
Uranium-Mediated Thiourea/Urea Conversion on Chelating Ligands

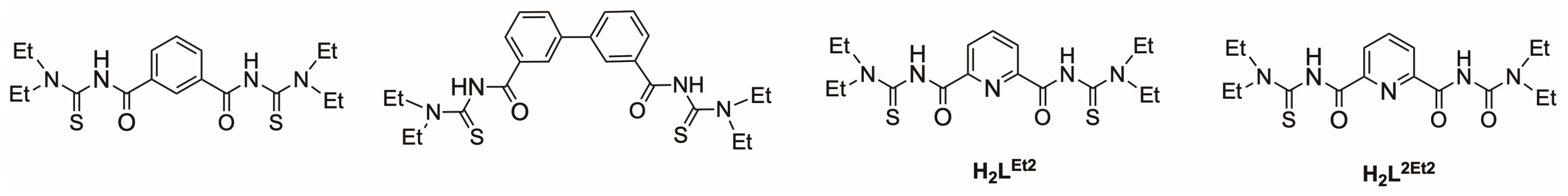
 , and
, and
Abstract

1. Introduction
2. Results and Discussion
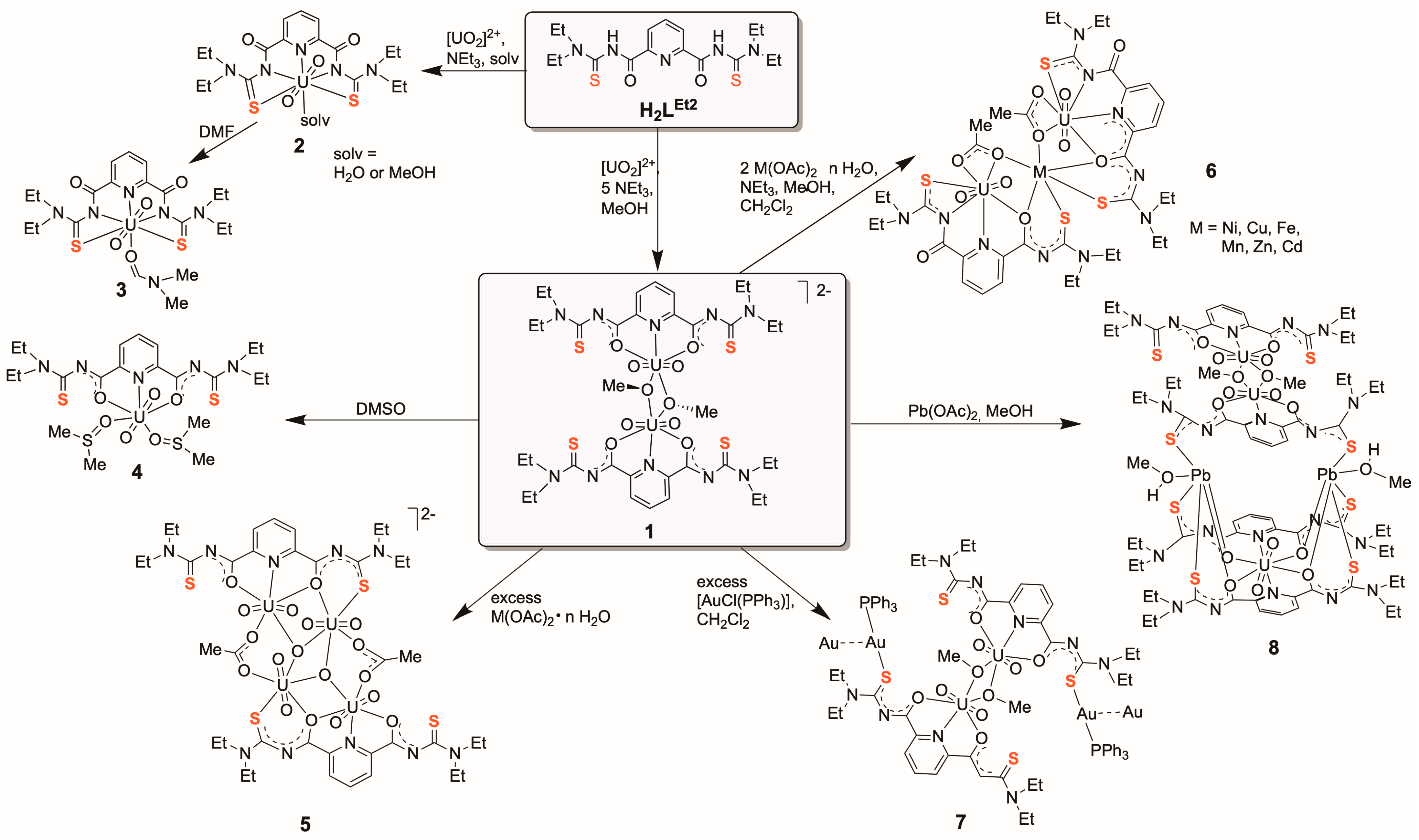
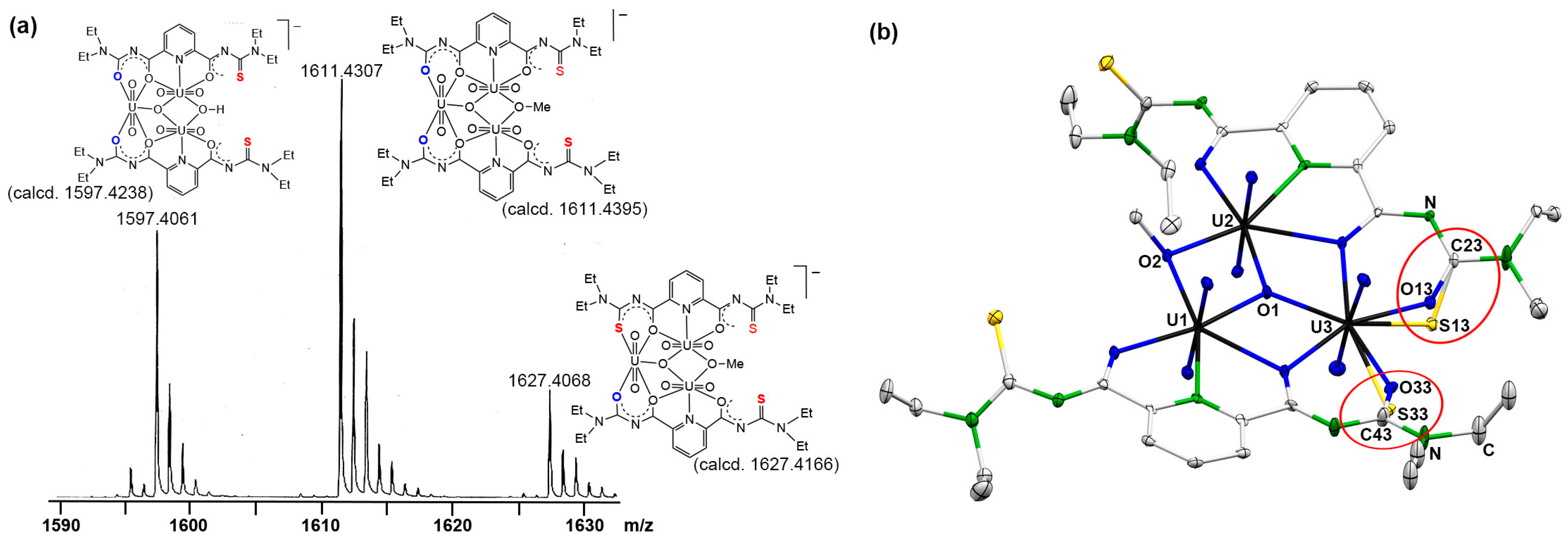
2.1. [(UO2)3(L2Et2)2(µ2-OR)(µ3-O)]− Complexes
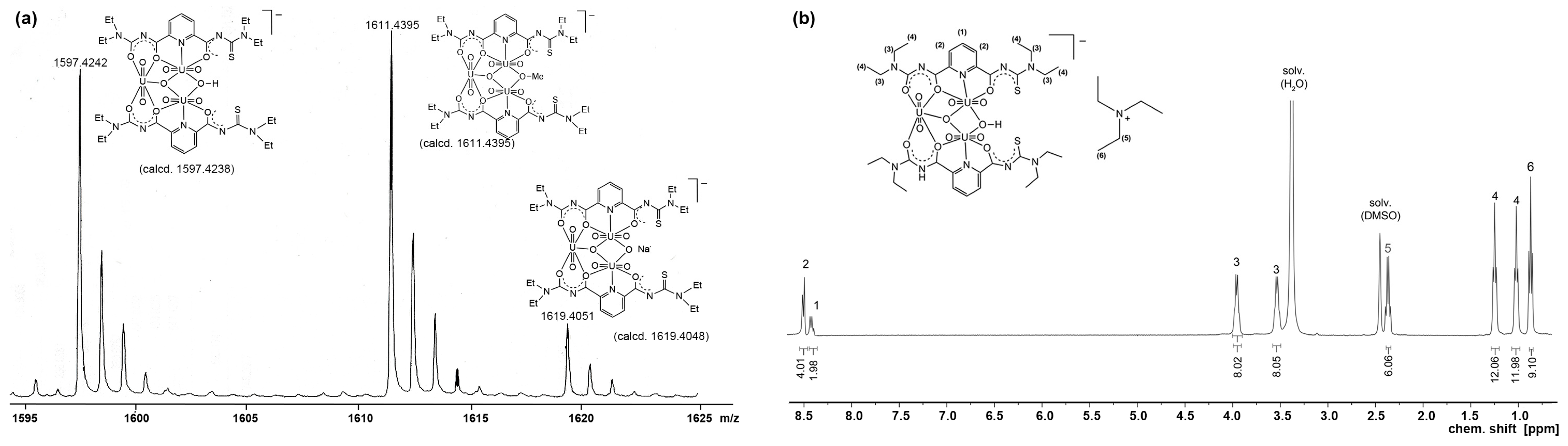
2.1.1. Complex Formation and Spectroscopy
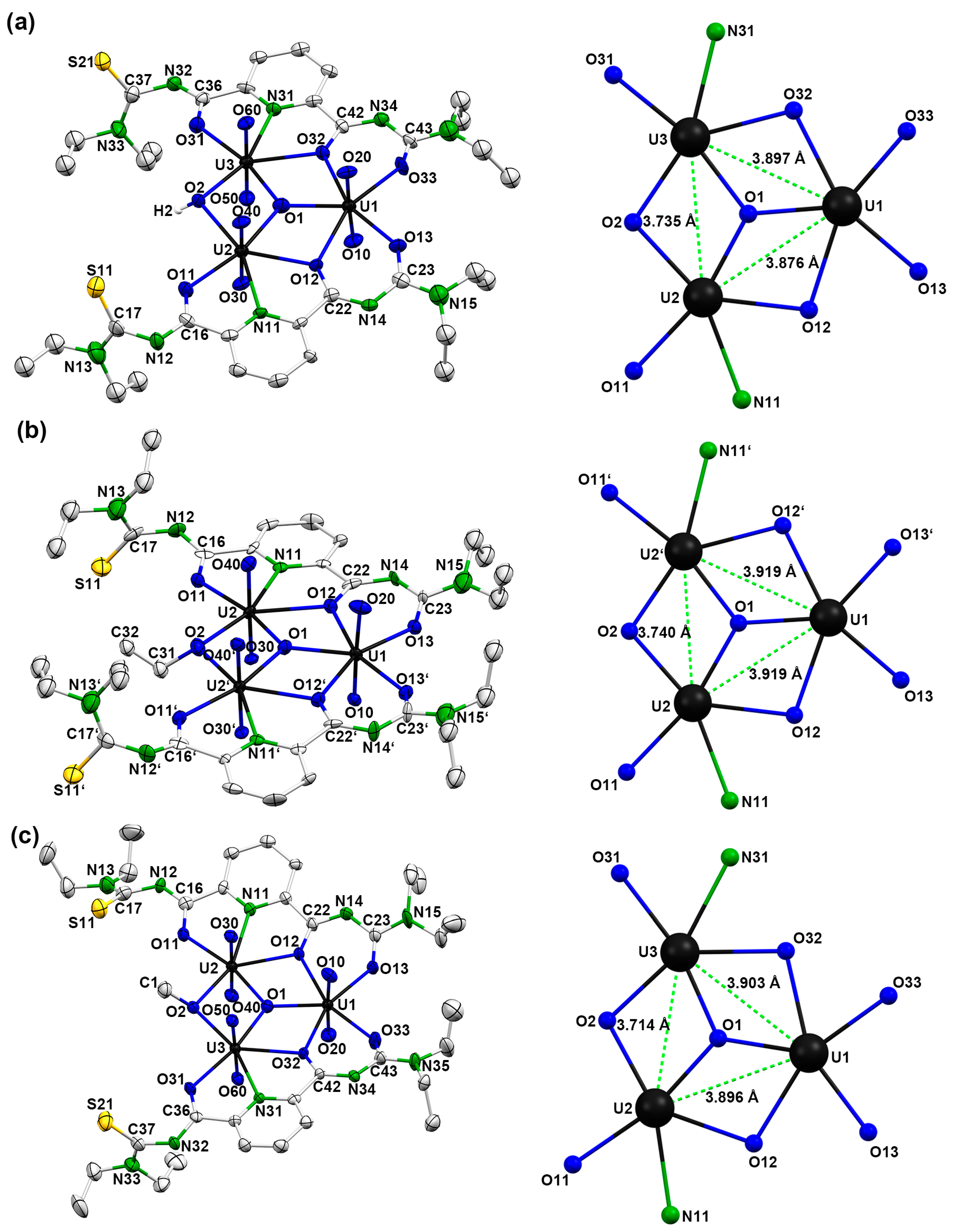
2.1.2. Single-Crystal X-Ray Crystallography
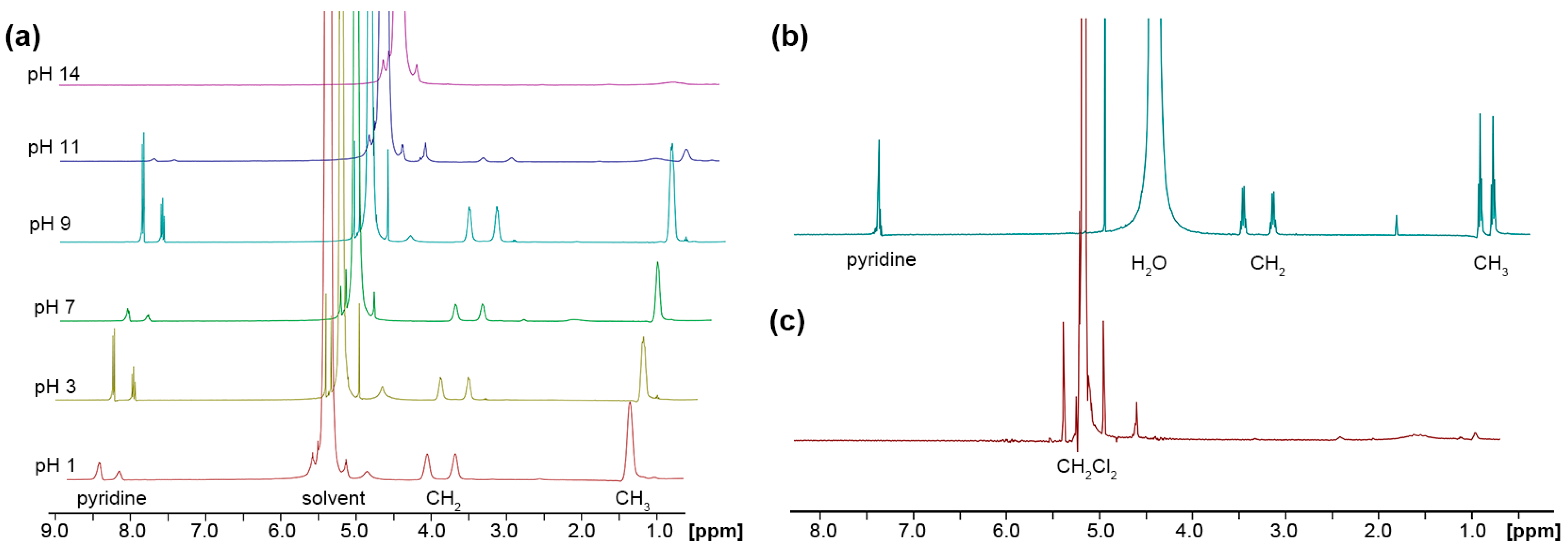
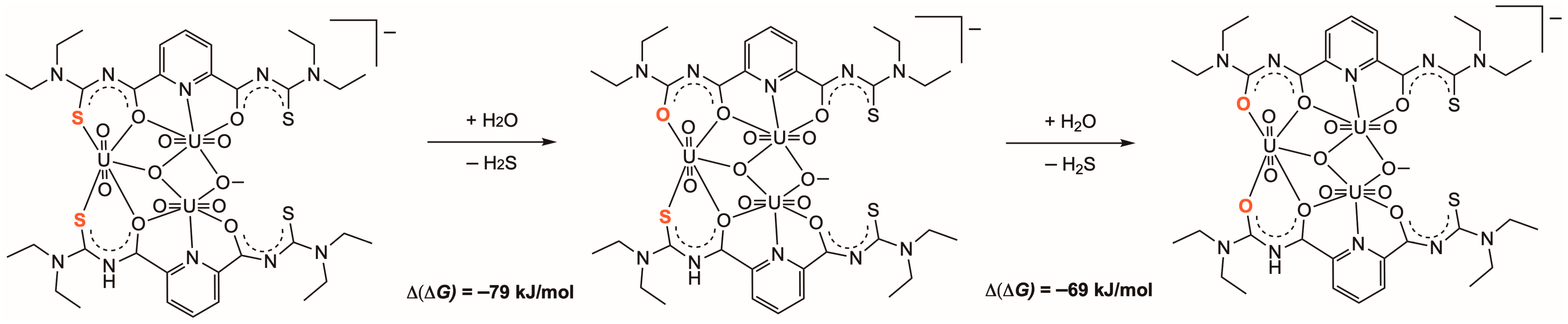
2.2. Hydrolysis of the Thiourea Units
2.3. DFT Calculations
3. Materials and Methods
3.1. Radiation Precaution
3.2. Syntheses
3.3. Spectroscopic and Analytical Methods
3.4. X-Ray Crystallography
3.5. Computational Chemistry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Albrecht-Schmitt, T.E. (Ed.) Organometallic and Coordination Chemistry of the Actinides; Springer: Berlin/Heidelberg, Germany, 2008; Volume 127. [Google Scholar]
- Teixeira Costa Peluzo, B.M.; Kraka, E. Uranium: The nuclear Fuel Cycle and Beyond. Int. J. Mol. Sci. 2022, 23, 4655. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.F.; Zhang, Z.; Rao, L.; Arnold, J. An overview and recent progress in the chemistry of uranium extraction from Seawater. Dalton Trans. 2018, 47, 639–644. [Google Scholar] [CrossRef]
- Loiseau, T.; Mihalcea, I.; Henry, N.; Volkringer, C. The crystal chemistry of uranium carboxylates. Coord. Chem. Rev. 2015, 266–267, 69–109. [Google Scholar] [CrossRef]
- Abraham, F.; Arab-Chapelet, B.; Rivenet, M.; Tamain, C.; Grandjean, S. Actinide Oxalates, solid state structures and applications. Coord. Chem. Rev. 2014, 266–267, 28–68. [Google Scholar] [CrossRef]
