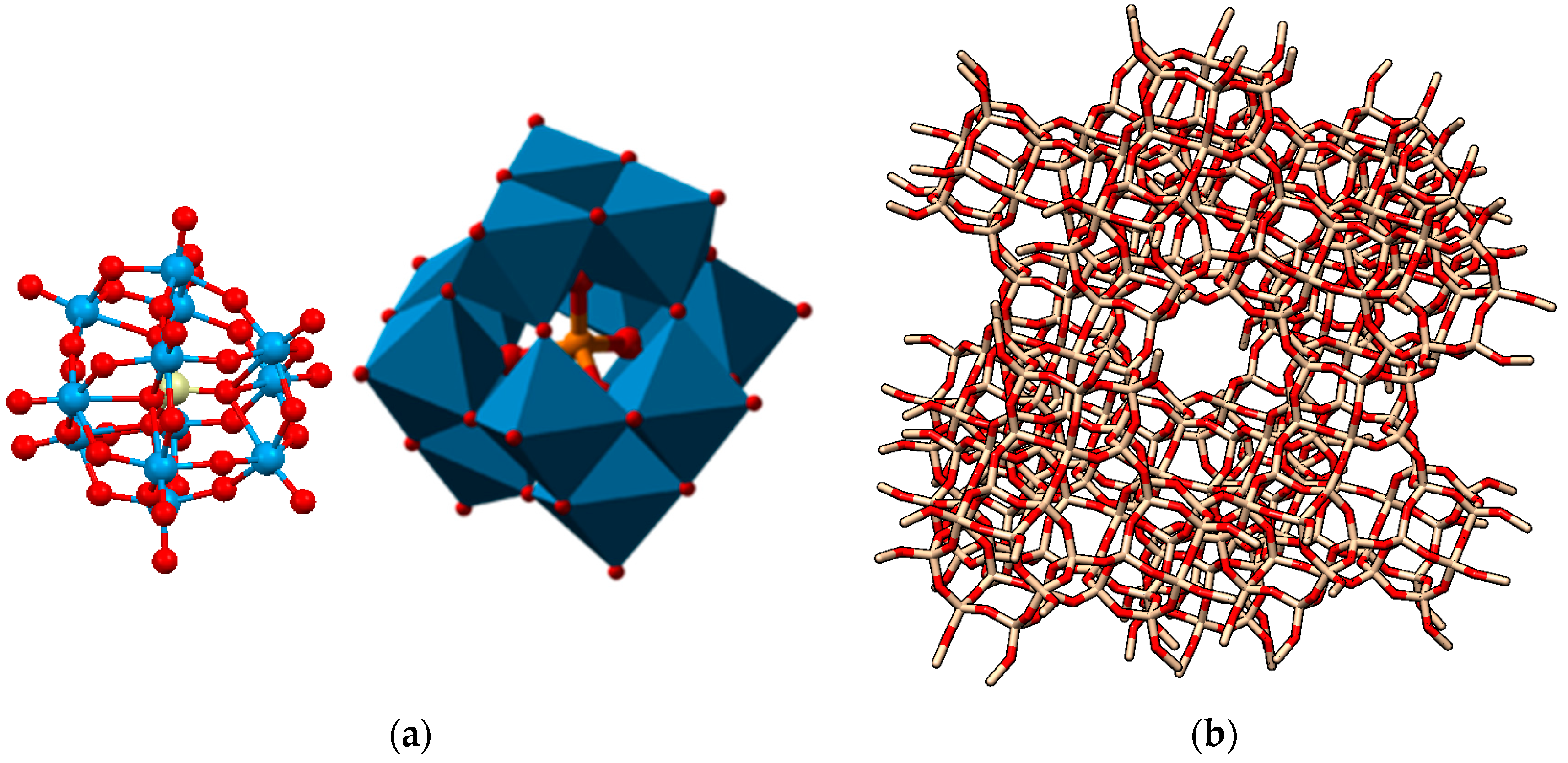
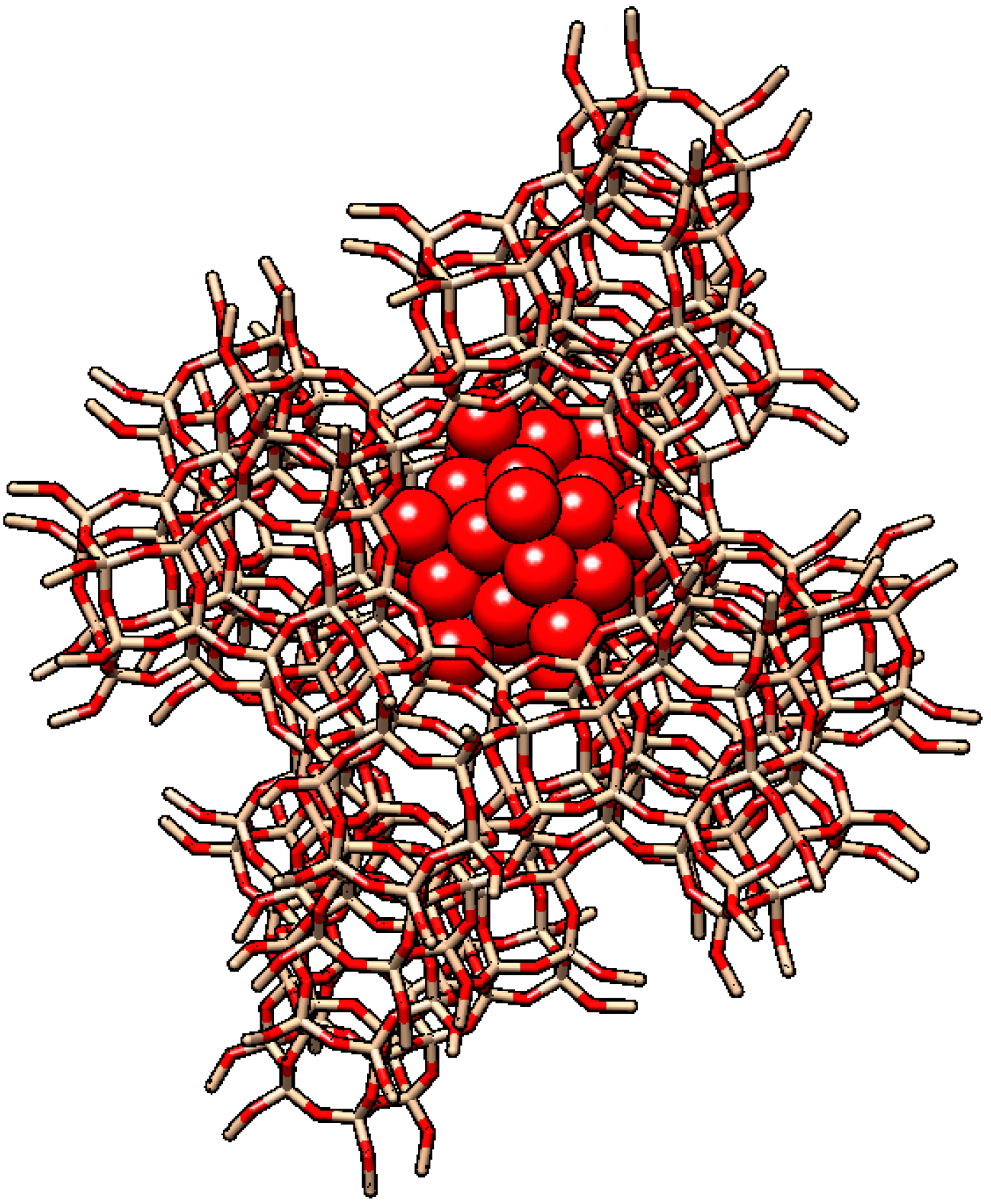
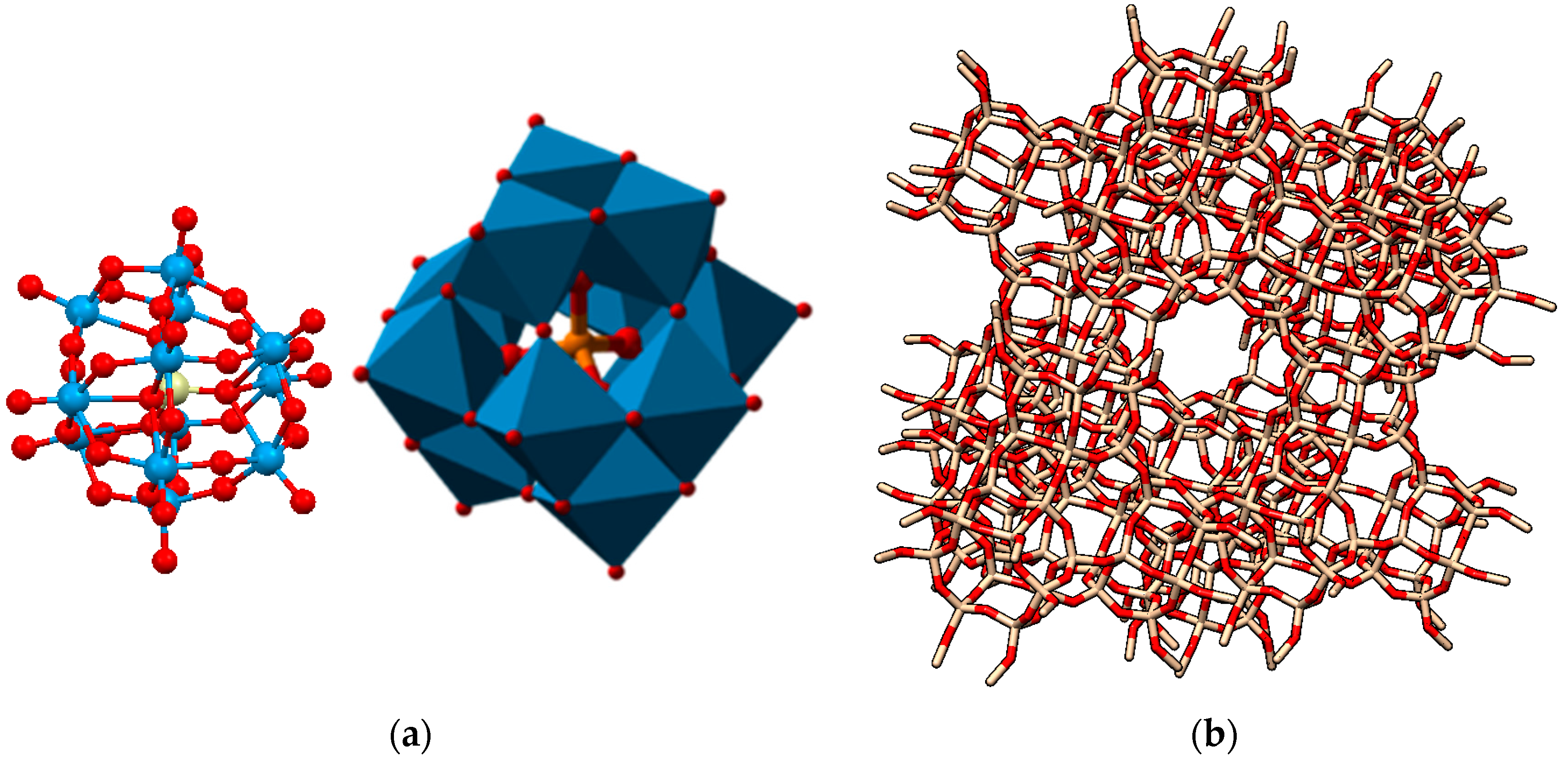
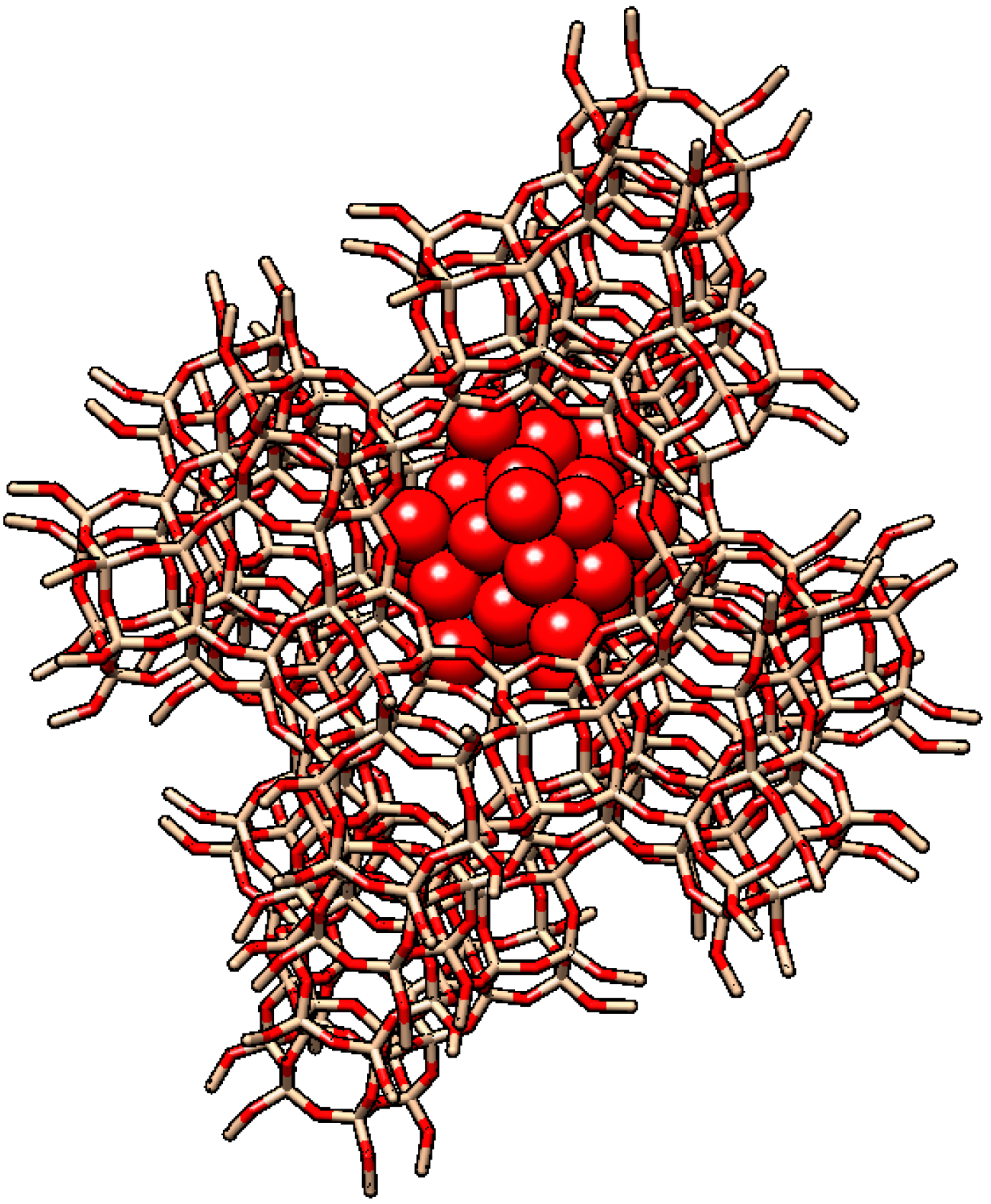
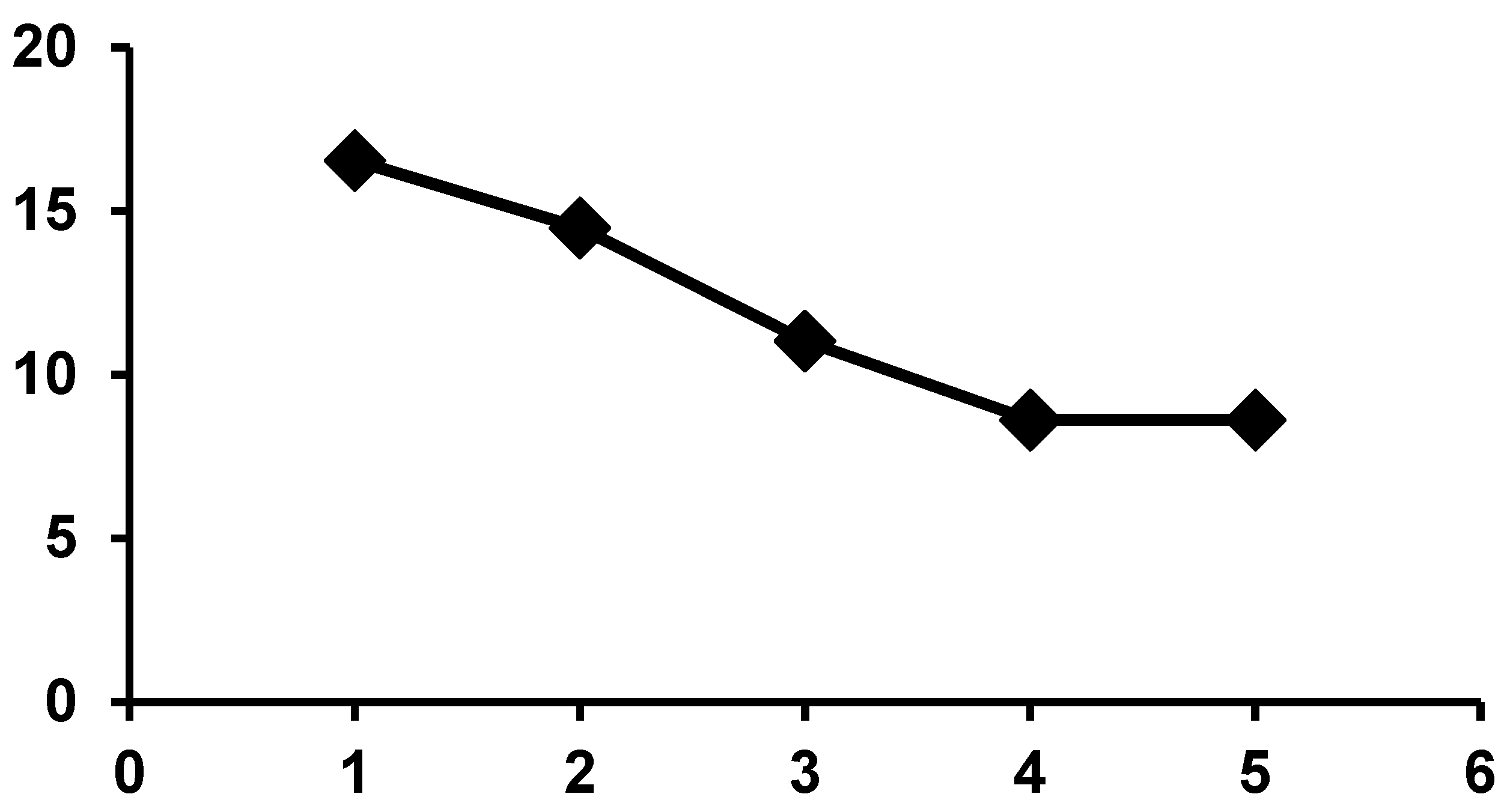
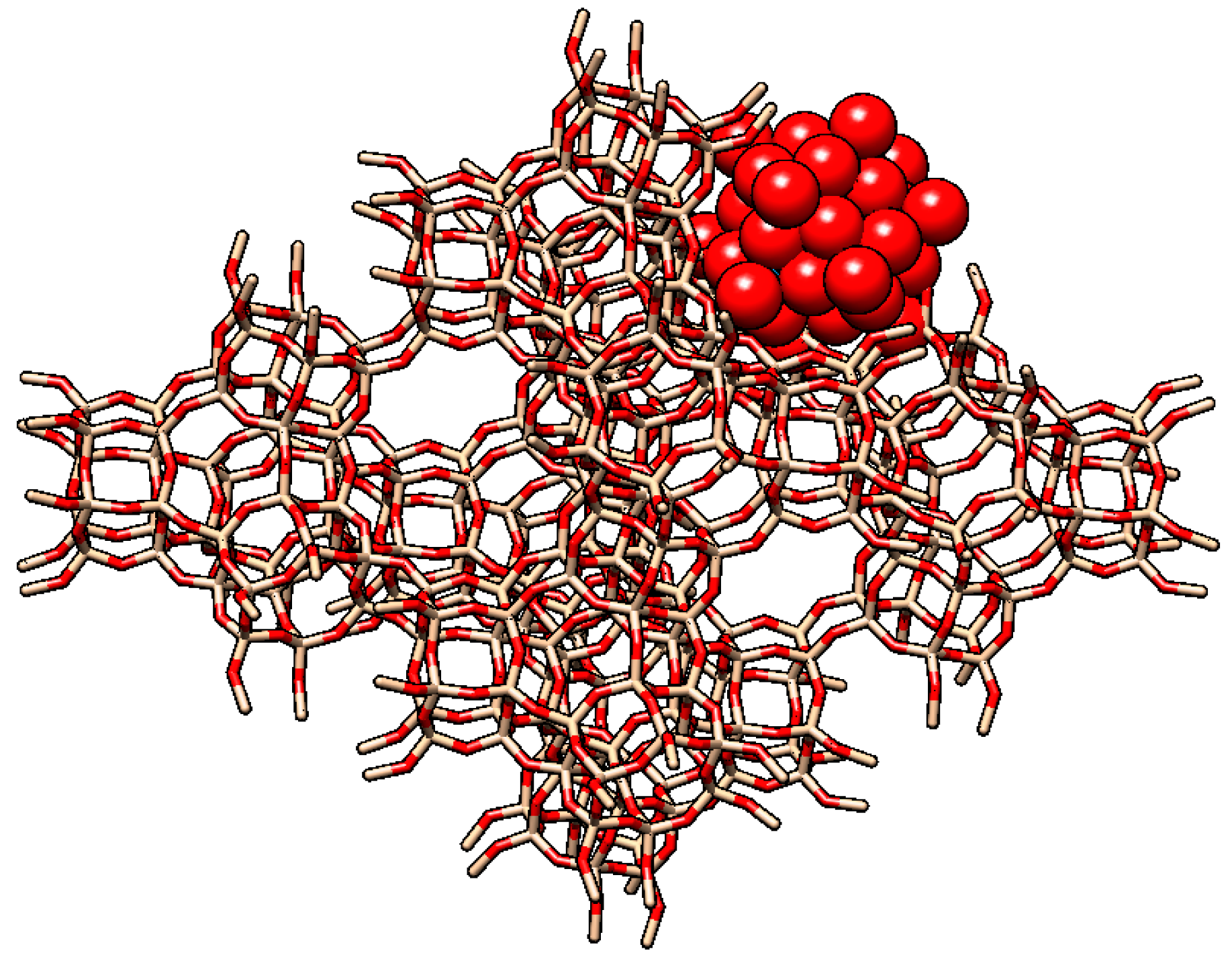
2.1. The Pioneering Works of Mukai et al. [12,13,14,15]
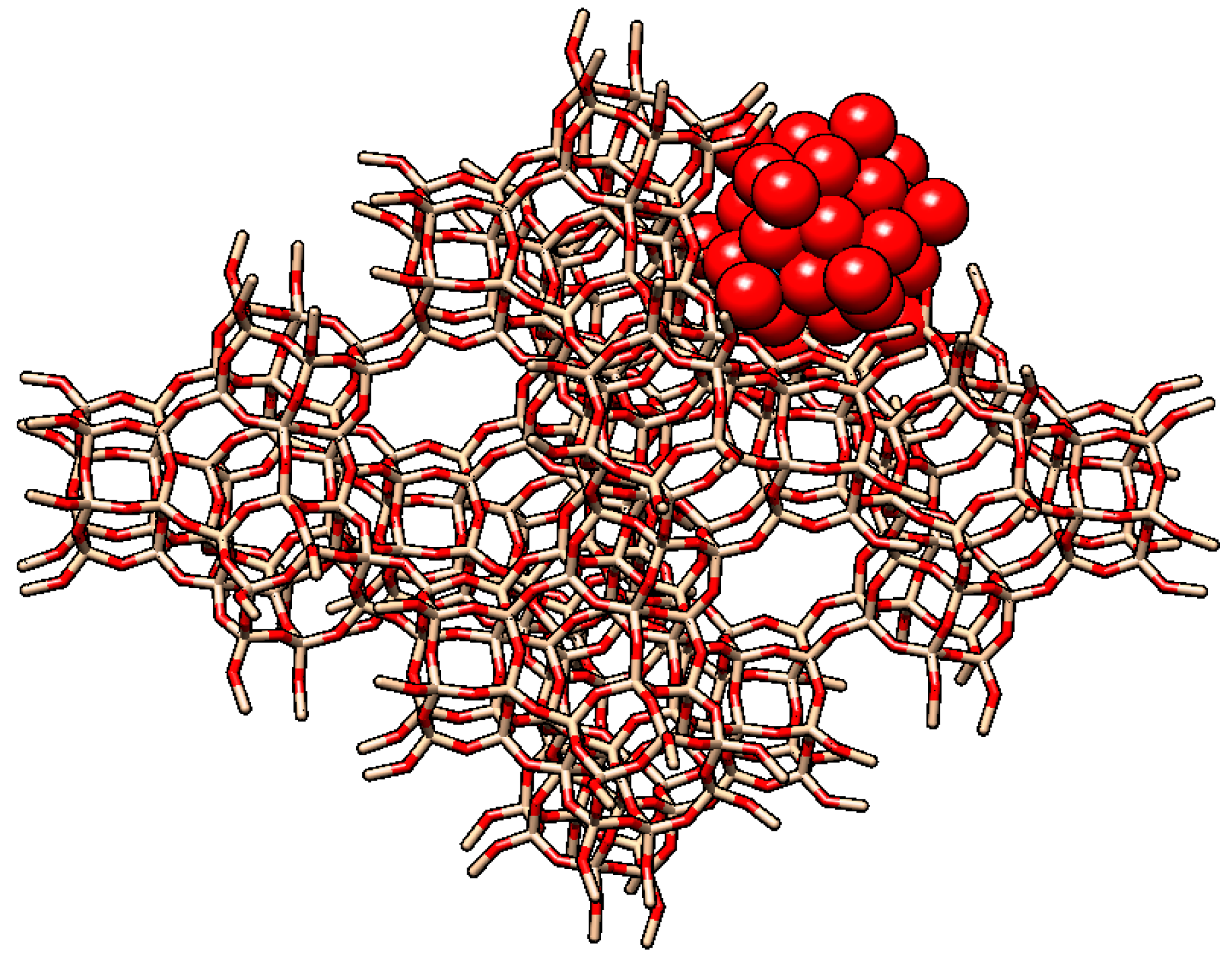
2.2. Other Reports of Syntheses of Encapsulated PMo12 in Y Zeolite
2.3. Synthesis of PMo12 on ZSM-5
2.4. Applications in Catalysis
3. Synthesis of Tungstic Keggin Species or their Salts in/on Zeolites
4. Synthesis of the Zeolite around the Polyoxometalate
