Electrochemical Quartz Microbalance for Studying Electrodeposited Pt Catalysts for Methanol Oxidation Reaction

 ,
,
Abstract
1. Introduction
2. Results and Discussions
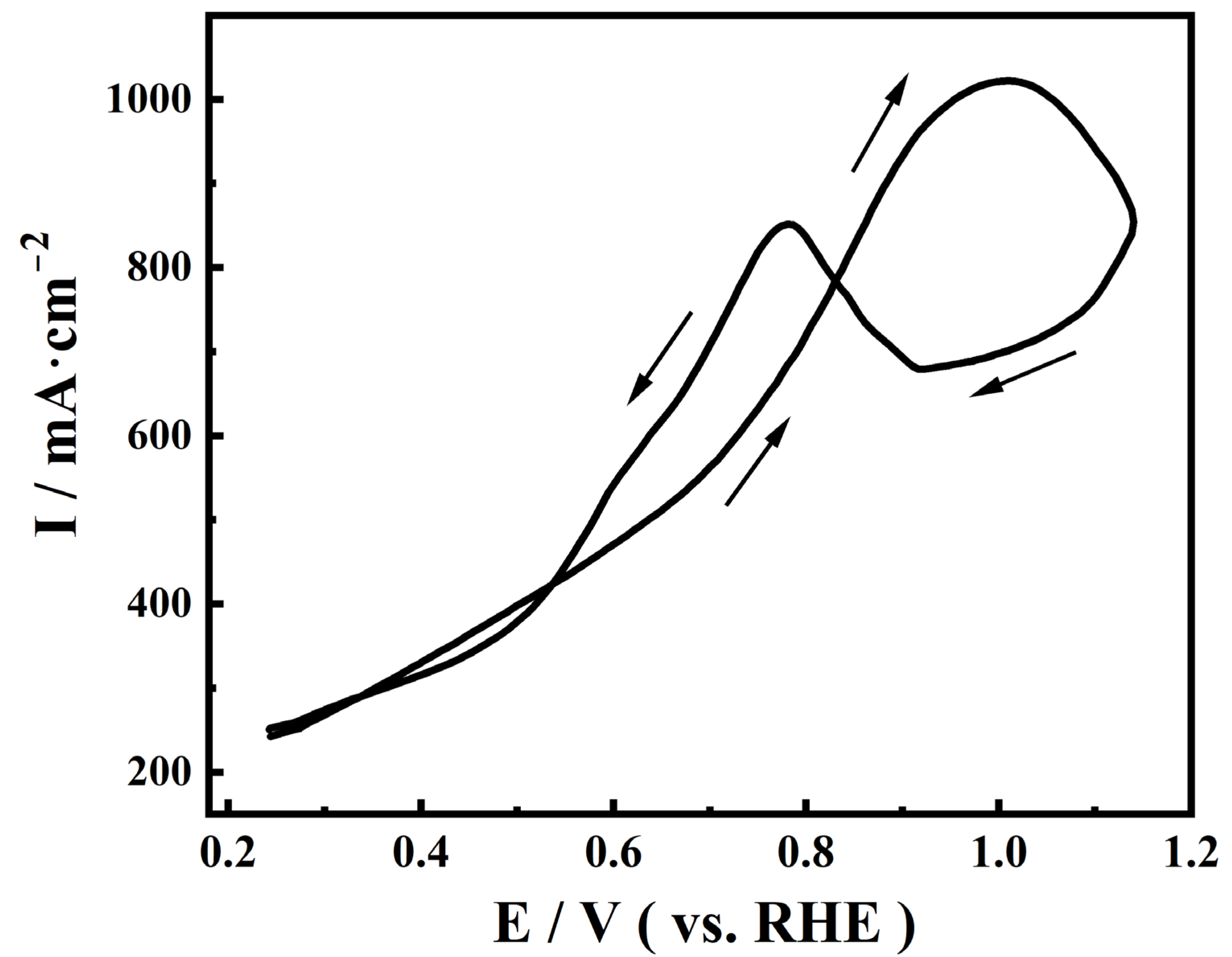
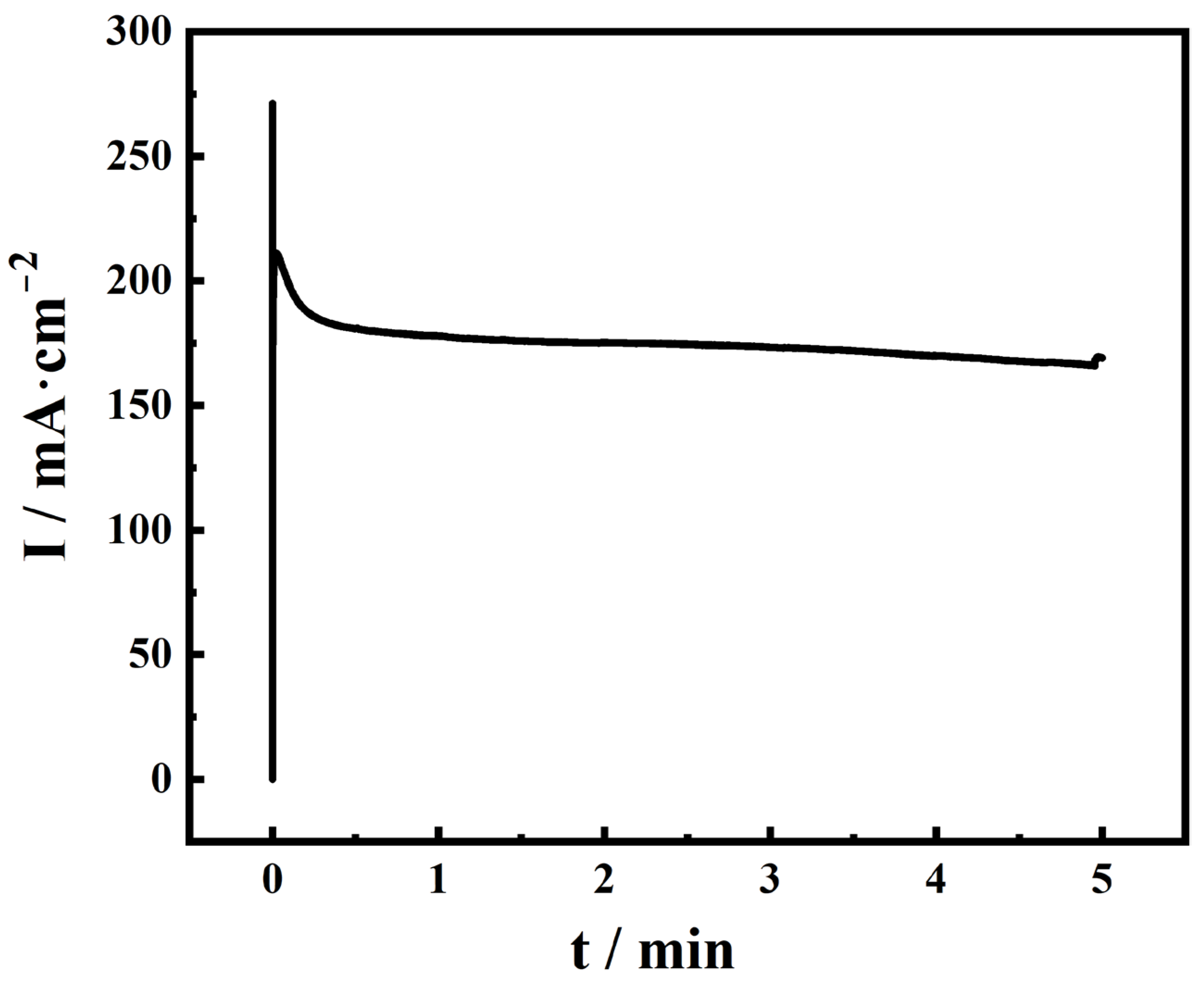
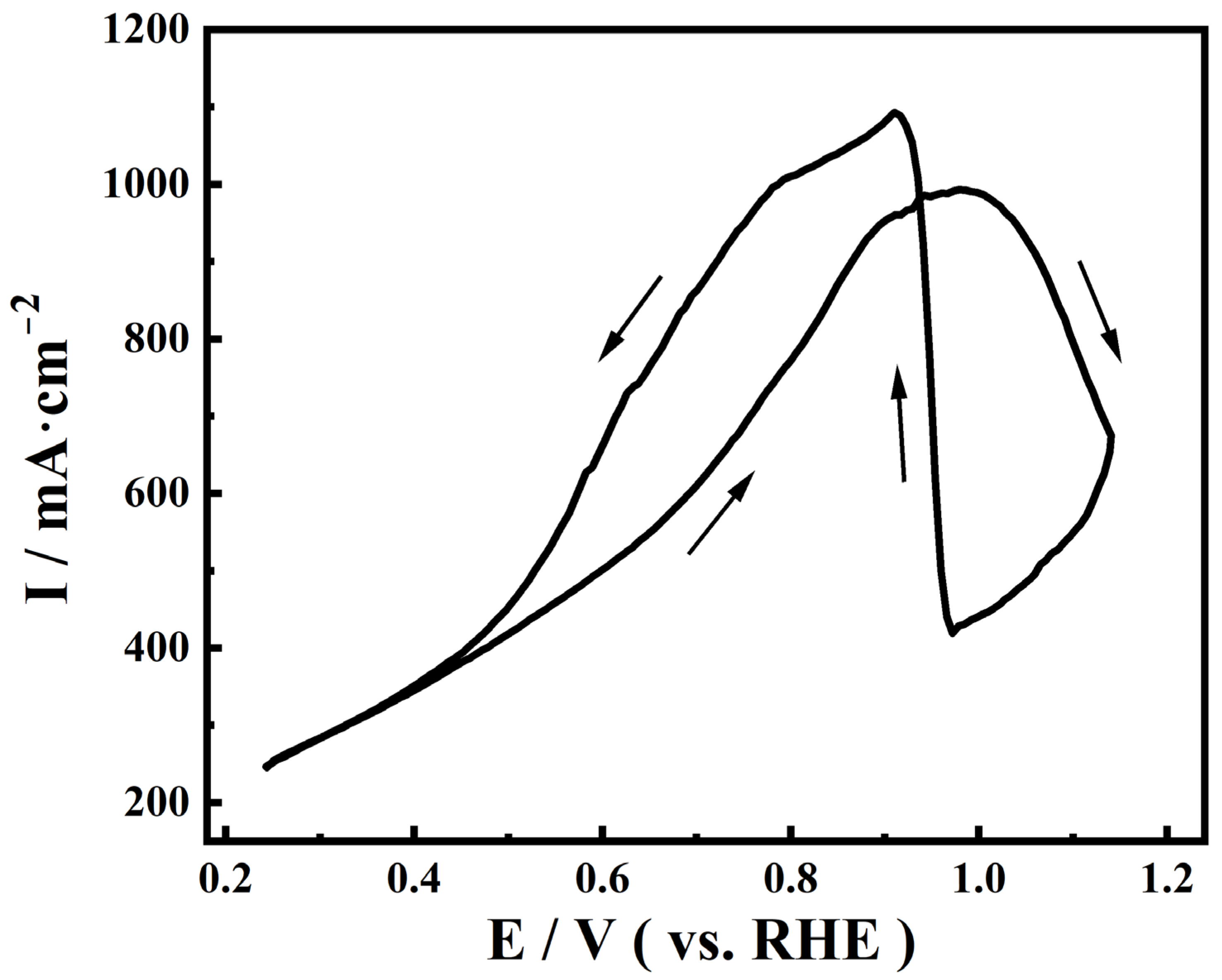
2.1. Performance of Electrodeposited Pt Catalysts on Diffusion Layer
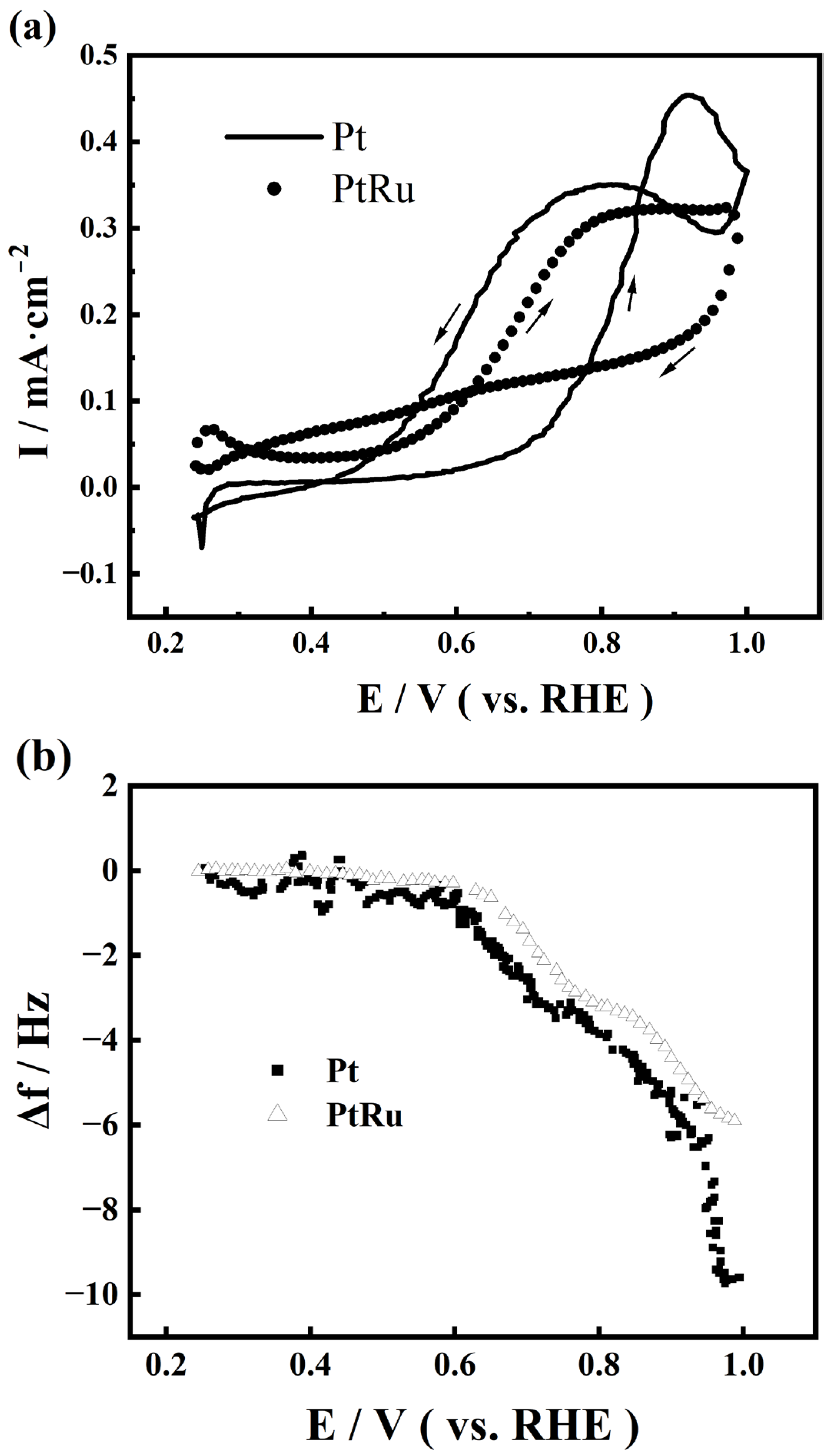
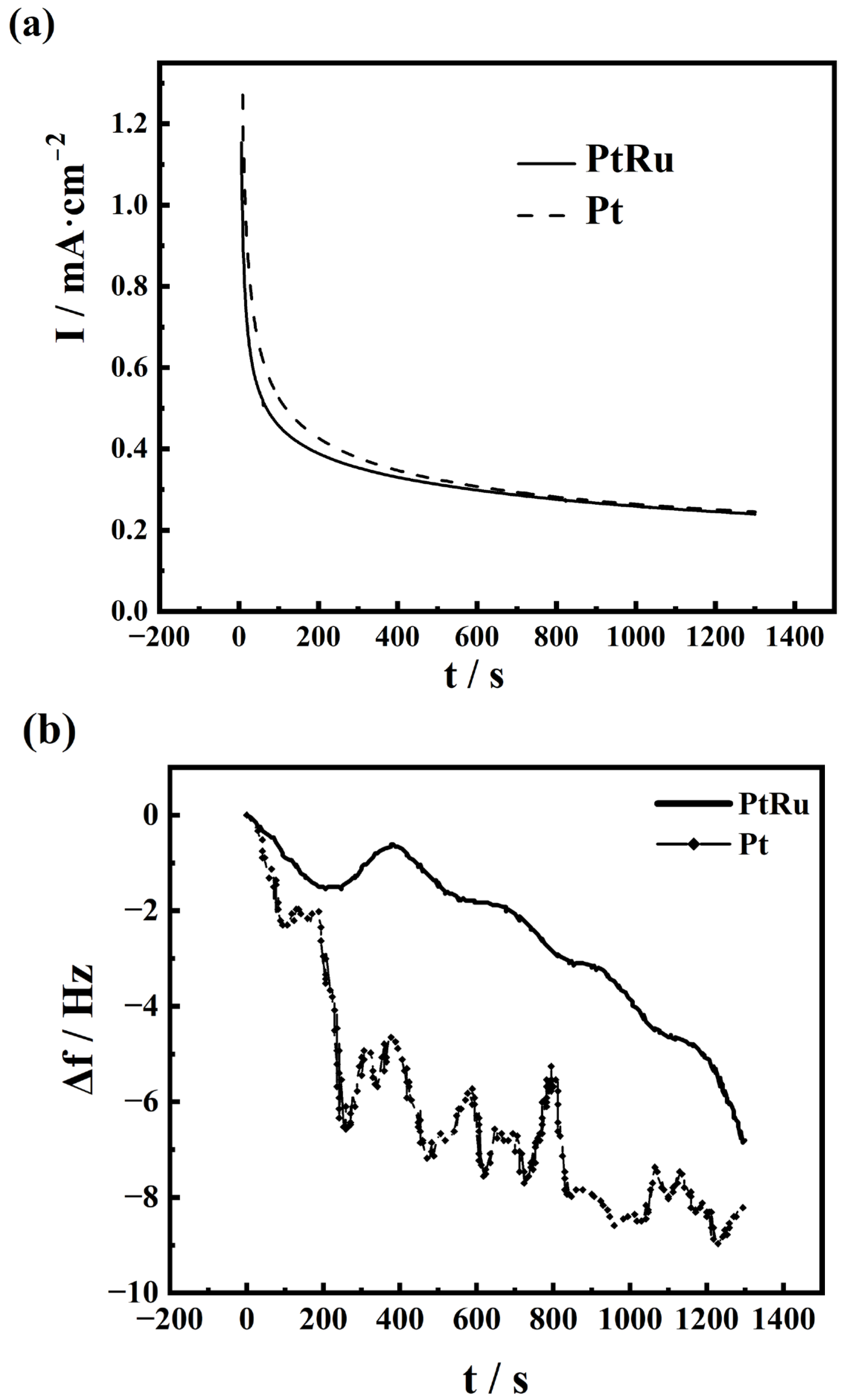
2.2. EQCM Analysis of Pt Catalysts
3. Materials and Methods
3.1. Reagent and Instrumentation
3.2. Materials Synthesis
3.3. Physical and Electrochemical Measurement
3.4. Test of Electrochemical Quartz Microbalance
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Poerwoprajitno, A.R.; Gloag, L.; Watt, J.; Cheong, S.; Tan, X.; Lei, H.; Tahini, H.A.; Henson, A.; Subhash, B.; Bedford, N.M.; et al. A single-Pt-atom-on-Ru-nanoparticle electrocatalyst for CO-resilient methanol oxidation. Nat. Catal. 2022, 5, 231–237. [Google Scholar] [CrossRef]
- Zhang, Z.; Ren, D.; Zhang, D.; Hu, T.; Zeng, C.; Xu, N.; Huo, Z.; Qiao, J. Electrolyte dependence for the electrochemical decarboxylation of medium-chain fatty acids (n-octanoic acid) into fuel on Pt electrode. Mater. Rep. Energy 2024, 4, 100244. [Google Scholar] [CrossRef]
- Tian, H.; Yu, Y.; Wang, Q.; Li, J.; Rao, P.; Li, R.; Du, Y.; Jia, C.; Luo, J.; Deng, P.; et al. Recent advances in two-dimensional Pt based electrocatalysts for methanol oxidation reaction. Int. J. Hydrogen Energy 2021, 46, 31202–31215. [Google Scholar] [CrossRef]
- Sarno, M.; Ponticorvo, E.; Scarpa, D. PtRh and PtRh/MoS2 nano-electrocatalysts for methanol oxidation and hydrogen evolution reactions. Chem. Eng. J. 2019, 377, 120600. [Google Scholar] [CrossRef]