- Hayes, C.; Leznoff, D.B. Actinide coordination and organometallic complexes with multidentate polyamide ligands. Coord. Chem. Rev. 2014, 266–269, 155–170. [Google Scholar] [CrossRef]
- Burns, C.J.; Eisen, M.S. Organoactinide Chemistry: Synthesis and Characterization. In The Chemistry of the Actinide and Transactinide Elements; Morss, L.R., Edelstein, N.M., Fuge, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 5. [Google Scholar]
- Ephritikhine, M. Molecular Actinide Compounds with soft chalcogen ligands. Coord. Chem. Rev. 2016, 319, 35–62. [Google Scholar] [CrossRef]
- Nief, F. Complexes containing bonds between group 3, lanthanide or actinide metals and non-first-row main group elements (excluding halogens). Coord. Chem. Rev. 1998, 178–180, 13–81. [Google Scholar] [CrossRef]
- Casellato, U.; Vidali, M.; Vigato, P.A. Actinide complexes with chelating ligands containing sulfur and amidic nitrogen donor atoms. Coord. Chem. Rev. 1979, 28, 231–277. [Google Scholar] [CrossRef]
- Abram, U.; Schulz Lang, E.; Bonfada, E. Thiosemicarbazonato Complexes of Uranium. Z. Anorg. Allg. Chem. 2002, 628, 1873–1878. [Google Scholar] [CrossRef]
- Garcia Santos, I.; Abram, U. Synthesis and structures of dioxouranium complexes with 2-pyridineformamide thiosemicarbazones. Inorg. Chem. Commun. 2004, 7, 440–442. [Google Scholar] [CrossRef]
- Gaunt, A.J.; Scott, B.L.; Neu, M.P. Homoleptic uranium(III) imidodiphosphinochalcogenides including the first structurally characterised molecular trivalent actinide–Se bond. Chem. Commun. 2005, 25, 3215–3217. [Google Scholar] [CrossRef]