5. Keggin Polyoxometalates Supported on Zeolites
5.1. Preliminary Studies
5.2. Applications in Refining (Hydroisomerization of Alkanes, Hydrodesulfurization)
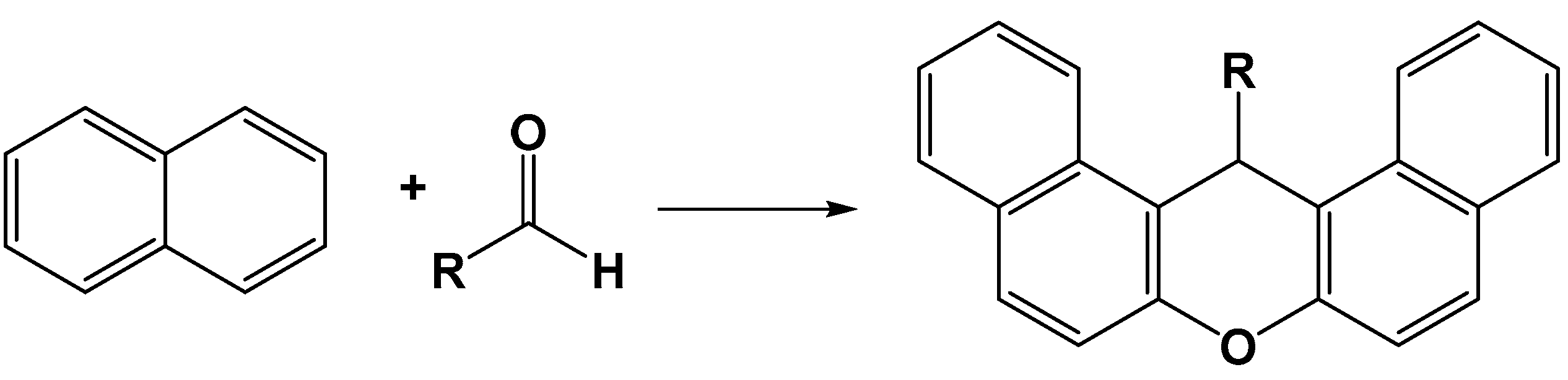
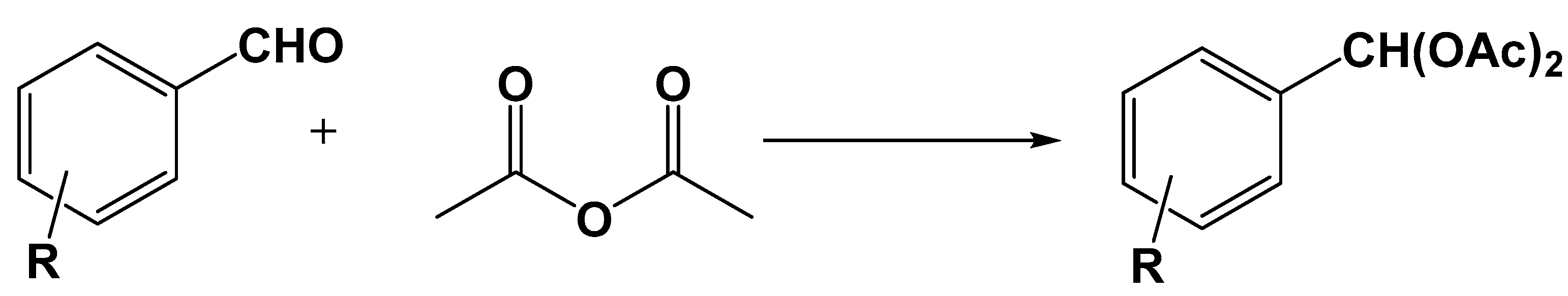
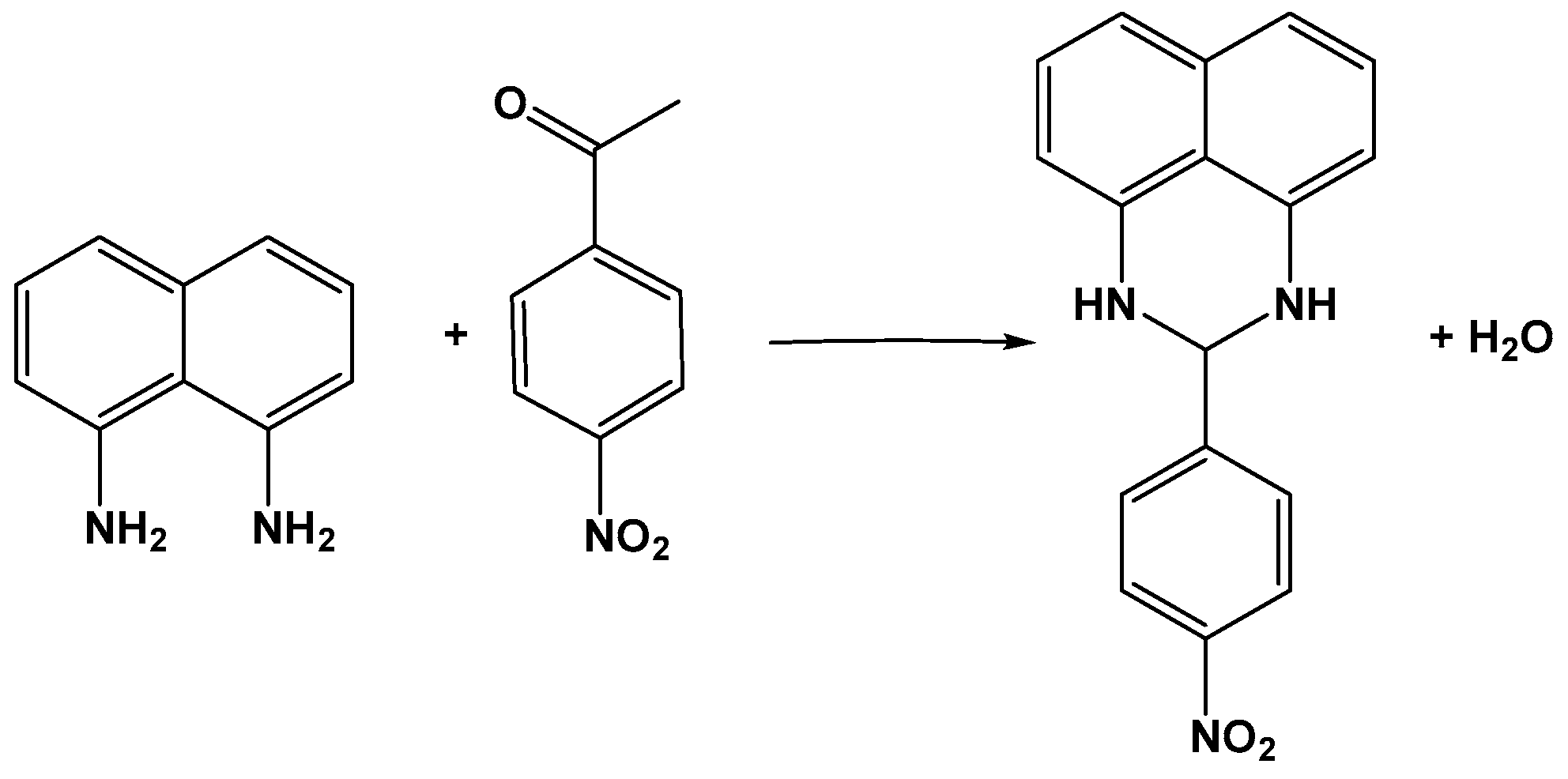
5.3. Applications in Organic Chemistry
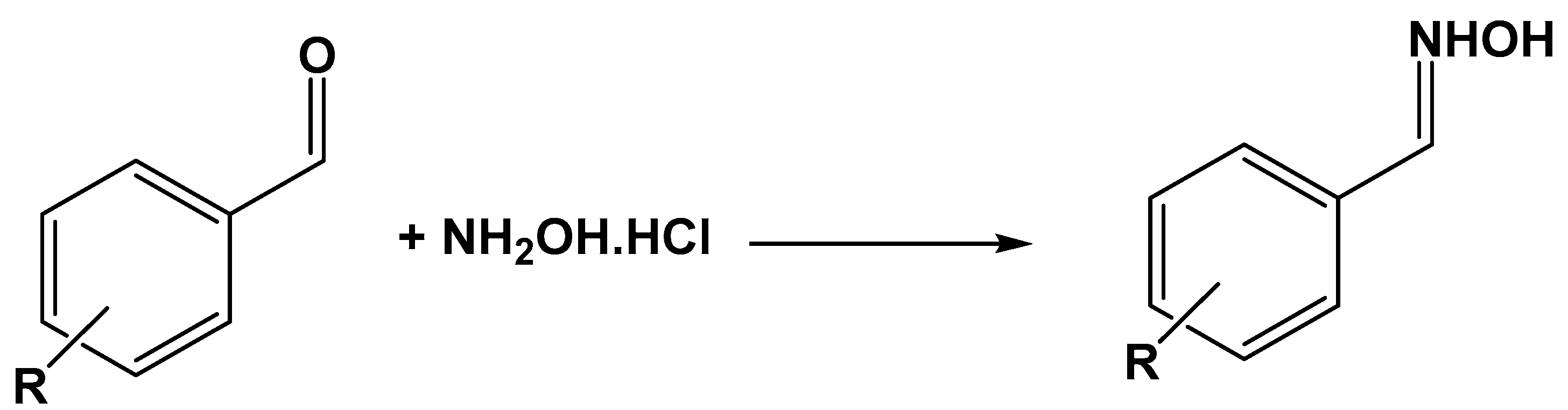
5.4. Applications in Photocatalysis
5.4.1. Systems without Titanium
5.4.2. Systems with Titanium
- A charge separation is created in the TiO2 species upon irradiation by the UV component of the light source.
- The electron from the conduction band of TiO2 is transferred to the polyoxometalate, which is reduced to a visibly active species PM12−.
- This reduced species is excited by the visible radiation leading to an excited PM12* species, which is able to transfer the excited electrons to the transition metal in the zeolite. This transfer is not direct but occurs via the Lewis acid sites of the zeolite.
- The excited species PM12* can also be formed by direct excitation of the polyoxometalate by the UV radiation followed by a reduction via the sacrificial electron donor.
- Finally, the substrate reacts with the transition metal.