- Gong, L.; Yang, Z.; Li, K.; Xing, W.; Liu, C.; Ge, J. Recent development of methanol electrooxidation catalysts for direct methanol fuel cell. J. Energy Chem. 2018, 27, 1618–1628. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, Q.; Ding, J.; Zhai, Y. Fe–N–C single atom catalysts for the electrochemical conversion of carbon, nitrogen and oxygen elements. Mater. Rep. Energy 2022, 2, 100141. [Google Scholar] [CrossRef]
- Zhang, J.; Qu, X.; Han, Y.; Shen, L.; Yin, S.; Li, G.; Jiang, Y.; Sun, S. Engineering PtRu bimetallic nanoparticles with adjustable alloying degree for methanol electrooxidation: Enhanced catalytic performance. Appl. Catal. B Environ. 2020, 263, 118345. [Google Scholar] [CrossRef]
- Huang, Z.; Xiao, Q.; Ding, T.; Xia, J.; Zhan, C.; Meng, X.; Pao, C.-W.; Hu, Z.; Huang, W.-H.; Wang, Y.; et al. Interfacial metal–coordinated bifunctional PtCo for practical fuel cells. Sci. Adv. 2025, 11, eadt4914. [Google Scholar] [CrossRef]
- Igarashi, H.; Fujino, T.; Watanabe, M. Hydrogen electro-oxidation on platinum catalysts in the presence of trace carbon monoxide. J. Electroanal. Chem. 1995, 391, 119–123. [Google Scholar] [CrossRef]
- van Spronsen, M.A.; Frenken, J.W.M.; Groot, I.M.N. Surface science under reaction conditions: CO oxidation on Pt and Pd model catalysts. Chem. Soc. Rev. 2017, 46, 4347–4374. [Google Scholar] [CrossRef]
- Martín, A.; Mitchell, S.; Mondelli, C.; Jaydev, S.; Pérez-Ramírez, J. Unifying views on catalyst deactivation. Nat. Catal. 2022, 5, 854–866. [Google Scholar] [CrossRef]
- Yaqoob, L.; Noor, T.; Iqbal, N. Recent progress in development of efficient electrocatalyst for methanol oxidation reaction in direct methanol fuel cell. Int. J. Energy Res. 2021, 45, 6550–6583. [Google Scholar] [CrossRef]
- Kocha, S.S. Chapter 3—Electrochemical Degradation: Electrocatalyst and Support Durability. In Polymer Electrolyte Fuel Cell Degradation; Mench, M.M., Kumbur, E.C., Veziroglu, T.N., Eds.; Academic Press: Boston, MA, USA, 2012; pp. 89–214. ISBN 978-0-12-386936-4. [Google Scholar]