- Gaunt, A.J.; Reilly, S.D.; Enriquez, A.E.; Scott, B.L.; Ibers, J.A.; Sekar, P.; Ingram, K.I.M.; Kaltsoyannis, N.; Neu, M.P. Experimental and Theoretical Comparison of Actinide and Lanthanide Bonding in M[N(EPR2)2]3 Complexes (M = U, Pu, La, Ce; E = S, Se, Te; R = Ph, iPr, H). Inorg. Chem. 2008, 47, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Gaunt, A.J.; Scott, B.L.; Neu, M.P. A Molecular Actinide–Tellurium Bond and Comparison of Bonding in [MIII{N(TePiPr2)2}3] (M = U, La). Angew. Chem. Int. Ed. 2006, 45, 1638–1641. [Google Scholar] [CrossRef] [PubMed]
- Ingram, K.I.M.; Kaltsoyannis, N.; Gaunt, A.J.; Neu, M.P. Covalency in the f-element–chalcogen bond: Computational studies of [M(N(EPH2)2)3] (M = La, U, Pu; E = O, S, Se, Te). J. Alloys Compd. 2007, 444–445, 369–375. [Google Scholar] [CrossRef]
- Katz, S.A. The Chemistry and Toxicology of Depleted Uranium. Toxics 2014, 2, 50–78. [Google Scholar] [CrossRef]
- Meyer, K.; Hartline, D.R. From Chemical Curiosities and Trophy Molecules to Uranium-Based Catalysis: Development for Uranium Catalysis as a New Facet in Molecular Uranium Chemistry. JACS Au 2021, 6, 698–709. [Google Scholar]
- King, D.M.; Liddle, S.T. Progress in molecular uranium-nitride chemistry. Coord. Chem. Rev. 2014, 266–267, 2–15. [Google Scholar] [CrossRef]
- Hayton, T.W. Recent developments in actinide–ligand multiple bonding. Chem. Commun. 2013, 49, 2956–2973. [Google Scholar] [CrossRef]
- Ephritikhine, M. Recent Advances in Organoactinide Chemistry As Exemplified by Cyclopentadienyl Compounds. Organometallics 2013, 32, 2464–2488. [Google Scholar] [CrossRef]
