

New Insights into the Catalytic Activity of Second Generation Hoveyda–Grubbs Complexes Having Phenyl Substituents on the Backbone




Abstract
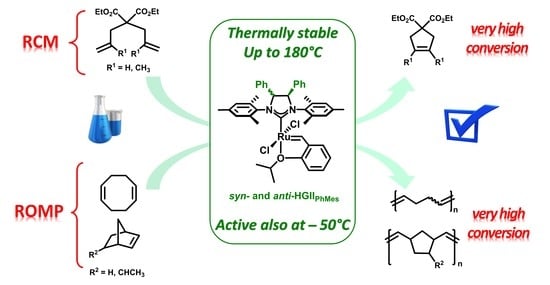
1. Introduction
2. Results and Discussion
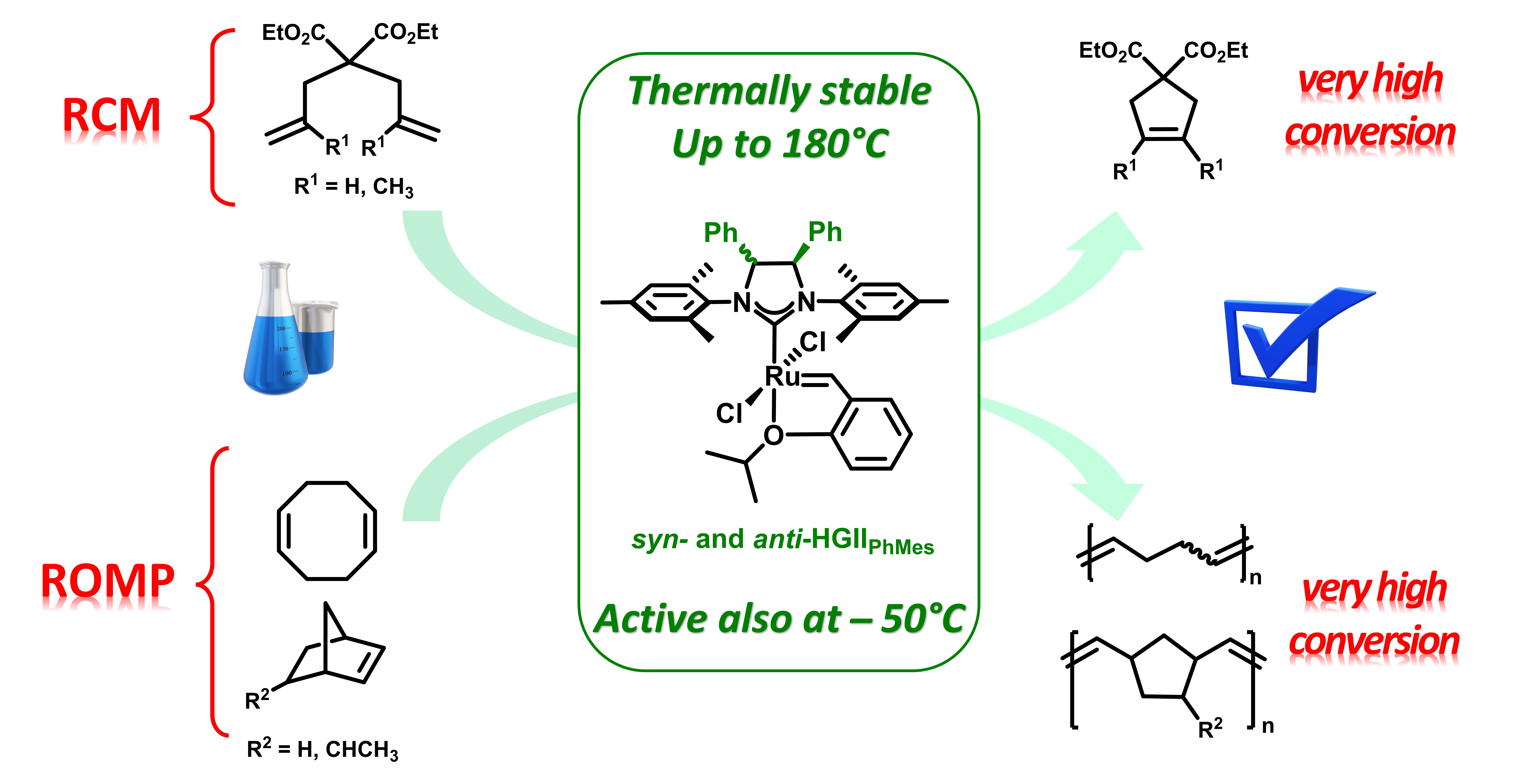
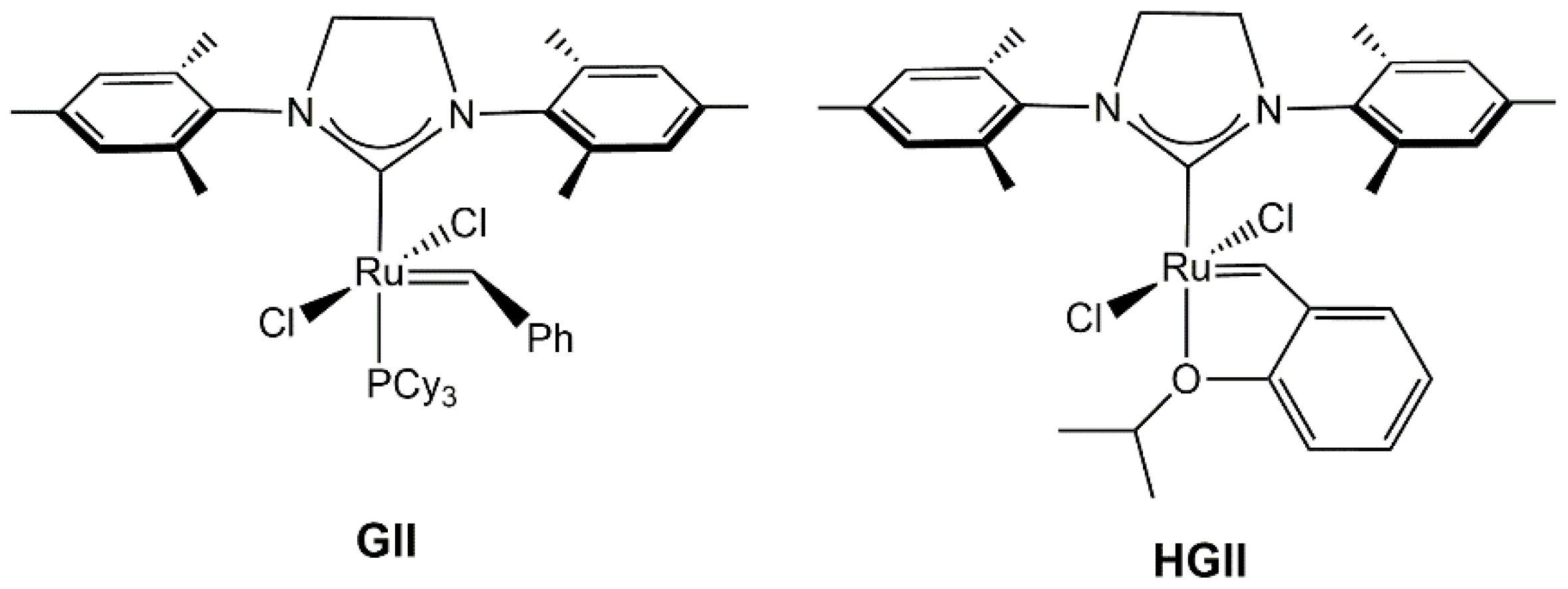
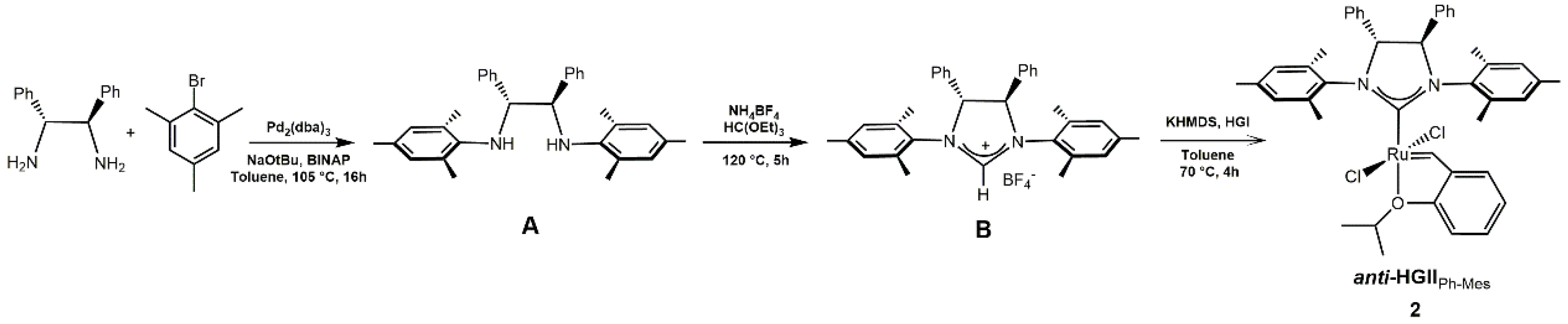
2.1. Synthesis and Characterization of Anti-HGIIPh-Mes Complex
2.2. Metathesis Reactions: Activity Studies
2.2.1. Ring Closing Metathesis Activity Studies

{kind=link}
{kind=link}
{kind=link}
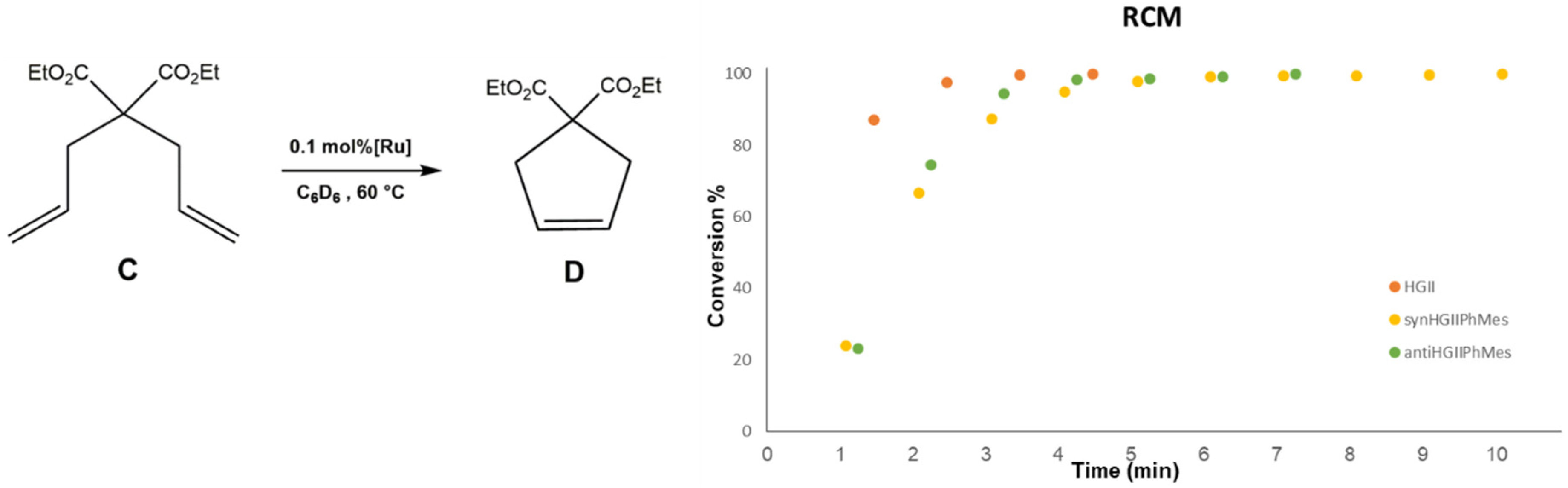{kind=link}
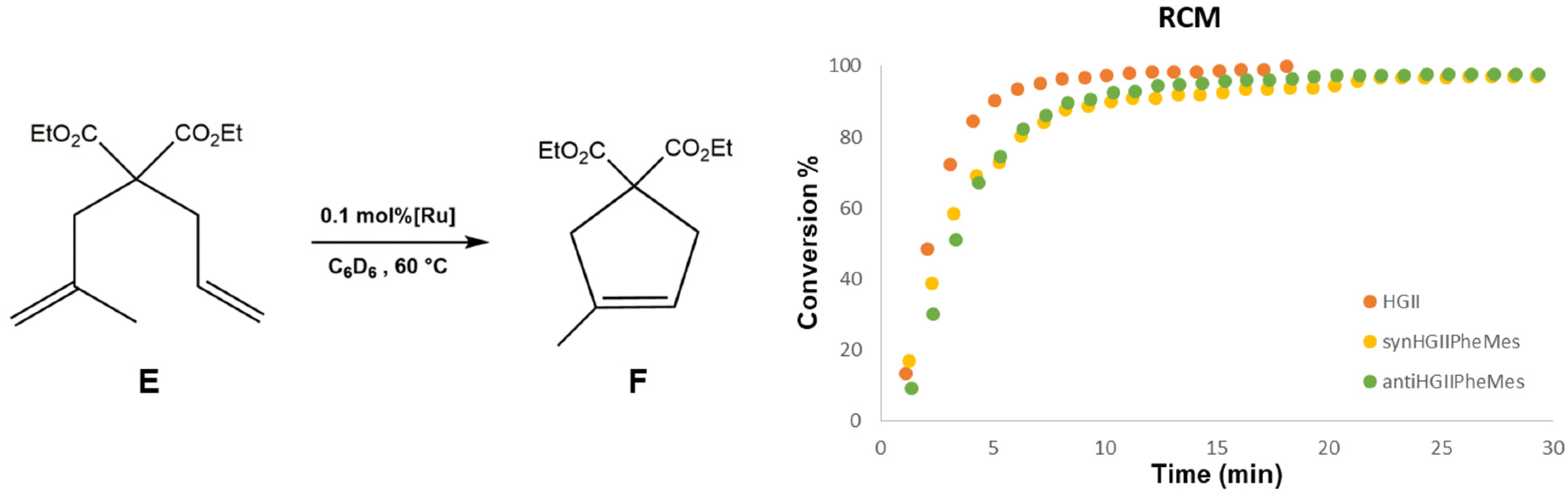{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Time (h) | Conversion % HGII | Conversion % syn-HGIIPh-Mes | Conversion % anti-HGIIPh-Mes |
| 1 | 20.0 [47] | 4.40 | 11.1 |
| 24 | 54.5 | 44.8 | 51.0 |
| 48 | 60.1 | 52.7 | 56.5 |
| 72 | 63.3 | 55.1 | 59.8 |
| 96 | 63.5 | 55.1 | 61.8 |
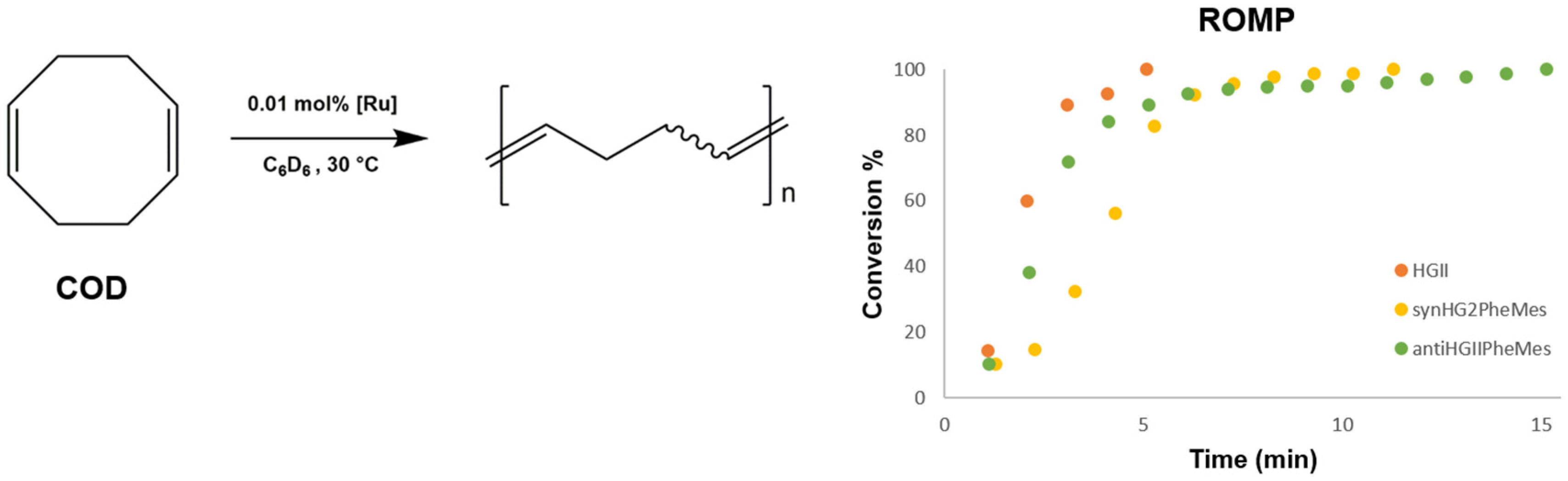
2.2.2. Ring-Opening Metathesis Polymerization Activity Studies
3. Experimental Part
3.1. Synthesis of 4R,5R-(1,3-Bis-mesityl)-(4,5-diphenyl-imidazolin-2-ylidene)-dichloro-(2-isopropoxybenzylidene)-ruthenium(II) (Anti-HGIIPh-Mes)
3.2. Ring Closing Metathesis
3.2.1. RCM of Diethyl-diallylmalonate (C)
3.2.2. RCM of Diethyl-allylmethallylmalonate (E)
3.2.3. RCM of Diethyl-dimethallylmalonate (G)
3.3. Ring Opening Metathesis Polymerization
3.3.1. ROMP of 1,5-Cyclooctadiene (COD)
3.3.2. ROMP of 2-Norbornene and 5-Ethyliden-2-Norbornene
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grela, K. Olefin Metathesis: Theory and Practice; John Wiley & Sons: Hoboken, NJ, USA, 2014; ISBN 978-1-118-71156-9. [Google Scholar]
