
Single-Crystal to Single-Crystal Reversible Transformations Induced by Thermal Dehydration in Keggin-Type Polyoxometalates Decorated with Copper(II)-Picolinate Complexes: The Structure Directing Role of Guanidinium


 ,
, 
Abstract
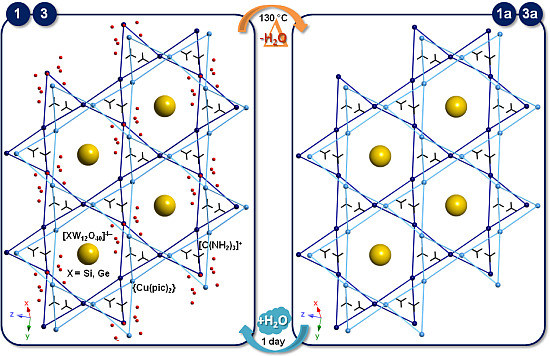
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Infrared Spectroscopy
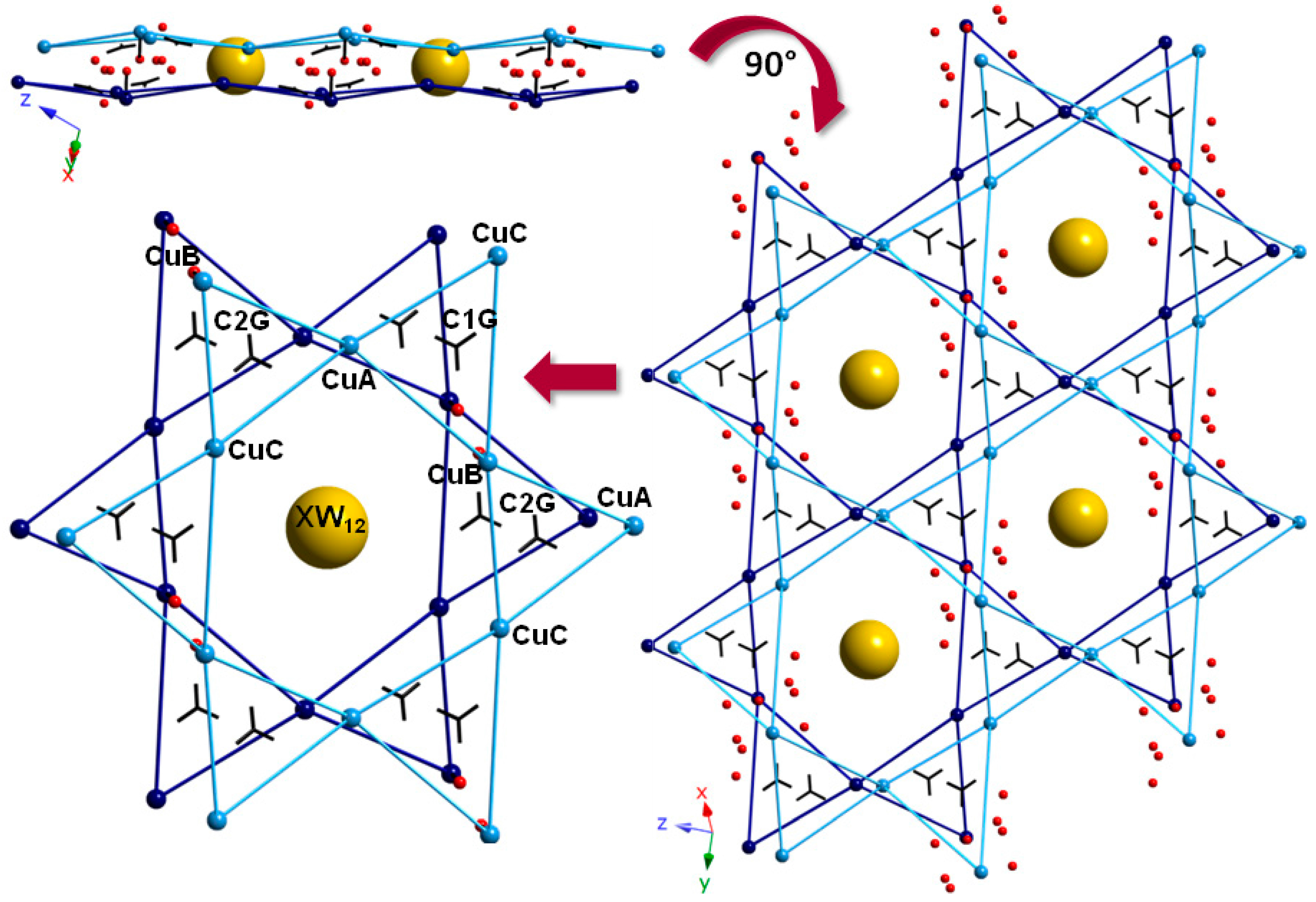
2.2. Crystal Structures of Compounds 1–3
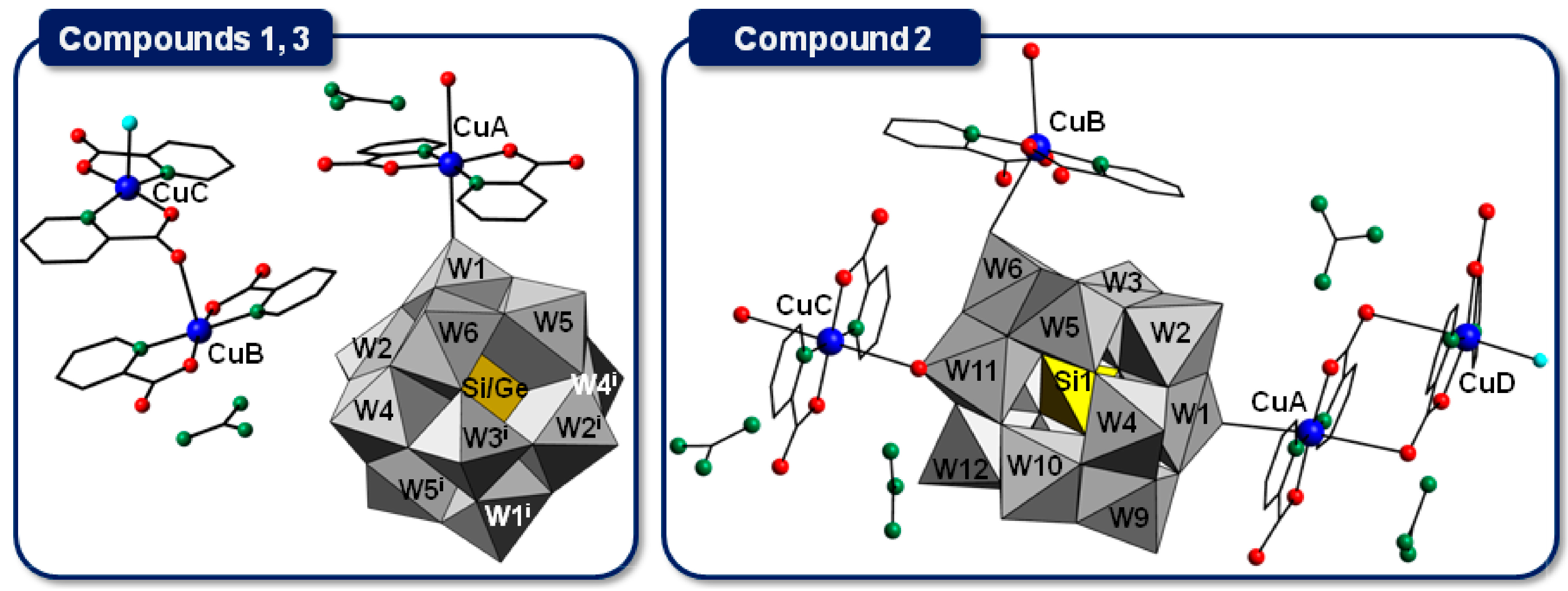
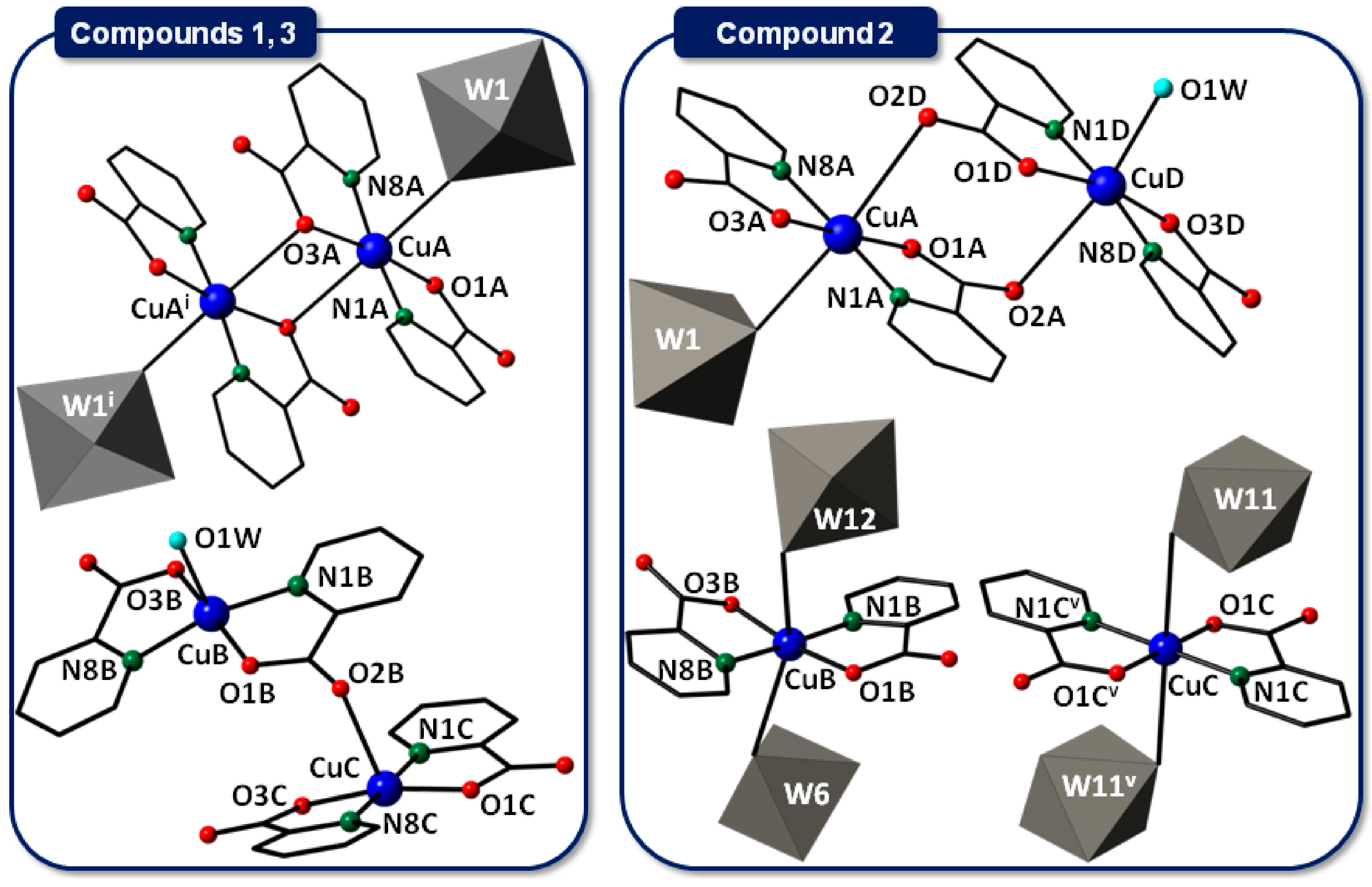
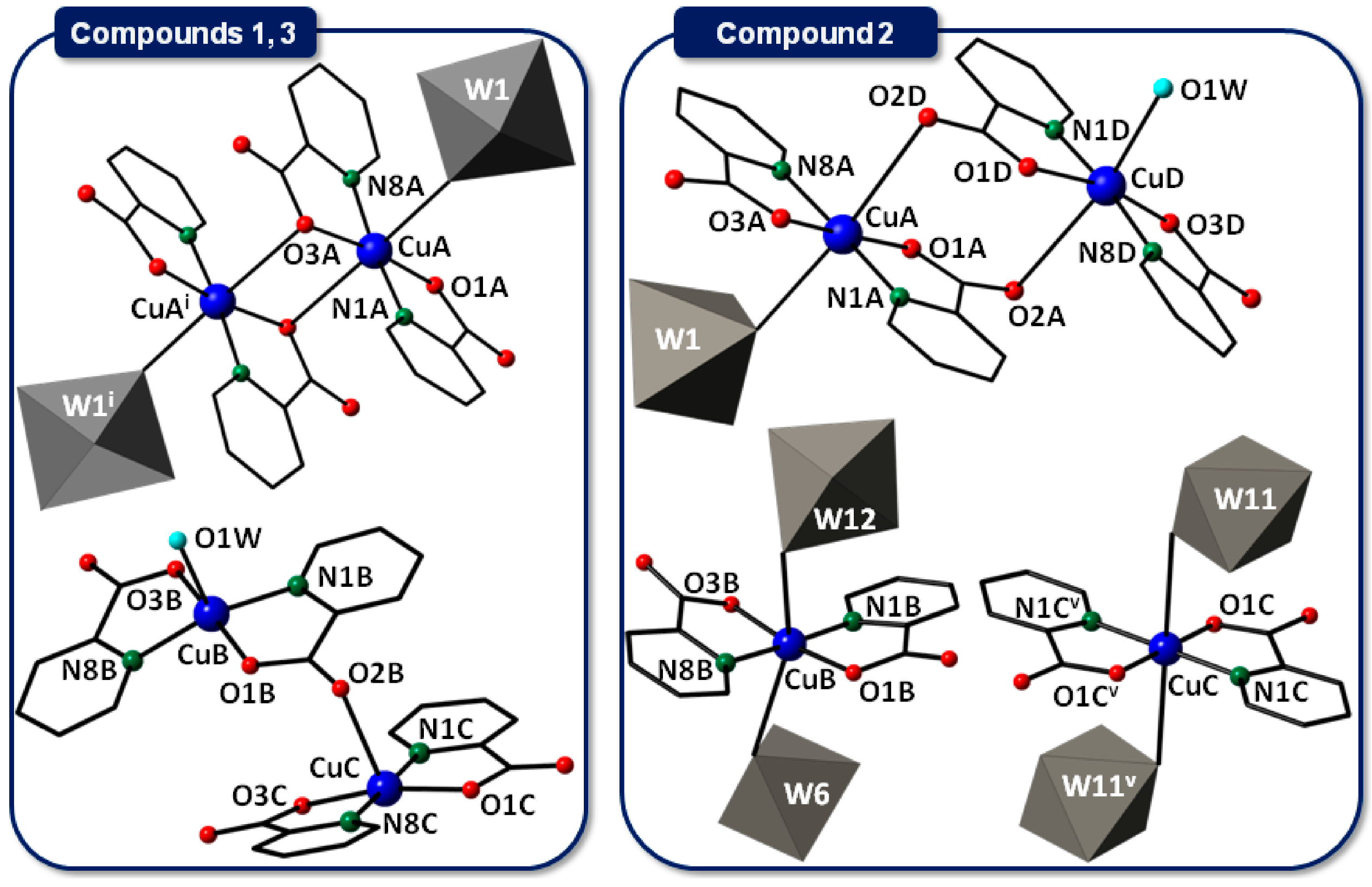
2.2.1. Copper(II)-Picolinate Complexes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | 1 | 1a | 3 | 3a | Bond | 2 |
|---|---|---|---|---|---|---|
| CuA–N1A | 1.964(8) | 1.959(9) | 1.965(8) | 1.956(16) | CuA–N1A | 1.98(2) |
| CuA–O1A | 1.956(7) | 1.962(7) | 1.958(7) | 1.972(14) | CuA–O1A | 1.91(2) |
| CuA–N8A | 1.965(9) | 1.953(8) | 1.954(8) | 1.961(16) | CuA–N8A | 1.96(2) |
| CuA–O3A | 1.973(7) | 1.970(7) | 1.963(7) | 1.975(14) | CuA–O3A | 1.97(2) |
| CuA–O1/ | 2.459(18)/ | 2.527(17)/ | 2.468(17)/ | 2.48(3)/ | CuA–O1 | 2.57(2) |
| –O1Z | 2.349(18) | 2.352(15) | 2.346(17) | 2.36(2) | CuA–O2Diii | 2.93(2) |
| CuA–O3Ai | 2.778(8) | 2.807(7) | 2.774(7) | 2.846(14) | ||
| CuB–N1B | 1.978(9) | 1.956(8) | 1.977(9) | 1.972(16) | CuB–N1B | 1.95(2) |
| CuB–O1B | 1.972(8) | 1.939(7) | 1.970(7) | 1.940(15) | CuB–O1B | 1.96(2) |
| CuB–N8B | 1.981(9) | 1.956(9) | 1.975(9) | 1.944(17) | CuB–N8B | 1.98(3) |
| CuB–O3B | 1.969(8) | 1.951(7) | 1.968(7) | 1.943(15) | CuB–O3B | 1.89(2) |
| CuB–O1W | 2.253(9) | – | 2.258(8) | – | CuB–O12/ | 2.77(2) |
| –O12Z | 2.98(5) | |||||
| CuB–O6iv | 2.77(2) | |||||
| CuC–N1C | 1.973(9) | 1.964(9) | 1.972(9) | 1.985(18) | CuC–N1C | 1.958(18) |
| CuC–O1C | 1.949(9) | 1.953(8) | 1.951(8) | 1.931(16) | CuC–O1C | 1.888(16) |
| CuC–N8C | 1.971(9) | 1.962(9) | 1.966(9) | 1.96(2) | CuC–N1Cv | 1.958(18) |
| CuC–O3C | 1.951(8) | 1.928(8) | 1.946(8) | 1.945(16) | CuC–O1Cv | 1.888(16) |
| CuC–O2Bii | 2.488(8) | 3.310(8) | 2.490(8) | 3.314(18) | CuC–O11 | 2.909(19) |
| CuC–O11v | 2.909(19) | |||||
| CuD–N1D | 1.96(3) | |||||
| CuD–O1D | 1.97(2) | |||||
| CuD–N8D | 1.97(2) | |||||
| CuD–O3D | 1.96(2) | |||||
| CuD–O1W | 2.29(3) | |||||
| CuD–O2Aiii | 2.97(3) | |||||
| CuA···CuAi | 3.572(2) | 3.554(2) | 3.568(2) | 3.617(3) | CuA···CuDiii | 5.305(6) |
| CuB···CuCii | 5.538(3) | 5.458(2) | 5.540(2) | 5.498(5) |
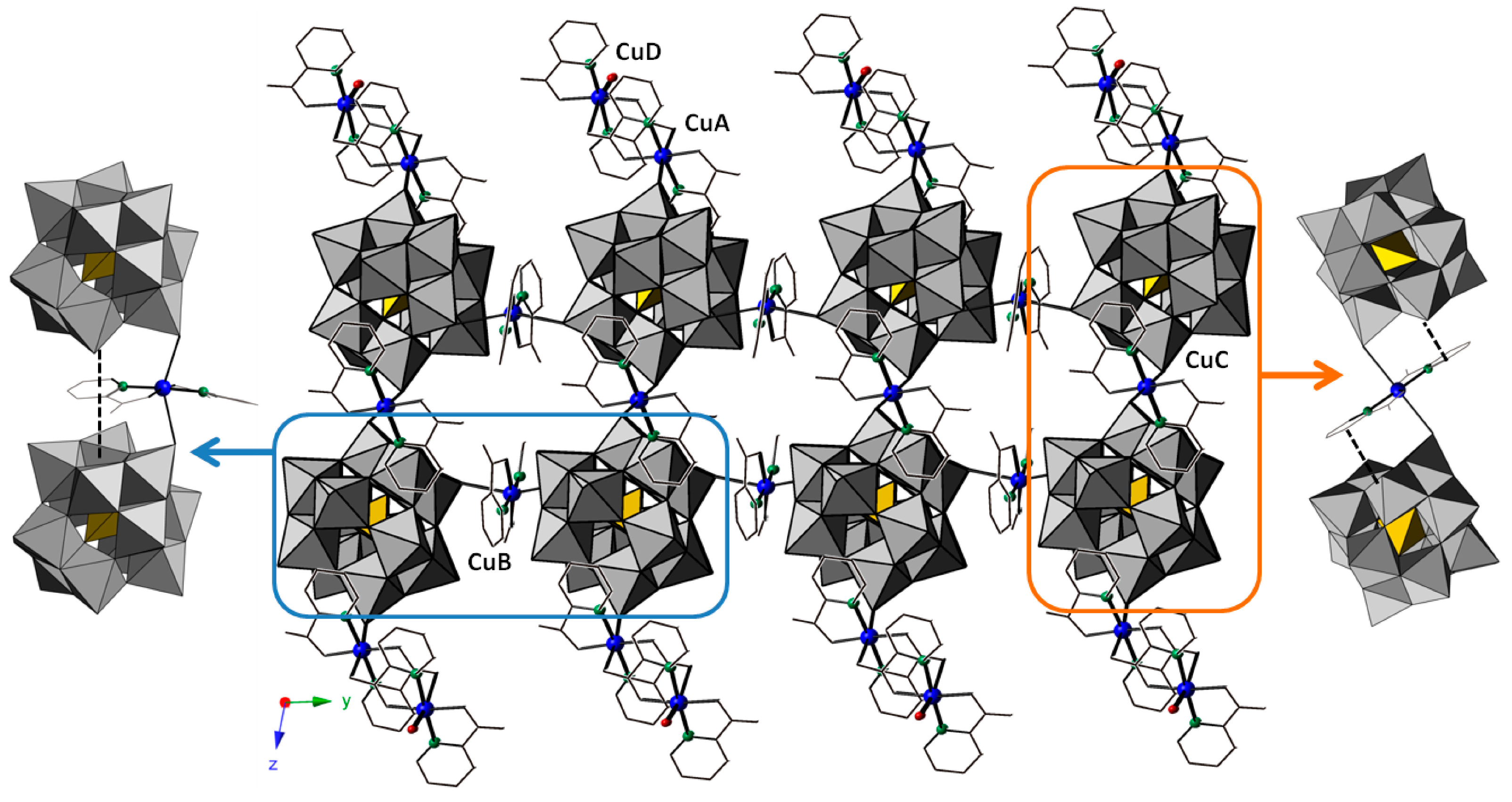
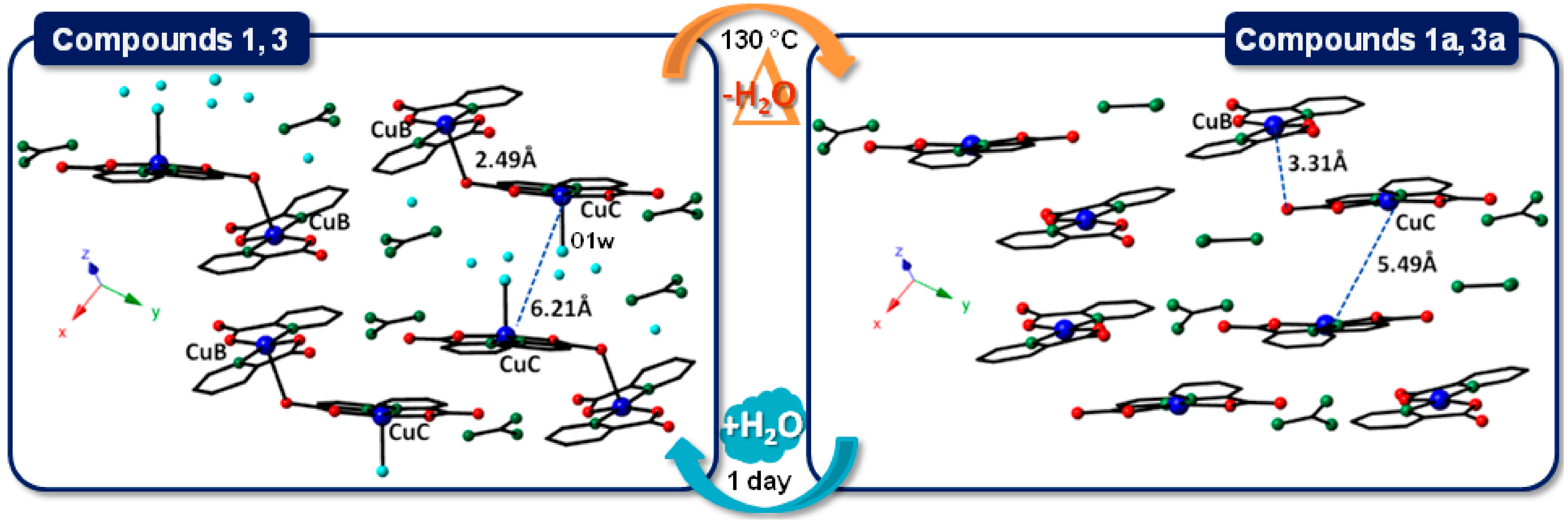
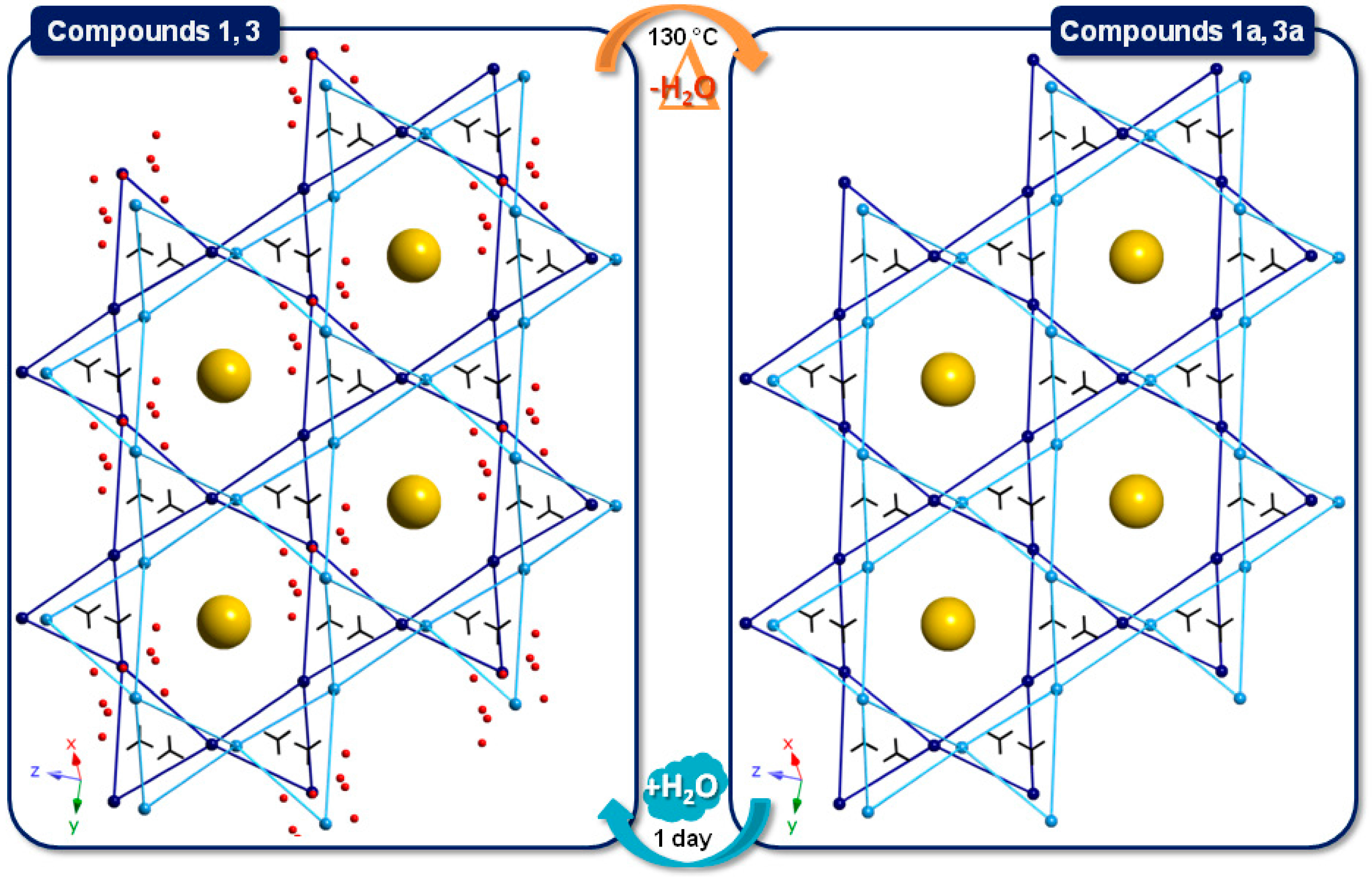
2.2.2. Crystal Packing of Compounds 1 and 3
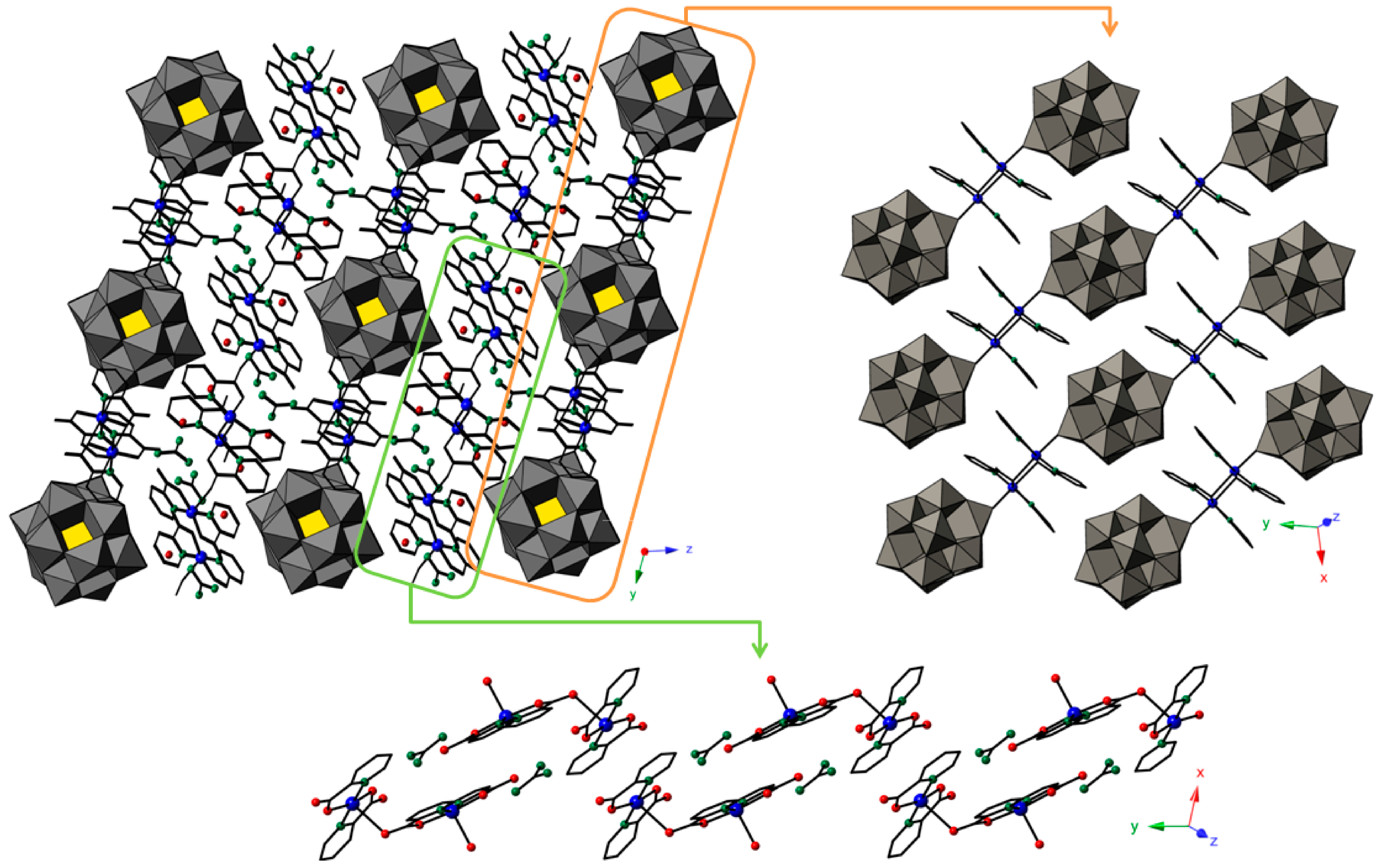
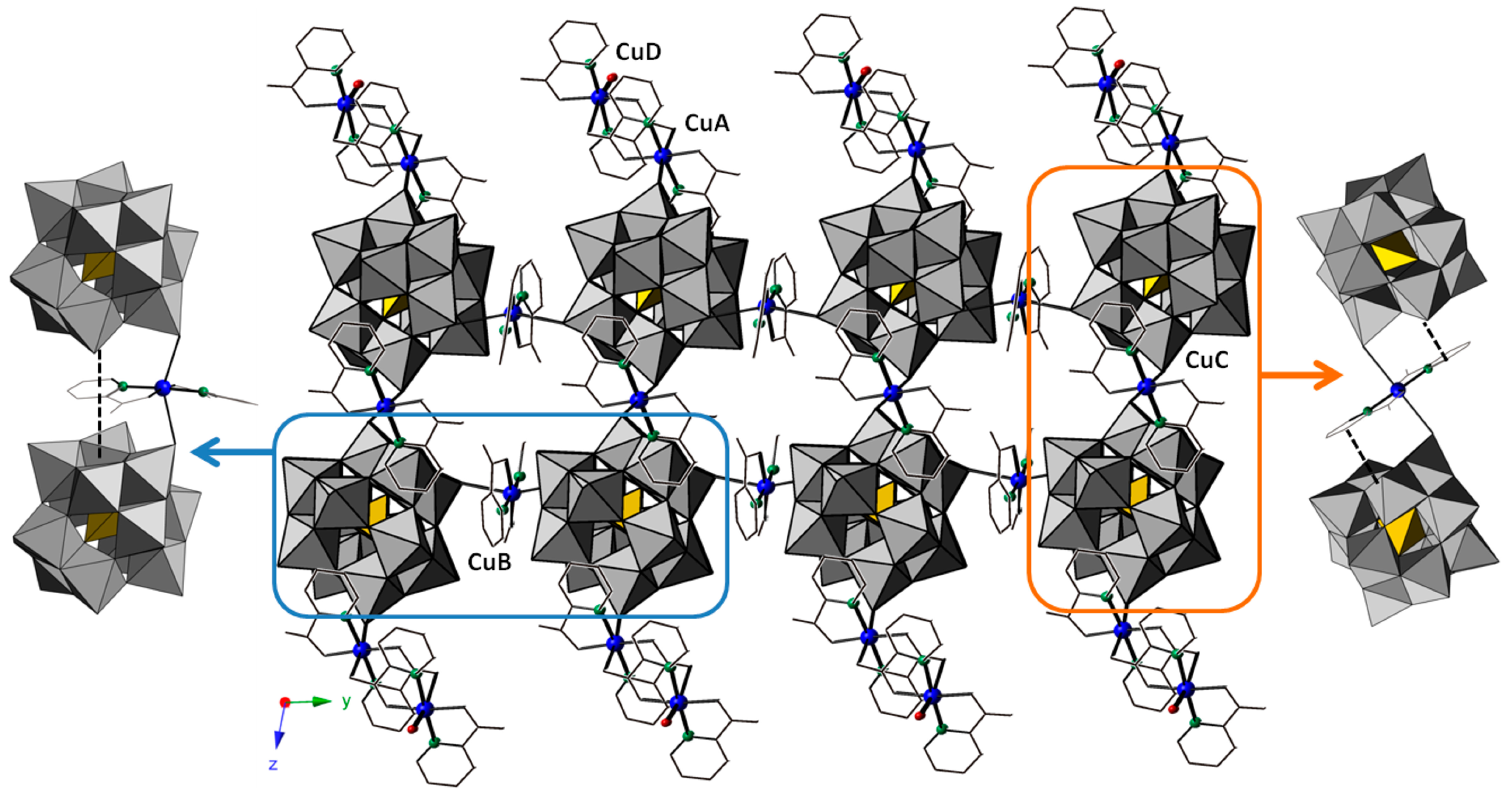
2.2.3. Crystal Packing of Compound 2
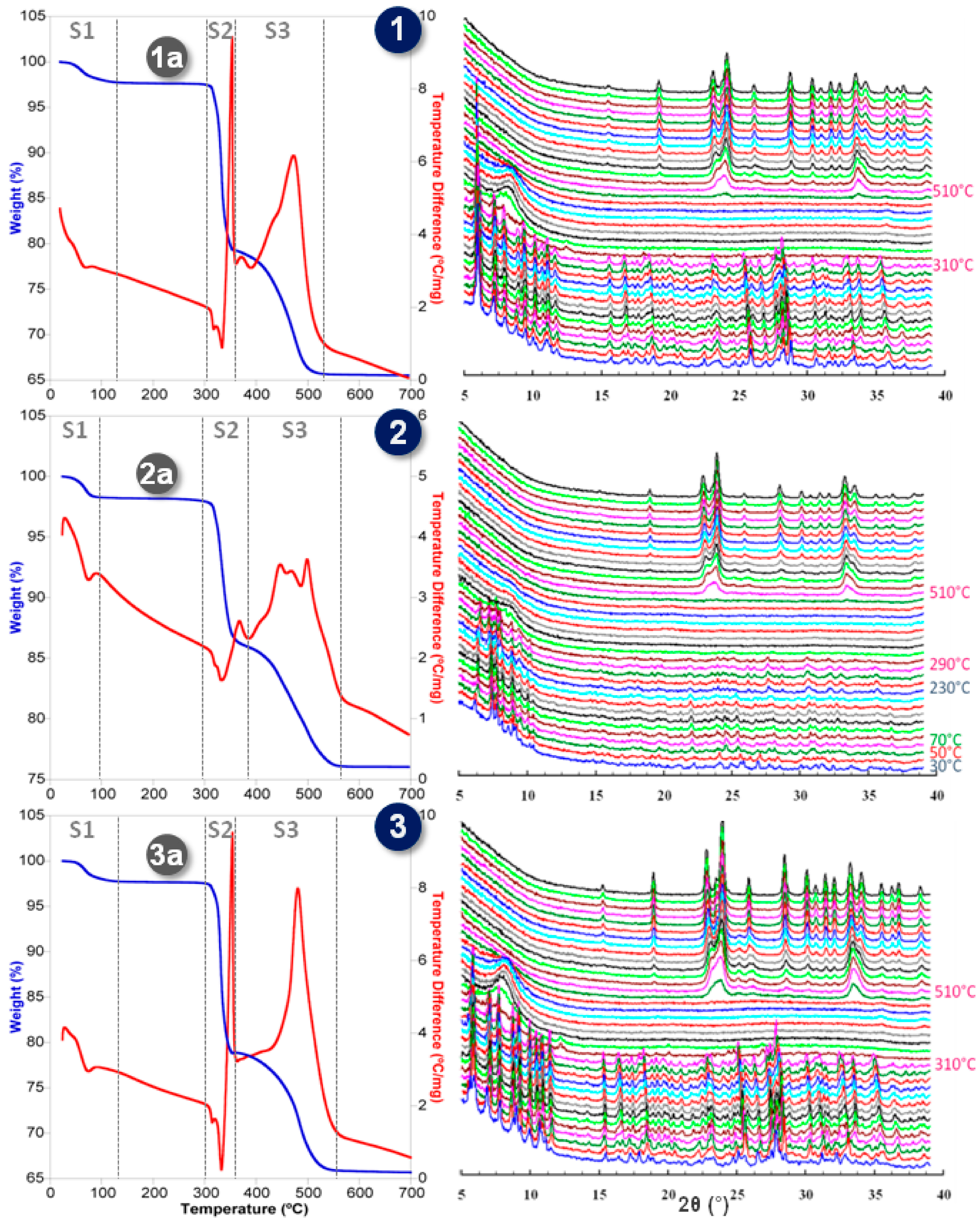
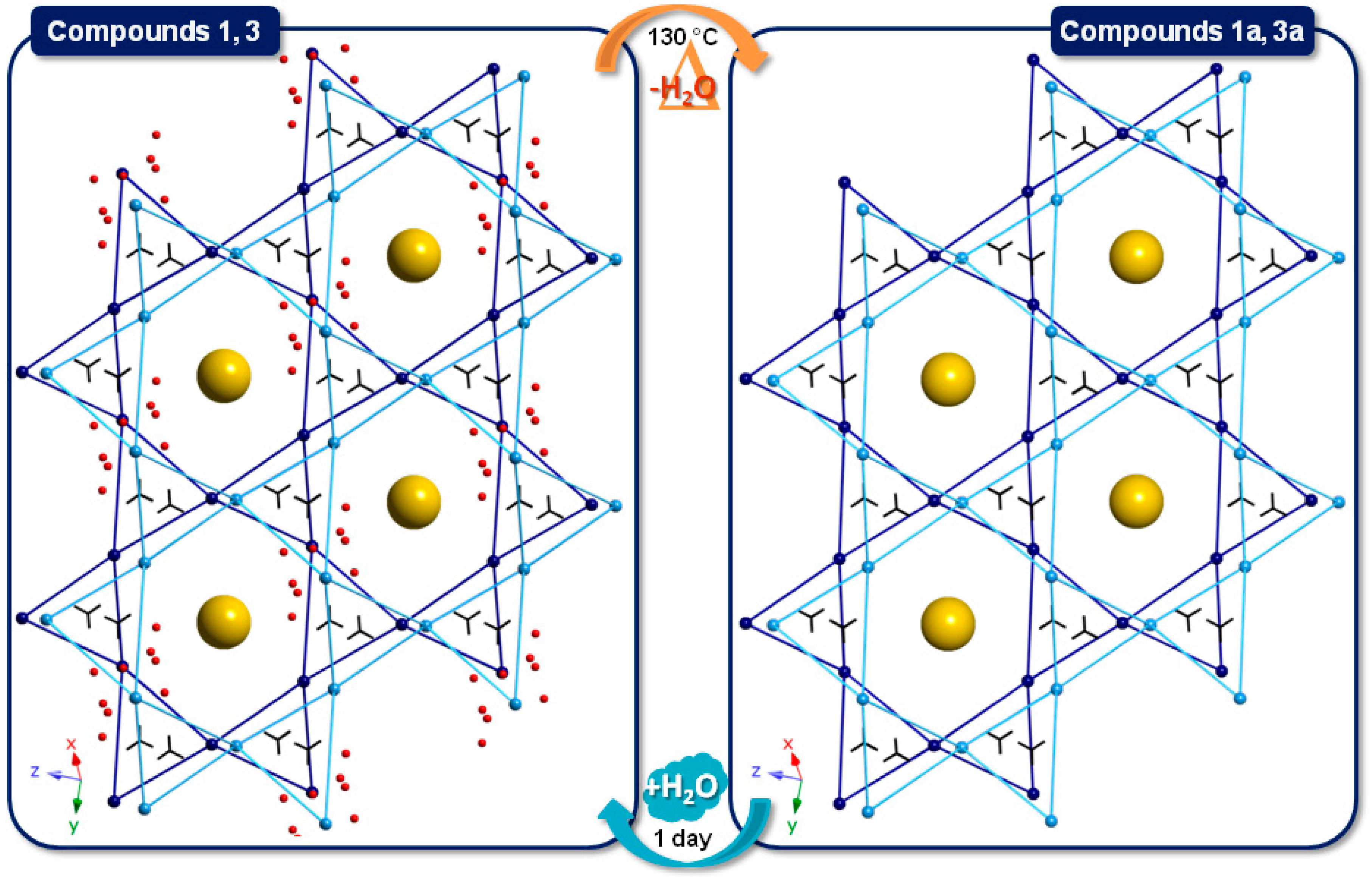
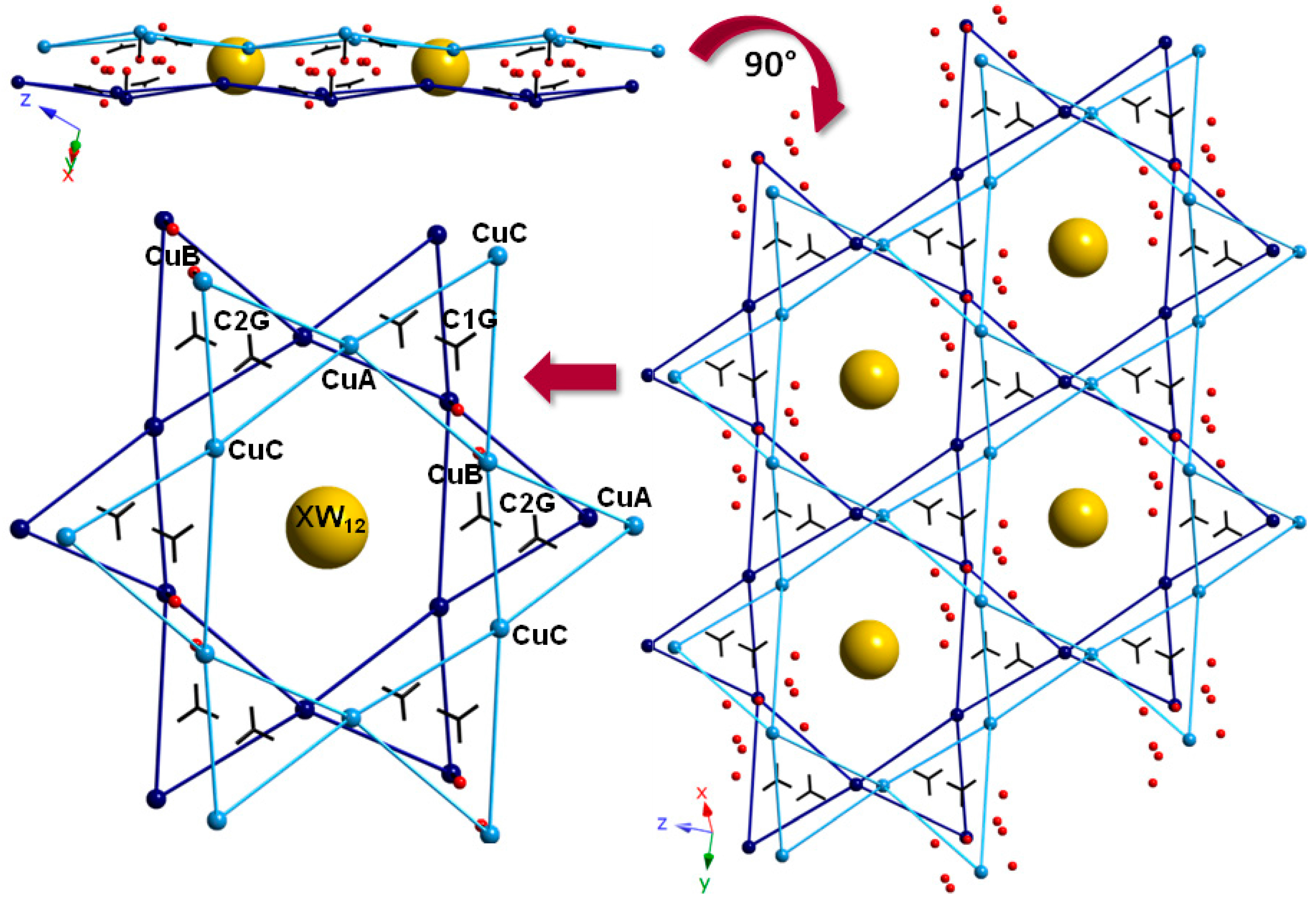
2.3. Thermostructural Behavior
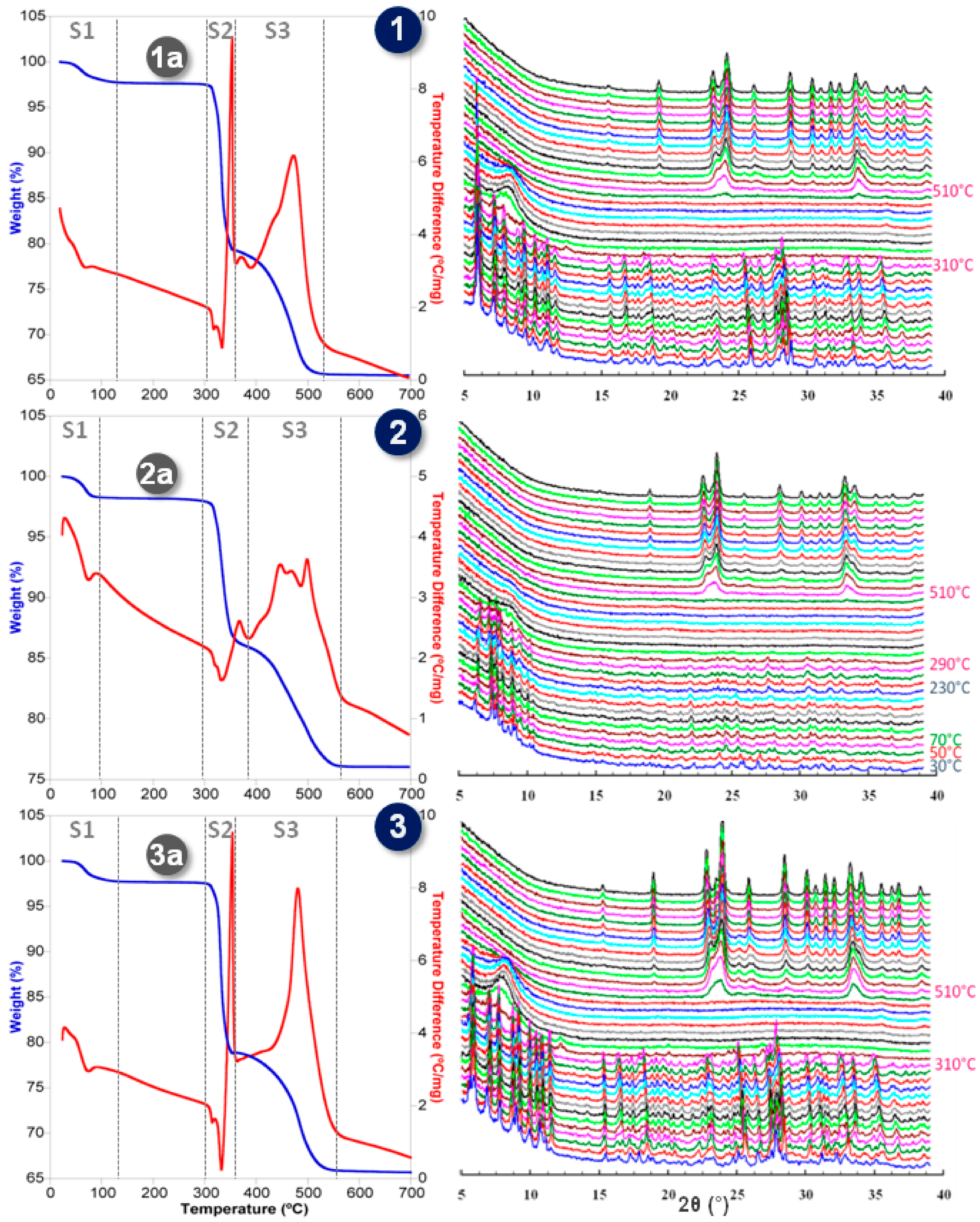

| Compounds | T (°C) | a (Ǻ) | b (Ǻ) | c (Ǻ) | α (°) | β (°) | γ (°) | V (Ǻ3) |
|---|---|---|---|---|---|---|---|---|
| 1 | r.t | 11.805(6) | 16.112(6) | 16.443(5) | 105.65(3) | 101.97(3) | 91.29(4) | 2936(2) |
