
Different Patterns of Pd-Promoted C-H Bond Activation in (Z)-4-Hetarylidene-5(4H)-oxazolones and Consequences in Photophysical Properties

 , , and
, , and 
Abstract

1. Introduction
2. Results and Discussion
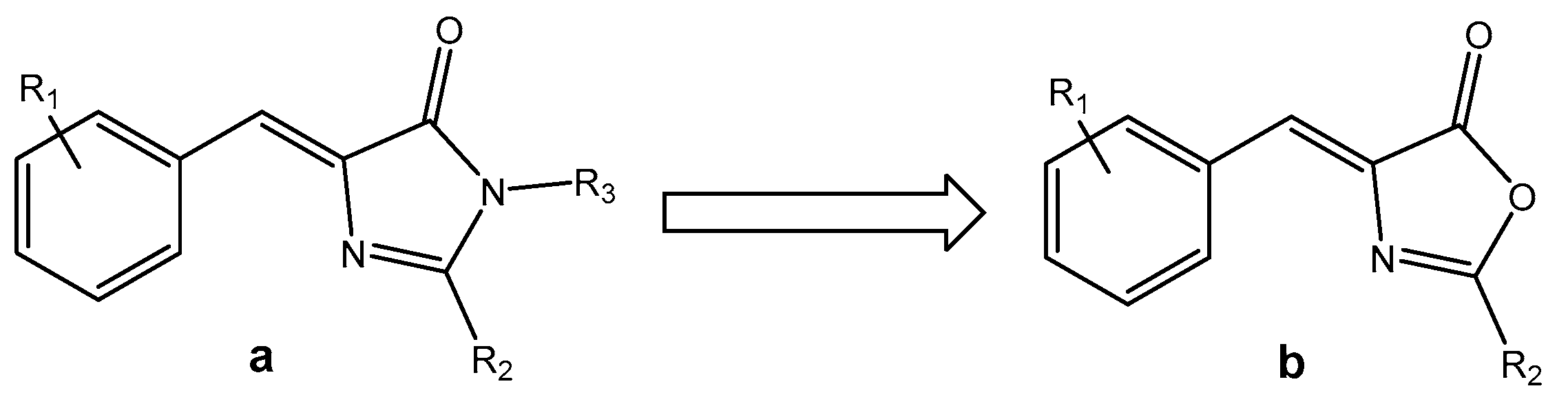
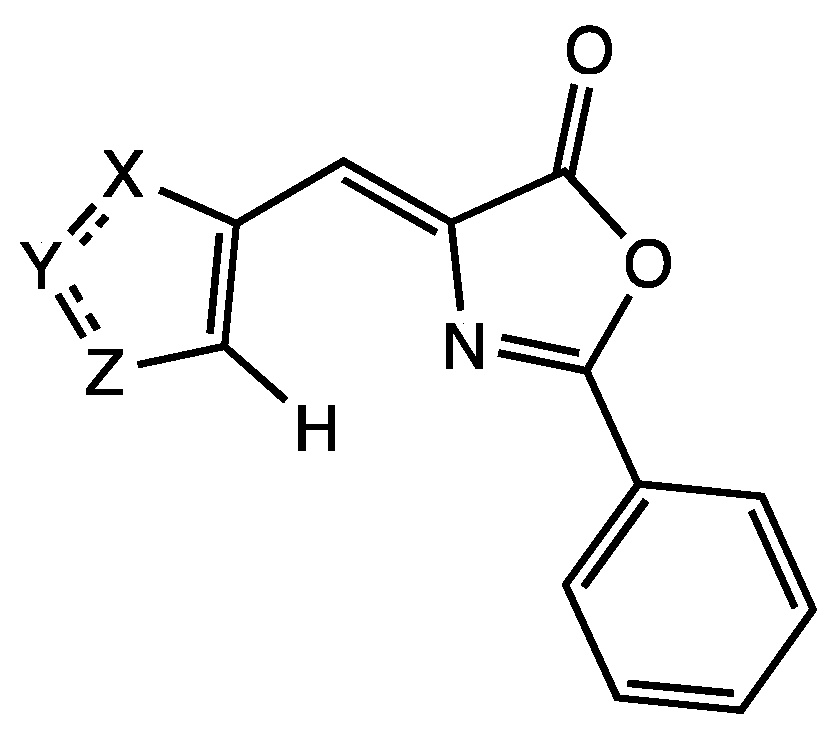
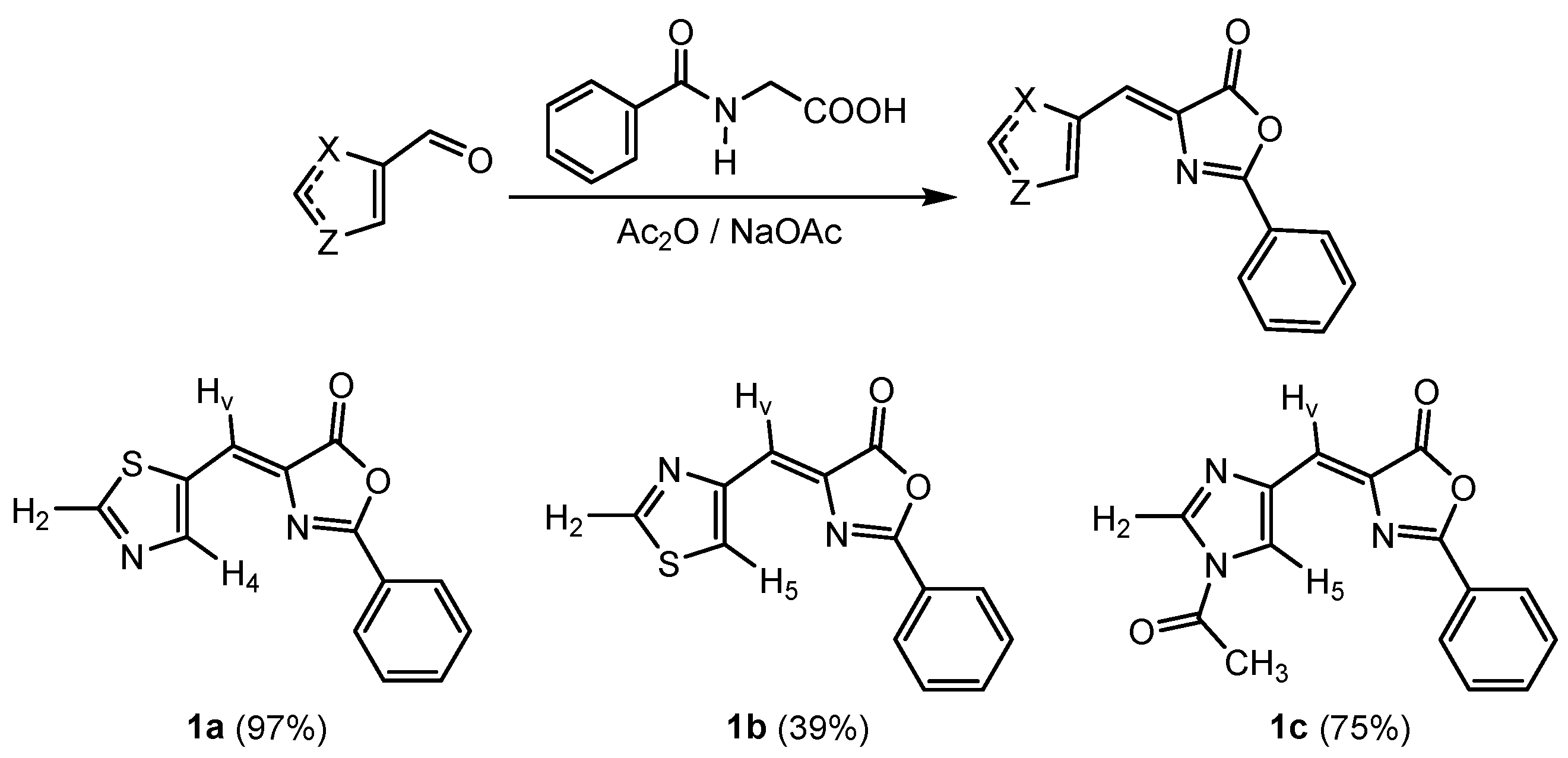
2.1. Synthesis and Characterization of Oxazolones from Heterocycles
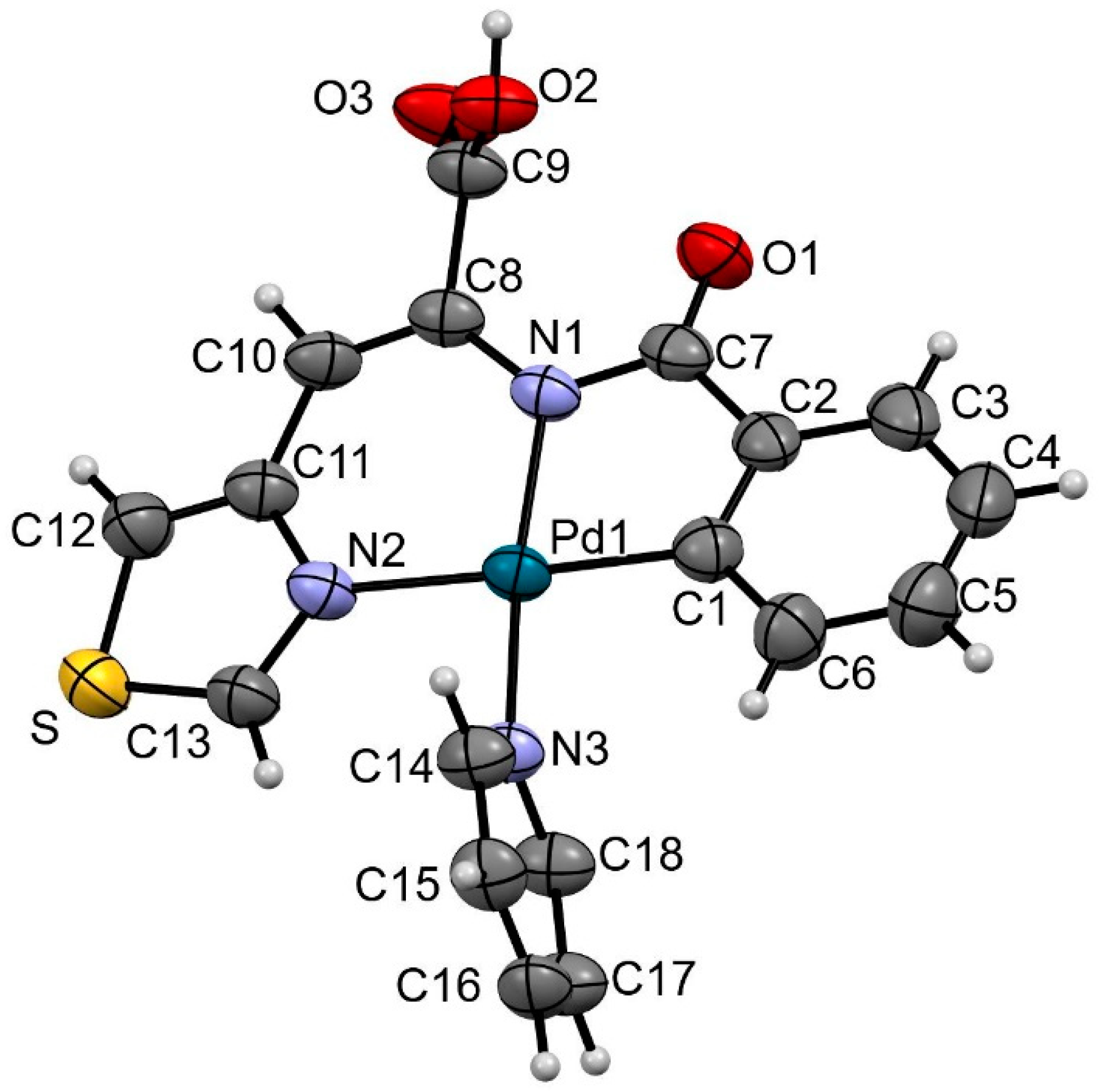
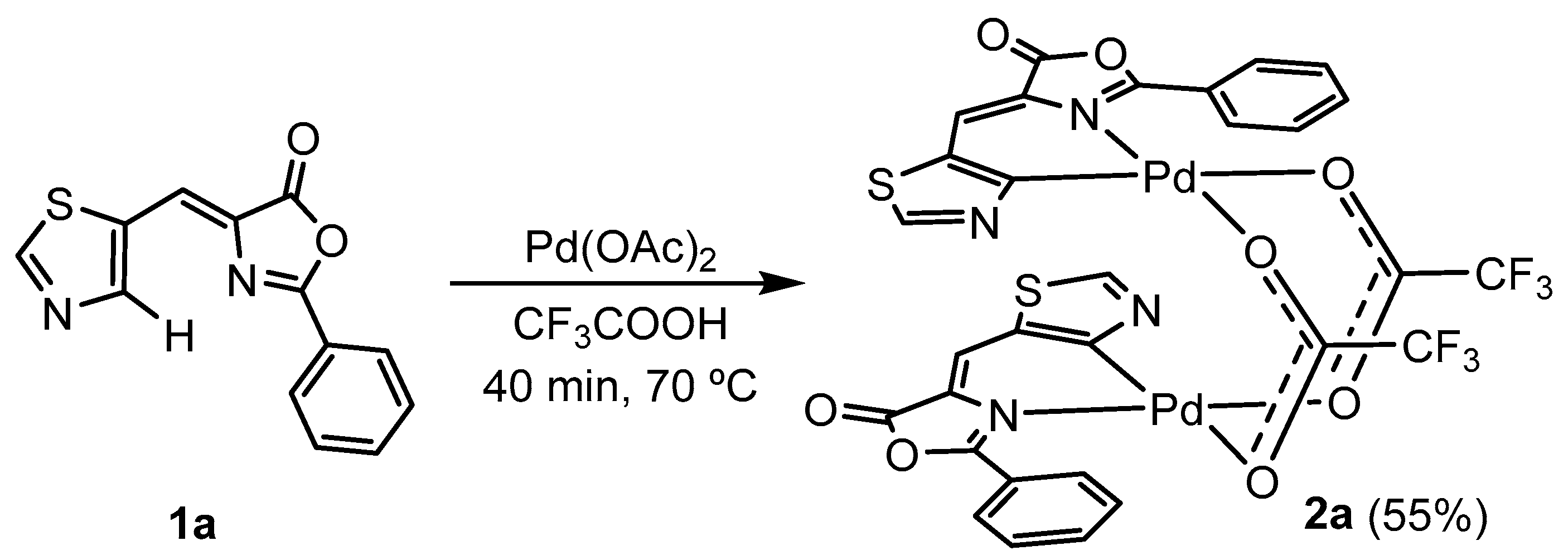
2.2. C-H Bond Activation Reactions: Synthesis and Characterization of the Orthometalated Dinuclear Pd Complex with Trifluoroacetate Bridges (2a) and the Tridentate Complex (2b)
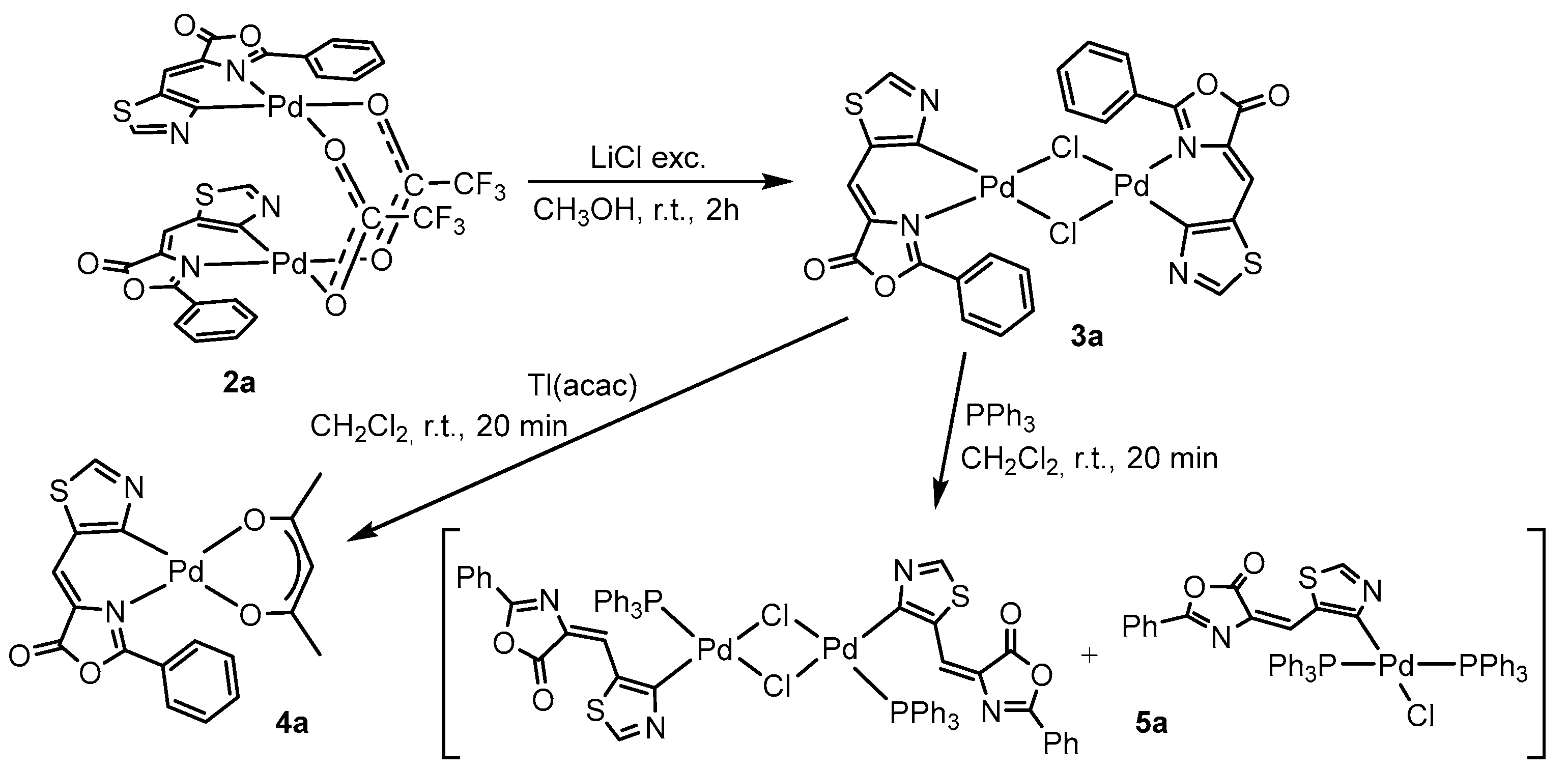
2.3. Reactivity of (2a), Synthesis of 3a–5a
2.4. Photophysical Properties
3. Materials and Methods
3.1. Synthesis and Characterization of Oxazolones from Heterocycles
3.2. Synthesis of Orthopalladated Derivatives 2a–5a and 2b
3.3. Spectroscopic and Analytical Methods
3.4. X-Ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valeur, B.; Berberan-Santos, M.N. Molecular Fluorescence, Principles and Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Edgar, A. Luminescent Materials. In Springer Handbook of Electronic and Photonic Materials; Kasap, S., Capper, P., Eds.; Springer Handbooks; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Kitai, A. Luminescent Materials and Applications, 1st ed.; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Kandarakis, I.S. Luminescence in medical image science. J. Lumin. 2016, 169, 553–558. [Google Scholar] [CrossRef]
