2. Results and Discussion
The cobalt(III) complexes (
4–
6) under investigation in this study were prepared according to
Figure 1A. Specifically, the cobalt(III) complexes
4–
6 were synthesised by reacting the appropriate
cis-dichlorobis-(polypyridyl)cobalt(III) chloride complex,
1a–
3a (where polypyridyl = 2,2′-bipyridine for
1a, 1,10-phenanthroline for
2a and 3,4,7,8-tetramethyl-1,10-phenanthroline for
3a) [
25,
26] with an equimolar amount of salicylic acid in methanol (dried over sodium sulphate) in the presence of excess silver(I) oxide. The crude product was precipitated using diethyl ether and converted to the hexafluorophosphate salt. This was sufficient to obtained
4 and
5 in their pure form as green solids. An additional purification step involving alumina column chromatography [dichloromethane: methanol (39:1)] was required to obtained
6 in its pure form as a green solid. All three cobalt(III) complexes were acquired in poor to reasonable overall yields (20–39%). The cobalt(III) complexes
4–
6 were characterised by
1H,
31P{
1H},
19F{
1H} NMR and infrared spectroscopy, high-resolution ESI mass spectrometry and elemental analysis (
Figures S1–S15). The aromatic region within the
1H NMR spectra of
4–
6 was shifted relative to the same region in the
1H NMR spectrum of salicylic acid, suggesting coordination of salicylate to cobalt (
Figures S1, S4, S7 and S16). Furthermore, the fact that only one set of signals was observed for the salicylate moiety in
4–
6 indicated that a single diastereoisomer was isolated in each case. The IR spectra for the cobalt(III) complexes
4–
6 displayed ν
asym(CO
2) and ν
sym(CO
2) stretching bands at 1620–1615 cm
−1 and 1427–1429 cm
−1, respectively (
Figures S10–S12). The difference between the ν
asym(CO
2) and ν
sym(CO
2) stretching bands for
4–
6 were between 186 and 192 cm
−1, which was too close to the 200 cm
−1 threshold to allow unambiguous assignment to a monodentate or bidentate binding mode for the carboxylate group (on the salicylate moiety) to the cobalt centre [
27,
28]. Based on the IR spectra for
4–
6 and structurally related cobalt(III) complexes it is likely that the binding mode for the salicylate moiety as a whole was bidentate (as depicted in
Figure 1A) [
14]. The high-resolution ESI mass spectra of
4–
6 (positive mode) displayed distinctive molecular ion peaks corresponding to the cationic component of
4–
6 with the appropriate isotopic pattern (
m/
z = 507.0869 a.m.u, [
4-PF
6]
+; 555.0868 a.m.u, [
5-PF
6]
+; 667.2122 a.m.u, [
6-PF
6]
+) (
Figures S13–S15). This provides further evidence for the formation of the desired cobalt(III) complexes
4–
6. The purity of the bulk solids obtained for
4–
6 was confirmed by elemental analysis (see SI). Crystals of
4 and
6 suitable for X-ray diffraction analysis were obtained by slow diffusion of diethyl ether into an acetonitrile solution of
4 and
6 (CCDC 2363763-2363764,
Figure 1B,C,
Table S1). Selected bond distances and bond angles are presented in
Tables S2 and S3. The cationic component of the complex exhibited a distorted octahedral geometry with the cobalt(III) centre coordinated to two polypyridyl ligands (2,2′-bipyridine for
4 and 3,4,7,8-tetramethyl-1,10-phenanthroline for
6) and a salicylate moiety via two hydroxyl groups. The cobalt(III) coordination sphere was consistent with the aforementioned spectroscopic and analytic data for
4 and
6. The average Co–O (1.867 Å for
4 and 1.872 Å for
6) and Co–N (1.933 Å for
4 and 1.942 Å for
6) bond distances were consistent with bond parameters for related octahedral cobalt(III) complexes [
26,
29,
30].
The lipophilicity of a given biological active agent is an important factor in determining its potential efficacy as it influences aqueous solubility and cell internalisation. The lipophilicity of
4–
6 was determined by measuring the extent to which it partitioned between octanol and water. The LogP values determined for
4–
6 varied from −1.57 ± 0.01 to −0.02 ± 0.01 (
Table S4). As expected, there was a clear correlation between the LogP value for
4–
6 and the bulkiness of the corresponding polypyridyl ligand within
4–
6. The higher lipophilicity of
6 (compared to
4 and
5) could translate to higher uptake and thus higher potency towards CSCs. UV-Vis spectroscopy and ESI mass spectrometry were performed to determine the stability of
6 (taken a representative member of the series) in biologically relevant solutions without and with a bioreductant. In DMSO, H
2O:DMSO (4:1) and PBS:DMSO (4:1), the absorbance and wavelengths of the bands associated to
6 (25 or 50 μM) remained unaltered over the course of 24 h at 37 °C, indicative of stability under these conditions (
Figures S17–S19). In PBS:DMSO (4:1) with 10-fold excess of ascorbic acid, a marked decrease in the absorption was observed for
6 (25 μM) over a period of 24 h at 37 °C (
Figure S20). The final UV trace for
6 at 24 h displayed a peak at ca. 280 nm with a shoulder at ca. 293 nm, consistent with free 3,4,7,8-tetramethyl-1,10-phenanthroline and salicylic acid, respectively (
Figure S21). The ESI mass spectrum of
6 (40 μM) in H
2O:DMSO (10:1) displayed a distinct signal corresponding to intact
6 (
m/
z = 667.4, [
6-PF
6]
+) after 24 h incubation (
Figure S22). In the presence of 10-fold excess of ascorbic acid, the signal corresponding to
6 completely disappeared after 24 h incubation and was replaced by signals corresponding to [3,4,7,8-tetramethyl-1,10-phenanthroline+H]
+ (
m/
z = 237.1), [salicylate]
− (
m/
z = 137.0), and [Co
II(3,4,7,8-tetramethyl-1,10-phenanthroline)
3]
2+ (
m/
z = 383.8) (
Figure S23). This suggests that the cobalt(III) centre in
6 undergoes reduction to cobalt(II), triggering ligand exchange. Taken together, the UV-Vis spectroscopy and ESI mass spectrometry studies showed that
6 could release 3,4,7,8-tetramethyl-1,10-phenanthroline and salicylate, as well as undergo structural rearrangement under biological reducing conditions, presumably via the reduction of the cobalt(III) centre to cobalt(II). Reduction to cobalt(II) is known to promote ligand substitution, which is supported by the detection of free 3,4,7,8-tetramethyl-1,10-phenanthroline and the cobalt(II) species [Co
II(3,4,7,8-tetramethyl-1,10-phenanthroline)
3]
2+ in the ESI mass spectrometry studies (
Figure S23). Prior to conducting cell-based studies, the stability of
4–
6 in mammary epithelial cell growth medium (MEGM) was investigated (
Figure S24). The UV-Vis trace of
4–
6 (50 μM) in MEGM:DMSO (4:1) displayed no changes over the course of 24 h at 37 °C. Therefore,
4–
6 were deemed suitably stable to progress to cell-based studies.