| 50 | 11.812(5) | 16.098(5) | 16.356(6) | 105.21(3) | 101.53(4) | 91.60(3) | 2958(2) | |
| 80 | 11.861(6) | 16.077(7) | 16.292(7) | 104.99(4) | 101.29(4) | 91.85(4) | 2982(2) | |
| 140 | 11.818(1) | 16.123(5) | 16.179(7) | 104.62(3) | 100.18(5) | 92.07(5) | 2926(2) | |
| 3 | r.t | 11.815(4) | 16.050(6) | 17.047(6) | 106.90(3) | 94.42(3) | 100.20(3) | 3016(2) |
| 50 | 11.81(1) | 16.05(2) | 17.06(2) | 106.91(9) | 94.40(7) | 100.09(8) | 3018(5) | |
| 80 | 11.88(3) | 16.35(6) | 16.55(5) | 106.0(3) | 92.9(2) | 101.3(3) | 3012(15) | |
| 140 | 11.75(3) | 15.89(4) | 16.21(4) | 103.4(2) | 91.8(2) | 100.8(2) | 2882(12) | |
| % of indexed reflections for cells at T > 50 °C: above 97% for 1 and below 57% for 3 | ||||||||
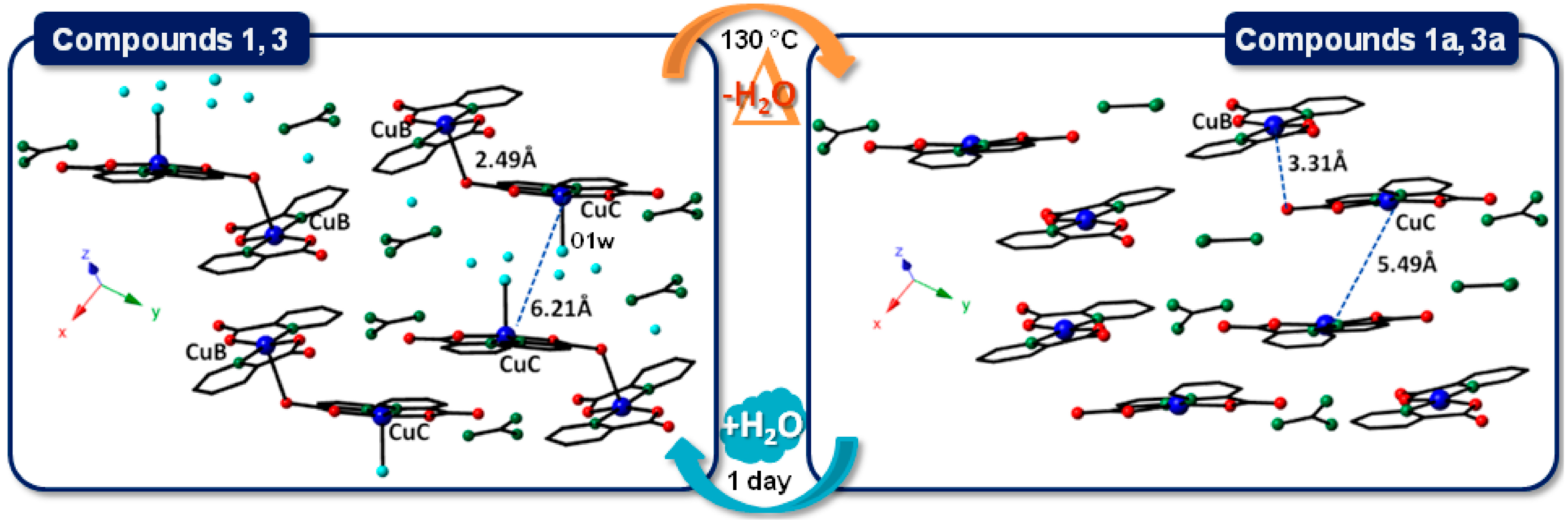
2.4. SCSC Transformations of Compounds 1 and 3 into the Anhydrous Phases 1a and 3a
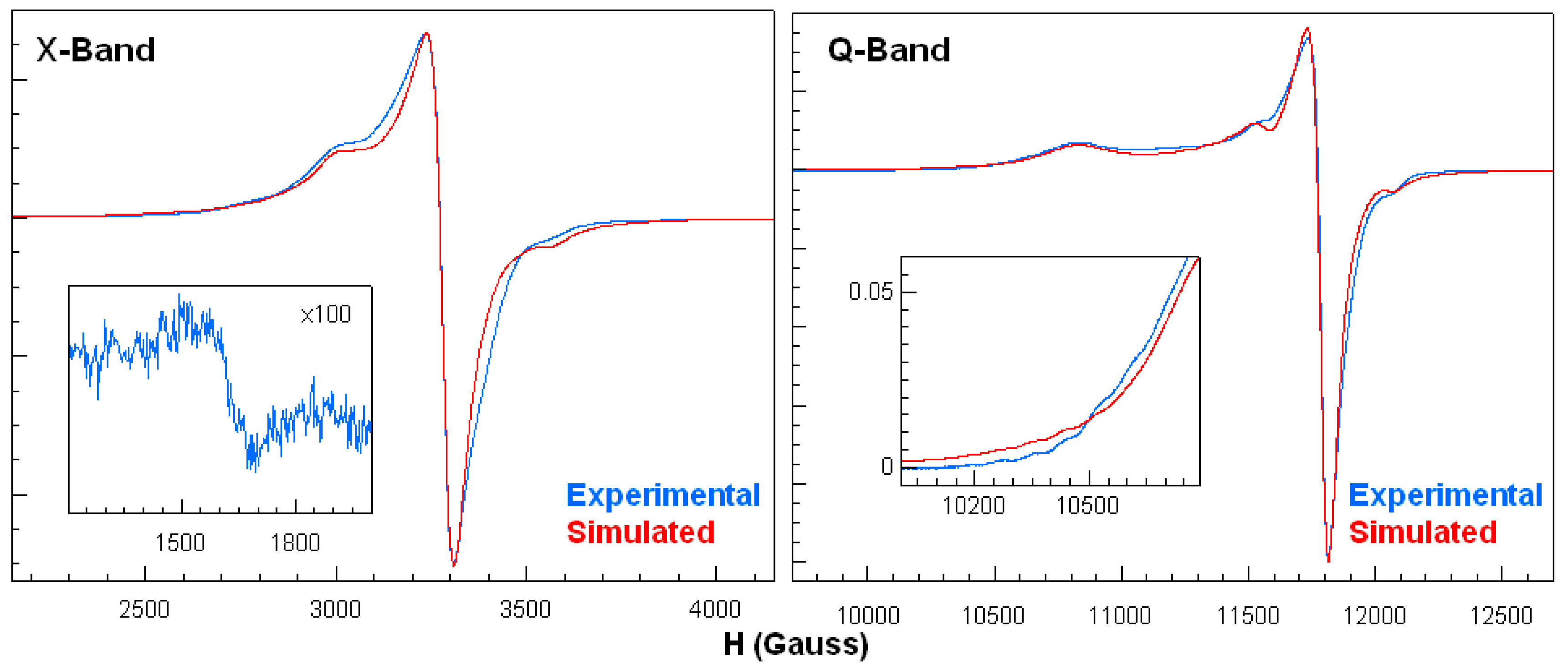
2.5. Electron Paramagnetic Resonance Spectroscopy for Compounds 1 and 1a
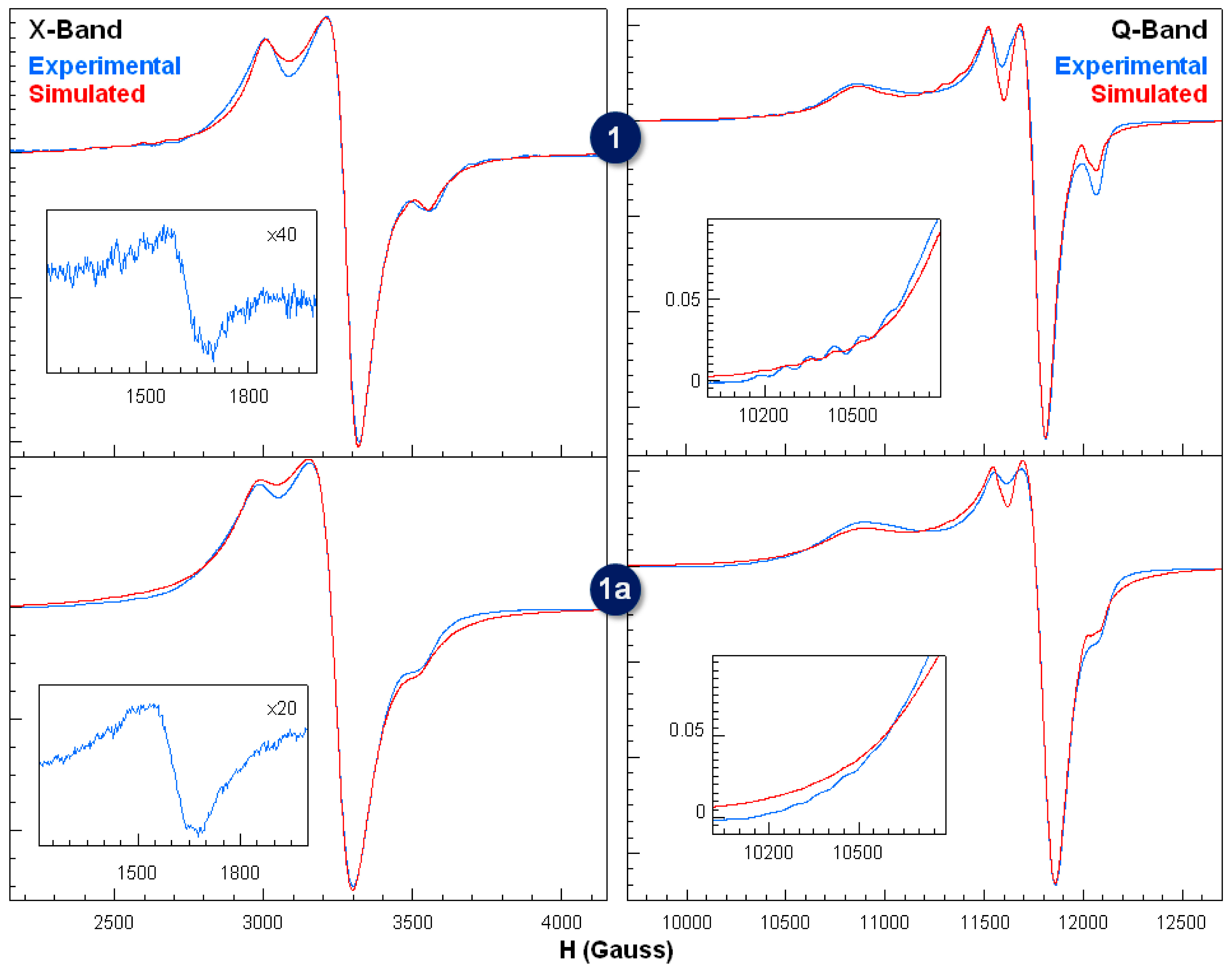
| Compounds | signal 1 | signal 2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| g1 | g2 | g⊥ | g3 = g|| | g⊥ | g|| | A | D | E | |
| 1 | 2.061(1) | 2.073(1) | 2.067(1) | 2.243(2) | 2.061(1) | 2.240(1) | 87(1) | 450(5) | 20(5) |
| 1a | 2.056(1) | 2.068(1) | 2.062(1) | 2.246(2) | 2.061(1) | 2.240(1) | 87(1) | 430(5) | 20(5) |
| 3 | 2.062(1) | 2.071(1) | 2.066(1) | 2.243(2) | 2.060(1) | 2.240(1) | 87(1) | 450(5) | 20(5) |
| 2 | - | - | 2.063(1) | 2.251(2) | 2.060(2) | 2.236(2) | 88(3) | 470(5) | - |
3. Experimental Section
3.1. Materials and Methods
3.2. Synthesis of [C(NH2)3]4[{SiW12O40}{Cu2(pic)4}]·[Cu2(pic)4(H2O)]2·6H2O (1) and [C(NH2)3]8[{SiW12O40}2{Cu(pic)2}3{Cu2(pic)4(H2O)}2]·8H2O (2)
3.3. Synthesis of [C(NH2)3]4[{GeW12O40}{Cu2(pic)4}]·[Cu2(pic)4(H2O)]2·6H2O (3)
3.4. X-ray Crystallography
| Parameters | 1 | 1a | 2 | 3 | 3a |
|---|---|---|---|---|---|
| Formula | C76H88Cu6N24 O72SiW12 | C76H72Cu6N24 O64SiW12 | C92H124Cu7N38 O118Si2W24 | C76H88Cu6Ge N24O72W12 | C76H72Cu6Ge N24O64W12 |
| Fw (g mol−1) | 5105.2 | 4961.1 | 8563.6 | 5149.7 | 5005.6 |
| Crystal system | triclinic | triclinic | triclinic | triclinic | triclinic |
| Space group | P–1 | P–1 | P–1 | P–1 | P–1 |
| a (Ǻ) | 11.7014(4) | 11.6039(3) | 11.9426(2) | 11.7110(3) | 11.6025(7) |
| b (Ǻ) | 15.9523(5) | 15.9379(5) | 12.8151(3) | 15.9628(6) | 15.9736(9) |
| c (Ǻ) | 17.0285(5) | 15.9984(5) | 30.0533(6) | 17.0341(5) | 15.9849(10) |
| α (°) | 107.102(3) | 104.292(3) | 101.508(2) | 107.057(3) | 104.371(5) |
| β (°) | 94.393(3) | 91.168(2) | 90.346(2) | 94.372(2) | 91.222(5) |
| γ (°) | 101.008(3) | 100.599(2) | 105.544(2) | 100.971(2) | 100.534(5) |
| V (Ǻ3) | 2952.2(2) | 2811.6(1) | 4333.6(2) | 2958.9(2) | 2814.5(3) |
| Z | 1 | 1 | 1 | 1 | 1 |
| ρcalcd (g cm−3) | 2.872 | 2.930 | 3.281 | 2.890 | 2.953 |
| μ (mm−1) | 23.180 | 13.447 | 16.822 | 13.025 | 24.412 |
| Reflections: | |||||
| Collected | 22568 | 19291 | 34946 | 19292 | 20323 |
| Unique | 11493 | 11071 | 17052 | 11018 | 10854 |
| Observed [ I > 2σ(I)] | 10892 | 10022 | 15317 | 10485 | 7649 |
| Rint | 0.032 | 0.021 | 0.023 | 0.022 | 0.049 |
| Parameters | 514 | 494 | 788 | 518 | 494 |