- Schumache, R. The Quartz Microbalance: A Novel Approach to the In-Situ Investigation of Interfacial Phenomena at the Solid/Liquid Junction. Angew. Chem. Int. Ed. 2010, 29, 329–343. [Google Scholar]
- Sauerbrey, G. The use of quartz oscillators for weighing thin layers and for microweighing. Z. Für Phys. C 1959, 155, 206–222. [Google Scholar]
- Ji, Y.; Yin, Z.-W.; Yang, Z.; Deng, Y.-P.; Chen, H.; Lin, C.; Yang, L.; Yang, K.; Zhang, M.; Xiao, Q.; et al. From bulk to interface: Electrochemical phenomena and mechanism studies in batteries via electrochemical quartz crystal microbalance. Chem. Soc. Rev. 2021, 50, 10743–10763. [Google Scholar] [CrossRef]
- Xie, Q.; Li, Z.; Deng, C.; Liu, M.; Zhang, Y.; Ma, M.; Xia, S.; Xiao, X.; Yin, D.; Yao, S. Electrochemical Quartz Crystal Microbalance Monitoring of the Cyclic Voltammetric Deposition of Polyaniline. A Laboratory Experiment for Undergraduates. J. Chem. Educ. 2007, 84, 681. [Google Scholar] [CrossRef]
- Nasirpouri, F. On the electrodeposition mechanism of Pb on copper substrate from a perchlorate solution studied by electrochemical quartz crystal microbalance. Ionics 2011, 17, 331–337. [Google Scholar] [CrossRef]
- Kugai, J.; Tanaka, S.; Seino, S.; Nakagawa, T.; Yamamoto, T.A.; Yamada, H. Electrochemical quartz crystal microbalance studies on specific adsorption of nanoparticle stabilizers on platinum surface. J. Electroanal. Chem. 2021, 897, 115596. [Google Scholar] [CrossRef]
- Wang, C.; Yang, Y.; Zhou, Z.; Li, Y.; Li, Y.; Hou, W.; Liu, S.; Tian, Y. Electrodeposited Poly(5-Amino-2-Naphthalenesulfonic Acid-co-o-Aminophenol) as the Electrode Material for Flexible Supercapacitor. Small 2024, 20, e2305994. [Google Scholar] [CrossRef]