The cytotoxicity of the cobalt(III) complexes
4–
6 towards bulk breast cancer cells (HMLER) and breast CSCs (HMLER-shEcad) grown in monolayers was determined using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. IC
50 values (the concentration required to decrease cell viability by half) were determined from dose-response curves (
Figures S25–S27) and are listed in
Table 1. All three cobalt(III) complexes
4–
6 displayed micromolar IC
50 values towards bulk breast cancer cells and breast CSCs. Cobalt(III) complex
6 was the most potent towards breast CSCs, whereas cobalt(III) complex
4 was the most selective for breast CSCs over bulk breast cancer cells (2.6-fold). Notably, cobalt(III) complex
6 displayed a significantly lower IC
50 value towards breast CSCs than salinomycin (
p < 0.05, 3.7-fold) and cisplatin (
p < 0.05, 4.9-fold) [
31,
32]. Salinomycin is a well-established anti-breast CSC agent and cisplatin is the gold-standard anticancer metallodrug used in the clinic [
33,
34]. Salicylic acid was non-toxic at the concentration tested towards bulk breast cancer cells and breast CSCs (IC
50 values > 100 μM,
Figure S28), suggesting that the potency of the cobalt(III) complexes
4–
6 can be attributed largely to the corresponding bis-(polypyridyl)cobalt(III) component. This assumption is supported by the fact that the corresponding
cis-dichlorobis-(polypyridyl)cobalt(III) hexafluorophosphate complexes
1b–
3b (
Figure 1A) displayed similar or greater potency towards bulk breast cancer cells and breast CSCs compared to
4–
6, respectively (
Figures S29–S31,
Table S5).
Given the strong potency of the cobalt(III) complexes
4–
6 towards breast CSCs grown in monolayers, their ability to inhibit the formation and viability of three-dimensional mammospheres (cultured from breast CSCs in serum-free, low attachment conditions) [
35] was determined. Upon treatment of single-cell suspensions of HMLER-shEcad cells with
4 (IC
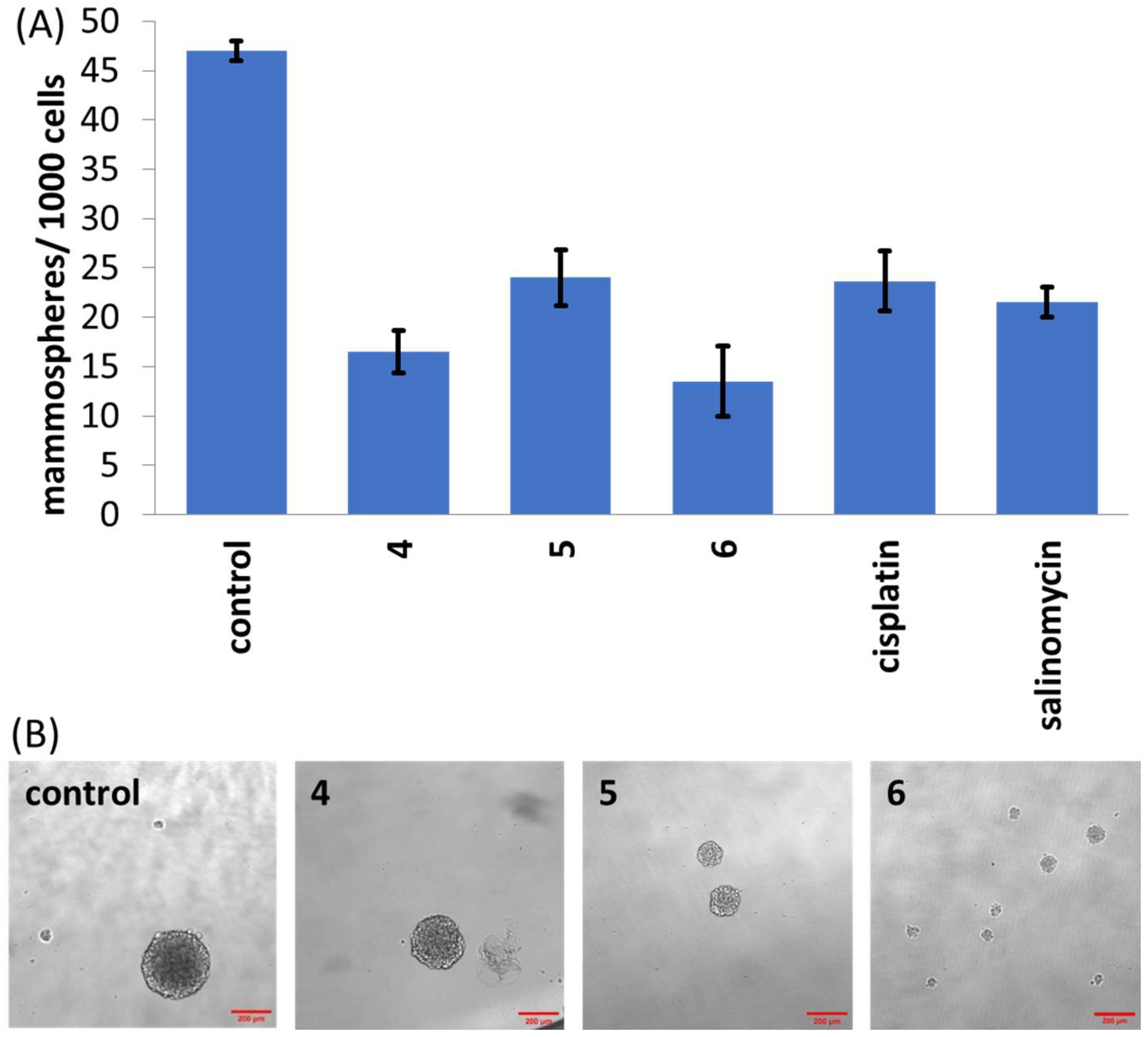
20 value for 5 days), the size of mammospheres formed was similar to the untreated control; however, the number of mammospheres formed was significantly lower (
p < 0.05), suggestive of moderate mammosphere inhibition (
Figure 2A,B). In contrast, treatment of single-cell suspensions of HMLER-shEcad cells with
5 and
6 (IC
20 value for 5 days) significantly reduced (
p < 0.05) the size and number of mammospheres formed compared to the untreated control (
Figure 2A,B). Notably, dosage with cobalt(III) complex
6 (IC
20 value for 5 days) led a 71% reduction in the number of mammospheres formed compared to the untreated control (
Figure 2A). Further, the mammosphere inhibitory effect of
6 was comparable to that of salinomycin and cisplatin (
