
Valence Tautomerism in Chromium Half-Sandwich Triarylmethylium Dyads †



Abstract
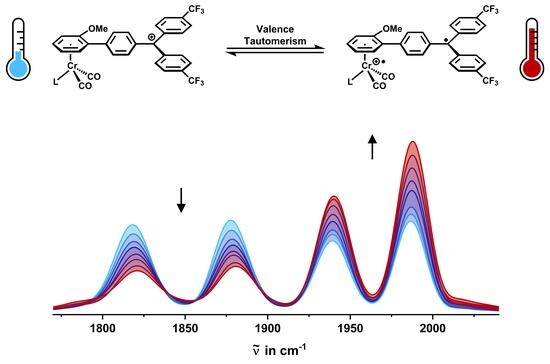
:
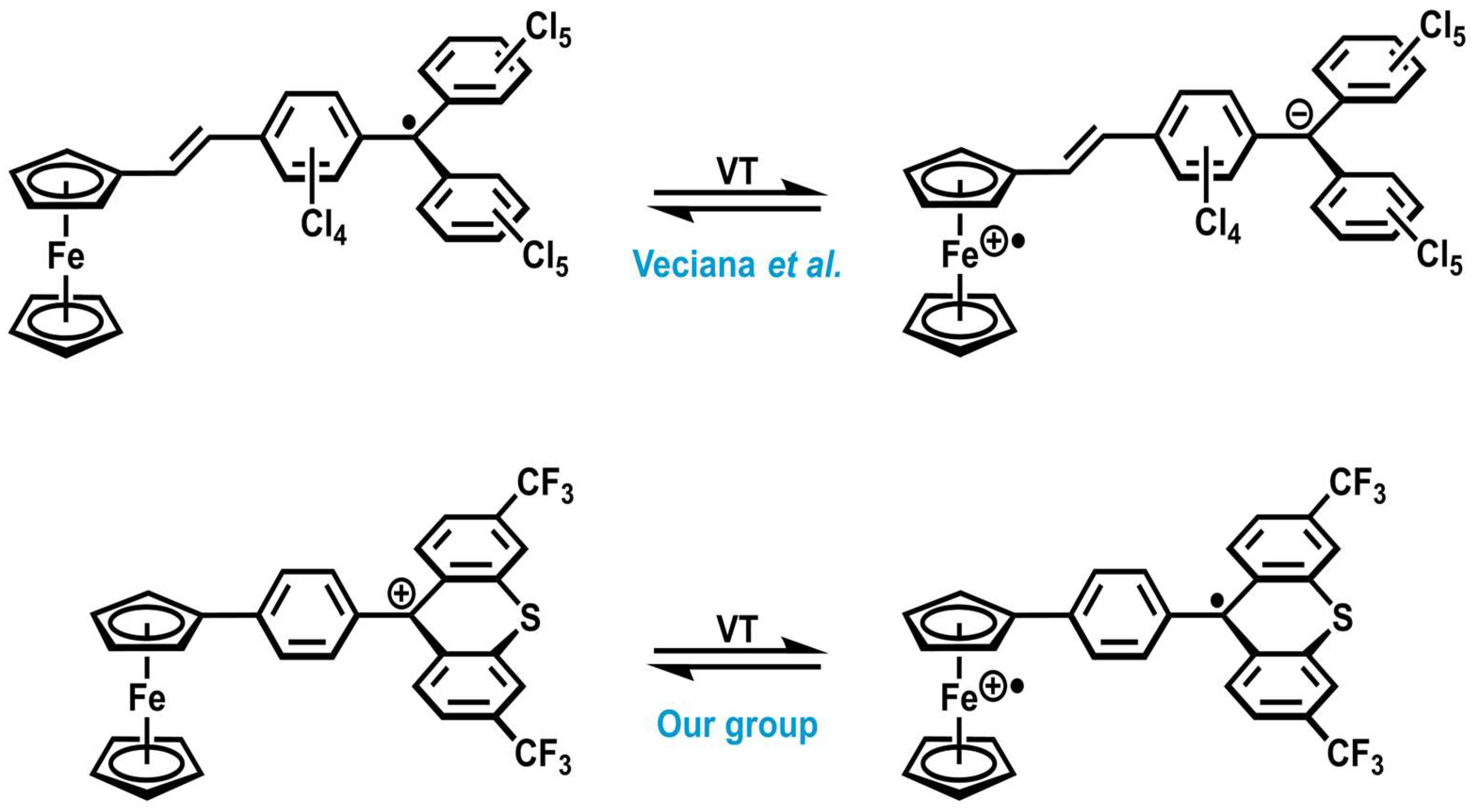