- Lee, W.-J.; Pyun, S.-I. Application of electrochemical quartz crystal microbalance technique to investigation of the interfacial reactions at metal electrode/electrolyte. Met. Mater. Int. 2000, 6, 331–343. [Google Scholar] [CrossRef]
- Lin, Z.; Yip, C.M.; Joseph, I.S.; Ward, M.D. Operation of an ultrasensitive 30-MHz quartz crystal microbalance in liquids. Anal. Chem. 1993, 65, 1546–1551. [Google Scholar] [CrossRef]
- Wu, T.-H.; Hardwick, L.; Scivetti, I.; Chen, J.-C.; Wang, J.-A.; Teobaldi, G.; Hu, C.-C. Quantitative Resolution of Complex Stoichiometric Changes During Electrochemical Cycling by Density Functional Theory Assisted, Electrochemical Quartz Crystal Microbalance. ACS Appl. Energy Mater. 2020, 3, 3347–3357. [Google Scholar] [CrossRef]
- Fenyun, Y.; Yulan, H.; Aimei, G.; Fan, Z.; Dong, S.; Weixin, C.; Honghong, C.; Xiaoping, Z.; Ronghua, Z. Investigation on the pseudocapacitive charge storage mechanism of MnO2 in various electrolytes by electrochemical quartz crystal microbalance (EQCM). Ionics 2019, 25, 2393–2399. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Marković, N.; Ross, P.N., Jr.; Cairns, E.J. Cairns Temperature-Dependent Methanol Electro-Oxidation on Well-Characterized Pt-Ru Alloys. J. Electrochem. Soc. 1994, 141, 1795–1803. [Google Scholar]
- Zhang, W.; Wang, X.; Tan, M.; Liu, H.; Ma, Q.; Xu, Q.; Pollet, B.G.; Su, H. Electrodeposited platinum with various morphologies on carbon paper as efficient and durable self-supporting electrode for methanol and ammonia oxidation reactions. Int. J. Hydrogen Energy 2023, 48, 2617–2627. [Google Scholar] [CrossRef]
- Huang, K.; Clausmeyer, J.; Luo, L.; Jarvis, K.; Crooks, R.M. Shape-controlled electrodeposition of single Pt nanocrystals onto carbon nanoelectrodes. Faraday Discuss. 2018, 210, 267–280. [Google Scholar] [CrossRef]