6. Other Polyoxometalates Supported on Zeolites
7. Conclusions
Conflicts of Interest
References
- Pope, M.T. Heteropoly and Isopoly Oxometalates; Springer-Verlag: Berlin, Germany, 1983. [Google Scholar]
- Coronado, E.; Gomez-Garcia, C.J. Polyoxometalate-based molecular materials. Chem. Rev. 1998, 98, 273–296. [Google Scholar] [CrossRef] [PubMed]
- Katsoulis, D.E. A survey of applications of polyoxometalates. Chem. Rev. 1998, 98, 359–388. [Google Scholar] [CrossRef] [PubMed]
- Kozhevnikov, I.V. Catalysis by heteropoly acids and multicomponent polyoxometalates in liquid-phase reactions. Chem. Rev. 1998, 98, 171–198. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, N.; Misono, M. Heterogeneous catalysis. Chem. Rev. 1998, 98, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Peters, F.; Pope, M.T.; Gatteschi, D. Polyoxometalates: Very large clusters-nanoscale magnets. Chem. Rev. 1998, 98, 239–272. [Google Scholar] [CrossRef] [PubMed]
- Rhule, J.T.; Hill, C.L.; Judd, D.A.; Schinazi, R.F. Polyoxometalates in medicine. Chem. Rev. 1998, 98, 327–358. [Google Scholar] [CrossRef] [PubMed]
- Sadakane, M.; Steckhan, E. Electrochemical properties of polyoxometalates as electrocatalysts. Chem. Rev. 1998, 98, 219–238. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, I.A. Homogeneous-phase electron-transfer reactions of polyoxometalates. Chem. Rev. 1998, 98, 113–170. [Google Scholar] [CrossRef] [PubMed]
- Keggin, J.F. Structure of the molecule of 12-phosphotungstic acid. Nature 1933, 131, 908–909. [Google Scholar] [CrossRef]
- Database of Zeolite Structures. Available online: http://www.iza-structure.org/databases/ (accessed on 2 May 2016).
- Mukai, S.R.; Lin, L.; Masuda, T.; Hashimoto, K. Key factors for the encapsulation of Keggin-type heteropoly acids in the supercages of Y-type zeolite. Chem. Eng. Sci. 2001, 56, 799–804. [Google Scholar] [CrossRef]
- Mukai, S.R.; Masuda, T.; Ogino, I.; Hashimoto, K. Preparation of encaged heteropoly acid catalyst by synthesizing 12-molybdophosphoric acid in the supercages of Y-type zeolite. Appl. Catal. A Gen. 1997, 165, 219–226. [Google Scholar] [CrossRef]
- Mukai, S.R.; Ogino, I.; Lin, L.; Kondo, Y.; Masuda, T.; Hashimoto, K. A stable “ship in the bottle” type 12-molybdophosphoric acid encaged Y-type zeolite catalyst for liquid phase reactions. React. Kinet. Catal. Lett. 2000, 69, 253–258. [Google Scholar] [CrossRef]
- Mukai, S.R.; Shimoda, M.; Lin, L.; Tamon, H.; Masuda, T. Improvement of the preparation method of “ship-in-the-bottle” type 12-molybdophosphoric acid encaged Y-type zeolite catalysts. Appl. Catal. A Gen. 2003, 256, 107–113. [Google Scholar] [CrossRef]
- Tran, M.H.; Ohkita, H.; Mizushima, T.; Kakuta, N. Hydrothermal synthesis of molybdenum oxide catalyst: Heteropoly acids encaged in US-Y. Appl. Catal. A Gen. 2005, 287, 129–134. [Google Scholar] [CrossRef]
- Wang, H.; Wang, R. Performance evalutaion of “ship-in-the-bottle” type heteropoly acid encaged Y-type zeolite as catalyst for oxidative desulfurization. Collect. Czechoslov. Chem. Commun. 2011, 76, 1595–1605. [Google Scholar] [CrossRef]
- Wei, R.; Guo, M.; Wang, J. Preparation, characterization and catalytic behavior of 12-molybdophosphoric acid encapsulated in the supercage of Cs+-exchanged Y zeolite. Chin. J. Chem. Eng. 2009, 17, 58–63. [Google Scholar] [CrossRef]
- Chen, L.; Wang, X.; Guo, X.; Guo, H.; Liu, H.; Chen, Y. In situ nanocrystalline HZSM-5 zeolites encaged heteropoly acid H3PMo12O40 and Ni catalyst for hydroconversion of n-octane. Chem. Eng. Sci. 2007, 62, 4469–4478. [Google Scholar] [CrossRef]
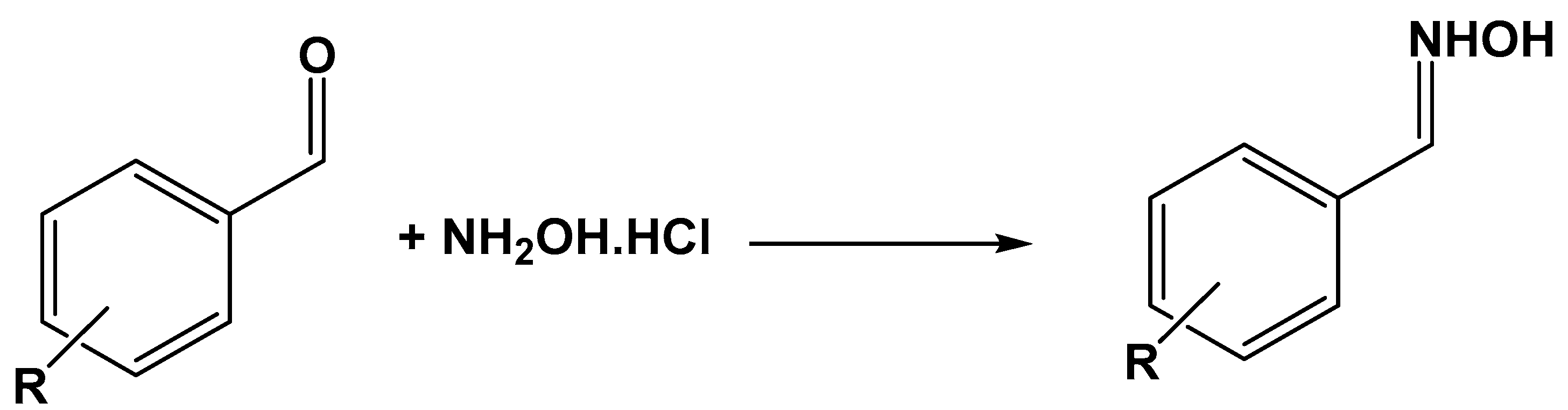
- Moghadam, M.; Tangestaninejad, S.; Mirkhani, V.; Mohammadpoor-Baltork, I.; Moosavifar, M. Host (nanocavity of dealuminated zeolite Y)-guest (12-molybdophosphoric acid) nanocomposite material: An efficient and reusable catalyst for oximation of aldehydes. Appl. Catal. A Gen. 2009, 358, 157–163. [Google Scholar] [CrossRef]
- Pourbeyram, S.; Moosavifar, M.; Hasanzadeh, V. Electrochemical characterization of the encapsulated polyoxometalates (POMs) into the zeolite. J. Electroanal. Chem. 2014, 714–715, 19–24. [Google Scholar] [CrossRef]