1. Introduction
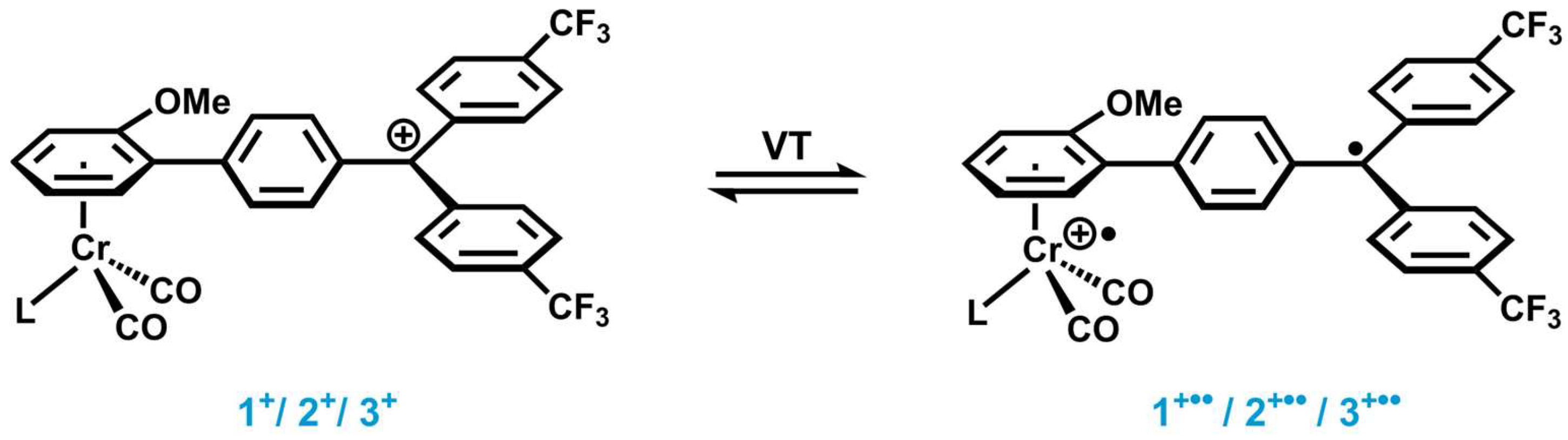
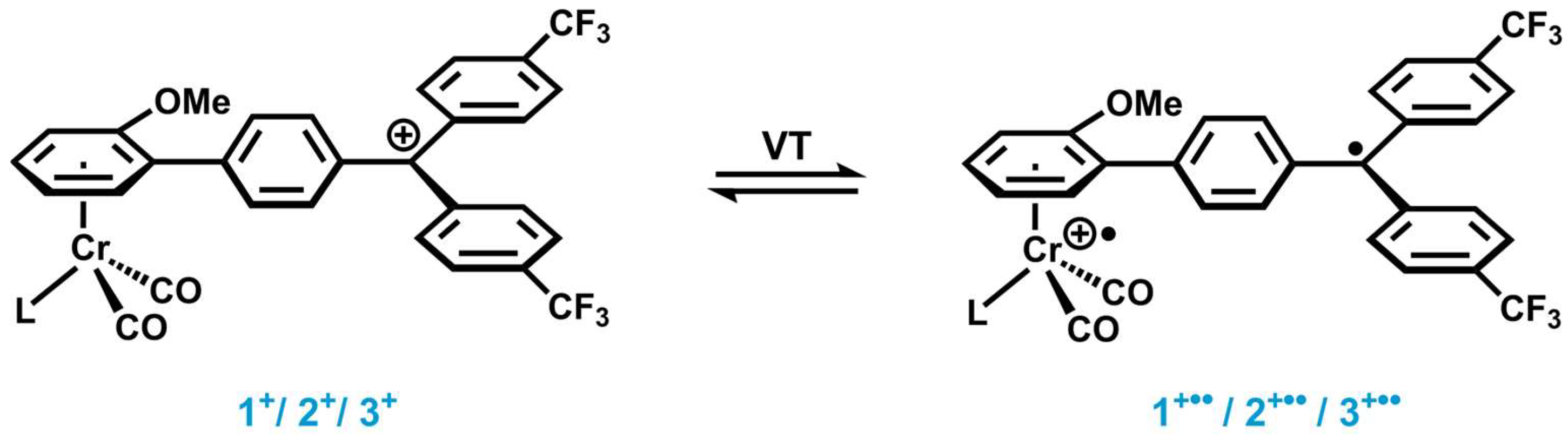