- Grubbs, R.H. Handbook of Metathesis; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2003; ISBN 978-3-527-61948-1. [Google Scholar]
- Schrock, R.R.; Hoveyda, A.H. Molybdenum and Tungsten Imido Alkylidene Complexes as Efficient Olefin-Metathesis Catalysts. Angew. Chem. Int. Ed. 2003, 42, 4592–4633. [Google Scholar] [CrossRef] [PubMed]
- Schrock, R.R. Multiple Metal–Carbon Bonds for Catalytic Metathesis Reactions (Nobel Lecture). Angew. Chem. Int. Ed. 2006, 45, 3748–3759. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, P.H.; Blechert, S. Alkene Metathesis: The Search for Better Catalysts. Dalton Trans. 2007, 24, 2479–2491. [Google Scholar] [CrossRef]
- Hoveyda, A.H.; Zhugralin, A.R. The Remarkable Metal-Catalysed Olefin Metathesis Reaction. Nature 2007, 450, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Higman, C.S.; Lummiss, J.A.M.; Fogg, D.E. Olefin Metathesis at the Dawn of Implementation in Pharmaceutical and Specialty-Chemicals Manufacturing. Angew. Chem. Int. Ed. 2016, 55, 3552–3565. [Google Scholar] [CrossRef]
- Hughes, D.L. Highlights of the Recent U.S. Patent Literature: Focus on Metathesis. Org. Process Res. Dev. 2016, 20, 1008–1015. [Google Scholar] [CrossRef]
- Vignon, P.; Vancompernolle, T.; Couturier, J.-L.; Dubois, J.-L.; Mortreux, A.; Gauvin, R.M. Cross-Metathesis of Biosourced Fatty Acid Derivatives: A Step Further Toward Improved Reactivity. ChemSusChem 2015, 8, 1143–1146. [Google Scholar] [CrossRef]
- Flick, A.C.; Leverett, C.A.; Ding, H.X.; McInturff, E.; Fink, S.J.; Helal, C.J.; O’Donnell, C.J. Synthetic Approaches to the New Drugs Approved During 2017. J. Med. Chem. 2019, 62, 7340–7382. [Google Scholar] [CrossRef]