- Moghadam, M.; tangestaninejad, S.; Mirkhani, V.; Mohammadpoor-Baltork, I.; Moosavifar, M. Host (nanocavity of dealuminated zeolite-Y)-guest (12-molybdophosphoric acid) nanocomposite material: An efficient and reusable catalyst for synthesis of 14-substituted-14-H-dibenzo [a,j] xanthenes under thermal and microwave irradiation conditions. C. R. Chim. 2011, 14, 489–495. [Google Scholar] [CrossRef]
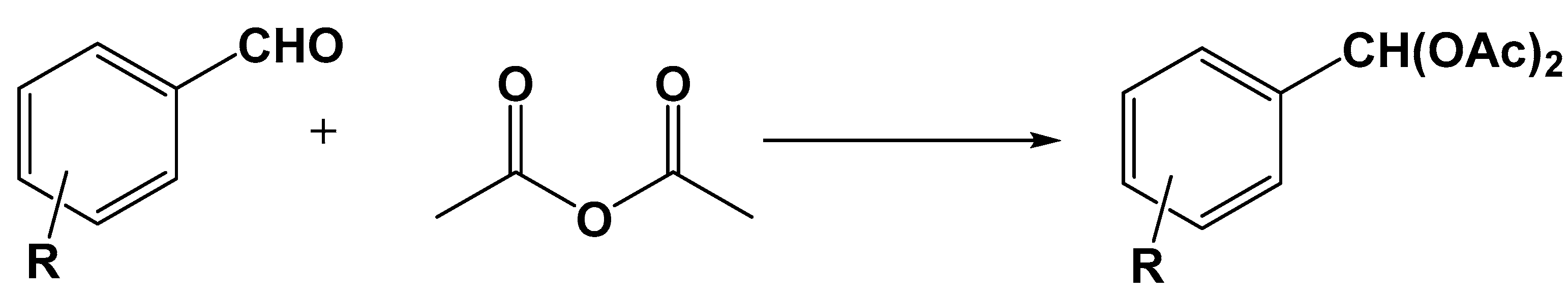
- Moosavifar, M.; Tangestaninejad, S.; Moghadam, M.; Mirkhani, V.; Mohammadpoor-Baltork, I. Host (nanocavity of zeolite-Y)-guest (molybdophosphoric acid) nanocomposite materials: An efficient catalyst for solvent-free synthesis and deprotection of 1,1-diacetates. C. R. Chim. 2011, 14, 953–956. [Google Scholar] [CrossRef]
- Ferreira, P.; Fonseca, I.M.; Ramos, A.M.; Vital, J.; Castanheiro, J.E. Esterification of glycerol with acetic acid over dodecamolybdophosphoric acid encaged in USY zeolite. Catal. Commun. 2009, 10, 481–484. [Google Scholar] [CrossRef]
- Vital, J.; Ramos, A.M.; Silva, I.F.; Castanheiro, J.E.; Blanco, M.N.; Caceres, C.; Vasquez, P.; Pizzio, L.; Thomas, H. Hydration of α-pinene over heteropolyacids encaged in USY zeolites. Stud. Surf. Sci. Catal. 2001, 135, 234–241. [Google Scholar]
- Castanheiro, J.E.; Ramos, A.M.; Fonseca, I.; Vital, J. The acid-catalyzed reaction of α-pinene over molybdophosphoric acid immobilised in dense polymeric membranes. Catal. Today 2003, 82, 187–193. [Google Scholar] [CrossRef]
- Sulikowski, B.; Haber, J.; Kubacka, A.; Pamin, K.; Olejniczak, Z.; Ptaszynski, J. Novel “ship-in-th-bottle” type catalyst: Evidence for encapsulation of 12-tungstophosphoric acid in the supercage of synthetic faujasite. Catal. Lett. 1996, 39, 27–31. [Google Scholar] [CrossRef]
- Jin, D.; Gao, J.; Hou, Z.; Guo, Y.; Lu, X.; Zhu, Y.; Zheng, X. Microwave assisted in situ synthesis of USY-encapsulated heteropoly acid (HPW-USY) catalysts. Appl. Catal. A Gen. 2009, 352, 259–264. [Google Scholar] [CrossRef]
- Anandan, S.; Ryu, S.Y.; Cho, W.J.; Yoon, M. Heteropolytungstic acid (H3PW12O40)-encapsulated into the titanium-exchanged HY (TiHY) zeolite: A novel photocatalyst for photoreduction of methyl orange. J. Mol. Catal. A Chem. 2003, 195, 201–208. [Google Scholar] [CrossRef]
- Anandan, S.; Yoon, M. Heteropolyacid-encapsulated TiHY zeolite as an inorganic photosynthetic reaction center mimicking the plant systems. J. Photochem. Photobiol. A Chem. 2003, 160, 181–184. [Google Scholar] [CrossRef]
- Yoon, M.; Chang, J.A.; Kim, Y.; Choi, Y.; Kim, K.; Lee, S.J. Heteropoly acid-incorporated TiO2 colloids as novel photocatalytic systems resembling the photosynthetic reaction center. J. Phys. Chem. B 2001, 105, 2539–2545. [Google Scholar] [CrossRef]
- Costa, A.A.; Braga, P.R.S.; de Macedo, J.L.; Dias, J.A.; Dias, S.C.L. Structural effects of WO3 incorporation on USY zeolite and application to free fatty acids esterification. Microporous Mesoporous Mater. 2012, 147, 142–148. [Google Scholar] [CrossRef]
- Zendehdel, M.; Mobinikhaledi, A.; Alikhani, H.; Jafari, N. Preparation of heteropoly acid/porous hybrid materials and investigation of their catalytic behavior in the synthesis of perimidine. J. Chin. Chem. Soc. 2010, 57, 683–689. [Google Scholar] [CrossRef]
- Sun, C.; Zhang, F.; Cao, J. A “build-bottle-around-ship” method to encapsulate ammonium molybdophosphate in zeolite Y. An efficient adsorbent for cesium. J. Colloid Interface Sci. 2015, 455, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Pamin, K.; Kubacka, A.; Olejniczak, Z.; Haber, J.; Sulikowski, B. Immobilization of dodecatungstophosphoric acid on dealuminated zeolite Y: A physicochemical study. Appl. Catal. A Gen. 2000, 194–195, 137–146. [Google Scholar] [CrossRef]
- Lefebvre, F. 31P MAS NMR Study of H3PW12O40 Supported on Silica. Formation of (Si–OH2+)(H2PW12O40−). Chem. Commun. 1992, 1992, 756–757. [Google Scholar] [CrossRef]
- Olejniczak, Z.; Sulikowski, B.; Kubacka, A.; Gasior, M. Heterogenization of 12-tungstophosphoric acid on stabilized zeolite Y. Top. Catal. 2000, 11, 391–400. [Google Scholar] [CrossRef]
- Sulikowski, B.; Rachwalik, R. Catalytic properties of heteropoly acid/zeolite hybrid materials: Toluene disproportionation and transalkylation with 1,2,4-trimethylbenzene. Appl. Catal. A Gen. 2003, 256, 173–182. [Google Scholar] [CrossRef]