| R(F) a [I > 2σ(I)] | 0.050 | 0.046 | 0.086 | 0.044 | 0.079 |
| wR(F2) a [all data] | 0.117 | 0.085 | 0.176 | 0.093 | 0.231 |
| GoF | 1.278 | 1.363 | 1.273 | 1.280 | 1.039 |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Reinoso, S.; Vitoria, P.; Gutiérrez-Zorrilla, J.M.; Lezama, L.; San Felices, L.; Beitia, J.I. Inorganic-Metalorganic Hybrids Based on Copper(II)-Monosubstituted Keggin Polyanions and Dinuclear Copper(II)-Oxalate Complexes. Synthesis, X-ray Structural Characterization, and Magnetic Properties. Inorg. Chem. 2005, 44, 9731–9742. [Google Scholar] [CrossRef] [PubMed]
- Reinoso, S.; Vitoria, P.; Gutiérrez-Zorrilla, J.M.; Lezama, L.; Madariaga, J.M.; San Felices, L.; Iturrospe, A. Coexistence of Five Different Copper(II)-Phenanthroline Species in the Crystal Packing of Inorganic-Metalorganic Hybrids Based on Keggin Polyoxometalates and Copper(II)-Phenanthroline-Oxalate Complexes. Inorg. Chem. 2007, 46, 4010–4021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, J.; Liu, Y.-Y.; Ma, J.-F. Five Polyoxometalate-Based Inorganic-Organic Hybrid Compounds Constructed by a Multidentate N-Donor Ligand: Syntheses, Structures, Electrochemistry, and Photocatalysis Properties. CrystEngComm 2013, 15, 3843–3853. [Google Scholar] [CrossRef]
- Aoki, S.; Kurashina, T.; Kasahara, Y.; Nishijima, T.; Nomiya, K. Polyoxometalate (POM)-Based, Multi-Functional, Inorganic-Organic, Hybrid Compounds: Syntheses and Molecular Structures of Silanol- and/or Siloxane Bond-Containing Species Grafted on Mono- and Tri-Lacunary Keggin POMs. Dalton Trans. 2011, 40, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhang, G.-Q.; Dang, D.-B.; Ma, P.-T.; Gao, H.; Niu, J.-Y. Assembly of Polyoxometalate-Based Inorganic-Organic Compounds from Silver-Schiff Base Building Blocks: Synthesis, Crystal Structures and Luminescent Properties. CrystEngComm 2011, 13, 4181–4187. [Google Scholar] [CrossRef]
- Dolbecq, A.; Dumas, E.; Mayer, C.R.; Mialane, P. Hybrid Organic-Inorganic Polyoxometalate Compounds: From Structural Diversity to Applications. Chem. Rev. 2010, 110, 6009–6048. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yu, Z.-T.; Yang, J.; Hua, W.; Liu, Y.-Y.; Ma, J.-F. First Three-Dimensional Inorganic-Organic Hybrid Material Constructed From an “Inverted Keggin” Polyoxometalate and a Copper(I)-Organic Complex. Inorg. Chem. 2011, 50, 8967–8972. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.M.; Wang, Y.S.; Wang, X.Q.; Korp, J.D.; Jacobson, A.J. Anion-Directed Crystallization of Coordination Polymers: Syntheses and Characterization of Cu4(2-pzc)4(H2O)8(Mo8O26)·2H2O and Cu3(2-pzc)4(H2O)2(V10O28H4)·6.5H2O (2-pzc = 2-Pyrazinecarboxylate). Inorg. Chem. 2001, 40, 1380–1385. [Google Scholar] [CrossRef] [PubMed]
- Hagrman, D.; Hagrman, P.J.; Zubieta, J. Solid-State Coordination Chemistry: The Self-Assembly of Microporous Organic-Inorganic Hybrid Frameworks Constructed from Tetrapyridylporphyrin and Bimetallic Oxide Chains or Oxide Clusters. Angew. Chem. 1999, 38, 3165–3168. [Google Scholar] [CrossRef]