- Shen, L.; Tu, F.; Shang, Z.; Ma, M.; Xia, Y.; Zhao, Z.; Zhao, L.; Wang, Z.; Shao, G. Surfactant-assisted synthesis of platinum nanoparticle catalysts for proton exchange membrane fuel cells. Int. J. Hydrogen Energy 2022, 47, 15001–15011. [Google Scholar] [CrossRef]
- Gorle, D.B.; Velacheri Kumman, V.; Kulandainathan, M.A. Highly efficient, large surface area and spherically shaped Pt particles deposited electrolytically synthesized graphene for methanol oxidation with impedance spectroscopy. Int. J. Hydrogen Energy 2017, 42, 16258–16268. [Google Scholar] [CrossRef]
- Lee, C.-K.; Jung, D.; Kim, C.-S.; Shin, D.-R. Methanol oxidation and electrochemical characteristics on electrodeposited Pt/WO3 catalyst. J. New Mater. Electrochem. Syst. 1999, 2, 125–129. [Google Scholar]
- Thompson, S.D.; Jordan, L.R.; Forsyth, M. Platinum electrodeposition for polymer electrolyte membrane fuel cells. Electrochim. Acta 2001, 46, 1657–1663. [Google Scholar] [CrossRef]
- Kamyabi, M.A.; Jadali, S.; Khangheshlaghi, L.S.; Heris, M.K.H. A high-performance Pt-based catalyst for the methanol oxidation reaction: Effect of electrodeposition mode and cocatalyst on electrocatalytic activity. New J. Chem. 2023, 47, 1209–1215. [Google Scholar] [CrossRef]
- Jin, C.; Huo, L.; Tang, J.; Li, S.; Jiang, K.; He, Q.; Dong, H.; Gong, Y.; Hu, Z. Precise Atomic Structure Regulation of Single-Atom Platinum Catalysts toward Highly Efficient Hydrogen Evolution Reaction. Small 2024, 20, e2309509. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Xu, C.; Shen, P.K.; Jiang, S.P. Effect of nitrogen-containing functionalization on the electrocatalytic activity of PtRu nanoparticles supported on carbon nanotubes for direct methanol fuel cells. Appl. Catal. B Environ. 2014, 158–159, 140–149. [Google Scholar] [CrossRef]