- Ronda, C. Challenges in Application of Luminescent Materials, a Tutorial Overview. Prog. Electromagn. Res. 2014, 147, 81–93. [Google Scholar] [CrossRef]
- Prodi, L.; Montalti, M.; Zaccheroni, N. Luminescence Applied in Sensor Science. Top. Curr. Chem. 2011, 300, 1–217. [Google Scholar]
- Feldmann, C.; Jüstel, T.; Ronda, C.R.; Schmidt, P.J. Inorganic Luminescent Materials: 100 Years of Research and Application. Adv. Funct. Mater. 2003, 13, 511–516. [Google Scholar] [CrossRef]
- Shimomura, O. Discovery of green fluorescent protein (GFP) (Nobel Lecture). Angew. Chem. Int. Ed. 2009, 48, 5590–5602. [Google Scholar] [CrossRef]
- Chalfie, M. GFP: Lighting Up Life (Nobel Lecture). Angew. Chem. Int. Ed. 2009, 48, 5603–5611. [Google Scholar] [CrossRef]
- Tsien, R.Y. Constructing and Exploiting the Fluorescent Protein Paintbox (Nobel Lecture). Angew. Chem. Int. Ed. 2009, 48, 5612–5626. [Google Scholar] [CrossRef]
- Remington, S.J. Fluorescent proteins: Maturation, photochemistry and photophysics. Curr. Opin. Struct. Biol. 2006, 16, 714–721. [Google Scholar] [CrossRef]
- Pakhomov, A.A.; Martynov, V.I. GFP Family: Structural Insights into Spectral Tuning. Chem. Biol. 2008, 15, 755–764. [Google Scholar] [CrossRef]
- Wachter, R.M. Chromogenic Cross-Link Formation in Green Fluorescent Protein. Acc. Chem. Res. 2007, 40, 120–127. [Google Scholar] [CrossRef]
- Mizuno, H.; Mal, T.K.; Tong, K.I.; Ando, R.; Furuta, T.; Ikura, M.; Miyawaki, A. Photo-Induced Peptide Cleavage in the Green-to-Red Conversion of a Fluorescent Protein. Mol. Cell. 2003, 12, 1051–1058. [Google Scholar]