- Hughes, D.; Wheeler, P.; Ene, D. Olefin Metathesis in Drug Discovery and Development—Examples from Recent Patent Literature. Org. Process Res. Dev. 2017, 21, 1938–1962. [Google Scholar] [CrossRef]
- Yu, M.; Lou, S.; Gonzalez-Bobes, F. Ring-Closing Metathesis in Pharmaceutical Development: Fundamentals, Applications, and Future Directions. Org. Process Res. Dev. 2018, 22, 918–946. [Google Scholar] [CrossRef]
- Davey, P.N.; Lovchik, M.A.; Goeke, A.; Lorincz, K.; Toth, F.; Ondi, L. Process Involving Cross Metathesis of Olefins. U.S. Patent No. 10,144,720, 2018. [Google Scholar]
- Mathys, M.; Kraft, P. Synthesis by Ring-Closing Alkyne Metathesis with Selective Hydrogenation, and Olfactory Comparison of (7E)- and (7Z)-Cyclohexadec-7-Enone (Aurelione®). Chem. Biodiver. 2014, 11, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Kraft, P.; Berthold, C. (4E,8Z)-12-Methyloxacyclotetradeca-4,8-Dien-2-One and Its 7a-Homologue: Conformationally Constrained Double-Unsaturated Macrocyclic Musks by Ring-Closing Alkyne Metathesis. Synthesis 2008, 2008, 543–550. [Google Scholar] [CrossRef]
- Schaetzer, J.; Edmunds, A.J.F.; Gaus, K.; Rendine, S.; De Mesmaeker, A.; Rueegg, W. Efficient Synthesis of Fused Bicyclic Ethers and Their Application in Herbicide Chemistry. Bioorg. Med. Chem. Lett. 2014, 24, 4643–4649. [Google Scholar] [CrossRef]
- Turczel, G.; Kovács, E.; Merza, G.; Coish, P.; Anastas, P.T.; Tuba, R. Synthesis of Semiochemicals via Olefin Metathesis. ACS Sust. Chem. Eng. 2019, 7, 33–48. [Google Scholar] [CrossRef]
- Daeuble, J.; Sparks, T.C.; Johnson, P.; Graupner, P.R. Modification of the Butenyl-Spinosyns Utilizing Cross-Metathesis. Bioorg. Med. Chem. 2009, 17, 4197–4205. [Google Scholar] [CrossRef]