Figure 2A). To decipher the ability of
4–
6 to reduce mammosphere viability, a colorimetric resazurin-based reagent, TOX8, was used (
Figure S32 and
Table 1). Cobalt(III) complex
4 was non-toxic within the concentration range tested (IC
50 > 133 μM), whereas cobalt(III) complexes
5 and
6 exhibited micromolar potency (
Figure S32 and
Table 1). The IC
50 value (concentration at which the mammosphere viability is reduced by half) for
6 was 8.1-fold and 11.1-fold lower than the IC
50 values for cisplatin and salinomycin, respectively [
36,
37]. Under identical conditions,
cis-dichlorobis-(3,4,7,8-tetramethyl-1,10-phenanthroline)cobalt(III) hexafluorophosphate
3b displayed micromolar potency (IC
50 = 2.30 ± 0.91 μM) towards mammospheres, while salicylic acid was non-toxic within the concentration range tested (IC
50 > 133 μM) (
Figure S33). This indicates that the presence of the bis-(3,4,7,8-tetramethyl-1,10-phenanthroline)cobalt(III) component in
6 is a major determinant of its mammosphere potency.
Table 1.
IC
50 values of the cobalt(III) complexes
4–
6, salicylic acid, cisplatin and salinomycin against HMLER and HMLER-shEcad cells and HMLER-shEcad mammospheres determined after 72 h or 120 h incubation (mean of two or three independent experiments ± SD).
1 Taken from references [
31,
32,
36,
37].
Table 1.
IC
50 values of the cobalt(III) complexes
4–
6, salicylic acid, cisplatin and salinomycin against HMLER and HMLER-shEcad cells and HMLER-shEcad mammospheres determined after 72 h or 120 h incubation (mean of two or three independent experiments ± SD).
1 Taken from references [
31,
32,
36,
37].
| Compound | HMLER
IC50 (μM) | HMLER-shEcad
IC50 (μM) | Mammosphere
IC50 (μM) |
|---|
| 4 | 34.71 ± 1.36 | 13.46 ± 1.20 | >100 |
| 5 | 2.81 ± 0.01 | 2.82 ± 0.02 | 11.76 ± 1.84 |
| 6 | 1.79 ± 0.06 | 1.15 ± 0.07 | 1.66 ± 0.49 |
| salicylic acid | >100 | >100 | >133 |
| cisplatin 1 | 2.57 ± 0.02 | 5.65 ± 0.30 | 13.50 ± 2.34 |
| salinomycin 1 | 11.40 ± 0.40 | 4.20 ± 0.30 | 18.50 ± 1.50 |
Figure 2.
(A) Quantification of mammosphere formation with HMLER-shEcad cells untreated and treated with 4, 5, 6, salinomycin or cisplatin (IC20 value, 5 days). Error bars represent standard deviations. (B) Representative bright-field images (×10) of HMLER-shEcad mammospheres in the absence and presence of 4, 5 or 6 (IC20 value, 5 days).
Figure 2.
(A) Quantification of mammosphere formation with HMLER-shEcad cells untreated and treated with 4, 5, 6, salinomycin or cisplatin (IC20 value, 5 days). Error bars represent standard deviations. (B) Representative bright-field images (×10) of HMLER-shEcad mammospheres in the absence and presence of 4, 5 or 6 (IC20 value, 5 days).