- Haber, J.; Pamin, K.; Matachowski, L.; Mucha, D. Catalytic performance of the dodecatungstophosphoric acid on different supports. Appl. Catal. A Gen. 2003, 256, 141–152. [Google Scholar] [CrossRef]
- Wang, J.; Lin, Z.; Han, S.Y.; Eum, M.; Lee, C.W. 12-tungstophosphoric acid supported on dealuminated USY as a catalyst for hydroisomerization of n-heptane. J. Ind. Eng. Chem. 2003, 9, 281–286. [Google Scholar]
- Wang, J.; Jiang, D.; Baeg, J.O.; Lee, C.W. Hydroisomerization of n-heptane over modified USY-supported H3PW12O40 catalysts: Effect of hydrothermal treatment for USY. J. Ind. Eng. Chem. 2004, 10, 454–459. [Google Scholar]
- Wei, R.P.; Wang, J.; Jiang, D.M.; Ren, X.Q. 12-Phosphotungstic acid catalysts supported on β zeolite for hydroisomerization of n-heptane. J. Mol. Catal. (China) 2005, 19, 251–254. [Google Scholar]
- Wei, R.; Wang, J.; Xiao, G. Hydroisomerization of n-heptane over Cr promoted Pt-bearing H3PW12O40 catalysts supported on dealuminated USY zeolite. Catal. Lett. 2009, 127, 360–367. [Google Scholar] [CrossRef]
- Wei, R.P.; Gu, Y.B.; Wang, J. Hydroisomerization of n-heptane over bimetal-bearing H3PW12O40 catalysts supported on dealuminated USY zeolite. Sci. China Ser. B Chem. 2008, 51, 120–127. [Google Scholar] [CrossRef]
- Gu, Y.B.; Wei, R.P.; Ren, X.Q.; Wang, J. Cs salts of 12-tungstophosphoric acid supported on dealuminated USY as catalysts for hydroisomerization of n-heptane. Catal. Lett. 2007, 113, 41–45. [Google Scholar] [CrossRef]
- Kostova, N.G.; Spojakina, A.A.; Dutkova, E.; Balaz, P. Mechanochemical approach for preparation of Mo-containing β-zeolite. J. Phys. Chem. Solids 2007, 68, 1169–1172. [Google Scholar] [CrossRef]
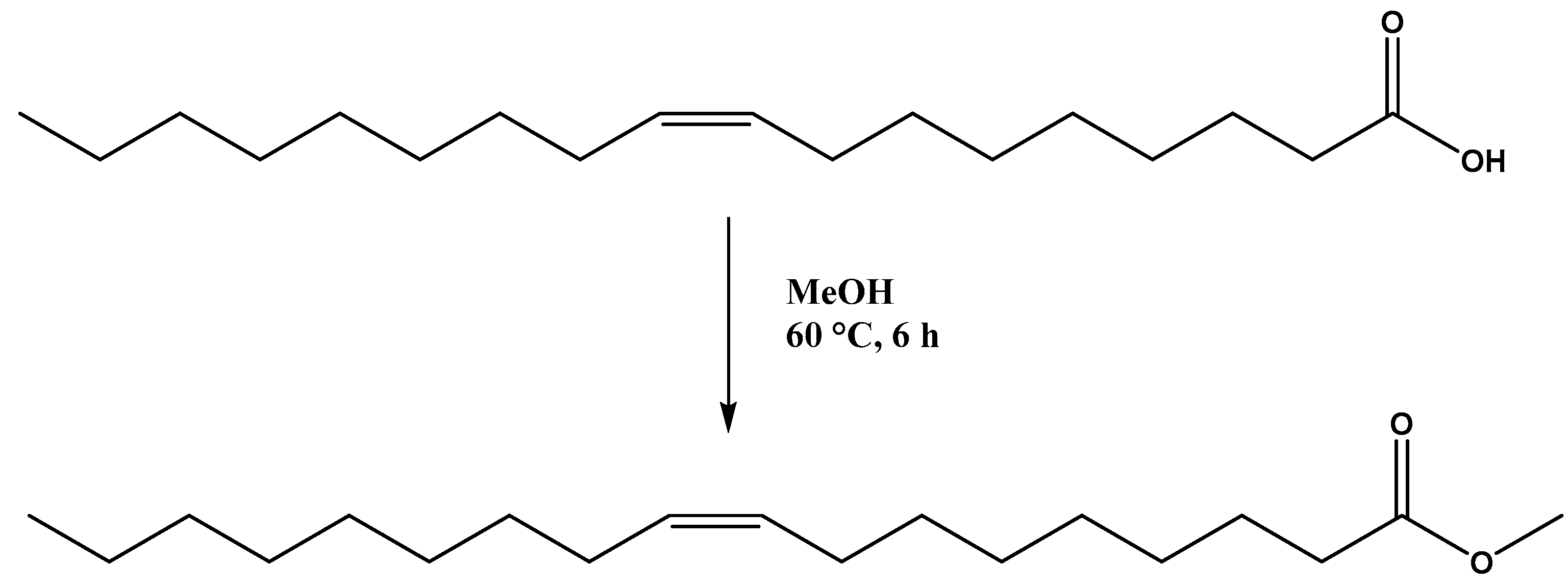
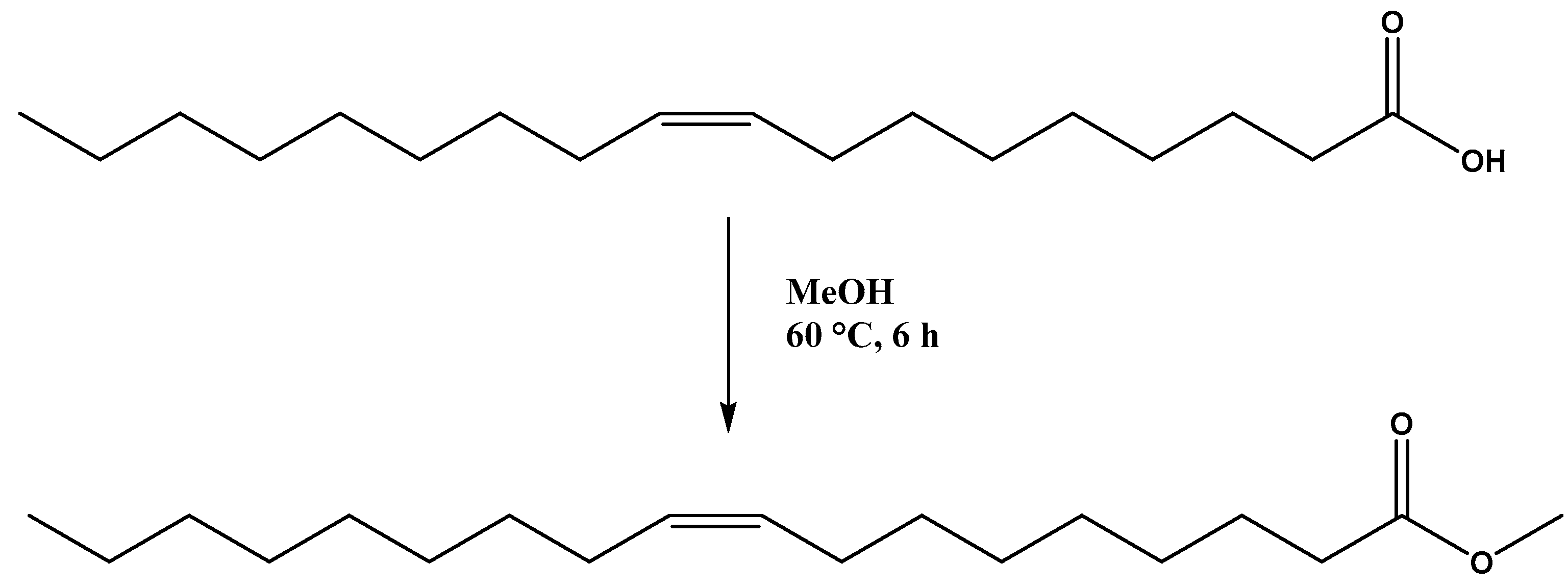
- Patel, A.; Narkhede, N. 12-tungstophosphoric acid anchored to zeolite Hβ: Synthesis, characterization, and biodiesel production by esterification of oleic acid with methanol. Energy Fuels 2012, 26, 6025–6032. [Google Scholar] [CrossRef]
- Narkhede, N.; Patel, A. Biodiesel production by esterification of oleic acid and transesterification of soybean oil using a new solid acid catalyst comprising 12-tungstosilicic acid and zeolite Hβ. Ind. Eng. Chem. Res. 2013, 52, 13637–13644. [Google Scholar] [CrossRef]
- Srinivas, M.; Sree, R.; Raveendra, G.; Kumar, C.R.; Prasad, P.S.S.; Lingaiah, N. Selective etherification of glycerol with tert-butanol over 12-tungstophosphoric acid catalysts supported on Y-zeolite. Indian J. Chem. 2014, 53A, 524–529. [Google Scholar]
- Zhang, F.; Wang, J.; Yuan, C.; Ren, X. Catalytic performances of heteropoly compounds supported on dealuminated ultra-stable Y zeolite for liquid-phase esterification. Sci. China Ser. B Chem. 2006, 49, 140–147. [Google Scholar] [CrossRef]
- Yuan, C.; Zhang, F.; Wang, J.; Ren, X. 12-Phosphotungstic acid and its Cs salt supported on various porous carriers as catalysts for the synthesis of fructone. Catal. Commun. 2005, 6, 721–724. [Google Scholar] [CrossRef]
- Zhang, F.; Yuan, C.; Wang, J.; Kong, Y.; Zhu, H.; Wang, C. Synthesis of fructone over dealuminated USY supported heteropoly acid and its salt catalysts. J. Mol. Catal. A Chem. 2006, 247, 130–137. [Google Scholar] [CrossRef]
- Zhang, F.; Yuan, C.; Ren, X.; Wang, J. Characterization of supported heteropoly acid (salt) and its catalytic behavior in synthesis of fructone. J. Chem. Ind. Eng. 2006, 57, 762–768. [Google Scholar]
- Yuan, C.; Zhang, F.; Wang, J.; Ren, X. Catalytic activity of Cs-salts of 12-phosphotungstic acid supported on dealuminated ultra-stable zeolite in synthesis of fructone-B. Petrochem. Technol. 2005, 34, 258–264. [Google Scholar]
- Zhang, F.M.; Wang, J.; Yuan, C.S.; Ren, X.Q. A pronounced catalytic activity of heteropoly compounds supported on dealuminated USY for liquid-phase esterification of acetic acid with n-butanol. Catal. Lett. 2005, 102, 171–174. [Google Scholar] [CrossRef]
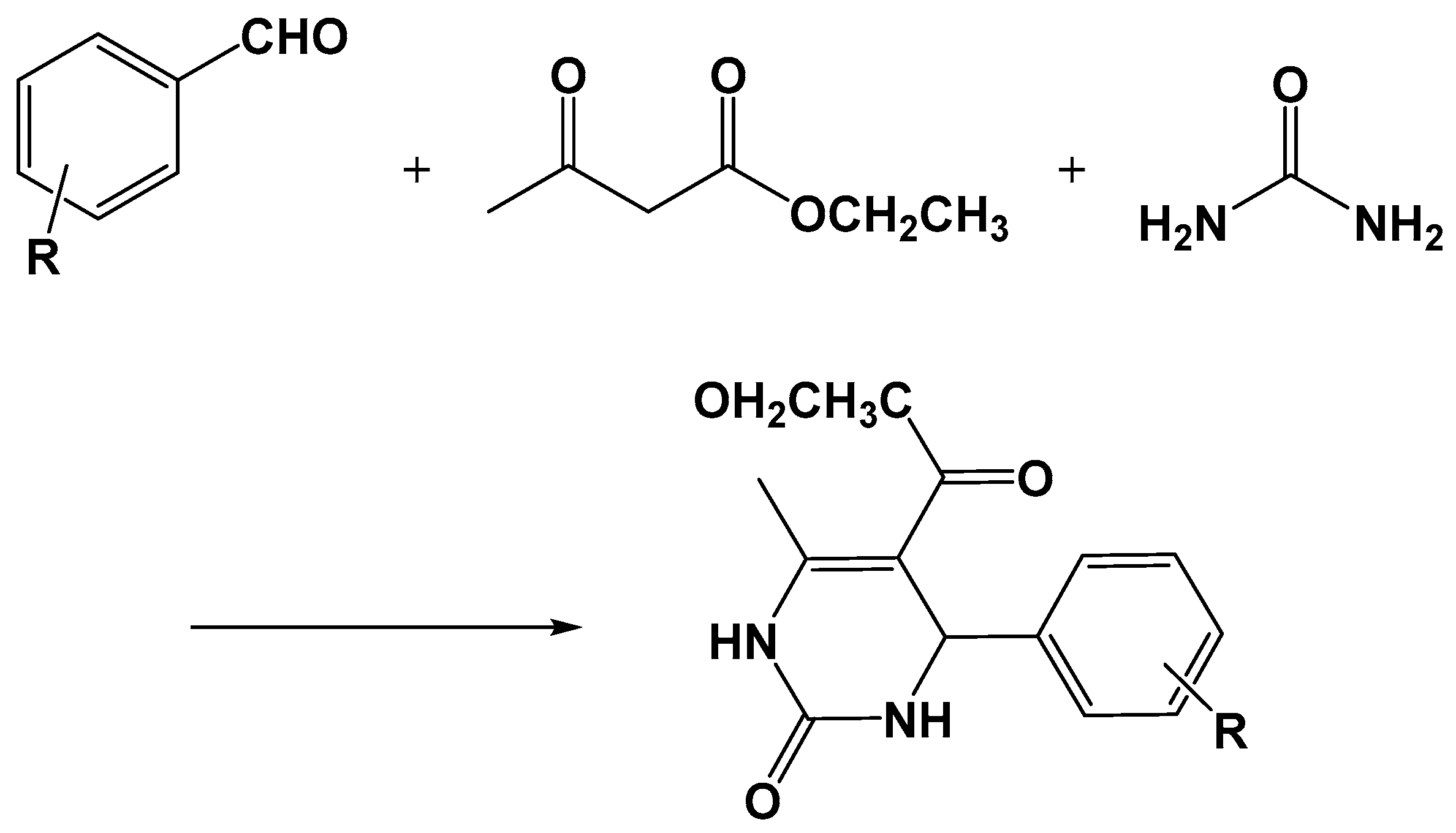
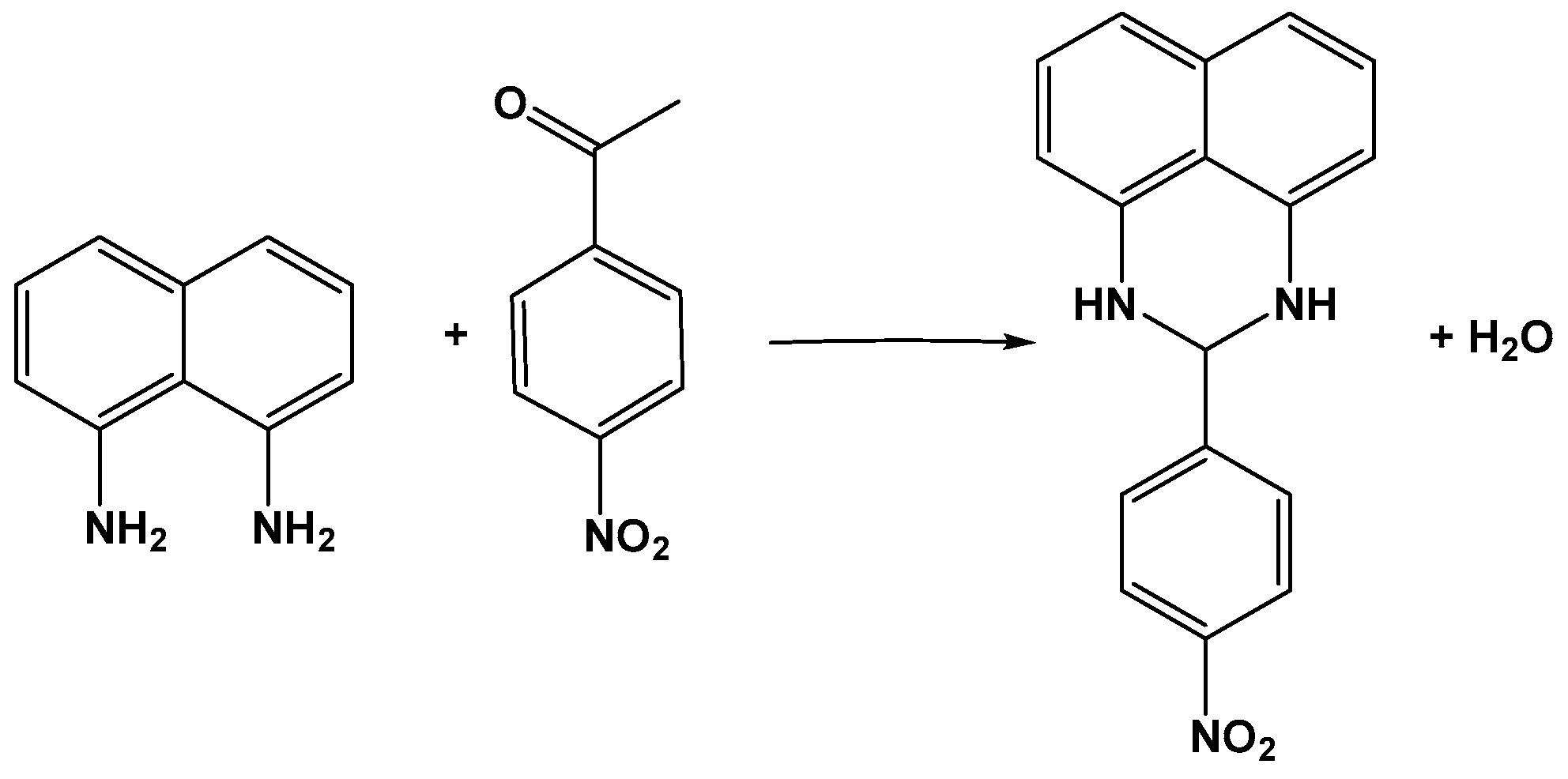
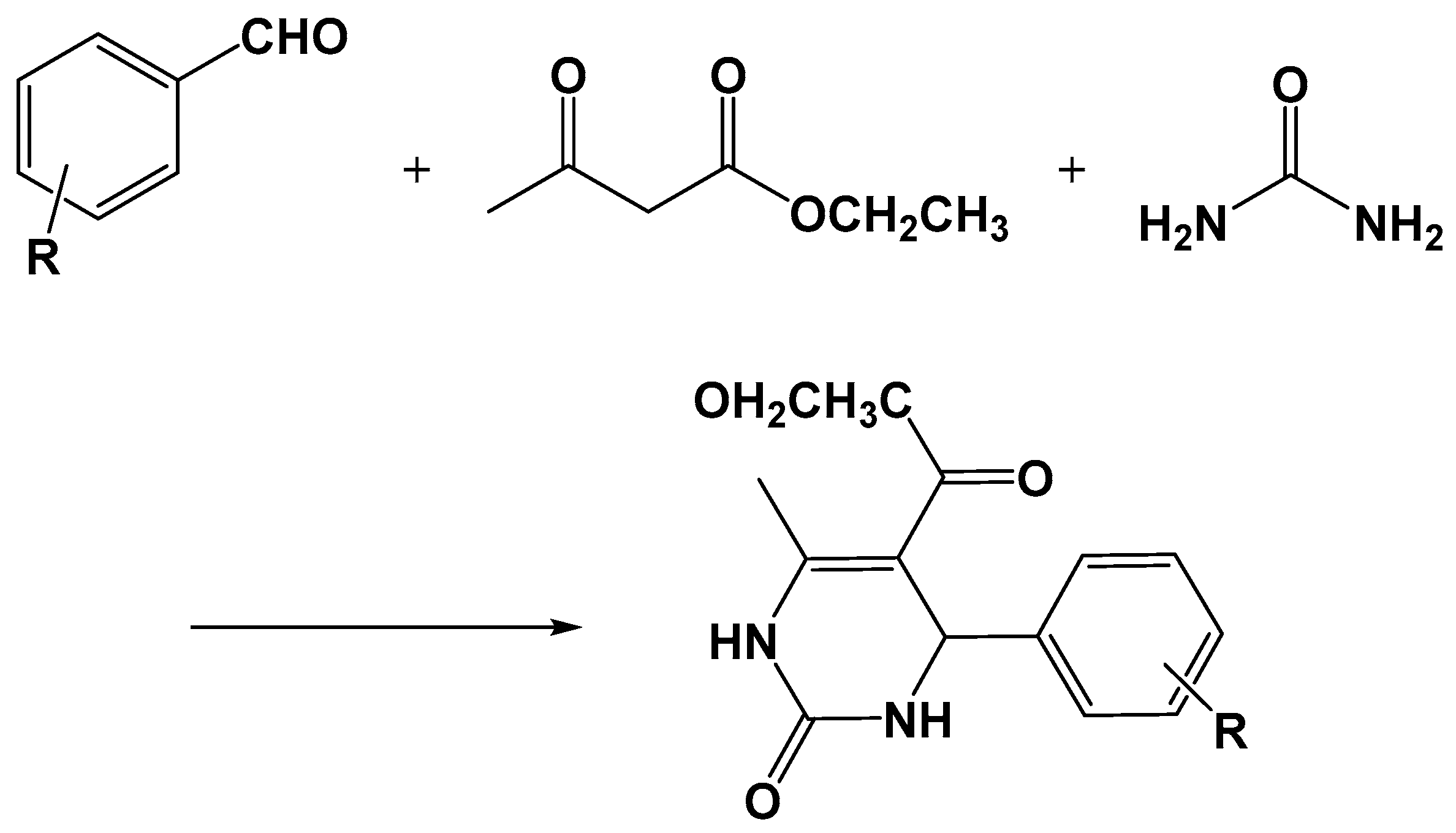
- Moosavifar, M. An appropriate one-pot synthesis of dihydropyrimidinones catalyzed by heteropoly acid supported on zeolite: An efficient and reusable catalyst for the Biginelli reaction. C. R. Chim. 2012, 15, 444–447. [Google Scholar] [CrossRef]