- Liddle, S.T. The Renaissance of Non-Aqueous Uranium Chemistry. Angew. Chem. Int. Ed. 2015, 54, 8604–8646. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.R.; Bart, S.C.; Meyer, K.; Cummins, C.C. Towards uranium catalysts. Nature 2008, 455, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Moro, F.; Mills, D.P.; Liddle, S.T.; van Slageren, J. The Inherent Single-Molecule Magnet Character of Trivalent Uranium. Angew. Chem. Int. Ed. 2013, 52, 3430–3433. [Google Scholar] [CrossRef]
- Liddle, S.T.; van Slageren, J. Improving f-element single molecule magnets. Chem. Soc. Rev. 2015, 44, 6655–6669. [Google Scholar] [CrossRef]
- Gardner, B.M.; Liddle, S.T. Small-Molecule Activation at Uranium(III). Eur. J. Inorg. Chem. 2013, 2013, 3753–3770. [Google Scholar] [CrossRef]
- Hohloch, S.; Garner, M.E.; Parker, B.F.; Arnold, J. New supporting ligands in actinide chemistry: Tetramethyltetraazaannulene complexes with thorium and uranium. Dalton Trans. 2017, 46, 13768–13782. [Google Scholar] [CrossRef]
- Andrews, M.B.; Cahill, C.L. Uranyl bearing hybrid materials: Synthesis, speciation and solid state structures. Chem. Rev. 2013, 113, 1121–1136. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.; Cahill, C.L. Synthesis, structure and fluorescent studies of novel uranium coordination polymers in the pyridine dicarboxylic acid system. Dalton Trans. 2006, 39, 4679–4690. [Google Scholar] [CrossRef] [PubMed]
- Carter, K.P.; Kalaj, M.; Cahill, C.L. Harnessing uranyl oxo atoms via halogen bonding interactions in molecular uranyl materials featuring 2,5-diiodobenzoic acid and N-donor capping ligands. Inorg. Chem. Front. 2017, 4, 65–78. [Google Scholar] [CrossRef]