Our previous work on a structurally related cobalt(III)-1,10-phenanthroline complex bearing diflunisal showed that its mechanism of action involved genomic DNA damage, downregulation of COX-2 expression and apoptosis [
14]. Therefore, we carried out cell-based studies to understand if the lead cobalt(III) complex
6 exhibits a similar mechanism of action. Upon incubation of HMLER-shEcad cells with
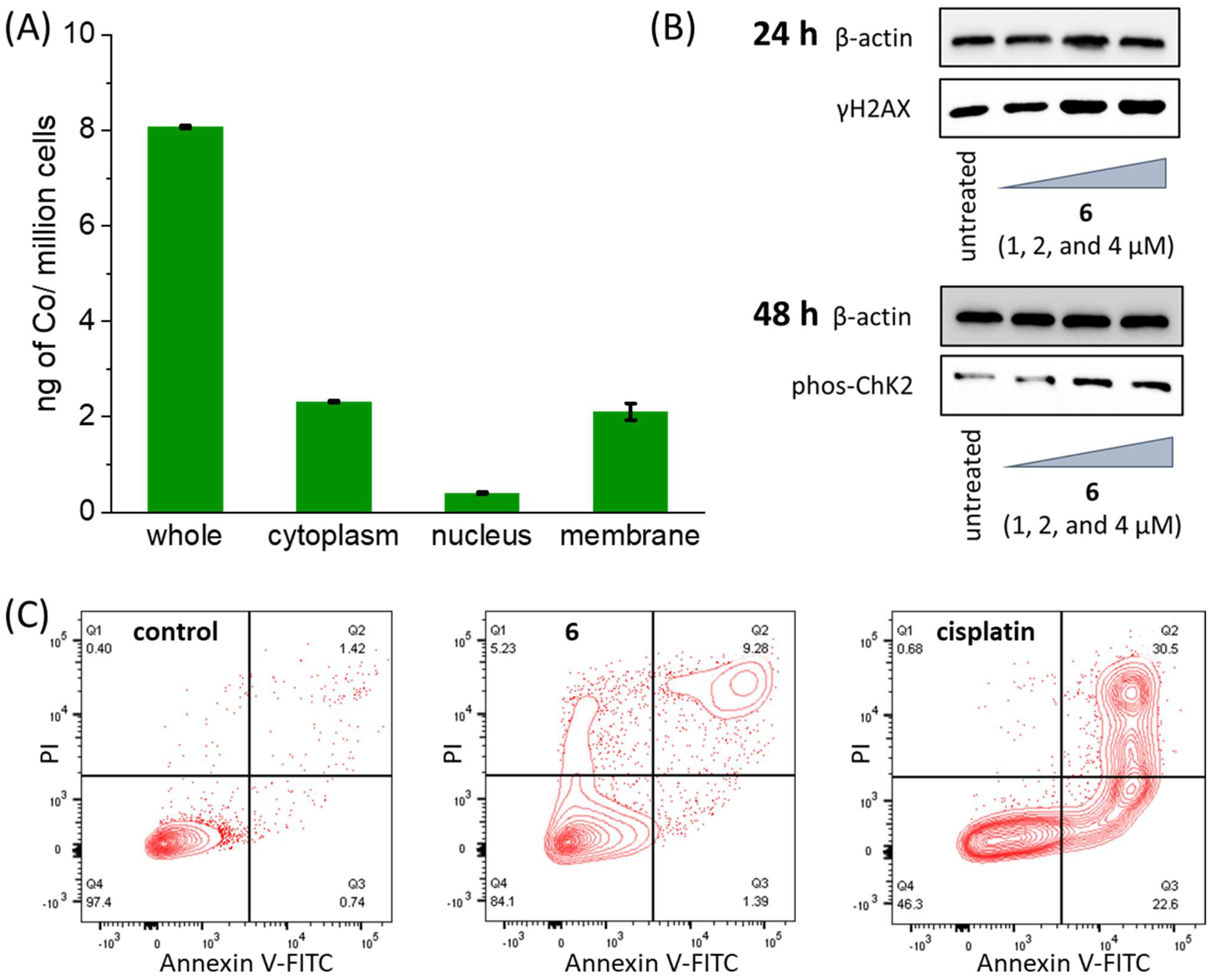
6 (10 μM for 24 h), a reasonable amount was internalised (8.08 ± 0.02 ng of Co/million cells) with an appreciable amount (5%, 0.40 ± 0.02 ng of Co/million cells) localised in the nucleus (
Figure 3A). Given that
6 is able to enter the nucleus, its ability to damage DNA and trigger the DNA damage response pathway was investigated by immunoblotting studies. HMLER-shEcad cells treated with
6 (1–4 μM for 24 h or 48 h) exhibited a clear increase in the expression of the phosphorylated forms of H2AX (γH2AX) and checkpoint kinase 2 (Chk2), indicative of DNA damage (
Figure 3B) [
38,
39]. Cobalt(III) complexes that are structurally similar to
6 are known to cleave DNA; therefore, we investigated the ability of
6 to mimic nuclease. Upon incubation of plasmid pUC19 DNA (100 ng) with
6 (0–100 μM for 24 h), a modest decrease in the amount of supercoiled DNA (form I) and a slight increase in the amount of nicked circular DNA (form II) was observed, indicative of limited DNA cleavage (
Figure S34A). In the presence of ascorbic acid (10-fold excess),
6 induced complete conversion of supercoiled DNA (form I) to nicked circular DNA (form II) at 10 μM and to several small fragments of DNA at 20 μM (
Figure S34B). To determine the oxidative mechanism by which
6 induces DNA cleavage, nuclease activity was probed in the presence of reactive oxygen species (ROS) scavengers (
tBuOH, KI, NaN
3, DMSO) and ascorbic acid (10-fold excess) (
Figure S34C). KI and NaN
3 displayed the greatest inhibitory effect, suggesting that hydrogen peroxide (H
2O
2) and singlet oxygen (
1O
2) are the major ROS intermediates formed during the DNA cleavage process. A similar result was previously observed for related cobalt(III) complexes [
15,
16]. In light of the oxidative mechanism by which
6 induces DNA cleavage, we sought to determine the ability of
6 to increase ROS levels in breast CSCs. To do this, the ROS detection agent 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate was used. HMLER-shEcad cells treated with
6 (2 × IC
50 value) displayed a significant increase in ROS levels compared to untreated control cells after short exposure times (0.5–6 h) (
Figure S35). The ROS levels were relatively lower upon prolonged exposure of
6 to HMLER-shEcad cells (16–24 h) (
Figure S35). This shows that
6 can increase intracellularly ROS levels in breast CSCs, particularly at short exposure times.