- Collado, S.; Pueyo, A.; Baudequin, C.; Bischoff, L.; Jiménez, A.I.; Cativiela, C.; Hourau, C.; Urriolabeitia, E.P. Orthopalladation of GFP-Like Fluorophores Through C–H Bond Activation: Scope and Photophysical Properties. Eur. J. Org. Chem. 2018, 2018, 6158–6166. [Google Scholar] [CrossRef]
- Ivashkin, P.E.; Yampolsky, I.V.; Lukyanov, K.A. Synthesis and properties of chromophores of fluorescent proteins. Russ. J. Bioorg. Chem. 2009, 35, 652–669. [Google Scholar] [CrossRef] [PubMed]
- Follenius-Wund, A.; Bourotte, M.; Schmitt, M.; Iyice, F.; Lami, H.; Bourguignon, J.J.; Haiech, J.; Pigault, C. Fluorescent derivatives of the GFP chromophore give a new insight into the GFP fluorescence process. Biophys. J. 2003, 85, 1839–1850. [Google Scholar]
- Rajbongshi, B.K.; Sen, P.; Ramanathan, G. Twisted intramolecular charge transfer in a model green fluorescent protein luminophore analog. Chem. Phys. Lett. 2010, 494, 295–300. [Google Scholar] [CrossRef]
- Martin, M.E.; Negri, F.; Olivucci, M. Origin, Nature, and Fate of the Fluorescent State of the Green Fluorescent Protein Chromophore at the CASPT2//CASSCF Resolution. J. Am. Chem. Soc. 2004, 126, 5452–5464. [Google Scholar] [CrossRef]
- Altoe’, P.; Bernardi, F.; Garavelli, M.; Orlandi, G.; Negri, F. Solvent effects on the vibrational activity and photodynamics of the green fluorescent protein chromophore: A quantum-chemical study. J. Am. Chem. Soc. 2005, 127, 3952–3963. [Google Scholar] [CrossRef]