- Lussier, A.J.; Lopez, R.A.K.; Burns, P.C. A Revised and Expanded Structure Hierarchy of Natural and Synthetic Hexavalent Uranium Compounds. Can. Min. 2016, 54, 177–283. [Google Scholar] [CrossRef]
- Zhang, Z.; Senchyk, G.; Liu, Y.; Spano, T.; Szymanowski, J.; Burns, P. Porous uranium diphosphonate frameworks with trinuclear units template by organic ammonium hydrolyzed from amine solvents. Inorg. Chem. 2017, 56, 13249–13256. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Spano, T.; Dembowski, M.; Kokot, A.; Szymanowski, J.; Burns, P.C. Sulfate-Centered Sodium-Icosahedron-Templated Uranyl Peroxide Phosphate Cages with Uranyl Bridged by μ-η1:η2 Peroxide. Inorg. Chem. 2016, 56, 1874–1880. [Google Scholar] [CrossRef] [PubMed]
- Serezhkina, L.B.; Grigor’ev, M.S.; Shimin, N.A.; Klepov, V.V.; Sereszhkin, V.N. First uranyl methacrylate complexes: Synthesis and structure. Russ. J. Inorg. Chem. 2015, 60, 672–683. [Google Scholar] [CrossRef]
- Serezhkina, L.B.; Vologzhanina, A.V.; Klepov, V.V.; Sereszhkin, V.N. Crystal Structure of R[UO2(CH3COO)3] (R = NH4+, K+, or Cs+). Crystallogr. Rep. 2010, 55, 773–779. [Google Scholar] [CrossRef]
- Carter, K.P.; Kalaj, M.; Kerridge, A.; Ridenour, J.A.; Cahill, C. How to Bend the Uranyl Cation via Crystal Engineering. Inorg. Chem. 2018, 57, 2714–2723. [Google Scholar] [CrossRef]
- Ridenour, J.A.; Cahill, C.L. Synthesis, structural analysis, and supramolecular assembly of a series of in situ generated uranyl-peroxide complexes with functionalized 2,2’-bipyridine and varied carboxylic acid ligands. New J. Chem. 2018, 42, 1816–1831. [Google Scholar] [CrossRef]
- Cambridge Crystallographic Database, Vers. 5.45. Available online: https://www.ccdc.cam.ac.uk/solutions/software/csd/ (accessed on 9 October 2024).