- Lu, Y.; Xu, Y.; Wang, E.B.; Lü, J.; Hu, C.W.; Xu, L. Novel Two-Dimensional Network Constructed from Polyoxomolybdate Chains Linked through Copper-Organonitrogen Coordination Polymer Chains: Hydrothermal Synthesis and Structure of [H2bpy][Cu(4,4‘-bpy)]2[HPCuMo11O39]. Cryst. Growth Des. 2005, 5, 257–260. [Google Scholar] [CrossRef]
- Shivaiah, V.; Nagaraju, M.; Das, S.K. Formation of a Spiral-Shaped Inorganic-Organic Hybrid Chain, [CuII(2,2‘-bipy)(H2O)2Al(OH)6Mo6O18]nn−: Influence of Intra- and Interchain Supramolecular Interactions. Inorg. Chem. 2003, 42, 6604–6606. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Yan, J.-H.; Duan, H.; Zhang, Z.-M.; Li, Y.-G.; Han, X.-B.; Shen, J.-Q.; Fu, H.; Wang, E.-B. Integration of Ln-Sandwich POMs into Molecular Porous Systems Leading to Self-Assembly of Metal-POM Framework Materials. Eur. J. Inorg. Chem. 2013, 4770–4774. [Google Scholar] [CrossRef]
- Kong, X.J.; Ren, Y.-P.; Zheng, P.-Q.; Long, Y.-X.; Long, L.-S.; Huang, R.-B.; Zheng, L.-S. Construction of Polyoxometalates-Based Coordination Polymers through Direct Incorporation between Polyoxometalates and the Voids in a 2D Network. Inorg. Chem. 2006, 45, 10702–10711. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.I.; Yohannes, E.; Doedens, R.J. A Novel Series of Materials Composed of Arrays of Vanadium Oxide Container Molecules, [V18O42(X)] (X = H2O, Cl−, Br−): Synthesis and Characterization of [M2(H2N(CH2)2NH2)5][(M(H2N(CH2)2NH2)2]2V18O42(X)]·9H2O (M = Zn, Cd). Inorg. Chem. 2003, 42, 3125–3129. [Google Scholar] [CrossRef] [PubMed]
- An, H.Y.; Wang, E.B.; Xiao, D.R.; Li, Y.G.; Su, Z.M.; Xu, L. Chiral 3D Architectures with Helical Channels Constructed from Polyoxometalate Clusters and Copper-Amino Acid Complexes. Angew. Chem. 2006, 45, 904–908. [Google Scholar] [CrossRef]
- Dai, L.M.; You, W.S.; Wang, E.B.; Wu, S.X.; Su, Z.M.; Du, Q.H.; Zhao, Y.; Fang, Y. Two Novel One-Dimensional α-Keggin-Based Coordination Polymers with Argentophilic {Ag3}3+/{Ag4}4+ Clusters. Cryst. Growth Des. 2009, 9, 2110–2116. [Google Scholar] [CrossRef]
- Darling, K.; Smith, T.M.; Vargas, J.; O’Connor, C.J.; Zubieta, J. Polyoxometalate Clusters as Building Blocks for Oxide Materials: Synthesis and Structure of a Three-dimensional Copper-Pyrazinetetrazolate / Keggin Assembly. Inorg. Chem. Commun. 2013, 32, 1–4. [Google Scholar] [CrossRef]
- Hao, X.-L.; Ma, Y.-Y.; Wang, Y.-H.; Zhou, W.-Z.; Li, Y.-G. New Organic-Inorganic Hybrid Assemblies based on Metal-bis(betaine) Coordination Complexes and Keggin-type Polyoxometalates. Inorg. Chem. Commun. 2014, 41, 19–24. [Google Scholar] [CrossRef]
- Li, S.; Ma, H.; Pang, H.; Zhang, Z.; Yu, Y.; Liu, H.; Yu, T. Tuning the Dimension of POM-Based Inorganic-Organic Hybrids from 3D Self-Penetrating Framework to 1D Poly-Pendant Chain via Changing POM Clusters and Introducing Secondary Spacers. CrystEngComm 2014, 16, 2045–2055. [Google Scholar] [CrossRef]
- Henry, N.; Costenoble, S.; Lagrenee, M.; Loiseau, T.; Abraham, F. Lanthanide-Based 0D and 2D Molecular Assemblies with the Pyridazine-3,6-dicarboxylate Linker. CrystEngComm 2011, 13, 251–258. [Google Scholar] [CrossRef]