- Czarnocki, S.J.; Czeluśniak, I.; Olszewski, T.K.; Malinska, M.; Woźniak, K.; Grela, K. Rational and Then Serendipitous Formation of Aza Analogues of Hoveyda-Type Catalysts Containing a Chelating Ester Group Leading to a Polymerization Catalyst Family. ACS Catal. 2017, 7, 4115–4121. [Google Scholar] [CrossRef]
- Leimgruber, S.; Trimmel, G. Olefin Metathesis Meets Rubber Chemistry and Technology. Monatsh. Chem. 2015, 146, 1081–1097. [Google Scholar] [CrossRef]
- Martinez, H.; Ren, N.; Matta, M.E.; Hillmyer, M.A. Ring-Opening Metathesis Polymerization of 8-Membered Cyclic Olefins. Polym. Chem. 2014, 5, 3507–3532. [Google Scholar] [CrossRef]
- Stadler, B.M.; Wulf, C.; Werner, T.; Tin, S.; de Vries, J.G. Catalytic Approaches to Monomers for Polymers Based on Renewables. ACS Catal. 2019, 9, 8012–8067. [Google Scholar] [CrossRef]
- Guadagno, L.; Longo, P.; Raimondo, M.; Naddeo, C.; Mariconda, A.; Sorrentino, A.; Vittoria, V.; Iannuzzo, G.; Russo, S. Cure behavior and mechanical properties of structural self-healing epoxy resins. J. Polym. Sci. Part B Polym. Phys. 2010, 48, 2413–2423. [Google Scholar] [CrossRef]
- Guadagno, L.; Raimondo, M.; Naddeo, C.; Longo, P.; Mariconda, A. Self-Healing Materials for Structural Applications. Polym. Eng. Sci. 2014, 54, 777–784. [Google Scholar] [CrossRef]
- Guadagno, L.; Raimondo, M.; Naddeo, C.; Longo, P.; Mariconda, A.; Binder, W.H. Healing Efficiency and Dynamic Mechanical Properties of Self-Healing Epoxy Systems. Smart Mater. Struct. 2014, 23, 045001. [Google Scholar] [CrossRef]
- Raimondo, M.; Longo, P.; Mariconda, A.; Guadagno, L. Healing Agent for the Activation of Self-Healing Function at Low Temperature. Adv. Comp. Mater. 2015, 24, 519–529. [Google Scholar] [CrossRef]
