From Stable ZnO and GaN Clusters to Novel Double Bubbles and Frameworks




Abstract
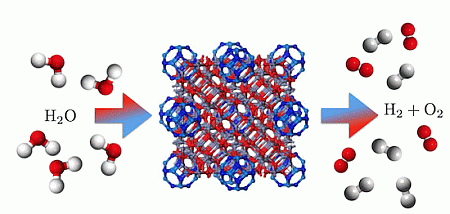
:
1. Introduction
2. Construction of Double Bubbles
High Symmetry Double Bubble Clusters as Secondary Building Units
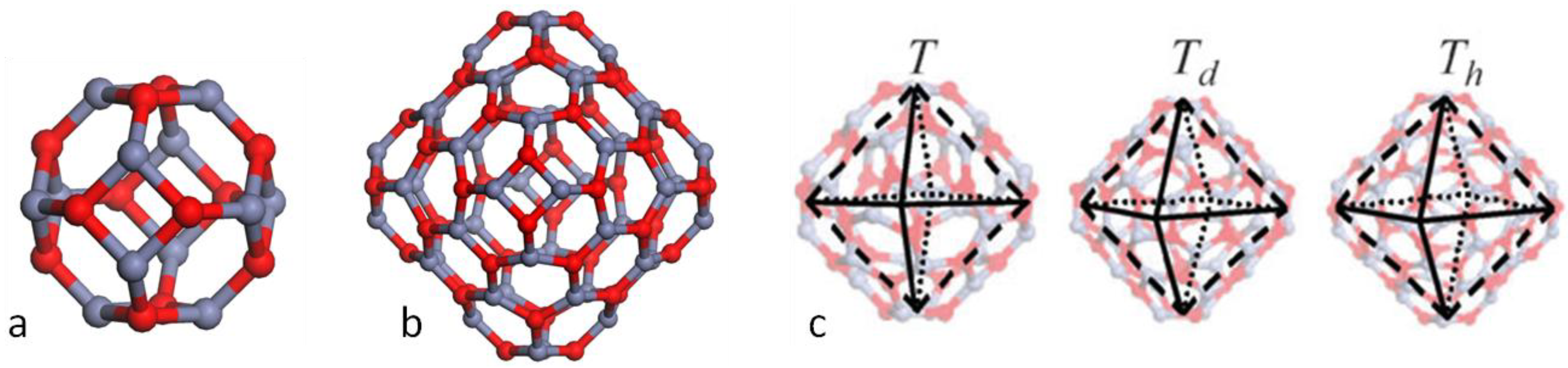
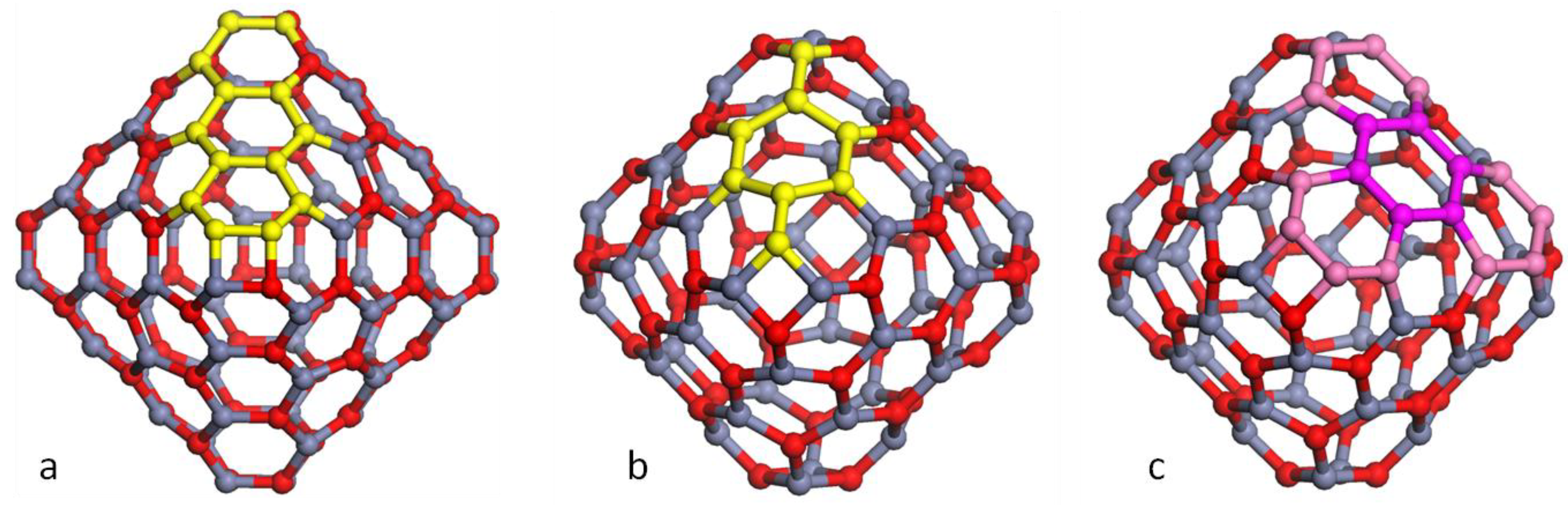
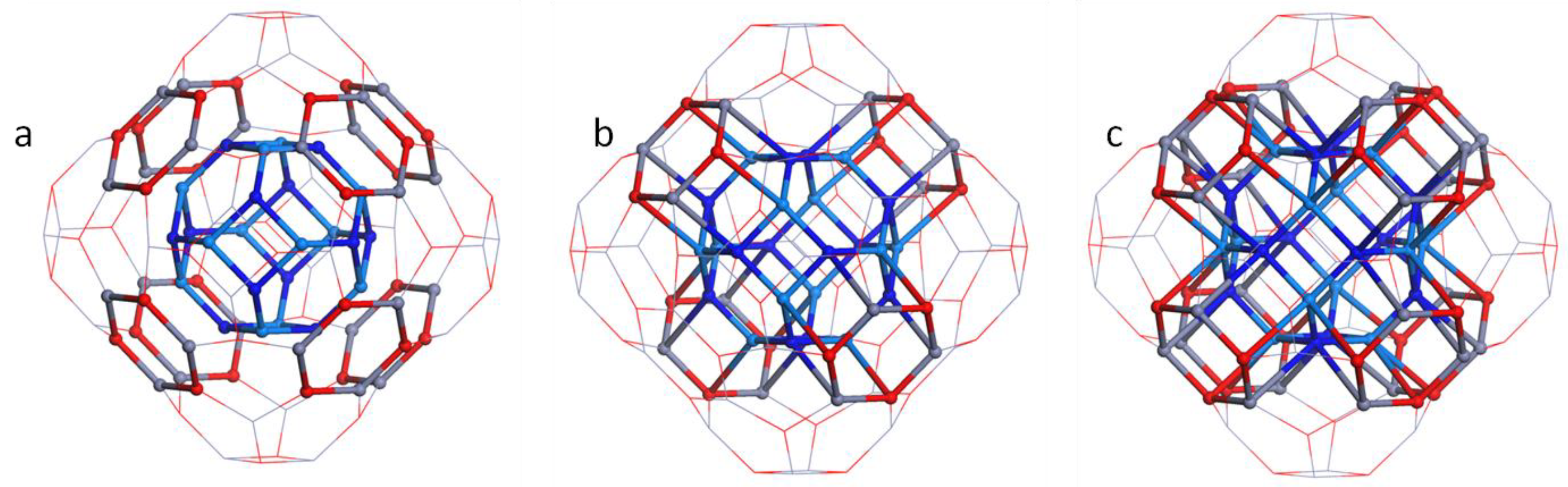

3. Results and Discussion
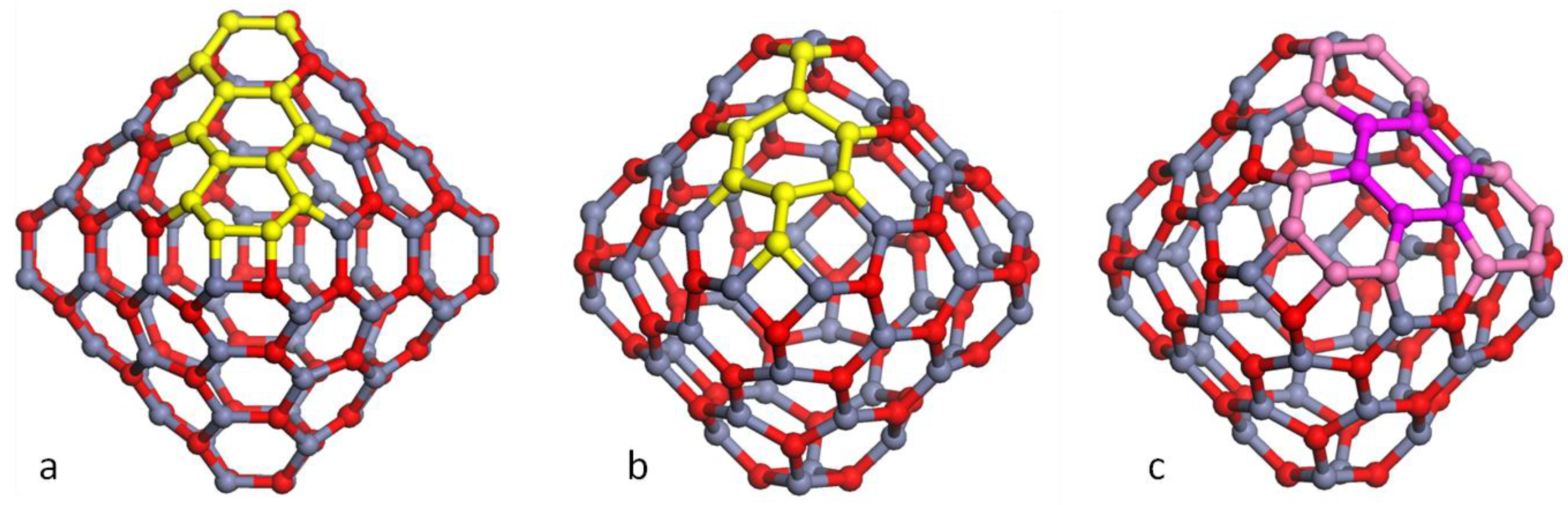
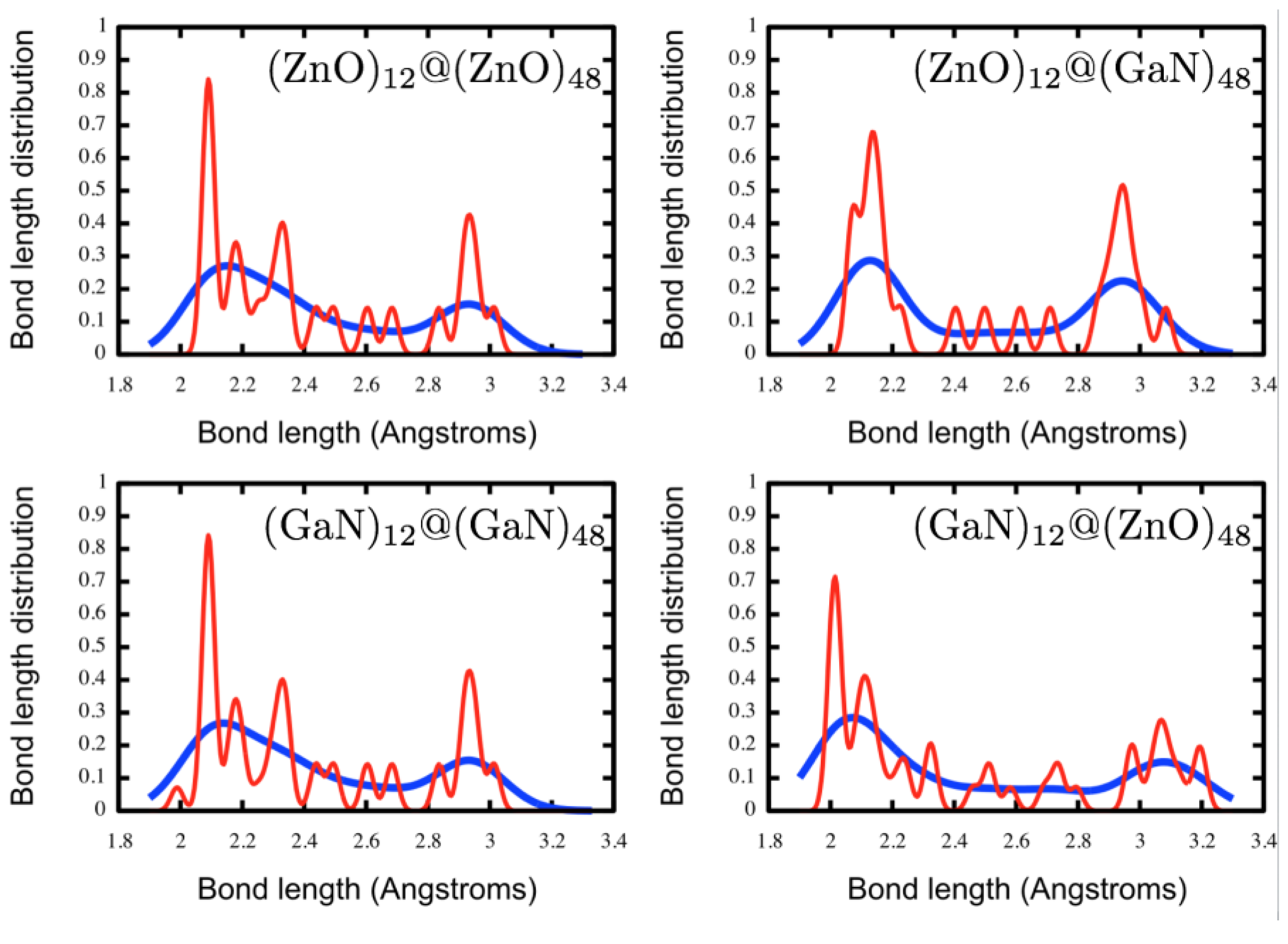
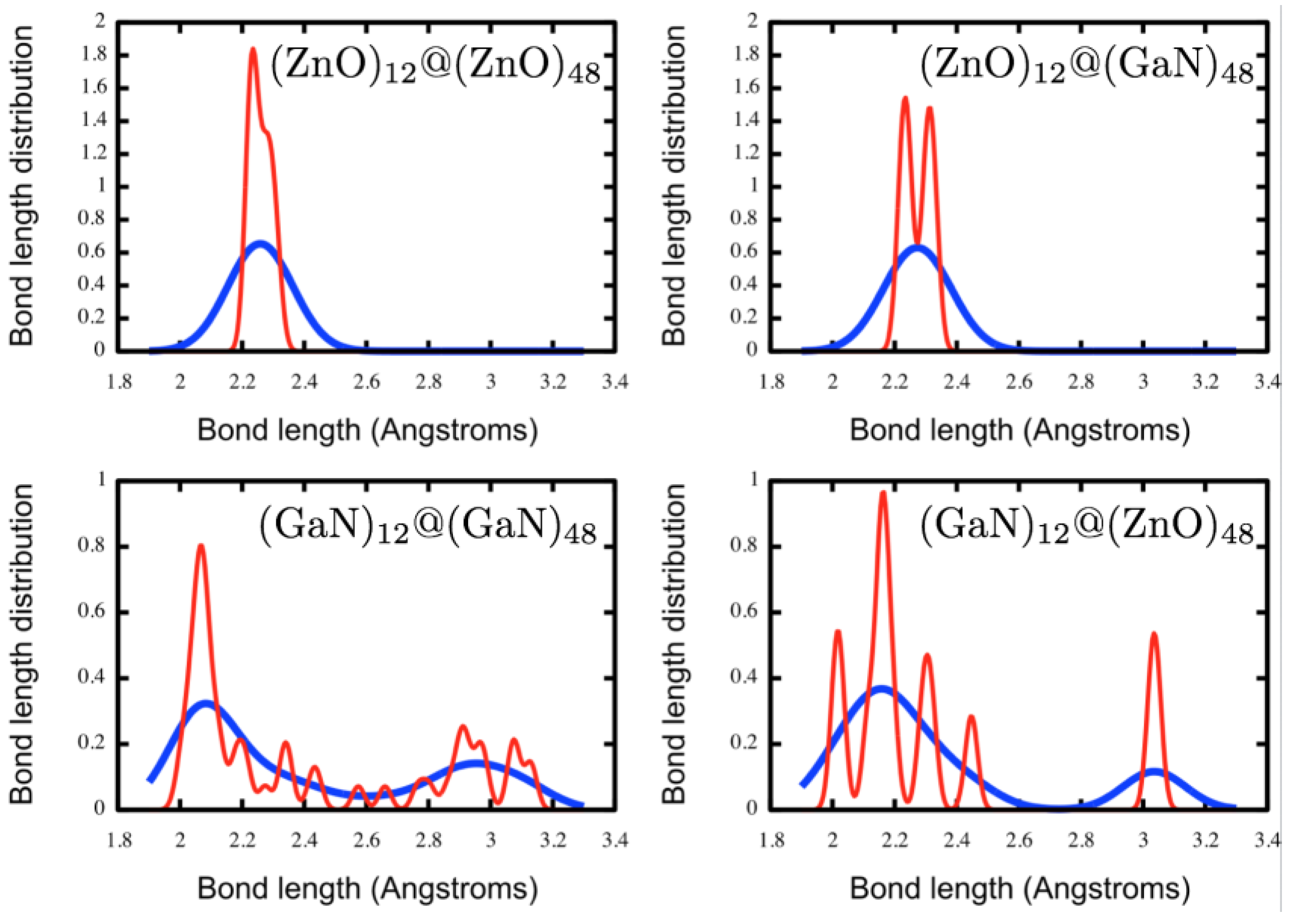
3.1. Double Bubble Clusters


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
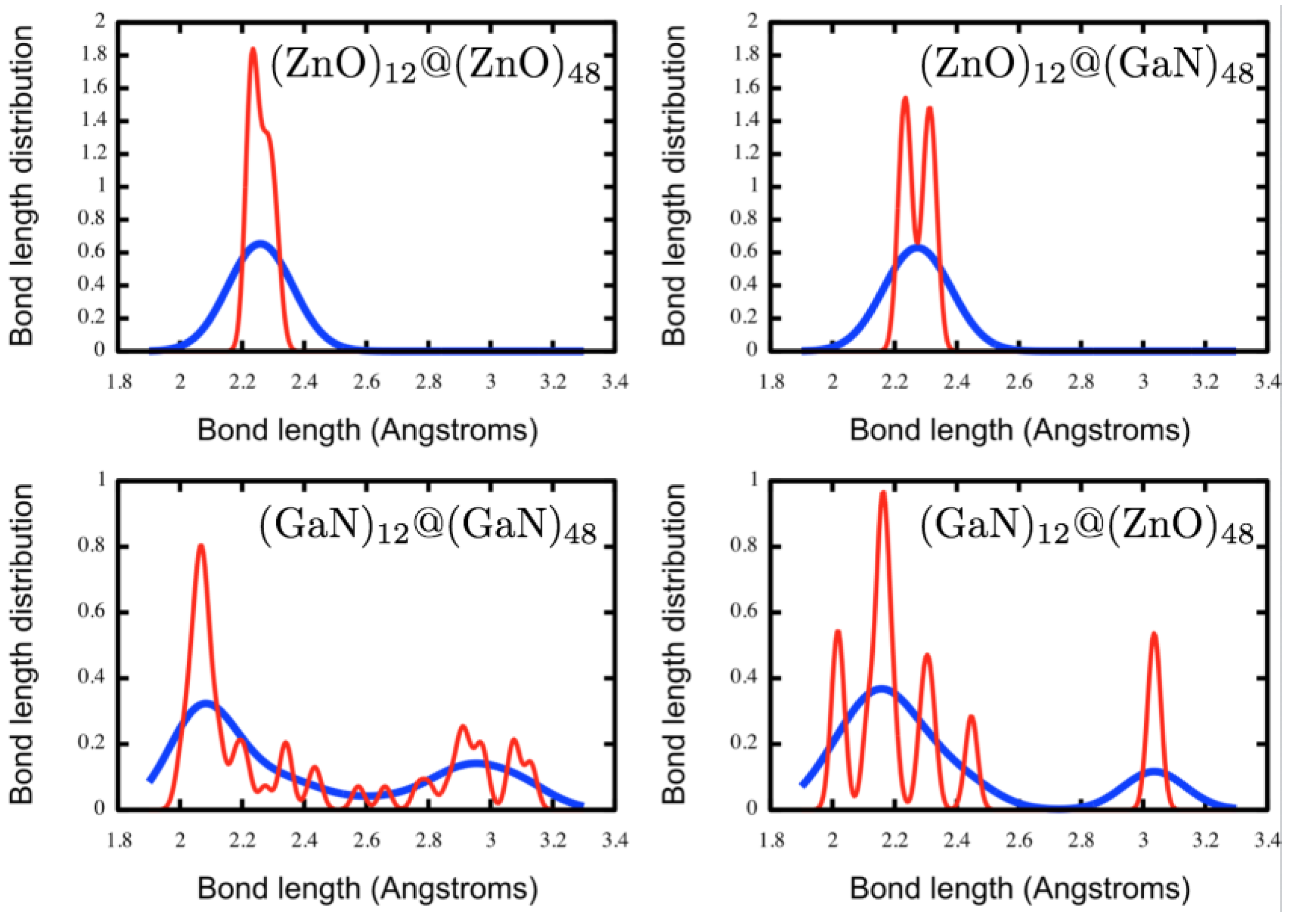{kind=link}
| System | Symmetry | Douter (Å) | Dinner (Å) | Dinter (Å) | |
|---|---|---|---|---|---|
| A | B | ||||
| (GaN)12@(ZnO)48 | C1 | 1.92 | 1.93 | 2.05 (0.1) | 3.08 (0.1) |
| (ZnO)12@(GaN)48 | C2 | 1.89 | 1.96 | 2.13 (0.1) | 2.94 (0.1) |
| (ZnO)12@(ZnO)48 | C2 | 1.93 | 1.95 | 2.10 (0.0) | 2.94 (0.2) |
| (GaN)12@(GaN)48 | C1 | 1.93 | 1.92 | 2.10 (0.1) | 2.94 (0.1) |

| System | COM difference (COMOuter–COMInner) | Normalised second moments of atom distribution | |
|---|---|---|---|
| Inner | Outer | ||