DNA lesions and oxidative stress, when left unmanaged, can lead to apoptosis [
40]. There are a number of morphological changes that occur during apoptosis; one of the most prominent features is cell membrane rearrangement. During this process, phosphatidylserine residues are translocated from the cell membrane interior to the cell membrane exterior [
41]. This can be detected by the phospholipid binding protein, Annexin V [
42]. Compromised cell membranes also enable propidium iodide internalisation [
42]. The FITC Annexin V-propidium iodide staining flow cytometry assay was used to determine if
6 is able induce changes to the HMLER-shEcad cell membrane representative of apoptosis. Treatment of HMLER-shEcad cells with
6 (4 × IC
50 value for 72 h) induced a significant population to express phosphatidylserine residues on their cell membrane exterior and take up propidium iodide compared to the untreated control sample (
Figure 3C). This suggests that
6 is able to prompt breast CSCs to undergo late-stage apoptosis. A similar result was observed for the treatment of HMLER-shEcad cells with the
bona fide apoptosis-inducer, cisplatin (25 μM for 72 h) (
Figure 3C). Immunoblotting studies indicated that HMLER-shEcad cells dosed with
6 (1–4 μM for 48 h) displayed higher levels of cleaved caspase 3, 7 and poly ADP ribose polymerase (PARP) compared to untreated cells (
Figure S36). This provides further proof that
6 induces breast CSC apoptosis [
43,
44,
45,
46,
47]. Cytotoxicity studies in the presence of the caspase inhibitor z-VAD-FMK (5 μM) [
48] showed that the potency of
6 towards HMLER-shEcad cells significantly decreased (IC
50 value = 2.58 ± 0.07 μM,
p < 0.05,
Figure S37), supporting the notion that
6 evokes caspase-dependent apoptosis of breast CSCs. Additional cytotoxicity studies were conducted with
6 in the presence of a necrosis inhibitor, IM-54, and a necroptosis inhibitor, necrostatin-1. Cytotoxicity studies in the presence of IM-54 (10 μM) and necrostatin-1 (20 μM) showed that the potency of
6 towards HMLER-shEcad cells increased (rather than decreased). The IC
50 value of
6 in the presence of IM-54 was 0.71 ± 0.08 μM and IC
50 value of
6 in the presence of necrostatin-1 was 0.57 ± 0.04 μM (
Figure S38). This suggests the
6 does not induce breast CSC death by necrosis or necroptosis. Taken together, the cell uptake, immunoblotting, flow cytometry and cytotoxicity studies indicate that
6 can enter breast CSC nuclei, induce nuclear DNA damage and prompt caspase-dependent apoptosis.
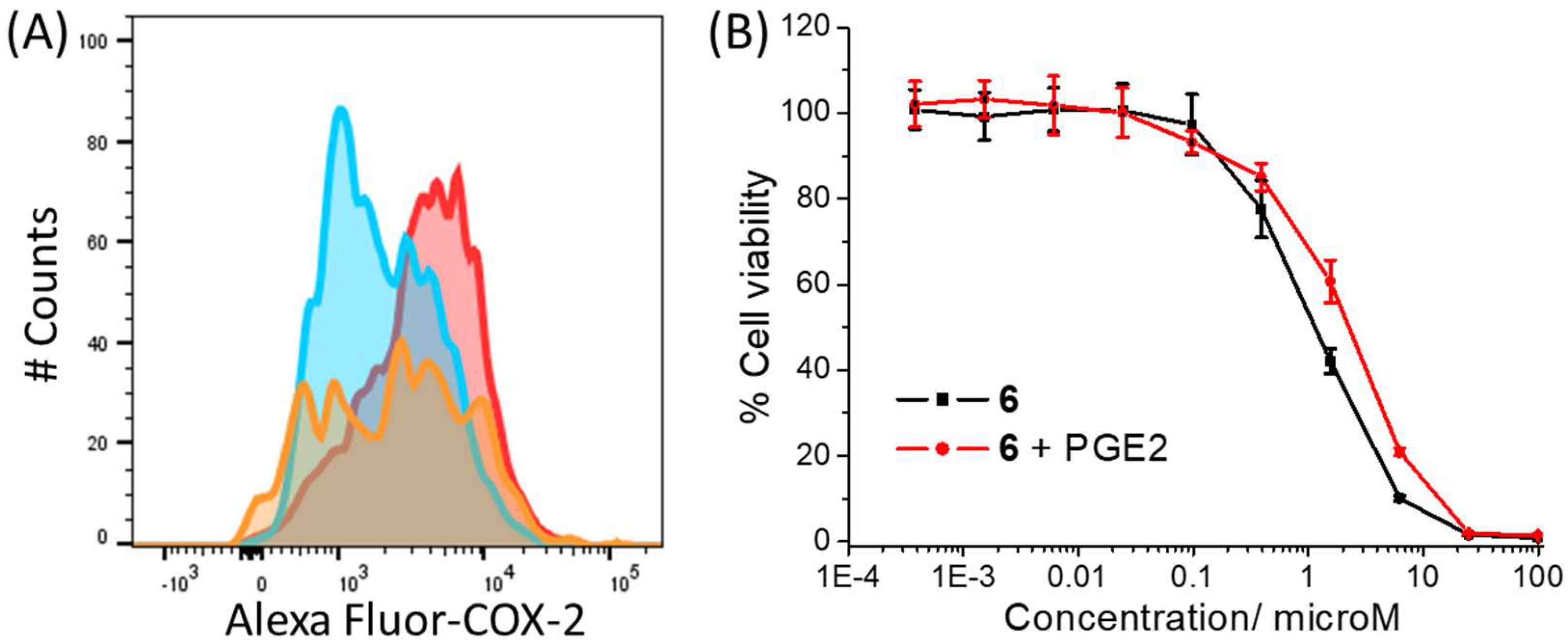
The ability of the cobalt(III) complex
6 to downregulate COX-2 expression was determined by flow cytometric studies. Specifically, HMLER-shEcad cells pre-treated with lipopolysaccharide (LPS) (2.5 μM for 24 h), to increase basal COX-2 levels, were treated with
6 (IC
50 value and 2 × IC
50 value for 48 h), and the COX-2 expression was determined by flow cytometry. These studies clearly showed the emergence a population with markedly lower COX-2 expression than untreated cells (
Figure 4A). This suggests that the mechanism of action of
6 involves COX-2 downregulation. As expected, HMLER-shEcad cells incubated with salicylic acid (40 μM for 48 h) also showed a marked decrease in COX-2 expression (
Figure S39). To further establish the link between COX-2 perturbation and the mechanism of action of
6, cytotoxicity studies were conducted with HMLER-shEcad cells in the presence of prostaglandin E2 (PGE2) (20 μM), the product of COX-2-mediated arachidonic acid processing [
49]. These studies showed that the potency of
6 decreased significantly (IC
50 value = 2.27 ± 0.18 μM,
p < 0.05,
Figure 4B) in the presence of PGE2, suggesting that
6 potentially induces breast CSC death through a COX-2-dependent pathway.