- Han, Z.; Wang, E.; You, W.; Liu, S.; Hu, C.; Xing, Y.; Jia, H.; Lin, Y. Synthesis and crystal structure of a novel compound: [H3PMo12O40][CO(NH2)2]3.5H2O. J. Mol. Struct. 2001, 595, 7–13. [Google Scholar] [CrossRef]
- Narkhede, N.; Patel, A. Greener, solvent free three component Biginelli reaction over different recyclable solid acid catalysts. J. Porous Mater. 2014, 21, 579–588. [Google Scholar] [CrossRef]
- Nandiwale, K.Y.; Sonar, S.K.; Niphadkar, P.S.; Joshi, P.N.; Deshpande, S.S.; Patil, V.S.; Bokade, V.V. Catalytic upgrading of renewable levulinic acid to ethyl levulinate biodiesel using dodecatungstophosphoric acid supported on desilicated H-ZSM-5 as catalyst. Appl. Catal. A Gen. 2013, 460–461, 90–98. [Google Scholar] [CrossRef]
- Marchena, C.L.; Frenzel, R.A.; Gomez, S.; Pierella, L.B.; Pizzio, L.R. Tungstophosphoric acid immobilized on ammonium Y and ZSM-5 zeolites: Synthesis, characterization and catalytic evaluation. Appl. Catal. B Environ. 2013, 130–131, 187–196. [Google Scholar] [CrossRef]
- Marchena, C.L.; Gomez, S.; Pierella, L.B.; Pizzio, L.R. Synthesis, characterization and catalytic evaluation of tungstophosphoric acid immobilized on Y zeolite. In Advanced Oxidation Technologies Sustainable Solutions for Environmental Treatments in Sustainable Energy Developments; CRC Press: Leiden, The Netherlands, 2014; Volume 9, pp. 43–57. [Google Scholar]
- Marchena, C.L.; Gomez, S.; Saux, C.; Pierella, L.B.; Pizzio, L.R. Tungstophosphoric acid heterogenized onto NH4ZSM-5 as an efficient and recyclable catalyst for the photodegradation of dyes. Quim. Nova 2015, 38, 518–525. [Google Scholar]
- Ozer, R.R.; Ferry, J.L. Photocatalytic oxidation of aqueous 1,2-dichlorobenzene be polyoxometalates supported on the NaY zeolite. J. Phys. Chem. B 2002, 106, 4336–4342. [Google Scholar] [CrossRef]
- Marchena, C.L.; Lerici, L.; Renzini, S.; Pierella, L.; Pizzio, L. Synthesis and characterization of a novel tungstosilicic acid immobilized on zeolites catalyst for the photodegradation of methyl orange. Appl. Catal. B Environ. 2016, 188, 23–30. [Google Scholar] [CrossRef]
- Rayalu, S.S.; Dubey, N.; Labhsetwar, N.K.; Kagne, S.; Devotta, S. UV and visibly active photocatalysts for water splitting reaction. Int. J. Hydrog. Energy 2007, 32, 2776–2783. [Google Scholar] [CrossRef]
- Chatti, R.; Rayalu, S.S.; Dubey, N.; Labhsetwar, N.; Devotta, S. Solar-based photoreduction of methyl orange using zeolite supported photocatalytic materials. Sol. Energy Mater. Sol. Cells 2007, 91, 180–190. [Google Scholar] [CrossRef]
- Dubey, N.; Labhsetwar, N.K.; Devotta, S.; Rayalu, S.S. Hydrogen evolution by water spliting using novel composite zeolite-based photocatalyst. Catal. Today 2007, 129, 428–434. [Google Scholar] [CrossRef]
- Dubey, N.; Rayalu, S.S.; Labhsetwar, N.K.; Naidu, R.R.; Chatti, R.V.; Devotta, S. Photocatalytic properties of zeolite-based materials for the photoreduction of methyl orange. Appl. Catal. A Gen. 2006, 303, 152–157. [Google Scholar] [CrossRef]
- Rayalu, S.S.; Dubey, N.; Chatti, R.V.; Joshi, M.V.; Labhsetwar, N.K.; Devotta, S. Effect of heteropolyacid and its route of incorporation on the photocatalytic properties of zeolite-based materials. Curr. Sci. 2007, 93, 1376–1382. [Google Scholar]
- Dubey, N.; Rayalu, S.S.; Labhsetwar, N.K.; Devotta, S. Visible light active zeolite-based photocatalysts for hydrogen evolution from water. Int. J. Hydrog. Energy 2008, 33, 5958–5966. [Google Scholar] [CrossRef]
- Chatti, R.V.; Dubey, N.; Joshi, M.V.; Labhsetwar, N.K.; Joshi, P.N.; Rayalu, S.S. Influence of zeolitic structure on photoreduction property and hydrogen evolution reaction. Int. J. Hydrog. Energy 2010, 35, 1911–1920. [Google Scholar] [CrossRef]
- Najafabadi, A.T.; Taghipour, F. Cobalt precursor role in the photocatalytic activity of the zeolite-supported TiO2-based photocatalysts under visible light: A promising tool toward zeolite-based core-shell photocatalysis. J. Photochem. Photobiol. A Chem. 2012, 248, 1–7. [Google Scholar] [CrossRef]