| (GaN)12@(ZnO)48 | 0.00, 0.11, 0.05 | 1.05, 1.01, 0.94 | 1.05, 1.01, 0.94 |
| (ZnO)12@(GaN)48 | 0.00, 0.00, 0.01 | 1.02, 1.00, 0.98 | 1.01, 1.00, 0.99 |
| (ZnO)12@(ZnO)48 | 0.00, 0.00, 0.04 | 1.09, 1.00, 0.91 | 1.05, 1.00, 0.95 |
| (GaN)12@(GaN)48 | 0.00, 0.01, −0.06 | 1.04, 1.01, 0.96 | 1.04, 1.01, 0.95 |

| System | EAssoc (kJ/mol) | Hf (kJ/mol) |
|---|---|---|
| (GaN)12@(ZnO)48 | −11.27 | 78.32 |
| (ZnO)12@(GaN)48 | −8.17 | 104.55 |
| (ZnO)12@(ZnO)48 | −9.38 | 68.50 |
| (GaN)12@(GaN)48 | −9.16 | 116.18 |

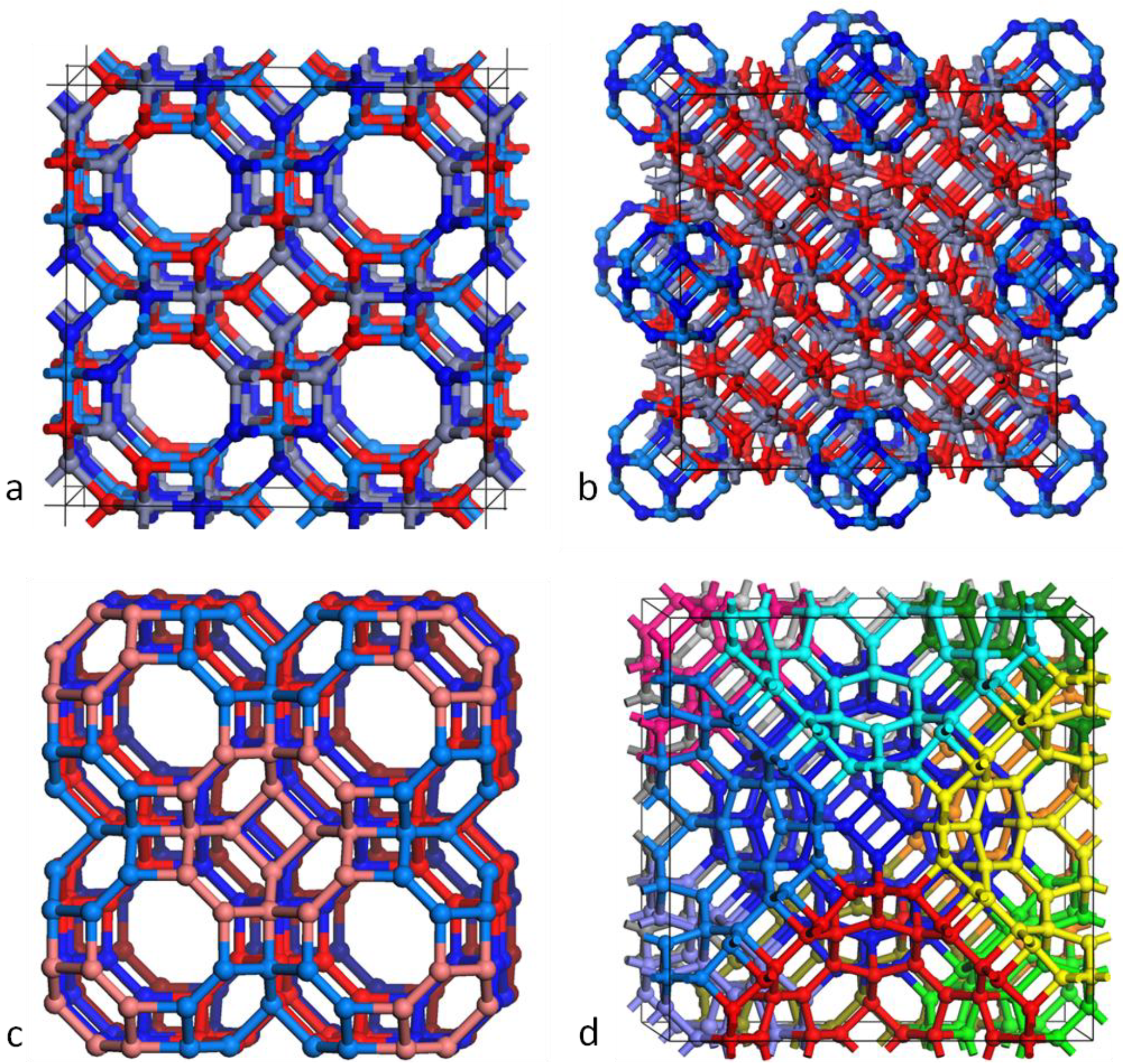
3.2. Double Bubble Frameworks
| System | Lattice parameter (Å) | Bulk modulus (GPa) | Douter (Å) | Dinner (Å) | Dinter (Å) | |
|---|---|---|---|---|---|---|
| A | B | |||||
| (GaN)12@(ZnO)48 | 19.26 | 78.84 | 1.96 | 1.94 | 2.08 (0.1) | 2.96 (0.2) |
| (ZnO)12@(GaN)48 | 18.84 | 77.88 | 1.90 | 2.01 | 2.27 (0.1) | - |
| (ZnO)12@(ZnO)48 | 19.26 | 69.78 | 1.94 | 2.00 | 2.26 (0.0) | - |
| (GaN)12@(GaN)48 | 18.94 | 101.92 | 1.93 | 1.94 | 2.18 (0.2) | 3.04 (0.1) |
| System | Lattice parameter, a (Å) | Lattice parameter, c (Å) | Bulk modulus (GPa) | u |
|---|---|---|---|---|
| ZnO | 3.251 | 5.204 | 146.136 | 0.382 |
| GaN | 3.187 | 2.760 | 188.367 | 0.378 |