4. Materials and Methods
4.1. General Procedures
All synthetic procedures were performed under normal atmospheric conditions.
1H,
31P{
1H} and
19F{
1H} NMR were recorded at room temperature on a Bruker Avance 400 spectrometer (
1H 400.0 MHz,
31P 162.0 MHz,
19F 376.5 MHz) with chemical shifts (δ, ppm) reported relative to the solvent peaks of the deuterated solvent. Fourier transform infrared (FTIR) spectra were recorded with an IRAffinity-1S Shimadzu spectrophotometer. UV-Vis absorption spectra were recorded on a Cary 3500 UV-Vis spectrophotometer. Inductively coupled plasma mass spectrometry (ICP-MS) were measured using a Thermo Scientific ICAP-Qc quadrupole ICP mass spectrometer. Elemental analysis of the compounds prepared was performed commercially by the University of Cambridge. The
cis-dichlorobis-(polypyridyl)cobalt(III) chloride complexes,
1a–
3a and the
cis-dichlorobis-(polypyridyl)cobalt(III) hexafluorophosphate complexes,
1b–
3b (where polypyridyl = 2,2′-bipyridine for
1a and
1b, 1,10-phenanthroline for
2a and
2b and 3,4,7,8-tetramethyl-1,10-phenanthroline for
3a and
3b) were prepared using reported protocols [
25,
26]. Salicylic acid, silver(I) oxide, sodium sulphate and sodium hexafluorophosphate were purchased from Sigma-Aldrich and used without further purification. Solvents were purchased from Fisher and used without further purification.
4.2. Synthesis of [Co(2,2′-Bipyridine)2(salicylate)]PF6 (4)
Salicylic acid (58 mg, 0.42 mmol), 1a (100 mg, 0.21 mmol) and silver(I) oxide (110 mg, 0.48 mmol) were dissolved in MeOH (50 mL) dried with Na2SO4. The solution was stirred at room temperature for 4 days. The silver salts were removed by filtration, yielding a dark green solution. The solution was concentrated to ca. 5 mL, and diethyl ether (100 mL) was added resulting in a green precipitate which was isolated and washed with diethyl ether (3 × 10 mL). The green residue was dissolved in DMSO (1 mL) and added to a saturated aqueous solution (1 mL) containing NaPF6. The resulting precipitate was collected and washed with water (3 × 10 mL) and dried to yield 4 as a green solid (27.5 mg, 20%); 1H NMR (400 MHz, CD3OD) δH 8.87 (dd, 1H), 8.84 (dd, 1H), 8.80 (dd, 1H), 8.75 (dd, 1H), 8.71 (dt, 1H), 8.68 (dd, 1H), 8.54 (td, 1H), 8.48 (td, 1H), 8.34–8.29 (m, 2H), 7.96 (ddd, 1H), 7.89 (ddd, 1H), 7.72–7.69 (dd, 2H), 7.57–7.50 (m, 3H), 7.04 (ddd, 1H), 6.77 (dd, 1H), 6.59 (ddd, 1H); 19F{1H}NMR (376 MHz, CD3OD) δ −74.86 (d, PF6); 31P{1H} NMR (162 MHz, CD3OD) δ −144.62 (sept, PF6); ATR-FTIR (solid, cm−1): 1619, 1588, 1519, 1466, 1449, 1427, 1331, 1222, 1135, 1030, 883, 835, 765, 717, 652, 595, 556, 469, 443, 386; ESI-MS Calcd. for C27H20CoN4O3 [M-PF6]+: 507.0867 a.m.u. Found [M-PF6]+: 507.0869 a.m.u.; Anal. Calcd. for C27H20CoF6N4O3P·H2O: C, 48.37; H, 3.31; N, 8.36. Found: C, 48.50; H, 3.17; N, 8.06.
4.3. Synthesis of [Co(1,10-Phenanthroline)2(salicylate)]PF6 (5)
Salicylic acid (58 mg, 0.42 mmol), 2a (100 mg, 0.19 mmol) and silver(I) oxide (165 mg, 0.72 mmol) were dissolved in MeOH (50 mL) dried with Na2SO4. The solution was stirred at room temperature for 4 days. The silver salts were removed by filtration, yielding a dark green solution. The solution was concentrated to ca. 5 mL, and diethyl ether (100 mL) was added resulting in a green precipitate which was isolated and washed with diethyl ether (3 × 10 mL). The green residue was dissolved in DMSO (1 mL) and added to a saturated aqueous solution (1 mL) containing NaPF6. The resulting precipitate was collected and washed with water (3 × 10 mL) and dried to yield 5 as a green solid (52.1 mg, 39%); 1H NMR (400 MHz, CD3OD) δH 9.15 (ddd, 2H), 9.11 (dd, 1H), 9.02 (dd, 1H), 8.85–8.82 (m, 2H), 8.47–8.43 (m, 2H), 8.38–8.34 (m, 2H), 8.28 (ddd, 2H), 7.81 (dd, 1H), 7.77–7.72 (m, 2H), 7.70–7.67 (m, 2H), 7.03 (ddd, 1H), 6.73 (d, 1H), 6.61 (ddd, 1H); 19F{1H}NMR (376 MHz, CD3OD) δ −74.84 (d, PF6); 31P{1H} NMR (162 MHz, CD3OD) δ −144.66 (sept, PF6); ATR-FTIR (solid, cm−1): 1615, 1607, 1588, 1468, 1448, 1429, 1345, 1329, 1242, 1143, 1036, 835, 765, 728, 686, 596, 555, 402; ESI-MS Calcd. for C31H20CoN4O3 [M-PF6]+: 555.0867 a.m.u. Found [M-PF6]+: 555.0868 a.m.u.; Anal. Calcd. for C31H20CoF6N4O3P·H2O: C, 51.83; H, 3.09; N, 7.80. Found: C, 51.86; H, 2.82; N, 7.59.