- Wang, X.; Qin, C.; Wang, E.; Li, Y.; Hao, N.; Hu, C.; Xu, L. Syntheses, Structures, and Photoluminescence of a Novel Class of d10 Metal Complexes Constructed from Pyridine-3,4-dicarboxylic Acid with Different Coordination Architectures. Inorg. Chem. 2004, 43, 1850–1856. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, J.; Beobide, G.; Castillo, O.; Luque, A.; Pérez-Yánez, S.; Román, P. Structure-Directing Effect of Organic Cations in the Assembly of Anionic In(III)/Diazinedicarboxylate Architectures. Cryst. Growth Des. 2012, 12, 1501–1512. [Google Scholar] [CrossRef]
- Pinar, A.B.; Gómez-Hortiguela, L.; McCusker, L.B.; Pérez-Pariente, J. Controlling the Aluminum Distribution in the Zeolite Ferrierite via the Organic Structure Directing Agent. Chem. Mater. 2013, 25, 3654–3661. [Google Scholar] [CrossRef]
- Van Bommel, K.J.C.; Friggeri, A.; Shinkai, S. Organic Templates for the Generation of Inorganic Materials. Angew. Chem. 2003, 42, 980–999. [Google Scholar] [CrossRef]
- Decker, R.; Schlickum, U.; Klappenberger, F.; Zoppellaro, G.; Klyatskaya, S.; Ruben, M.; Barth, J.V.; Brune, H. Using Metal-Organic Templates to Steer the Growth of Fe and Co Nanocluster. Appl. Phys. Lett. 2008, 93, 243102 / 1–243102 / 3. [Google Scholar] [CrossRef]
- Abrahams, B.F.; Hawley, A.; Haywood, M.G.; Hudson, T.A.; Robson, R.; Slizys, D.A. Serendipity and Design in the Generation of New Coordination Polymers: An Extensive Series of Highly Symmetrical Guanidinium-Templated, Carbonate-Based Networks with the Sodalite Topology. J. Am. Chem. Soc. 2004, 126, 2894–2904. [Google Scholar] [CrossRef] [PubMed]
- Reinoso, S.; Dickman, M.H.; Kortz, U. Selective Crystallization of Dimeric vs. Monomeric Dimethyltin-Containing Tungstoarsenates(III) and -antimonates(III) with the Guanidinium Cation. Eur. J. Inorg. Chem. 2009, 947–953. [Google Scholar] [CrossRef]
- Piedra-Garza, L.F.; Reinoso, S.; Dickman, M.H.; Sanguineti, M.M.; Kortz, U. The First 3-Dimensional Assemblies of Organotin-Functionalized Polyanions. Dalton Trans. 2009, 6231–6234. [Google Scholar] [CrossRef]
- Reinoso, S.; Bassil, B.S.; Barsukova, M.; Kortz, U. pH-Controlled Assemblies of Dimethyltin-Functionalized 9-Tungstophosphates with Guanidinium as Structure-Directing Cation. Eur. J. Inorg. Chem. 2010, 2537–2542. [Google Scholar] [CrossRef]
- Iturrospe, A.; Artetxe, B.; Reinoso, S.; San Felices, L.; Vitoria, P.; Lezama, L.; Gutiérrez-Zorrilla, J.M. Copper(II) Complexes of Tetradentate Pyridyl Ligands Supported on Keggin Polyoxometalates: Single-Crystal to Single-Crystal Transformations Promoted by Reversible Dehydration Processes. Inorg. Chem. 2013, 52, 3084–3093. [Google Scholar] [CrossRef] [PubMed]
- Iturrospe, A.; San Felices, L.; Reinoso, S.; Artetxe, B.; Lezama, L.; Gutiérrez-Zorrilla, J.M. Reversible Dehydration in Polyoxometalate-Based Hybrid Compounds: A Study of Single-Crystal to Single-Crystal Transformations in Keggin-Type Germanotungstates Decorated with Copper(II) Complexes of Tetradentate N-Donor Ligands. Cryst. Growth Des. 2014, 14, 2318–2328. [Google Scholar] [CrossRef]
- Wéry, A.S.J.; Gutiérrez-Zorrilla, J.M.; Luque, A.; Ugalde, M.; Román, P. Phase Transitions in Metavanadates. Polymerization of Tetrakis(tert-Butylammonium)-cyclo-Tetrametavanadate. Chem. Mater. 1996, 8, 408–413. [Google Scholar] [CrossRef]