- Megley, C.M.; Dickson, L.A.; Maddalo, S.L.; Chandler, G.J.; Zimmer, M. Photophysics and dihedral freedom of the chromophore in yellow, blue, and green fluorescent protein. J. Phys. Chem. B 2009, 113, 302–308. [Google Scholar] [CrossRef]
- Wu, L.; Burgess, K. Syntheses of Highly Fluorescent GFP-Chromophore Analogues. J. Am. Chem. Soc. 2008, 130, 4089–4096. [Google Scholar] [CrossRef]
- Baranov, M.S.; Lukyanov, K.A.; Borissova, A.O.; Shamir, J.; Kosenkov, D.; Slipchenko, L.V.; Tolbert, L.M.; Yampolsky, I.V.; Solntsev, K.M. Conformationally Locked Chromophores as Models of Excited-State Proton Transfer in Fluorescent Proteins. J. Am. Chem. Soc. 2012, 134, 6025–6032. [Google Scholar] [CrossRef]
- Laga, E.; Dalmau, D.; Arregui, S.; Crespo, O.; Jiménez, A.I.; Pop, A.; Silvestru, C.; Urriolabeitia, E.P. Fluorescent Orthopalladated Complexes of 4-Aryliden-5(4H)-oxazolones from the Kaede Protein: Synthesis and Characterization. Molecules 2021, 26, 1238. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, D.; Crespo, O.; Matxain, J.M.; Urriolabeitia, E.P. Fluorescence Amplification of Unsaturated Oxazolones Using Palladium: Photophysical and Computational Studies. Inorg. Chem. 2023, 62, 9792–9806. [Google Scholar] [CrossRef] [PubMed]
- Dumitras, D.; Dalmau, D.; García-Orduña, P.; Pop, A.; Silvestru, A.; Urriolabeitia, E.P. Orthopalladated imidazolones and thiazolones: Synthesis, photophysical properties and photochemical reactivity. Dalton Trans. 2024, 53, 8948–8967. [Google Scholar] [CrossRef] [PubMed]