4.4. Synthesis of [Co(3,4,7,8-Tetramethyl-1,10-Phenanthroline)2(salicylate)]PF6 (6)
Salicylic acid (22 mg, 0.16 mmol), 3a (50 mg, 0.08 mmol) and silver(I) oxide (100 mg, 0.44 mmol) were dissolved in MeOH (50 mL) dried with Na2SO4. The solution was stirred at room temperature for 4 days. The silver salts were removed by filtration, yielding a dark green solution. The solution was concentrated to ca. 5 mL, and diethyl ether (100 mL) was added resulting in a green precipitate which was isolated and washed with diethyl ether (3 × 10 mL). The green residue was dissolved in DMSO (1 mL) and added to a saturated aqueous solution (1 mL) containing NaPF6. The resulting precipitate was collected and washed with water (3 × 10 mL) and dried. The crude product was further purified by Al2O3 column chromatography [DCM:MeOH (39:1)] to yield 6 as a green solid (15.0 mg, 23%); 1H NMR (400 MHz, CD3OD) δH 8.82 (s, 1H), 8.71 (s, 1H), 8.56 (dd, 2H), 8.46 (dd, 2H), 7.74 (dd, 1H), 7.41 (s, 1H), 7.30 (s, 1H), 7.03 (ddd, 1H), 6.70 (dd, 1H), 6.59 (ddd, 1H), 3.04 (d, 6H), 2.83 (d, 6H), 2.67 (d, 6H), 2.26 (d, 6H); 19F{1H}NMR (376 MHz, CD3OD) δ −74.94 (d, PF6); 31P{1H} NMR (162 MHz, CD3OD) δ −144.76 (sept, PF6); ATR-FTIR (solid, cm−1): 1620, 1607, 1588, 1534, 1462, 1448, 1428, 1328, 1314, 1247, 1137, 1017, 831, 755, 716, 678, 630, 554, 487, 449; ESI-MS Calcd. for C39H36CoN4O3 [M-PF6]+: 667.2119 a.m.u. Found [M-PF6]+: 667.2122 a.m.u.; Anal. Calcd. for C39H36CoF6N4O3P·2H2O: C, 55.20; H, 4.75; N, 6.60. Found: C, 55.01; H, 4.25; N, 6.71.
4.5. X-ray Crystallography
Crystals were mounted in inert oil on glass fibres and transferred to a Bruker Apex 2000 CCD area detector diffractometer. Data were collected using graphite-monochromated Mo-Kα radiation (λ = 0.71073) at 150(2) K. Scan type ϖ. Absorption corrections based on multiple scans were applied using SADAB [
50] or spherical harmonics implemented in SCALE3 ABSPACK scaling algorithm [
51]. The structures were solved by direct methods and refined on
F2 using the program SHELXT-2016 [
52]. All non-hydrogen atoms were refined anisotropically. The CCDC deposition numbers 2363763 and 2363764 contain the supplementary crystallographic data. This data can be obtained free of charge via The Cambridge Crystallography Data Centre.
4.6. Measurement of Water-Octanol Partition Coefficient (LogP)
The LogP values for 4–6 were determined using the shake-flask method and UV-vis spectroscopy. The 1-octanol used in this experiment was pre-saturated with water. An aqueous solution of 4–6 (500 μL, 100 μM) was incubated with 1-octanol (500 μL) in a 1.5 mL tube. The tube was shaken at room temperature for 24 h. The two phases were separated by centrifugation, and the 4–6 content in each phase was determined by UV-vis spectroscopy.
4.7. Cell Culture
The human mammary epithelial cell lines, HMLER and HMLER-shEcad, were kindly donated by Prof. R. A. Weinberg (Whitehead Institute, MIT, Cambridge, MA, USA). HMLER and HMLER-shEcad cells were maintained in Mammary Epithelial Cell Growth Medium (MEGM) with supplements and growth factors (BPE, hydrocortisone, hEGF, insulin and gentamicin/amphotericin-B). The cells were grown at 310 K in a humidified atmosphere containing 5% CO2.
4.8. Cytotoxicity Studies: MTT Assay
Exponentially growing cells were seeded at a density of approximately 5 × 103 cells per well in 96-well flat-bottomed microplates and allowed to attach for 24 h prior to the addition of compounds. Various concentrations of the test compounds (0.0004–100 μM) were added and incubated for 72 h at 37 °C (total volume 200 μL). Stock solutions of the compounds were prepared as 10 mM DMSO solutions and diluted using cell media. The final concentration of DMSO in each well was ≤1 %. After 72 h, 20 μL of MTT (4 mg ml−1 in PBS) was added to each well, and the plates were incubated for an additional 4 h at 37 °C. The media/MTT mixture was eliminated, and DMSO (100 μL per well) was added to dissolve the formazan precipitates. The optical density was measured at 550 nm using a 96-well multiscanner autoreader. Absorbance values were normalised to (DMSO-containing) control wells and plotted as concentration of compound versus % cell viability. IC50 values were interpolated from the resulting dose dependent curves. The reported IC50 values are the average of three independent experiments (n = 18).
4.9. Tumorsphere Formation and Viability Assay
HMLER-shEcad cells (5 × 103) were plated in ultralow-attachment 96-well plates (Corning) and incubated in MEGM supplemented with B27 (Invitrogen), 20 ng mL−1 EGF and 4 μg mL−1 heparin (Sigma) for 5 days. Studies were also conducted in the presence of 4–6, cisplatin and salinomycin (0–133 μM). Mammospheres treated with 4–6, cisplatin and salinomycin (IC20 value, 5 days) were counted and imaged using an inverted microscope. The viability of the mammospheres was determined by addition of a resazurin-based reagent, TOX8 (Sigma). After incubation for 16 h, the fluorescence of the solutions was read at 590 nm (λex = 560 nm). Viable mammospheres reduce the amount of the oxidised TOX8 form (blue) and concurrently increased the amount of the fluorescent TOX8 intermediate (red), indicating the degree of mammosphere cytotoxicity caused by the test compound. Fluorescence values were normalised to DMSO-containing controls and plotted as concentration of test compound versus % mammospheres viability. IC50 values were interpolated from the resulting dose dependent curves. The reported IC50 values are the average of two independent experiments, each consisting of two replicates per concentration level (overall n = 4).