| System | HF/atom (kJ/mol) |
|---|---|
| (GaN)12@(ZnO)48 | 13.17 |
| (ZnO)12@(GaN)48 | 21.46 |
| (ZnO)12@(ZnO)48 | 18.54 |
| (GaN)12@(GaN)48 | 27.71 |
4. Computational Detail
4.1. Interatomic Potentials Calculations
4.2. Density Functional Theory Calculations
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar]
- Vispute, R.D.; Talyansky, V.; Choopun, S.; Sharma, R.P.; Venkatesan, T.; He, M.; Tang, X.; Halpern, J.B.; Spencer, M.G.; Li, Y.X.; et al. Heteroepitaxy of ZnO on GaN and its implications for fabrication of hybrid optoelectronic devices. Appl. Phys. Lett. 1998, 73, 348–350. [Google Scholar] [CrossRef]
- Nakamura, S.; Pearton, S.; Fasol, G. The Blue Laser Diode: The Complete Story; Springer-Verlag: Berlin, Germany, 2000. [Google Scholar]
- Yu, Q.-X.; Xu, B.; Wu, Q.-H.; Liao, Y.; Wang, G.-Z.; Fang, R.-C.; Lee, H.-Y.; Lee, C.-T. Optical properties of ZnO/GaN heterostructure and its near-ultraviolet light-emitting diode. Appl. Phys. Lett. 2003, 83, 4713. [Google Scholar] [CrossRef]
- Zhu, H.; Shan, C.-X.; Yao, B.; Li, B.-H.; Zhang, J.-Y.; Zhang, Z.-Z.; Zhao, D.-X.; Shen, D.-Z.; Fan, X.-W.; Lu, Y.-M.; et al. Ultralow-threshold laser realized in zinc oxide. Adv. Mater. 2009, 21, 1613–1617. [Google Scholar] [CrossRef]