- Wang, M.; Song, X.; Yang, Q.; Hua, H.; Dai, S.; Hu, C.; Wei, D. Pt nanoparticles supported on graphene three-dimensional network structure for effective methanol and ethanol oxidation. J. Power Sources 2015, 273, 624–630. [Google Scholar] [CrossRef]
- Li, M.; Fang, Y.; Zhang, G.; Cui, P.; Yang, Z.; He, J. Carbon-supported Pt5P2 nanoparticles used as a high-performance electrocatalyst for the methanol oxidation reaction. J. Mater. Chem. A 2020, 8, 10433–10438. [Google Scholar] [CrossRef]
- Budi, S.; Pathoni, A.S.; Auliya, A.; Winarsih, S.; Fauzi, M.H.; Yusmaniar; Suliasih, B.A.; Syafei, H. Efficient stabilizing agent-free synthesis of gold nanoparticles via square-wave pulse deposition for enhanced catalytic performance in ethanol electrooxidation. Mater. Rep. Energy 2024, 4, 100294. [Google Scholar] [CrossRef]
- Calderón, J.C.; García, G.; Calvillo, L.; Rodríguez, J.L.; Lázaro, M.J.; Pastor, E. Electrochemical oxidation of CO and methanol on Pt–Ru catalysts supported on carbon nanofibers: The influence of synthesis method. Appl. Catal. B Environ. 2015, 165, 676–686. [Google Scholar] [CrossRef]
- Gloaguen, F.; Léger, J.-M.; Lamy, C. An electrochemical quartz crystal microbalance study of the hydrogen underpotential deposition at a Pt electrode. J. Electroanal. Chem. 1999, 467, 186–192. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, B.; Guo, W.; Wang, L.; Chen, J.; Pan, H.; Sun, W. Toward Electrocatalytic Methanol Oxidation Reaction: Longstanding Debates and Emerging Catalysts. Adv. Mater. 2023, 35, 2211099. [Google Scholar] [CrossRef]
- Duan, Y.; Wang, L.-L.; Zheng, W.-X.; Zhang, X.-L.; Wang, X.-R.; Feng, G.-J.; Yu, Z.-Y.; Lu, T.-B. Oxyanion Engineering on RuO2 for Efficient Proton Exchange Membrane Water Electrolysis. Angew. Chem. Int. Ed. 2024, 63, e202413653. [Google Scholar] [CrossRef]
- Wang, Z.-B.; Yin, G.-P.; Lin, Y.-G. Synthesis and characterization of PtRuMo/C nanoparticle electrocatalyst for direct ethanol fuel cell. J. Power Sources 2007, 170, 242–250. [Google Scholar] [CrossRef]