4.10. Cellular Uptake and Fractionation Studies
To measure the cellular uptake of 6, ca. 1 million HMLER-shEcad cells were treated with 6 (10 μM) at 37 °C for 24 h. After incubation, the media was removed, and the cells were washed with PBS (2 mL × 3) and harvested. The number of cells was counted at this stage using a haemocytometer. This mitigated any cell death induced by 6 at the administered concentration and experimental cell loss. Half of the cellular pellet was dissolved in 65% HNO3 (250 μL) overnight. Half of the cellular pellet was used to determine the cobalt content in the cytoplasmic, nuclear, and membrane fractions. The Thermo Scientific NE-PER Nuclear and Cytoplasmic Extraction Kit was used to extract and separate the cytoplasmic, nuclear and membrane fractions. The fractions were dissolved in 65% HNO3 (250 μL final volume) overnight. All samples were diluted 17-fold with water and analysed using inductively coupled plasma mass spectrometry (ICP-MS, Thermo Scientific ICAP-Qc quadrupole ICP mass spectrometer). Cobalt levels are expressed as mass of Co (ng) per million cells. Results are presented as the mean of three determinations for each data point.
4.11. Immunoblotting Analysis
HMLER-shEcad cells (5 × 103 cells) were incubated with 6 (1–4 μM) for 24 or 48 h at 37 °C. Cells were washed with PBS, scraped into SDS-PAGE loading buffer (64 mM Tris-HCl (pH6.8)/9.6% glycerol/2% SDS/5% β-mercaptoethanol/0.01% Bromophenol Blue) and incubated at 95 °C for 10 min. Whole-cell lysates were resolved by 4–20% sodium dodecylsulphate polyacylamide gel electrophoresis (SDS-PAGE; 200 V for 25 min), followed by electro transfer to polyvinylidene difluoride membrane, PVDF (350 mA for 1 h). Membranes were blocked in 5% (w/v) non-fat milk in PBST (PBS/0.1% Tween 20) and incubated with the appropriate primary antibodies (Cell Signalling Technology). After incubation with horseradish peroxidase-conjugated secondary antibodies (Cell Signalling Technology), immune complexes were detected with the ECL detection reagent (Bio-Rad) and analysed using a chemiluminescence imager (Bio-Rad ChemiDoc Imaging System).
4.12. DNA Cleavage Studies
Plasmid DNA (pUC19) was purchased from Invitrogen. The DNA cleavage activity of 6 was determined by monitoring the conversion of supercoiled plasmid DNA (form I) to nicked circular DNA (form II) in Tris-HCl buffer (5 mM, pH 7.4), using agarose-gel electrophoresis. To probe the effect of compound concentration on cleavage, solutions containing DNA (100 ng) and 6 (0–100 μM) without and with ascorbic acid (10 equivalents), with a total reaction volume of 20 μL, were incubated at 37 °C for 24 h. To determine the oxidative cleavage mechanism, solutions containing DNA (100 ng), 6 (10 μM) with ascorbic acid (10 equivalents) and various radical scavenges (10 mM or 40 mM of KI, DMSO, tBuOH, and NaN3), with a total reaction volume of 20 μL, were incubated at 37 °C for 24 h. After incubation, loading buffer (5 μL, containing 0.25% bromophenol blue, 0.25% xylene cyanol and 60% glycerol) was added, and reaction mixtures were immediately loaded onto a 1% agarose gel containing ethidium bromide (1.0 mg mL−1). The DNA fragments were separated by applying 60 V for 2 h in Tris-acetate EDTA (TAE) buffer. The DNA bands were analysed under UV light using an imager (Bio-Rad ChemiDoc Imaging System).
4.13. Intracellular ROS Assay
HMLER-shEcad cells (5 × 103) were seeded in each well of a 96-well plate. After incubating the cells overnight, they were treated with 6 (2 × IC50 value for 0.5–24 h) and incubated with 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (20 μM) for 30 min. The intracellular ROS level was determined by measuring the fluorescence of the solutions in each well at 529 nm (λex = 504 nm).
4.14. Annexin V-Propidium Iodide Assay
HMLER-shEcad cells were seeded in 6-well plates (at a density of 5 × 105 cells/mL), and the cells were allowed to attach overnight. The cells were incubated without and with 6 (4 × IC50 value for 72 h) or cisplatin (25 μM for 72 h) at 37 °C. Cells were then harvested by trypsinisation. The FITC Annexin V/Dead Cell Apoptosis Kit was used. The manufacturer’s (Thermo Fisher Scientific) protocol was followed to carry out this experiment. Briefly, harvested untreated and treated cells (1 × 106) were suspended in 1× Annexin binding buffer (100 μL) (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4); then, 5 μL FITC Annexin V and 1 μL PI (100 μg/mL) were added to each sample and incubated at room temperature for 15 min, after which more 1× Annexin binding buffer (400 μL) was added while gently mixing. The cells were analysed using a FACSCanto II flow cytometer (BD Biosciences) (10,000 events per sample were acquired) at the University of Leicester FACS Facility. The FL1 channel was used to assess Annexin V binding, and the FL2 channel was used to assess PI uptake. Cell populations were analysed using the FlowJo software 10.5.3 (Tree Star).
4.15. COX-2 Expression Assay
HMLER-shEcad cells were seeded in 6-well plates (at a density of 5 × 105 cells/mL), and the cells were allowed to attach overnight. The cells were treated with lipopolysaccharide (LPS) (2.5 μM for 24 h) and then treated with 6 (IC50 value and 2 × IC50 value) or salicylic acid (40 μM) and incubated for a further 48 h. The cells were then harvested by trypsinization, fixed with 4% paraformaldehyde (at 37 °C for 10 min), permeabilised with ice-cold methanol (for 30 min), and suspended in PBS (200 μL). The Alexa Fluor® 488 nm labelled anti-COX-2 antibody (5 μL) was then added to the cell suspension and incubated in the dark for 1 h. The cells were then washed with PBS (1 mL) and analysed using a FACSCanto II flow cytometer (BD Biosciences) (10,000 events per sample were acquired) at the University of Leicester FACS Facility. The FL1 channel was used to assess COX-2 expression. Cell populations were analysed using the FlowJo software (Tree Star).





{kind=link}
{kind=link}
{kind=link}
{kind=link}