- Erlenmeyer, E. Ueber die Condensation der Hippursäure mit Phtalsäureanhydrid und mit Benzaldehyd. Justus Liebigs Ann. Der Chem. 1893, 275, 1–8. [Google Scholar]
- Plöchl, J. Ueber Phenylglycidasäure (Phenyloxacrylsäure). Chem. Ber. 1883, 16, 2815. [Google Scholar] [CrossRef]
- Plöchl, J. Ueber einige Derivate der Benzoylimidozimmtsäure. Chem. Ber. 1884, 17, 1616. [Google Scholar] [CrossRef]
- Carter, H.E. Azlactones. Org. React. 1946, 3, 198–237. [Google Scholar]
- Filler, R. Advances in Heterocyclic Chemistry; Katritzky, A.R., Ed.; Academic Press: New York, NY, USA, 1954; Volume 4, p. 75. [Google Scholar]
- Rao, Y.S.; Filler, R. Geometric Isomers of 2-Aryl(Aralkyl)-4-arylidene(alkylidene)-5(4H)-oxazolones. Synthesis 1975, 12, 749–764. [Google Scholar] [CrossRef]
- Rao, Y.S.; Filler, R. Oxazoles. In The Chemistry of Heterocyclic Compounds; Turchi, I.J., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1986; Volume 3, pp. 363–691. [Google Scholar]
- Prokof’ev, E.P.; Karpeiskaya, E.I. The Proton Coupled 13C NMR Direct Determination of Z-, E-Configuration of 4-Benzyliden-2-Phenyl(Methyl)- Δ2-Oxazolin-5-Ones and Products of Their Solvolysis. Tetrahedron Lett. 1979, 20, 737–740. [Google Scholar] [CrossRef]
- Vogeli, U.; von Phillipsborn, W. Vicinal C,H Spin Coupling in Substituted Alkenes. Stereochemical Significance and Structural Effects. Org. Magn. Reson. 1975, 7, 617–627. [Google Scholar] [CrossRef]