- Maria, L.; Santos, I.C.; Santos, I. Uranium(iii) complexes supported by hydrobis(mercaptoimidazolyl)borates: Synthesis and oxidation chemistry. Dalton Trans. 2016, 47, 10601–10612. [Google Scholar] [CrossRef]
- Kelley, S.P.; Smetana, V.; Nuss, J.S.; Dixon, D.A.; Vasiliu, M.; Mudring, A.-V.; Rogers, R.D. Dehydration of UO2Cl2·3H2O and Nd(NO3)3·6H2O with a Soft Donor Ligand and Comparison of Their Interactions through X-ray Diffraction and Theoretical Investigation. Inorg. Chem. 2020, 59, 2861–2869. [Google Scholar] [CrossRef]
- Noufele, C.N.; Hagenbach, A.; Abram, U. Uranyl Complexes with Aroylbis(N,N-dialkylthioureas. Inorg. Chem. 2018, 57, 12255–12269. [Google Scholar] [CrossRef]
- Noufele, C.N.; Schulze, D.; Roca Jungfer, M.; Hagenbach, A.; Abram, U. Bimetallic uranium complexes with 2,6-dipicolinoylbis(N,N-dialkylthioureas). Molecules 2024, 29, 5001. [Google Scholar] [CrossRef]
- Nguyen, H.H.; Jegathesh, J.J.; Takiden, A.; Hauenstein, D.; Pham, C.T.; Le, C.D.; Abram, U. 2,6-Dipicolinoylbis(N,N-dialkylthioureas) as versatile building blocks for oligo- and polynuclear architectures. Dalton Trans. 2016, 45, 10771–10779. [Google Scholar] [CrossRef]
- Pham, C.T.; Nguyen, H.H.; Hagenbach, A.; Abram, U. Iron(III) Metallacryptand and Metallacryptate Assemblies Derived from Aroylbis(N,N-diethylthioureas). Inorg. Chem. 2017, 56, 11406–11416. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.T.; Roca Jungfer, M.; Abram, U. Indium(III) {2}-Metallacryptates Assembled from 2,6-Dipicolinoyl-bis(N,N-diethylthiourea. New J. Chem. 2020, 44, 3672–3680. [Google Scholar] [CrossRef]
- Jesudas, J.J.; Pham, C.T.; Hagenbach, A.; Abram, U.; Nguyen, H.H. Trinuclear ‘CoIILnIIICoII’ Complexes (Ln = La, Ce, Nd, Sm, Gd, Dy, Er and Yb) with 2,6-Dipicolinoyl-bis(N,N-diethylthiourea)—Synthesis, Structures and Magnetism. Inorg. Chem. 2020, 58, 386–395. [Google Scholar] [CrossRef]
- Sucena, S.F.; Demirer, T.I.; Baitullina, A.; Hagenbach, A.; Grewe, J.; Spreckelmeyer, S.; März, J.; Barkleit, A.; daSilva Maia, P.I.; Nguyen, H.H.; et al. Gold-based Coronands as Hosts for M3+ Metal Ions: Ring Size Matters. Molecules 2023, 28, 5421. [Google Scholar] [CrossRef] [PubMed]
- Santos dos Santos, S.; Schwade, V.D.; Schulz Lang, E.; Pham, C.T.; Roca Jungfer, M.; Abram, U.; Nguyen, H.H. Organotellurium(II) and -(IV) Compounds with Picolinoylbis(thioureas): From Simple 1:1 Adducts to Multimetallic Aggregates. Eur. J. Inorg. Chem. 2024, 27, e202400344. [Google Scholar] [CrossRef]
- Douglass, I.B.; Dains, F.B. Some Derivatives of Benzoyl and Furoyl Isothiocyanates and their Use in Synthesizing Heterocyclic Compounds. J. Am. Chem. Soc. 1934, 56, 719–721. [Google Scholar] [CrossRef]
- Lintvedt, R.L.; Heeg, M.J.; Ahmad, N.; Glick, M.D. Uranyl complexes of beta-polyketonates. Crystal and molecular structure of a mononuclear uranyl 1,3,5-triketonate and a novel trinuclear uranyl 1,3,5-triketonate with a trigonal-planar bridging oxide. Inorg. Chem. 1982, 21, 2350–2356. [Google Scholar] [CrossRef]
- Charpin, P.; Lance, M.; Nierlich, M.; Vigner, D.; Livet, J.; Musikas, C. Structures de complexes d’azoture d’uranyle et de tétraalkylammonium. (I) Triazoture d’uranyle et de tétraéthylammonium. (II) μ3-oxo-azoture d’uranyle et de tétraméthylammonium. Acta Cryst. C 1986, 42, 1691–1694. [Google Scholar] [CrossRef]
- Gatto, C.C.; Schulz Lang, E.; Kupfer, A.; Hagenbach, A.; Abram, U. Mono-, Di- and Trinuclear Dioxo Complexes of Uranium Containing Hydrazonato and Azomethine Ligands. Z. Anorg. Allg. Chem. 2004, 630, 1286–1295. [Google Scholar] [CrossRef]
- Back, D.F.; Manzoni de Oliveira, G.; Schulz Lang, E. Reversible Transamination of Alanine with Pyridoxal (Vitamin B6) in the Presence of the UO22+ Ion: Synthesis and X-ray Characterization of [(UO2PmHpyr)3(μ3-O)]Cl·3H2O (PmHpyr = Pyridoxaminylpiruvate Anion). Z. Anorg. Allg. Chem. 2007, 633, 729–733. [Google Scholar] [CrossRef]