2. Results and Discussion
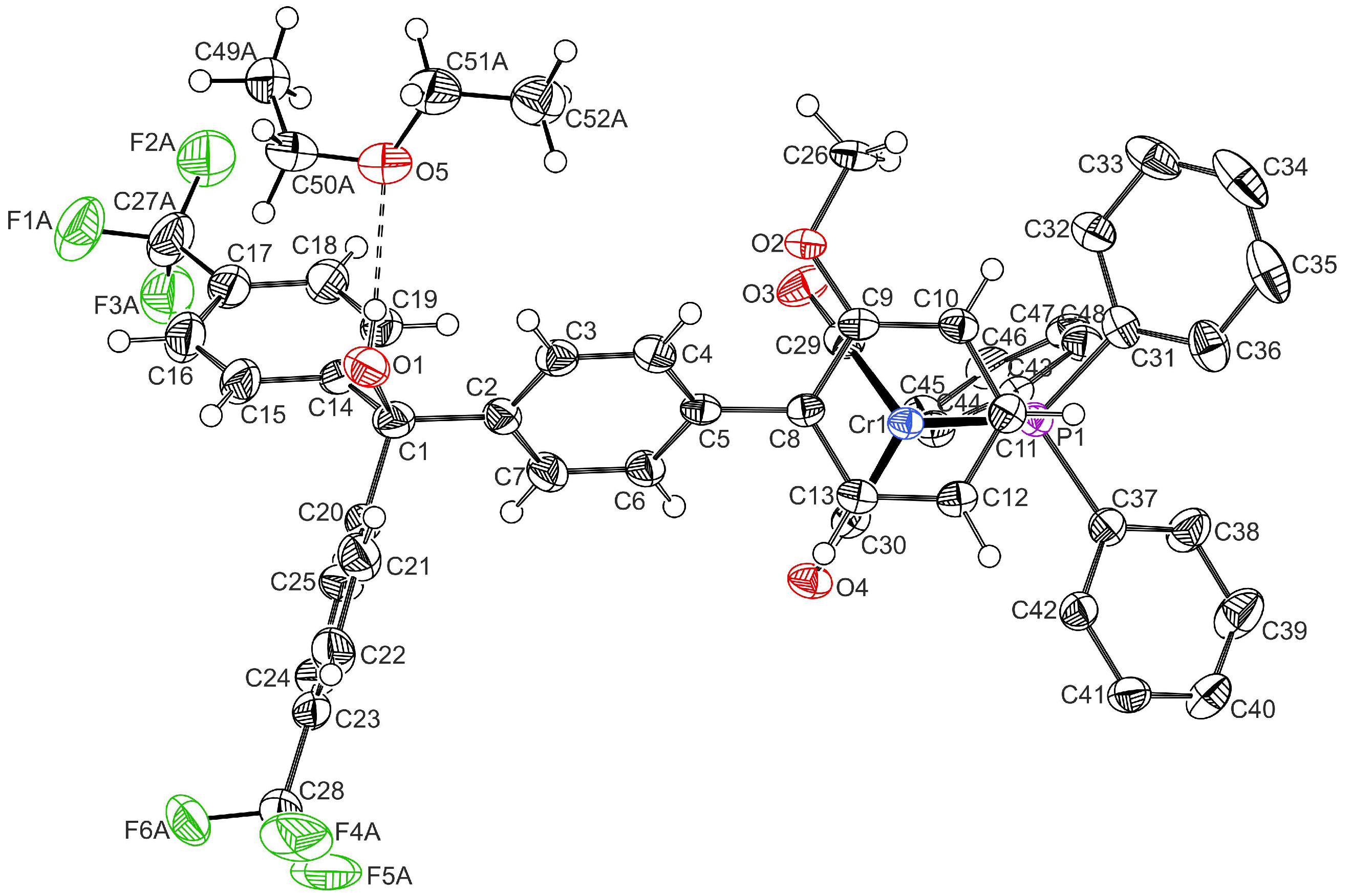