- Wang, Z.-B.; Zuo, P.; Yin, G.-P. Effect of W on activity of Pt–Ru/C catalyst for methanol electrooxidation in acidic medium. J. Alloys Compd. 2009, 479, 395–400. [Google Scholar] [CrossRef]
- Jiang, S.; Zhu, L.; Ma, Y.; Wang, X.; Liu, J.; Zhu, J.; Fan, Y.; Zou, Z.; Hu, Z. Direct immobilization of Pt–Ru alloy nanoparticles on nitrogen-doped carbon nanotubes with superior electrocatalytic performance. J. Power Sources 2010, 195, 7578–7582. [Google Scholar] [CrossRef]
- Li, B.; Higgins, D.C.; Zhu, S.; Li, H.; Wang, H.; Ma, J.; Chen, Z. Highly active Pt–Ru nanowire network catalysts for the methanol oxidation reaction. Catal. Commun. 2012, 18, 51–54. [Google Scholar] [CrossRef]
- Biegler, T.; Rand, D.A.J.; Woods, R. Limiting oxygen coverage on platinized platinum; Relevance to determination of real platinum area by hydrogen adsorption. J. Electroanal. Chem. Interfacial Electrochem. 1971, 29, 269–277. [Google Scholar] [CrossRef]
- Chen, W.; Cao, J.; Yang, J.; Cao, Y.; Zhang, H.; Jiang, Z.; Zhang, J.; Qian, G.; Zhou, X.; Chen, D.; et al. Molecular-level insights into the electronic effects in platinum-catalyzed carbon monoxide oxidation. Nat. Commun. 2021, 12, 6888. [Google Scholar] [CrossRef]
- Qiao, W.; Huang, X.; Feng, L. Advances of PtRu-Based Electrocatalysts for Methanol Oxidation. Chin. J. Struct. Chem. 2022, 41, 2207016–2207034. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, J.; Wang, J.; Wang, Q.; Wang, Y.; Wang, K.; Wang, Z.; Gu, M.; Tang, Z.; Lim, J.; et al. Single-atom catalyst for high-performance methanol oxidation. Nat. Commun. 2021, 12, 5235. [Google Scholar] [CrossRef]
- Moses, O.A.; Chen, W.; Adam, M.L.; Wang, Z.; Liu, K.; Shao, J.; Li, Z.; Li, W.; Wang, C.; Zhao, H.; et al. Integration of data-intensive, machine learning and robotic experimental approaches for accelerated discovery of catalysts in renewable energy-related reactions. Mater. Rep. Energy 2021, 1, 100049. [Google Scholar]