- Jean-Louis Hérisson, P.; Chauvin, Y. Catalyse de transformation des oléfines par les complexes du tungstène. II. Télomérisation des oléfines cycliques en présence d’oléfines acycliques. Makromol. Chem. 1971, 141, 161–176. [Google Scholar] [CrossRef]
- Vougioukalakis, G.C.; Grubbs, R.H. Ruthenium-Based Heterocyclic Carbene-Coordinated Olefin Metathesis Catalysts. Chem. Rev. 2010, 110, 1746–1787. [Google Scholar] [CrossRef]
- Samojłowicz, C.; Bieniek, M.; Grela, K. Ruthenium-Based Olefin Metathesis Catalysts Bearing N-Heterocyclic Carbene Ligands. Chem. Rev. 2009, 109, 3708–3742. [Google Scholar] [CrossRef]
- Ogba, O.M.; Warner, N.C.; O’Leary, D.J.; Grubbs, R.H. Recent Advances in Ruthenium-Based Olefin Metathesis. Chem. Soc. Rev. 2018, 47, 4510–4544. [Google Scholar] [CrossRef]
- Paradiso, V.; Costabile, C.; Grisi, F. NHC Backbone Configuration in Ruthenium-Catalyzed Olefin Metathesis. Molecules 2016, 21, 117. [Google Scholar] [CrossRef]
- Stewart, I.C.; Ung, T.; Pletnev, A.A.; Berlin, J.M.; Grubbs, R.H.; Schrodi, Y. Highly Efficient Ruthenium Catalysts for the Formation of Tetrasubstituted Olefins via Ring-Closing Metathesis. Org. Lett. 2007, 9, 1589–1592. [Google Scholar] [CrossRef]
- Berlin, J.M.; Campbell, K.; Ritter, T.; Funk, T.W.; Chlenov, A.; Grubbs, R.H. Ruthenium-Catalyzed Ring-Closing Metathesis to Form Tetrasubstituted Olefins. Org. Lett. 2007, 9, 1339–1342. [Google Scholar] [CrossRef]
- Troiano, R.; Costabile, C.; Grisi, F. Alternating Ring-Opening Metathesis Polymerization Promoted by Ruthenium Catalysts Bearing Unsymmetrical NHC Ligands. Catalysts 2023, 13, 34. [Google Scholar] [CrossRef]