- Vologzhanina, A.V.; Serezhkina, L.B.; Neklyudova, N.A.; Serezkhin, V.N. Synthesis and characterisation of a trinuclear uranyl complex: Crystal structure of (CN3H6)5[(UO2)3O(OH)2(CH3COO)(C2O4)3]. Inorg. Chim. Acta 2009, 362, 4921–4925. [Google Scholar] [CrossRef]
- Rodriguez-Dieguez, A.; Mota, A.J.; Seco, J.M.; Palacios, M.A.; Romerosa, A.; Colacio, E. Influence of metal ions, coligands and reaction conditions on the structural versatility and properties of 5-pyrimidyl-tetrazolate containing complexes. Dalton Trans. 2009, 43, 9578–9586. [Google Scholar] [CrossRef]
- Szabo, Z.; Furo, I.; Csöregh, I. Combinatorial Multinuclear NMR and X-ray Diffraction Studies of Uranium(VI)-Nucleotide Complexes. J. Am. Chem. Soc. 2005, 127, 15236–15247. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.; Nakaguchi, M.; Morimoto, K. Synthesis, Structures, and Proton Self-Exchange Reaction of μ3-Oxido/Hydroxido Bridged Trinuclear Uranyl(VI) Complexes with Tridentate Schiff-Base Ligands. Inorg. Chem. 2017, 56, 4057–4064. [Google Scholar] [CrossRef] [PubMed]
- Schaper, G.; Wenzel, M.; Hennersdorf, F.; Lindoy, L.F.; Weigand, J.J. Saccharified Uranyl Ions: Self-Assembly of UO22+ into Trinuclear Anionic Complexes by the Coordination of Glucosamine-Derived Schiff Bases. Chem.-Eur. J. 2021, 27, 8484–8491. [Google Scholar] [CrossRef]
- Lu, H.; Zheng, Z.; Qiu, J.; Qian, Y.; Wang, J.-Q.; Lin, J. Unveiling the new function of uranyl molecular clusters as fluorometric sensors for UV and X-ray dosimetry. Dalton Trans. 2022, 51, 3041–3045. [Google Scholar] [CrossRef]
- Gangguly, T.; Chakraborty, A.B.; Majumdar, A. Transition Metal Mediated Hydrolysis of C-S Bonds: An Overview of a New Reaction Strategy. Org. Inorg. Au 2023, 3, 332–349. [Google Scholar] [CrossRef]
- Jones, W.D.; Vivic, D.A.; Martin Chin, R.; Roache, J.H.; Myers, A.W. Homogeneous models of thiophene HDS reactions. Selectivity in thiophene C-S cleavage and thiophene reactions with dinuclear metal complexes. Polyhedron 1997, 16, 3115–3128. [Google Scholar] [CrossRef]
- Angelici, R.J. An overview of modeling studies in HDS, HDN and HDO catalysis. Polyhedron 1997, 16, 3073–3088. [Google Scholar] [CrossRef]
- Bianchini, C.; Meli, A. Hydrogenation, Hydrogenolysis, and Desulfurization of Thiophenes by Soluble Metal Complexes: Recent Achievements and Future Directions. Acc. Chem. Res. 1998, 31, 109–116. [Google Scholar] [CrossRef]
- Duayne Whitehurst, D.; Isoda, T.; Mochida, I. Present State of the Art and Future Challenges in the Hydrodesulfurization of Polyaromatic Sulfur Compounds. In Advanced Catalysis; Eley, D.D., Haag, W.O., Gates, B., Knözinger, H., Eds.; Academic Press: Cambridge, MA, USA, 1998; Volume 42, pp. 345–471. [Google Scholar]
- Brunet, S.; Mey, D.; Pérot, G.; Bouchy, C.; Diehl, F. On the hydrodesulfurization of FCC gasoline: A review. Appl. Catal. A Gen. 2005, 278, 143–172. [Google Scholar] [CrossRef]
- Garcia-Valenzuela, J.A. Simple Thiourea Hydrolysis or Intermediate Complex Mechanism? Taking up the Formation of Metal Sulfides from Metal-Thiourea Alkaline Solutions. Comments Inorg. Chem. 2017, 73, 99–115. [Google Scholar] [CrossRef]
- Beyer, L.; Hoyer, E.; Liebscher, J.; Hartmann, H. Komplexbildung mit N-Acyl-thioharnstoffen. Z. Chem. 1981, 21, 81–91. [Google Scholar] [CrossRef]
- Beyer, L.; Hoyer, E.; Hennig, H.; Kirmse, R.; Hartmann, H.; Liebscher, J. Synthese und Charakterisierung neuartiger Übergangsmetall-chelate von 1,1-Dialkyl-3-benzoyl-thioharnstoffen. J. Prakt. Chem. 1975, 317, 829–839. [Google Scholar] [CrossRef]
- Koch, K.R. New chemistry with old ligands: N-alkyl- and N, N-dialkyl-N′-acyl(aroyl)thioureas in co-ordination, analytical and process chemistry of the platinum group metals. Coord. Chem. Rev. 2001, 216−217, 473–488. [Google Scholar] [CrossRef]