- Shevlin, S.A.; Guo, Z.X.; van Dam, H.J.J.; Sherwood, P.; Catlow, C.R.A.; Sokol, A.A.; Woodley, S.M. Structure, optical properties and defects in nitride (III–V) nanoscale cage clusters. Phys. Chem. Chem. Phys. 2008, 10, 1944–1959. [Google Scholar]
- Catlow, C.R.A.; French, S.A.; Sokol, A.A.; Al-Sunaidi, A.A.; Woodley, S.M. Zinc oxide: A case study in contemporary computational solid state chemistry. J. Comput. Chem. 2008, 29, 2234–2249. [Google Scholar]
- Catlow, C.R.A.; Bromley, S.T.; Hamad, S.; Mora-Fonz, M.; Sokol, A.A.; Woodley, S.M. Modelling nano-clusters and nucleation. Phys. Chem. Chem. Phys. 2010, 12, 786–811. [Google Scholar] [CrossRef]
- Watkins, M.B.; Shevlin, S.A.; Sokol, A.A.; Slater, B.; Catlow, C.R.A.; Woodley, S.M. Bubbles and microporous frameworks of silicon carbide. Phys. Chem. Chem. Phys. 2009, 11, 3186–3200. [Google Scholar] [CrossRef]
- Woodley, S.M.; Watkins, M.B.; Sokol, A.A.; Shevlin, S.A.; Catlow, C.R.A. Construction of nano- and microporous frameworks from octahedral bubble clusters. Phys. Chem. Chem. Phys. 2009, 11, 3176–3185. [Google Scholar] [CrossRef]