- Monsigny, L.; Kajetanowicz, A.; Grela, K. Ruthenium Complexes Featuring Unsymmetrical N-Heterocyclic Carbene Ligands–Useful Olefin Metathesis Catalysts for Special Tasks. Chemical Rec. 2021, 21, 3648–3661. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.K.; Grubbs, R.H. Olefin Metathesis Catalyst: Stabilization Effect of Backbone Substitutions of N-Heterocyclic Carbene. Org. Lett. 2008, 10, 2693–2696. [Google Scholar] [CrossRef] [PubMed]
- Grisi, F.; Mariconda, A.; Costabile, C.; Bertolasi, V.; Longo, P. Influence of Syn and Anti Configurations of NHC Backbone on Ru-Catalyzed Olefin Metathesis. Organometallics 2009, 28, 4988–4995. [Google Scholar] [CrossRef]
- Costabile, C.; Mariconda, A.; Cavallo, L.; Longo, P.; Bertolasi, V.; Ragone, F.; Grisi, F. The Pivotal Role of Symmetry in the Ruthenium-Catalyzed Ring-Closing Metathesis of Olefins. Chem. Eur. J. 2011, 17, 8618–8629. [Google Scholar] [CrossRef]
- Perfetto, A.; Costabile, C.; Longo, P.; Bertolasi, V.; Grisi, F. Probing the Relevance of NHC Ligand Conformations in the Ru-Catalysed Ring-Closing Metathesis Reaction. Chem. Eur. J. 2013, 19, 10492–10496. [Google Scholar] [CrossRef]
- Perfetto, A.; Costabile, C.; Longo, P.; Grisi, F. Ruthenium Olefin Metathesis Catalysts with Frozen NHC Ligand Conformations. Organometallics 2014, 33, 2747–2759. [Google Scholar] [CrossRef]
- Borguet, Y.; Zaragoza, G.; Demonceau, A.; Delaude, L. Ruthenium Catalysts Bearing a Benzimidazolylidene Ligand for the Metathetical Ring-Closure of Tetrasubstituted Cycloolefins. Dalton Trans. 2015, 44, 9744–9755. [Google Scholar] [CrossRef]
- Longo, P.; Mariconda, A.; Calabrese, E.; Raimondo, M.; Naddeo, C.; Vertuccio, L.; Russo, S.; Iannuzzo, G.; Guadagno, L. Development of a New Stable Ruthenium Initiator Suitably Designed for Self-Repairing Applications in High Reactive Environments. J. Ind. Eng. Chem. 2017, 54, 234–251. [Google Scholar] [CrossRef]