- Rodenstein, A.; Odendal, J.A.; Kirmse, R.; Koch, K.R. Synthesis and molecular structure of the first binuclear {(bis-N-isophthaloyl ureato)copper(II)pyridine}2 complex derived from the {(bis-N-isophthaloyl-selenoureato)copper(II)}2 complex by O for Se atom exchange. Coord. Chem. Commun. 2011, 14, 99–102. [Google Scholar] [CrossRef]
- Sheldrick, G. SADABS, Vers. 2014/5; University of Göttingen: Göttingen, Germany, 2014. [Google Scholar]
- Coppens, P. The Evaluation of Absorption and Extinction in Single-Crystal Structure Analysis. In Crystallographic Computing; Muksgaard: Copenhagen, Denmark, 1979. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Bennett, L.; Melchers, B.; Proppe, B. High-Performance Computing at ZEDAT; Freie Universität: Berlin, Germany, 2020; Available online: https://refubium.fu-berlin.de/handle/fub188/26993 (accessed on 24 October 2024).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11-18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Hay, P.J. Ab initio studies of excited states of polyatomic molecules including spin-orbit and multiplet effects: The electronic states of UF6. J. Chem. Phys. 1983, 79, 5469–5482. [Google Scholar] [CrossRef]
- Spitznagel, G.W.; Clark, T.; von Ragué Schleyer, P.; Hehre, W.J. An evaluation of the performance of diffuse function-augmented basis sets for second row elements, Na-Cl. J. Comput. Chem. 1987, 8, 1109–1116. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; von Ragué Schleyer, P. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Neese, F. All-Electron Scalar Relativistic Basis Sets for the Actinides. J. Chem. Theory Comput. 2011, 7, 677–684. [Google Scholar] [CrossRef]
- Shamov, G.A.; Schreckenbach, G.; Vo, T.N. A Comparative Relativistic DFT and Ab Initio Study on the Structure and Thermodynamics of the Oxofluorides of Uranium(IV), (V) and (VI). Chem. Eur. J. 2007, 13, 4932–4947. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef]
- Rappoport, D.; Furche, F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| U1–O1 | U1–O12 | U1–O13 | U1–O32 | U1–O33 | U2–O1 | U2–O11 | U2–N11 | U2–O12 | U2–O2 | |
| 9 | 2.196(9) | 2.532(7) | 2.340(8) | 2.507(9) | 2.308(8) | 2.212(8) | 2.345(8) | 2.538(8) | 2.481(8) | 2.388(8) |
| 10 | 2.205(15) | 2.535(10) | 2.342(10) | - | - | 2.510(11) | 2.341(11) | 2.560(12) | 2.535(10) | 2.361(10) |
| 11 | 2.202(5) | 2.498(5) | 2.342(5) | 2.492(5) | 2.331(6) | 2.225(6) | 2.351(5) | 2.542(7) | 2.504(5) | 2.368(5) |
| U3–O1 | U3–O31 | U3–N31 | U3–O32 | U3–O2 | C16–O11 | C22–O12 | C23–O13 | C36–O31 | C42–O32 | |
| 9 | 2.341(8) | 2.316(8) | 2.531(9) | 2.486(8) | 2.396(7) | 1.28(1) | 1.28(1) | 1.26(1) | 1.29(1) | 1.31(1) |
| 10 | - | - | - | - | - | 1.297(19) | 1.290(18) | 1.265(18) | - | - |
| 11 | 2.227(6) | 2.349(5) | 2.542(6) | 2.515(4) | 2.349(5) | 1.295(10) | 1.286(9) | 1.259(10) | 1.286(9) | 1.283(9) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noufele, C.N.; Roca Jungfer, M.; Hagenbach, A.; Nguyen, H.H.; Abram, U. Uranium-Mediated Thiourea/Urea Conversion on Chelating Ligands. Inorganics 2024, 12, 295. https://doi.org/10.3390/inorganics12110295
Noufele CN, Roca Jungfer M, Hagenbach A, Nguyen HH, Abram U. Uranium-Mediated Thiourea/Urea Conversion on Chelating Ligands. Inorganics. 2024; 12(11):295. https://doi.org/10.3390/inorganics12110295
Chicago/Turabian StyleNoufele, Christelle Njiki, Maximilian Roca Jungfer, Adelheid Hagenbach, Hung Huy Nguyen, and Ulrich Abram. 2024. "Uranium-Mediated Thiourea/Urea Conversion on Chelating Ligands" Inorganics 12, no. 11: 295. https://doi.org/10.3390/inorganics12110295
APA StyleNoufele, C. N., Roca Jungfer, M., Hagenbach, A., Nguyen, H. H., & Abram, U. (2024). Uranium-Mediated Thiourea/Urea Conversion on Chelating Ligands. Inorganics, 12(11), 295. https://doi.org/10.3390/inorganics12110295