- Carrasco, J.; Illas, F.; Bromley, S.T. Ultralow-density nanocage-based metal-oxide polymorphs. Phys. Rev. Lett. 2007, 99, 235502. [Google Scholar]
- Hamad, S.; Catlow, C.R.A.; Spanó, E.; Matxain, J.M.; Ugalde, J.M. Structure and properties of ZnS nanoclusters. J. Phys. Chem. B 2005, 109, 2703–2709. [Google Scholar]
- Al-Sunaidi, A.A.; Sokol, A.A.; Catlow, C.R.A.; Woodley, S.M. Structures of zinc oxide nanoclusters: As found by revolutionary algorithm techniques. J. Phys. Chem. C 2008, 112, 18860–18875. [Google Scholar] [CrossRef]
- Behrman, E.C.; Foehrweiser, R.K.; Myers, J.R.; French, B.R.; Zandler, M.E. Possibility of stable spheroid molecules of ZnO. Phys. Rev. A 1994, 49, R1543–R1546. [Google Scholar] [CrossRef]
- Jensen, F.; Toftlund, H. Structure and stability of C24 and B12N12 isomers. Chem. Phys. Lett. 1993, 201, 89–96. [Google Scholar] [CrossRef]
- Golberg, D.; Bando, Y.; Stephan, O.; Kurashima, K. Octahedral boron nitride fullerenes formed by electron beam irradiation. Appl. Phys. Lett. 1998, 73, 2441–2443. [Google Scholar] [CrossRef]
- Woodley, S.M. Applications of Evolutionary Computation in Chemistry; Springer: Berlin, Germany, 2004; Volume 110. [Google Scholar]