- Guadagno, L.; Longo, P.; Mariconda, A.; Calabrese, E.; Raimondo, M.; Naddeo, C.; Vertuccio, L.; Russo, S.; Iannuzzo, G. Grubbs-Hoveyda Type Catalyst for Metathesis Reactions in Highly Reactive Environments. Eur. Patent No. 16188680A, 2016. [Google Scholar]
- Calabrese, E.; Longo, P.; Naddeo, C.; Mariconda, A.; Vertuccio, L.; Raimondo, M.; Guadagno, L. Design of Self-Healing Catalysts for Aircraft Application. Int. J. Struct. Integr. 2018, 9, 723–736. [Google Scholar] [CrossRef]
- Ritter, T.; Hejl, A.; Wenzel, A.G.; Funk, T.W.; Grubbs, R.H. A Standard System of Characterization for Olefin Metathesis Catalysts. Organometallics 2006, 25, 5740–5745. [Google Scholar] [CrossRef]
- Perfetto, A.; Bertolasi, V.; Costabile, C.; Paradiso, V.; Caruso, T.; Longo, P.; Grisi, F. Methyl and Phenyl Substituent Effects on the Catalytic Behavior of NHC Ruthenium Complexes. RSC Adv. 2016, 6, 95793–95804. [Google Scholar] [CrossRef]
- Ambrosio, C.; Paradiso, V.; Costabile, C.; Bertolasi, V.; Caruso, T.; Grisi, F. Stable Ruthenium Olefin Metathesis Catalysts Bearing Symmetrical NHC Ligands with Primary and Secondary N-Alkyl Groups. Dalton Trans. 2018, 47, 6615–6627. [Google Scholar] [CrossRef] [PubMed]
- Paradiso, V.; Bertolasi, V.; Costabile, C.; Caruso, T.; Dąbrowski, M.; Grela, K.; Grisi, F. Expanding the Family of Hoveyda–Grubbs Catalysts Containing Unsymmetrical NHC Ligands. Organometallics 2017, 36, 3692–3708. [Google Scholar] [CrossRef]
- Seiders, T.J.; Ward, D.W.; Grubbs, R.H. Enantioselective Ruthenium-Catalyzed Ring-Closing Metathesis. Org. Lett. 2001, 3, 3225–3228. [Google Scholar] [CrossRef]