- Ritchie, C.; Streb, C.; Thiel, J.; Mitchell, S.G.; Miras, H.N.; Long, D.-L.; Boyd, T.; Peacock, R.D.; McGlone, T.; Cronin, L. Reversible Redox Reactions in an Extended Polyoxometalate Framework Solid. Angew. Chem. 2008, 47, 6881–6884. [Google Scholar] [CrossRef]
- Thiel, J.; Ritchie, C.; Streb, C.; Long, D.-L.; Cronin, L. Heteroatom-Controlled Kinetics of Switchable Polyoxometalate Frameworks. J. Am. Chem. Soc. 2009, 131, 4180–4181. [Google Scholar] [CrossRef] [PubMed]
- Uehara, K.; Mizuno, N. Heterolytic Dissociation of Water Demonstrated by Crystal-to-Crystal Core Interconversion from (μ-Oxo)divanadium to Bis(μ-hydroxo)divanadium Substituted Polyoxometalates. J. Am. Chem. Soc. 2011, 133, 1622–1625. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.-X.; Zhao, W.-F.; Xu, X.; Tang, J.; Wu, C.D. From 1D to 3D Single-Crystal-to-Single-Crystal Structural Transformations Based on Linear Polyanion [Mn4(H2O)18WZnMn2(H2O)2(ZnW9O34)2]4−. Inorg. Chem. 2011, 50, 12387–12389. [Google Scholar] [CrossRef] [PubMed]
- Uchida, S.; Takahashi, E.; Mizuno, N. Porous Ionic Crystals Modified by Post-Synthesis of K2[Cr3O(OOCH)6(etpy)3]2[α-SiW12O40]·8H2O through Single-Crystal-to-Single-Crystal Transformation. Inorg. Chem. 2013, 52, 9320–9326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-Z.; Gu, W.; Liu, X.; Dong, Z.; Li, B. Solid-State Photopolymerization of a Photochromic Hybrid Based on Keggin Tungstophosphates. CrystEngComm 2008, 10, 652–654. [Google Scholar] [CrossRef]
- Zhang, L.-Z.; Gu, W.; Dong, Z.; Liu, X.; Li, B. Phase Transformation of a Rare-Earth Anderson Polyoxometalate at Low Temperature. CrystEngComm 2008, 10, 1318–1320. [Google Scholar] [CrossRef]
- Reinoso, S.; Dickman, M.H.; Praetorius, A.; Kortz, U. Low-Temperature Phase of Hexaguanidinium Heptamolybdate Monohydrate. Acta Crystallogr. 2008, E64, m614–m615. [Google Scholar]
- Barats-Damatov, D.; Shimon, L.J.W.; Feldman, Y.; Bendikov, T.; Neumann, R. Solid-State Crystal-to-Crystal Phase Transitions and Reversible Structure-Temperature Behavior of Phosphovanadomolybdic Acid, H5PV2Mo10O40. Inorg. Chem. 2015, 4, 628–634. [Google Scholar] [CrossRef]
- Chen, C.L.; Goforth, A.M.; Smith, M.D.; Su, C.Y.; zur Loye, H.-C. [Co2(ppca)2(H2O)(V4O12)0.5]: A Framework Material Exhibiting Reversible Shrinkage and Expansion through a Single-Crystal-to-Single-Crystal Transformation Involving a Change in the Cobalt Coordination Environment. Angew. Chem. 2005, 44, 6673–6677. [Google Scholar] [CrossRef]
- Vittal, J.J. Supramolecular Structural Transformations Involving Coordination Polymers in the Solid State. Coord. Chem. Rev. 2007, 251, 1781–1795. [Google Scholar] [CrossRef]
- Abeysinghe, D.; Smith, M.D.; Yeon, J.; Morrison, G.; zur Loye, H.-C. Observation of Multiple Crystal-to-Crystal Transitions in a New Reduced Vanadium Oxalate Hybrid Material, Ba3[(VO)2(C2O4)5(H2O)6]·(H2O)3, Prepared via a Mild, Two-Step Hydrothermal Method. Cryst. Growth Des. 2014, 14, 4749–4758. [Google Scholar] [CrossRef]