- Woodley, S.M. Prediction of crystal structures using evolutionary algorithms and related techniques. In Applications of Evolutionary Computation in Chemistry; Springer-Verlag: Berlin, Germany, 2004; Volume 110, pp. 95–132. [Google Scholar]
- Foster, M.D.; Friedrichs, O.D.; Bell, R.G.; Paz, F.A.A.; Klinowski, J. Structural evaluation of systematically enumerated hypothetical uninodal zeolites. Angew. Chem. 2003, 115, 4026–4029. [Google Scholar] [CrossRef]
- Zwijnenburg, M.A.; Illas, F.; Bromley, S.T. Apparent scarcity of low-density polymorphs of inorganic solids. Phys. Rev. Lett. 2010, 104, 175503. [Google Scholar] [CrossRef]
- Enyashin, A.N.; Gemming, S.; Bar-Sadan, M.; Popovitz-Biro, R.; Hong, S.Y.; Prior, Y.; Tenne, R.; Seifert, G. Structure and stability of molybdenum sulfide fullerenes. Angew. Chem. Int. Ed. 2007, 46, 623–627. [Google Scholar] [CrossRef]
- Parilla, P.A.; Dillon, A.C.; Jones, K.M.; Riker, G.; Schulz, D.L.; Ginley, D.S.; Heben, M.J. The first true inorganic fullerenes? Nature 1999, 397, 114. [Google Scholar] [CrossRef]
- Kasuya, A.; Sivamohan, R.; Barnakov, Y.A.; Dmitruk, I.M.; Nirasawa, T.; Romanyuk, V.R.; Kumar, V.; Mamykin, S.V.; Tohji, K.; Jeyadevan, B.; et al. Ultra-stable nanoparticles of CdSe revealed from mass spectrometry. Nat. Mater. 2004, 3, 99–102. [Google Scholar] [CrossRef]