- Cuesta, A. Study of adsorbed water on Pt during methanol oxidation by ATR-SEIRAS (surface-enhanced infrared absorption spectroscopy). J. Electroanal. Chem. 2006, 587, 329–330. [Google Scholar] [CrossRef]
- Ye, F.; Xu, C.; Liu, G.; Yuan, M.; Wang, Z.; Du, X.; Lee, J.K. Effect of pulse electrodeposition parameters on electrocatalytic the activity of methanol oxidation and morphology of Pt/C catalyst for direct methanol fuel cells. Energy Convers. Manag. 2018, 160, 85–92. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zong, B.; Pan, X.; Wei, B.; Zhang, L.; Feng, X.; Hou, C.; Yan, H.; Xie, W.; Liu, G.; Ye, F. Electrochemical Quartz Microbalance for Studying Electrodeposited Pt Catalysts for Methanol Oxidation Reaction. Inorganics 2025, 13, 111. https://doi.org/10.3390/inorganics13040111
Zong B, Pan X, Wei B, Zhang L, Feng X, Hou C, Yan H, Xie W, Liu G, Ye F. Electrochemical Quartz Microbalance for Studying Electrodeposited Pt Catalysts for Methanol Oxidation Reaction. Inorganics. 2025; 13(4):111. https://doi.org/10.3390/inorganics13040111
Chicago/Turabian StyleZong, Bangfeng, Xiaojun Pan, Bo Wei, Lifang Zhang, Xiangxiong Feng, Chenggong Hou, Hai Yan, Wenju Xie, Guicheng Liu, and Feng Ye. 2025. "Electrochemical Quartz Microbalance for Studying Electrodeposited Pt Catalysts for Methanol Oxidation Reaction" Inorganics 13, no. 4: 111. https://doi.org/10.3390/inorganics13040111
APA StyleZong, B., Pan, X., Wei, B., Zhang, L., Feng, X., Hou, C., Yan, H., Xie, W., Liu, G., & Ye, F. (2025). Electrochemical Quartz Microbalance for Studying Electrodeposited Pt Catalysts for Methanol Oxidation Reaction. Inorganics, 13(4), 111. https://doi.org/10.3390/inorganics13040111