- Roiban, G.D.; Serrano, E.; Soler, T.; Aullón, G.; Grosu, I.; Cativiela, C.; Martínez, M.; Urriolabeitia, E.P. Regioselective Orthopalladation of (Z)-2-Aryl-4-Arylidene-5(4H)-Oxazolones: Scope, Kinetico-Mechanistic, and Density Functional Theory Studies of the C–H Bond Activation. Inorg. Chem. 2011, 50, 8132–8143. [Google Scholar] [CrossRef] [PubMed]
- Roiban, D.; Serrano, E.; Soler, T.; Grosu, I.; Cativiela, C.; Urriolabeitia, E.P. Unexpected [2 + 2] C–C bond coupling due to photocycloaddition on orthopalladated (Z)-2-aryl-4-arylidene-5(4H)-oxazolones. Chem. Commun. 2009, 4681–4683. [Google Scholar] [CrossRef] [PubMed]
- Serrano, E.; Juan, A.; García-Montero, A.; Soler, T.; Jiménez-Márquez, F.; Cativiela, C.; Gomez, M.V.; Urriolabeitia, E.P. Stereoselective Synthesis of 1,3-Diaminotruxillic Acid Derivatives: An Advantageous Combination of C–H–ortho–Palladation and On-Flow [2 + 2]–Photocycloaddition in Microreactors. Chem. Eur. J. 2016, 22, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Carrera, C.; Denisi, A.; Cativiela, C.; Urriolabeitia, E.P. Functionalized 1,3-Diaminotruxillic Acids by Pd-Mediated C–H Activation and [2 + 2]–Photocycloaddition of 5(4H)–Oxazolones. Eur. J. Inorg. Chem. 2019, 2019, 3481–3489. [Google Scholar] [CrossRef]
- Guy Orpen, A.; Brammer, L.; Allen, F.H.; Kennard, O.; Watson, D.G.; Taylor, R. Supplement. Tables of bond lengths determined by X-ray and neutron diffraction. Part 2. Organometallic compounds and co-ordination complexes of the d- and f-block metals. J. Chem. Soc. Dalton Trans. 1989, S1–S83. [Google Scholar]