 | ||||
|---|---|---|---|---|
| a Run | Catalyst | Monomer | Amount of Polymer (g) | Conversion % |
| 1 | HGII | 2-norbornene | 0.991 | 95.3 |
| 2 | syn-HGIIPh-Mes | 2-norbornene | 0.908 | 87.4 |
| 3 | anti-HGIIPh-Mes | 2-norbornene | 0.997 | 95.9 |
| 4 | HGII | ENB | 1.21 | 91.7 |
| 5 | syn-HGIIPh-Mes | ENB | 1.08 | 81.2 |
| 6 | anti-HGIIPh-Mes | ENB | 0.670 | 50.4 |
| b 7 | HGII | ENB | 0.736 | 55.4 |
| b 8 | syn-HIIPh-Mes | ENB | 1.01 | 76.0 |
| b 9 | anti-HGIIPh-Mes | ENB | 0.585 | 44.0 |
| c 10 | HGII | ENB | 1.31 | 98.7 |
| c 11 | syn-HGIIPh-Mes | ENB | 1.08 | 81.7 |
| c 12 | anti-HGIIPh-Mes | ENB | 1.32 | >99 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Amato, A.; Mariconda, A.; Longo, P. New Insights into the Catalytic Activity of Second Generation Hoveyda–Grubbs Complexes Having Phenyl Substituents on the Backbone. Inorganics 2023, 11, 244. https://doi.org/10.3390/inorganics11060244
D’Amato A, Mariconda A, Longo P. New Insights into the Catalytic Activity of Second Generation Hoveyda–Grubbs Complexes Having Phenyl Substituents on the Backbone. Inorganics. 2023; 11(6):244. https://doi.org/10.3390/inorganics11060244
Chicago/Turabian StyleD’Amato, Assunta, Annaluisa Mariconda, and Pasquale Longo. 2023. "New Insights into the Catalytic Activity of Second Generation Hoveyda–Grubbs Complexes Having Phenyl Substituents on the Backbone" Inorganics 11, no. 6: 244. https://doi.org/10.3390/inorganics11060244
APA StyleD’Amato, A., Mariconda, A., & Longo, P. (2023). New Insights into the Catalytic Activity of Second Generation Hoveyda–Grubbs Complexes Having Phenyl Substituents on the Backbone. Inorganics, 11(6), 244. https://doi.org/10.3390/inorganics11060244