- Tian, Y.; Allan, P.K.; Renouf, C.L.; He, X.; McCormick, L.J.; Morris, R.E. Synthesis and Structural Characterization of a Single-Crystal to Single-Crystal Transformable Coordination Polymer. Dalton Trans. 2014, 43, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, L.; Morsali, A.; Marandi, F.; Pantenburg, I.; Tehrani, A.A. Dynamic Crystal-to-Crystal Transformation of 1D to 2D Lead(II) Coordination Polymers by De- and Rehydration with No Change in the Morphology of Nano-Particles. New J. Chem. 2014, 38, 3375–3378. [Google Scholar] [CrossRef]
- Hanson, K.; Calin, N.; Bugaris, D.; Scancella, M.; Sevov, S.C. Reversible Repositioning of Zinc Atoms within Single Crystals of a Zinc Polycarboxylate with an Open-Framework Structure. J. Am. Chem. Soc. 2004, 126, 10502–10503. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, S.; Uemura, K. Dynamic Porous Properties of Coordination Polymers Inspired by Hydrogen Bonds. Chem. Soc. Rev. 2005, 34, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, S.; Matsuda, R. Chemistry of Coordination Space of Porous Coordination Polymers. Coord. Chem. Rev. 2007, 251, 2490–2509. [Google Scholar] [CrossRef]
- Stamatatos, T.C.; Efthymiou, C.G.; Stoumpos, C.C.; Perlepes, S.P. Adventures in the Coordination Chemistry of Di-2-pyridyl Ketone and Related Ligands: From High-Spin Molecules and Single-Molecule Magnets to Coordination Polymers, and from Structural Aesthetics to an Exciting New Reactivity Chemistry of Coordinated Ligands. Eur. J. Inorg. Chem. 2009, 3361–3368. [Google Scholar] [CrossRef]
- Huang, D.; Wang, W.; Zhang, X.; Chen, C.; Chen, F.; Liu, Q.; Liao, D.; Li, L.; Sun, L. Synthesis, Structural Characterizations and Magnetic Properties of a Series of Mono-, Di- and Polynuclear Manganese Pyridinecarboxylate Compounds. Eur. J. Inorg. Chem. 2004, 1454–1464. [Google Scholar] [CrossRef]
- Biswas, C.; Mukherjee, P.; Drew, M.G.B.; Gómez-García, C.J.; Clemente-Juan, J.M.; Ghosh, A. Anion-Directed Synthesis of Metal-Organic Frameworks Based on 2-Picolinate Cu(II) Complexes: A Ferromagnetic Alternating Chain and Two Unprecedented Ferromagnetic Fish Backbone Chains. Inorg. Chem. 2007, 46, 10771–10780. [Google Scholar] [CrossRef] [PubMed]
- Pope, M.T.; Müller, A. Polyoxometalate Chemistry: An Old Field with New Dimensions in Several Disciplines. Angew. Chem. 1991, 30, 34–48. [Google Scholar] [CrossRef]
- Hervé, G.; Tézé, A.; Contant, R. General Principles of the Synthesis of Polyoxometalates in Aqueous Solution. In Polyoxometalate Molecular Science; Borrás-Almenar, J.J., Coronado, E., Müller, A., Pope, M.T., Eds.; Kluwer: Dordrecht, The Netherlands, 2003; NATO Science Series II; Volume 98, pp. 33–54. [Google Scholar]
- Zhang, C.-J.; Pang, H.-J.; Tang, Q.; Chen, Y.-G. A Feasible Route to Approach 3D POM-Based Hybrids: Utilizing Substituted or Reduced Keggin Anions with High Charge Density. Dalton Trans. 2012, 41, 9365–9372. [Google Scholar] [CrossRef] [PubMed]
- San Felices, L.; Vitoria, P.; Gutiérrez-Zorrilla, J.M.; Lezama, L.; Reinoso, S. Hybrid Inorganic-Metalorganic Compounds Containing Copper(II)-Monosubstituted Keggin Polyanions and Polymeric Copper(I) Complexes. Inorg. Chem. 2006, 45, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Żurowska, B.; Mroziński, J.; Ciunik, Z. One-Dimensional Copper(II) Compound with a Double Out-of-Plane Carboxylato-Bridge—Another Polymorphic Form of Cu(pyridine-2-carboxylate)2. Polyhedron 2007, 26, 1251–1258. [Google Scholar] [CrossRef]