- Sierra, S.; Gomez, M.V.; Jiménez, A.I.; Pop, A.; Silvestru, C.; Marín, M.L.; Boscá, F.; Sastre, G.; Gómez-Bengoa, E.; Urriolabeitia, E.P. Stereoselective, Ruthenium-Photocatalyzed Synthesis of 1,2-Diaminotruxinic Bis-amino Acids from 4-Arylidene-5(4H)-oxazolones. J. Org. Chem. 2022, 87, 3529–3545. [Google Scholar] [CrossRef]
- Forniés, J.; Martínez, F.; Navarro, R.; Urriolabeitia, E.P. Synthesis and reactivity of acetylacetonato-Cγ complexes of MII (M = Pd or Pt): X-ray crystal structure of [Pd(C6F5)(OOCPh)(bipy)]. J. Organomet. Chem. 1995, 495, 185–194. [Google Scholar] [CrossRef]
- Pearson, R. Antisymbiosis and the trans effect. Inorg. Chem. 1973, 12, 712–713. [Google Scholar] [CrossRef]
- Roiban, D.; Serrano, E.; Soler, T.; Contel, M.; Grosu, I.; Cativiela, C.; Urriolabeitia, E.P. Ortho-Palladation of (Z)-2-Aryl-4-Arylidene-5(4H)-Oxazolones. Structure and Functionalization. Organometallics. 2010, 29, 1428–1435. [Google Scholar] [CrossRef]
- Würth, C.; Grabolle, M.; Pauli, J.; Spieles, M.; Resch-Genger, U. Comparison of Methods and Achievable Uncertainties for the Relative and Absolute measurement of Photoluminescence Quantum Yields. Anal. Chem. 2011, 83, 3431–3439. [Google Scholar] [CrossRef]
- Juanhuix, J.; Gil-Ortiz, F.; Cuní, G.; Colldelram, C.; Nicolás, J.; Lidón, J.; Boter, E.; Ruget, C.; Ferrer, S.; Benach, J. Developments in optics and performance at BL13-XALOC, the macromolecular crystallography beamline at the Alba synchrotron. J. Synchr. Rad. 2014, 21, 679–689. [Google Scholar] [CrossRef] [PubMed]
- SAINT Software Reference Manuals, Version V8.40B in APEX4; Bruker Analytical Xray Systems, Inc.: Madison, WI, USA, 2016.