- Botti, S.; Marques, M.A.L. Identification of fullerene-like cdse nanoparticles from optical spectroscopy calculations. Phys. Rev. B 2007, 75, 035311. [Google Scholar] [CrossRef]
- Diogo, H.P.; da Piedade, M.E.M.; Dennis, T.J.S.; Hare, J.P.; Kroto, H.W.; Taylor, R.; Walton, D.R.M. Walton, D.R.M. Enthalpies of formation of buckminsterfullerene (C60) and of the parent ions C60+, C602+, C603+ and C60−. J. Chem. Soc. Faraday Trans. 1993, 89, 3541–3544. [Google Scholar] [CrossRef]
- Curl, R.F.; Haddon, R.C. On the formation of the fullerenes. Philos. Trans. 1993, 343, 19–32. [Google Scholar] [CrossRef]
- Gale, J.D.; Rohl, A.L. The general utility lattice program (gulp). Mol. Simul. 2003, 29, 291–341. [Google Scholar] [CrossRef]
- Whitmore, L.; Sokol, A.A.; Catlow, C.R.A. Surface structure of zinc oxide (1010), using an atomistic, semi-infinite treatment. Surf. Sci. 2002, 498, 135–146. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar]
- Blum, V.; Gehrke, R.; Hanke, F.; Havu, P.; Havu, V.; Ren, X.; Reuter, K.; Scheffler, M. Ab initio molecular simulations with numeric atom-centered orbitals. Comput. Phys. Commun. 2009, 180, 2175–2196. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Farrow, M.R.; Buckeridge, J.; Catlow, C.R.A.; Logsdail, A.J.; Scanlon, D.O.; Sokol, A.A.; Woodley, S.M. From Stable ZnO and GaN Clusters to Novel Double Bubbles and Frameworks. Inorganics 2014, 2, 248-263. https://doi.org/10.3390/inorganics2020248
Farrow MR, Buckeridge J, Catlow CRA, Logsdail AJ, Scanlon DO, Sokol AA, Woodley SM. From Stable ZnO and GaN Clusters to Novel Double Bubbles and Frameworks. Inorganics. 2014; 2(2):248-263. https://doi.org/10.3390/inorganics2020248
Chicago/Turabian StyleFarrow, Matthew R., John Buckeridge, C. Richard A. Catlow, Andrew J. Logsdail, David O. Scanlon, Alexey A. Sokol, and Scott M. Woodley. 2014. "From Stable ZnO and GaN Clusters to Novel Double Bubbles and Frameworks" Inorganics 2, no. 2: 248-263. https://doi.org/10.3390/inorganics2020248
APA StyleFarrow, M. R., Buckeridge, J., Catlow, C. R. A., Logsdail, A. J., Scanlon, D. O., Sokol, A. A., & Woodley, S. M. (2014). From Stable ZnO and GaN Clusters to Novel Double Bubbles and Frameworks. Inorganics, 2(2), 248-263. https://doi.org/10.3390/inorganics2020248