- Najafabadi, A.T.; Taghipour, F. Physicochemical impact of zeolites as the support for photocatalytic hydrogen production using solar-activated TiO2-based nanoparticles. Energy Convers. Manag. 2014, 82, 106–113. [Google Scholar] [CrossRef]
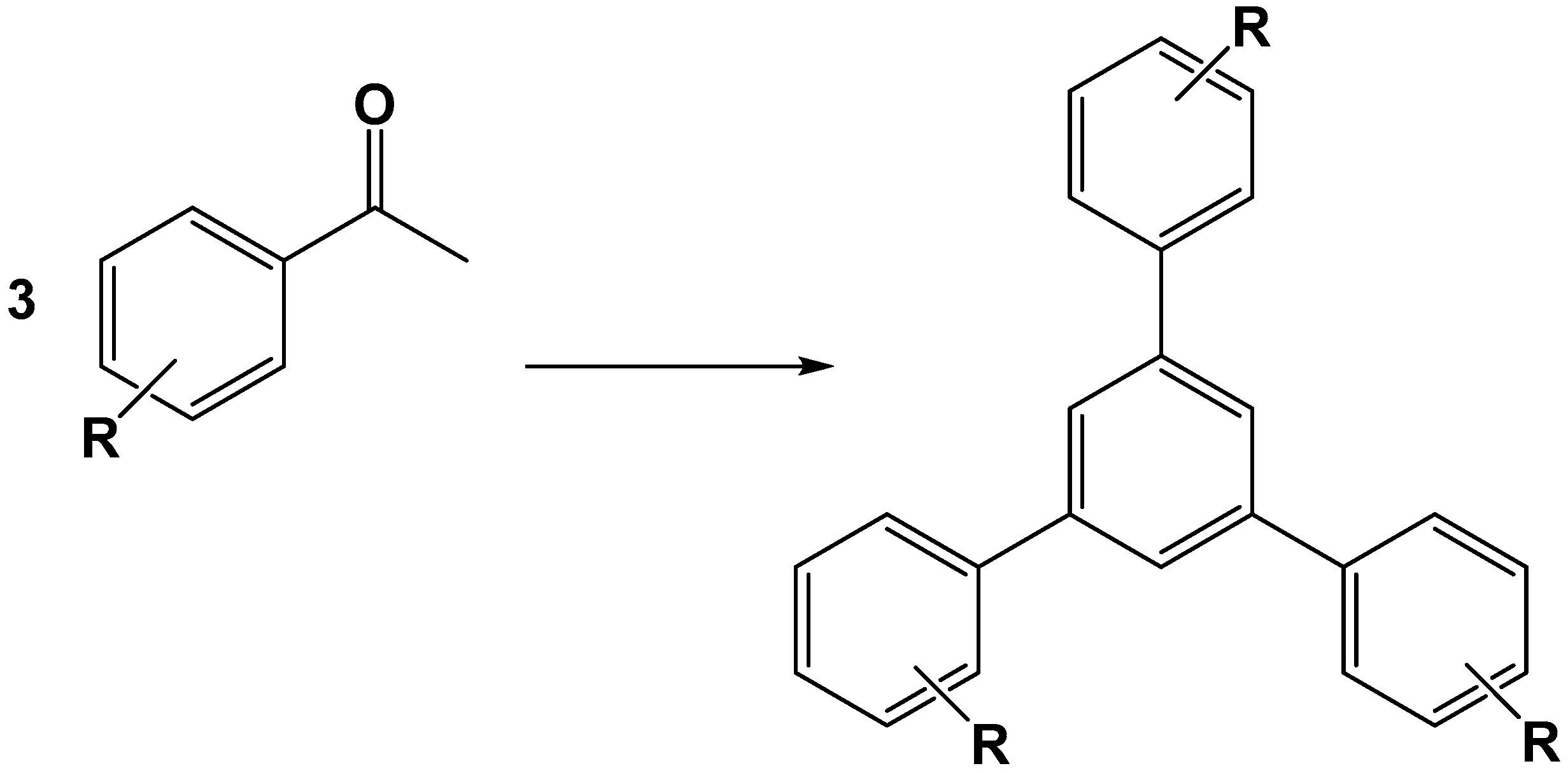
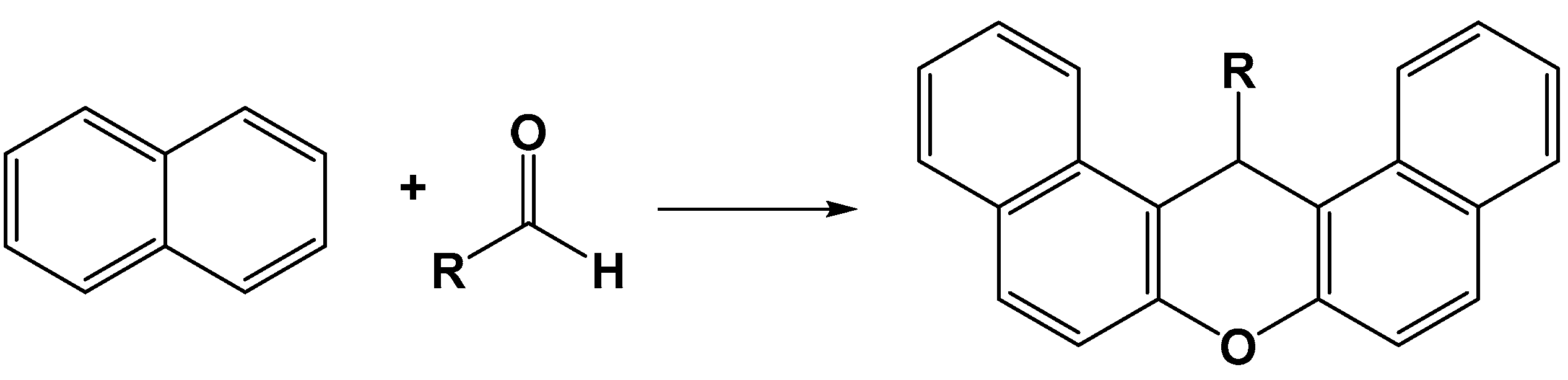
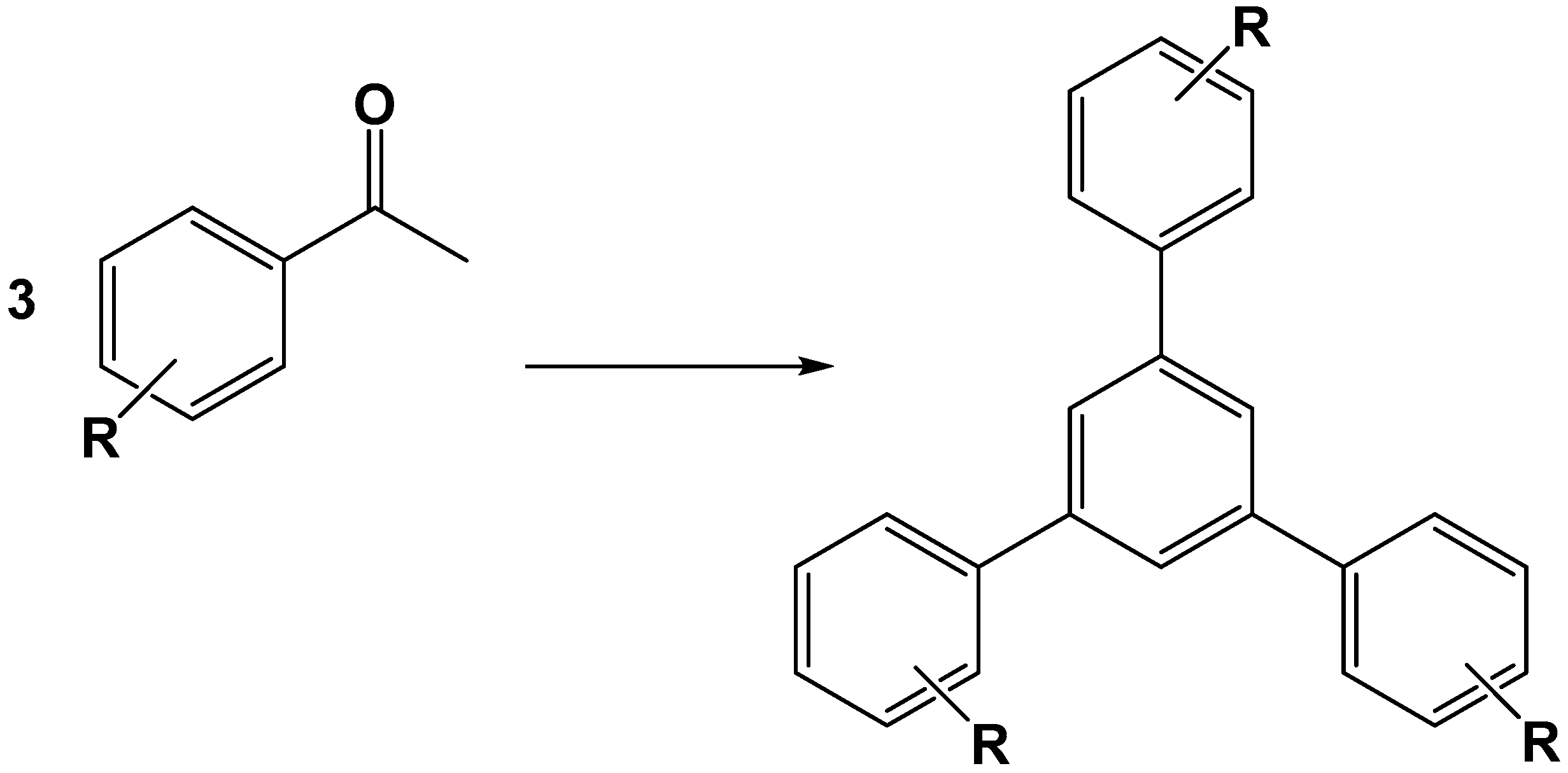
- Tayebee, R.; Jarrahi, M. H6P2W18O62/nanoclinoptilolite as an efficient nanohybrid catalyst in the cyclotrimerization of aryl methyl ketones under solvent-free conditions. RSC Adv. 2015, 5, 21206–21214. [Google Scholar] [CrossRef]
- Alberti, A. On the crystal structure of the zeolite heulandite. Tschermaks Mineral. Petrogr. Mitt. 1972, 18, 129–146. [Google Scholar] [CrossRef]
- Narkhede, N.; Patel, A. Efficient synthesis of biodiesel over a recyclable catalyst comprising a monolacunary silicotungstate and zeolite Hβ. RSC Adv. 2014, 4, 64379–64387. [Google Scholar] [CrossRef]
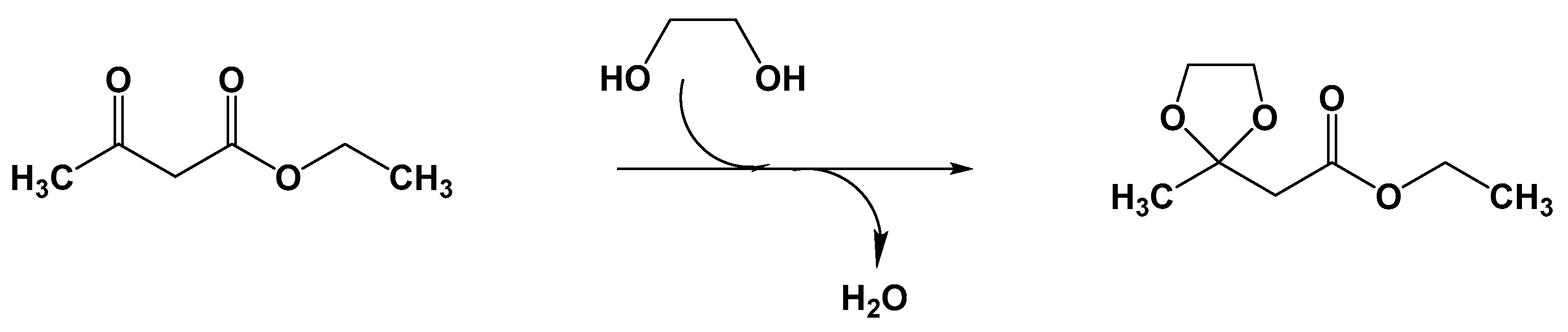
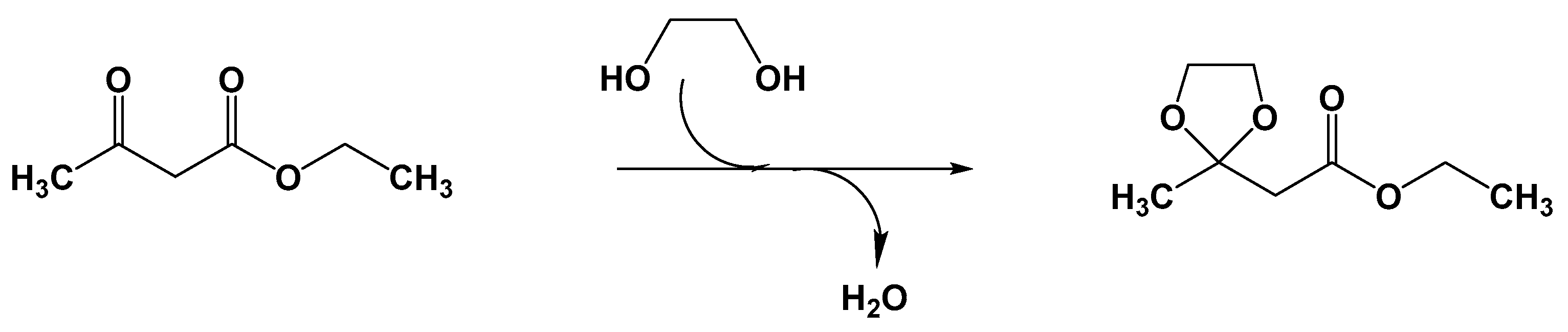
- Narkhede, N.; Patel, A. Sustainable valorisation of glycerol via acetalization as well as carboxylation reactions over silicotungstates anchored to zeolite Hβ. Appl. Catal. A Gen. 2016, 515, 154–163. [Google Scholar] [CrossRef]
- Singh, S.; Narkhede, N.; Patel, A. Aerobic oxidation of alcohols and alkenes over a novel lacunary phosphomolybdate anchored to zeolite Hβ. RSC Adv. 2015, 5, 36270–36278. [Google Scholar] [CrossRef]
- Dai, C.; Zhang, A.; Li, J.; Liu, M.; Song, C.; Guo, X. Synthesis of yolk-shell HPW@Hollow silicalite-1 for esterification reaction. Chem. Commun. 2014, 50, 4846–4848. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
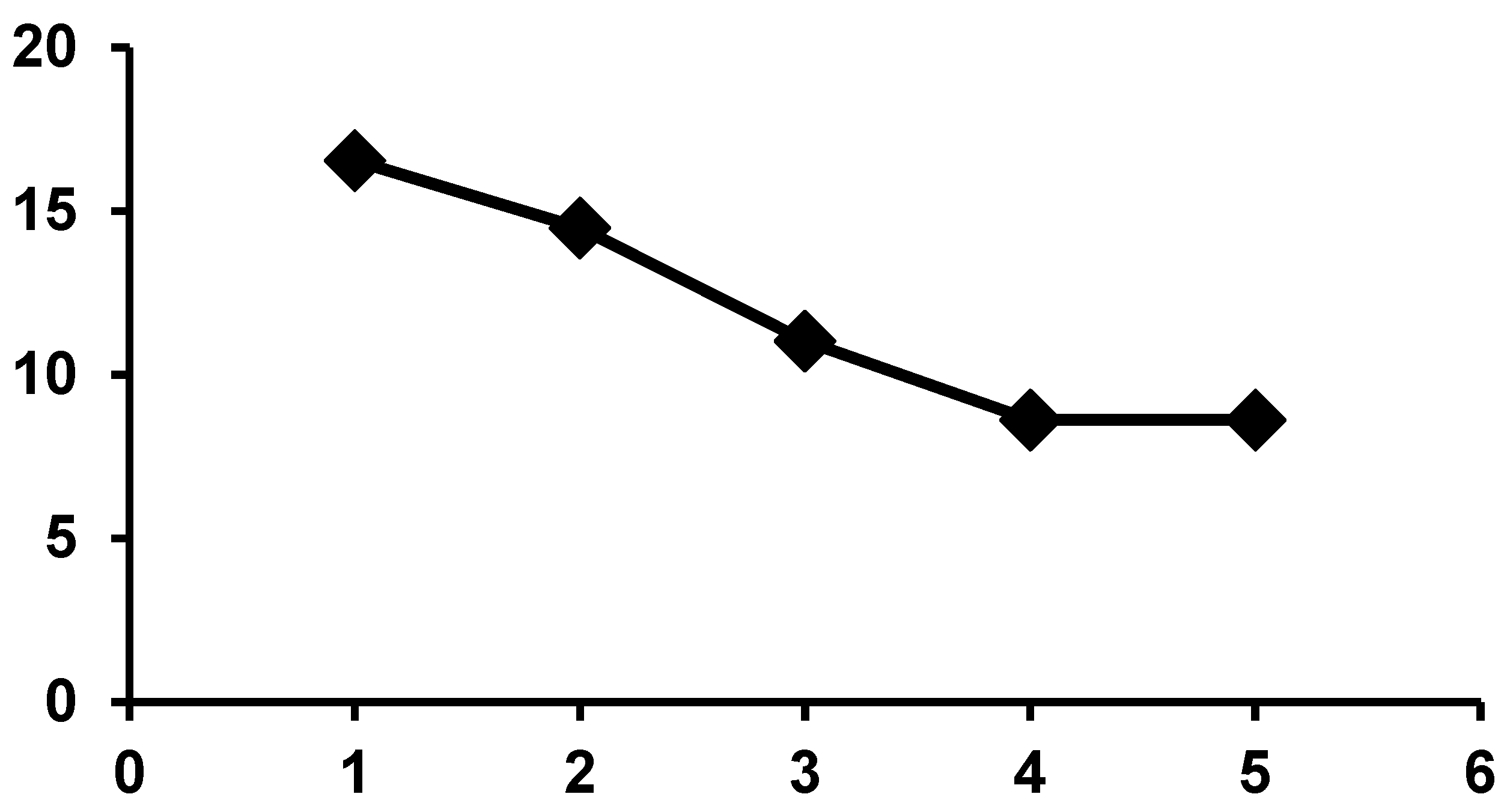
| Catalyst | Conversion (%) |
|---|---|
| no catalyst | 4.9 |
| H3PMo12O40 | 9.0 |
| CsH2PMo12O40 | 8.3 |
| Cs2.5H0.5PMo12O40 | 7 |
| Catalyst | Conversion (%) | Selectivity for Isomerization (%) |
|---|---|---|
| Pt/Hβ | 27.9 | 99.0 |
| Pt-5% PW12/Hβ | 32.2 | 96.1 |
| Pt-10% PW12/Hβ | 47.1 | 83.8 |
| Pt-15% PW12/Hβ | 34.5 | 70.0 |
| Pt/Hβ-650(5) | 14.8 | 100 |
| Pt-10% PW12/Hβ-300(5) | 21.1 | 95.0 |
| Pt-10% PW12/Hβ-550(5) | 25.3 | 91.1 |
| Pt-10% PW12/Hβ-650(5) | 39.6 | 88.5 |
| Pt-10% PW12/Hβ-700(5) | 20.1 | 92.4 |
| Pt-10% PW12/Hβ-650(4) | 19.3 | 96.0 |
| Pt-10% PW12/Hβ-650(6) | 21.0 | 91.1 |
| Pt-10% PW12/Hβ-650(7) | 20.3 | 93.8 |
| Catalyst | R–C(=O)H, R = | R′C(=O)CH2C(=O)CH3, R′ = | NH2–C(=X)NH2, X = | Reaction Time (min) | Yield (%) |
|---|---|---|---|---|---|
| PW12/Hβ | C6H5 | –OC2H5 | O | 15 | 98 |
| SiW12/Hβ | C6H5 | –OC2H5 | O | 15 | 95 |
| PW12/Hβ | C6H5 | –OC2H5 | S | 17 | 90 |
| SiW12/Hβ | C6H5 | –OC2H5 | S | 20 | 92 |
| PW12/Hvβ | C6H5 | –OCH3 | O | 14 | 92 |
| SiW12/Hβ | C6H5 | –OCH3 | O | 12 | 94 |
| PW12/Hβ | C6H5 | –OCH3 | S | 15 | 97 |
| SiW12/Hβ | C6H5 | –OCH3 | S | 15 | 96 |
| PW12/Hβ | 4-ClC6H4 | –OC2H5 | O | 30 | 89 |
| SiW12/Hβ | 4-ClC6H4 | –OC2H5 | O | 28 | 90 |
| PW12/Hβ | 4-NO2C6H4 | –OC2H5 | O | 32 | 82 |
| SiW12/Hβ | 4-NO2C6H4 | –OC2H5 | O | 30 | 92 |
| Substrate | Conversion (%) | Selectivity (%) | TON |
|---|---|---|---|
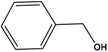 | 25.5 | 90 | 9551 |
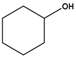 | 21 | >99 | 8590 |
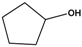 | 20 | >99 | 8171 |
 | 9 | >99 | 3745 |
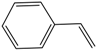 | 57 | 72 | 20,975 |
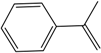 | 59 | 70 | 22,099 |
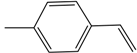 | 50 | 67 | 18,728 |
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lefebvre, F. Synthesis, Characterization and Applications in Catalysis of Polyoxometalate/Zeolite Composites. Inorganics 2016, 4, 13. https://doi.org/10.3390/inorganics4020013
Lefebvre F. Synthesis, Characterization and Applications in Catalysis of Polyoxometalate/Zeolite Composites. Inorganics. 2016; 4(2):13. https://doi.org/10.3390/inorganics4020013
Chicago/Turabian StyleLefebvre, Frédéric. 2016. "Synthesis, Characterization and Applications in Catalysis of Polyoxometalate/Zeolite Composites" Inorganics 4, no. 2: 13. https://doi.org/10.3390/inorganics4020013
APA StyleLefebvre, F. (2016). Synthesis, Characterization and Applications in Catalysis of Polyoxometalate/Zeolite Composites. Inorganics, 4(2), 13. https://doi.org/10.3390/inorganics4020013