- Żurowska, B.; Mroziński, J.; Ślepokura, K. Structure and Magnetic Properties of a Double Out-of-Plane Carboxylato-Bridged Cu(II) Compound with Pyridine-2-carboxylate. Polyhedron 2007, 26, 3379–3387. [Google Scholar] [CrossRef]
- Woodward, P.M.; Sleight, A.W.; Vogt, T.J. Structure Refinement of Triclinic Tungsten Trioxide. Phys. Chem. Solids 1995, 56, 1305–1315. [Google Scholar] [CrossRef]
- Schofield, P.F.; Knight, K.S.; Redfern, S.A.T.; Cressey, G. Distortion Characteristics Across the Structural Phase Transition in (Cu1–xZnx)WO4. Acta Crystallogr. 1997, B53, 102–112. [Google Scholar] [CrossRef]
- Tézé, A.; Hervé, G.; Finke, R.G.; Lyon, D.K. α-, β-, and γ-Dodecatungstosilicic Acids: Isomers and Related Lacunary Compounds. Inorg. Synth. 1990, 27, 85–96. [Google Scholar]
- Hervé, G.; Tézé, A. Study of α and β-Enneatungstosilicates and Germanates. Inorg. Chem. 1977, 16, 2115–2117. [Google Scholar] [CrossRef]
- CrysAlisPro Software System, version 171.36.24 ed; Agilent Technologies UK Ltd.: Oxford, UK, 2012.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Crystallogr. 2009, D65, 148–155. [Google Scholar]
- Farrugia, L.J. WinGX Suite for Small-Molecule Single-Crystal Crystallography. J. Appl. Crystallogr. 1999, 32, 837–836. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pache, A.; Reinoso, S.; Felices, L.S.; Iturrospe, A.; Lezama, L.; Gutiérrez-Zorrilla, J.M. Single-Crystal to Single-Crystal Reversible Transformations Induced by Thermal Dehydration in Keggin-Type Polyoxometalates Decorated with Copper(II)-Picolinate Complexes: The Structure Directing Role of Guanidinium. Inorganics 2015, 3, 194-218. https://doi.org/10.3390/inorganics3020194
Pache A, Reinoso S, Felices LS, Iturrospe A, Lezama L, Gutiérrez-Zorrilla JM. Single-Crystal to Single-Crystal Reversible Transformations Induced by Thermal Dehydration in Keggin-Type Polyoxometalates Decorated with Copper(II)-Picolinate Complexes: The Structure Directing Role of Guanidinium. Inorganics. 2015; 3(2):194-218. https://doi.org/10.3390/inorganics3020194
Chicago/Turabian StylePache, Aroa, Santiago Reinoso, Leire San Felices, Amaia Iturrospe, Luis Lezama, and Juan M. Gutiérrez-Zorrilla. 2015. "Single-Crystal to Single-Crystal Reversible Transformations Induced by Thermal Dehydration in Keggin-Type Polyoxometalates Decorated with Copper(II)-Picolinate Complexes: The Structure Directing Role of Guanidinium" Inorganics 3, no. 2: 194-218. https://doi.org/10.3390/inorganics3020194
APA StylePache, A., Reinoso, S., Felices, L. S., Iturrospe, A., Lezama, L., & Gutiérrez-Zorrilla, J. M. (2015). Single-Crystal to Single-Crystal Reversible Transformations Induced by Thermal Dehydration in Keggin-Type Polyoxometalates Decorated with Copper(II)-Picolinate Complexes: The Structure Directing Role of Guanidinium. Inorganics, 3(2), 194-218. https://doi.org/10.3390/inorganics3020194