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D.J. SADABS 2016/2. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed]
- CrysAlis RED, CCD Camera Data Reduction Program; Rigaku Oxford Diffraction: Oxford, UK, 2019.
- Sheldrick, G.M. SHELXS-86, Phase annealing in SHELX-90: Direct methods for larger structures. Acta Crystallogr. 1990, A46, 467. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXS 97 and SHELXL 97, Program for Crystal Structure Solution and Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Sect. C Struct. Chem. 2015, C71, 3. [Google Scholar]


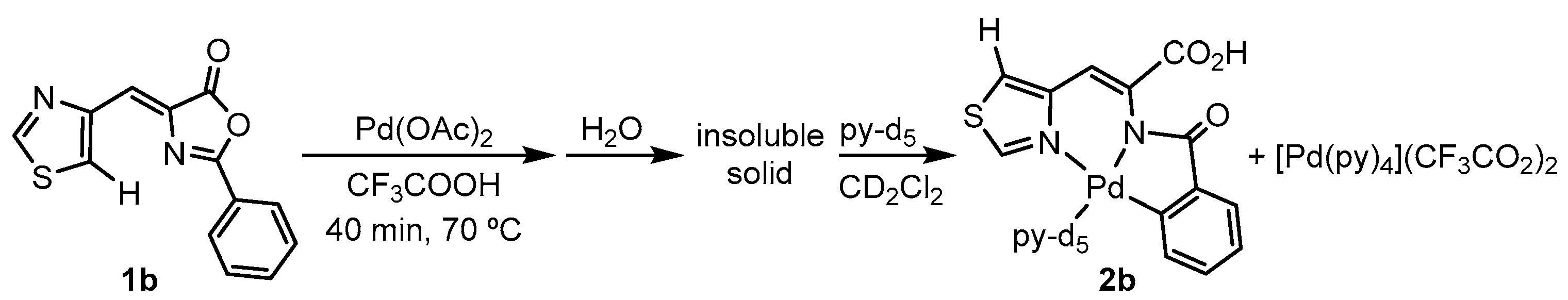

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λabs.max (nm) | λexc.max (nm) | λemis.max (nm) |
|---|---|---|---|
| 1a | 378 | 378 | 434 |
| 1b | 386 | ||
| 1c | 373 | ||
| 2a | 425, 449 | 451 | 474 |
| 4a | 406 | 420 | 512 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez, M.; Dalmau, D.; Crespo, O.; García-Orduña, P.; Lahoz, F.; Martín, A.; Urriolabeitia, E.P. Different Patterns of Pd-Promoted C-H Bond Activation in (Z)-4-Hetarylidene-5(4H)-oxazolones and Consequences in Photophysical Properties. Inorganics 2024, 12, 271. https://doi.org/10.3390/inorganics12100271
Martínez M, Dalmau D, Crespo O, García-Orduña P, Lahoz F, Martín A, Urriolabeitia EP. Different Patterns of Pd-Promoted C-H Bond Activation in (Z)-4-Hetarylidene-5(4H)-oxazolones and Consequences in Photophysical Properties. Inorganics. 2024; 12(10):271. https://doi.org/10.3390/inorganics12100271
Chicago/Turabian StyleMartínez, Miguel, David Dalmau, Olga Crespo, Pilar García-Orduña, Fernando Lahoz, Antonio Martín, and Esteban P. Urriolabeitia. 2024. "Different Patterns of Pd-Promoted C-H Bond Activation in (Z)-4-Hetarylidene-5(4H)-oxazolones and Consequences in Photophysical Properties" Inorganics 12, no. 10: 271. https://doi.org/10.3390/inorganics12100271
APA StyleMartínez, M., Dalmau, D., Crespo, O., García-Orduña, P., Lahoz, F., Martín, A., & Urriolabeitia, E. P. (2024). Different Patterns of Pd-Promoted C-H Bond Activation in (Z)-4-Hetarylidene-5(4H)-oxazolones and Consequences in Photophysical Properties. Inorganics, 12(10), 271. https://doi.org/10.3390/inorganics12100271