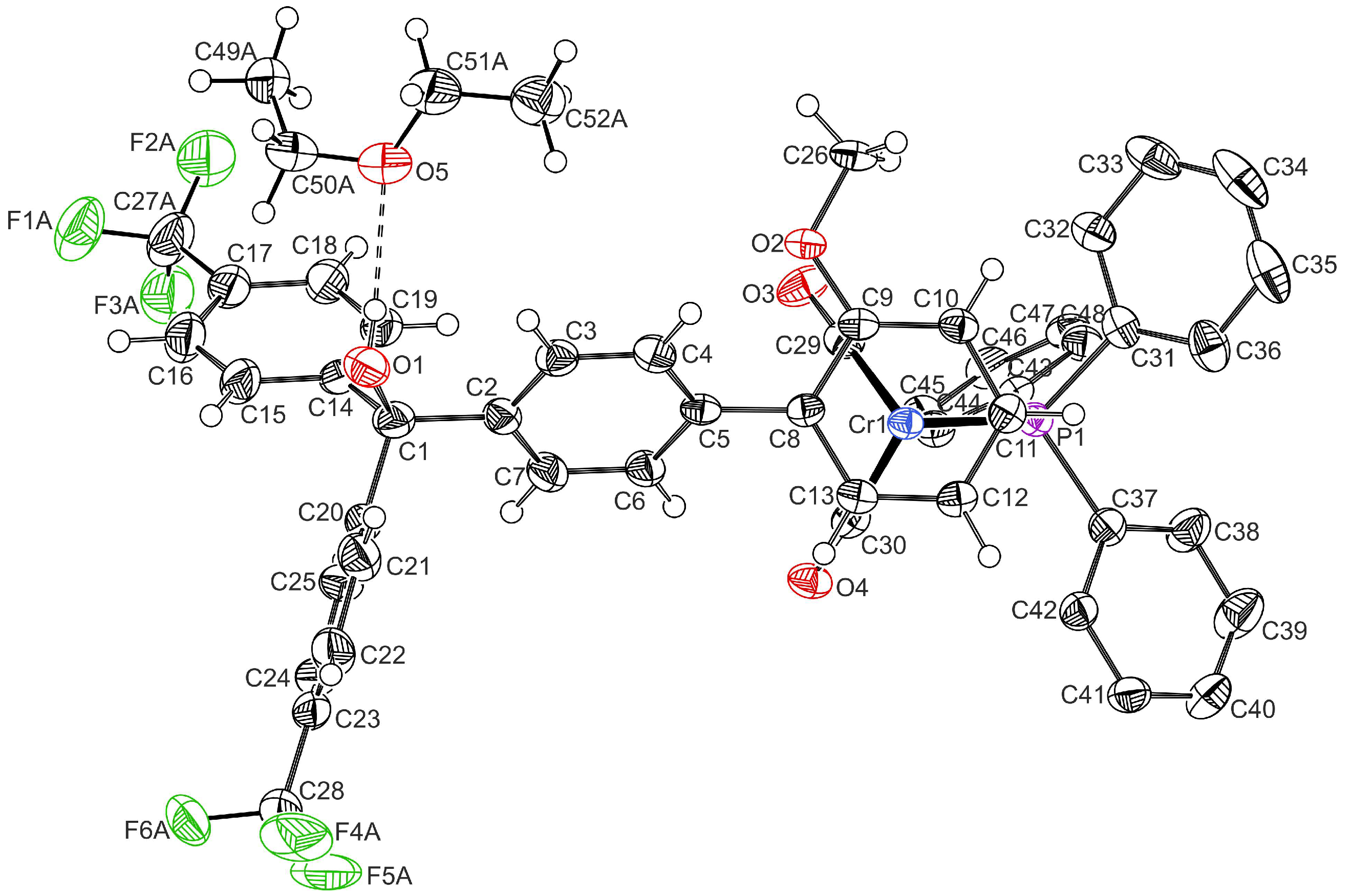
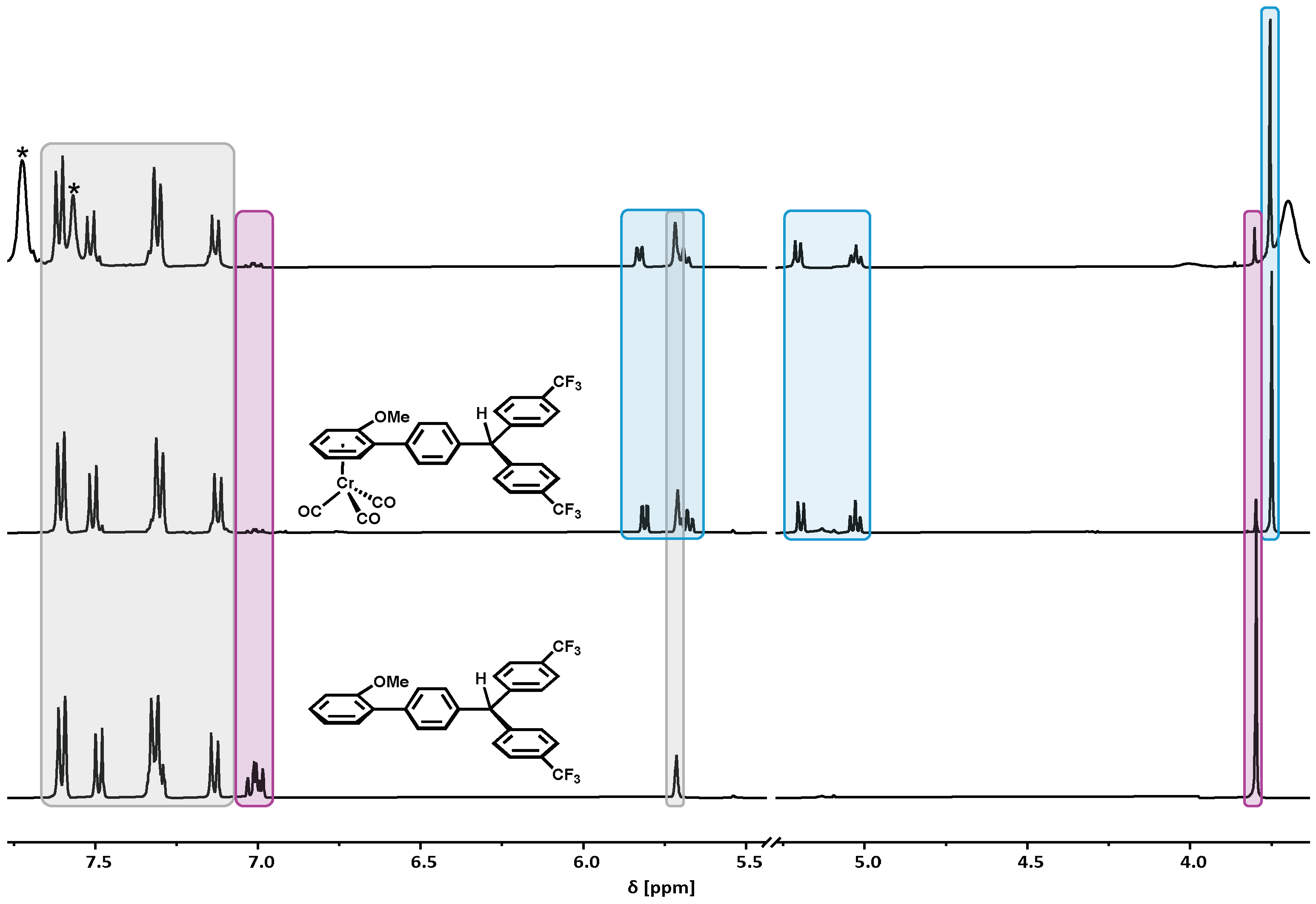
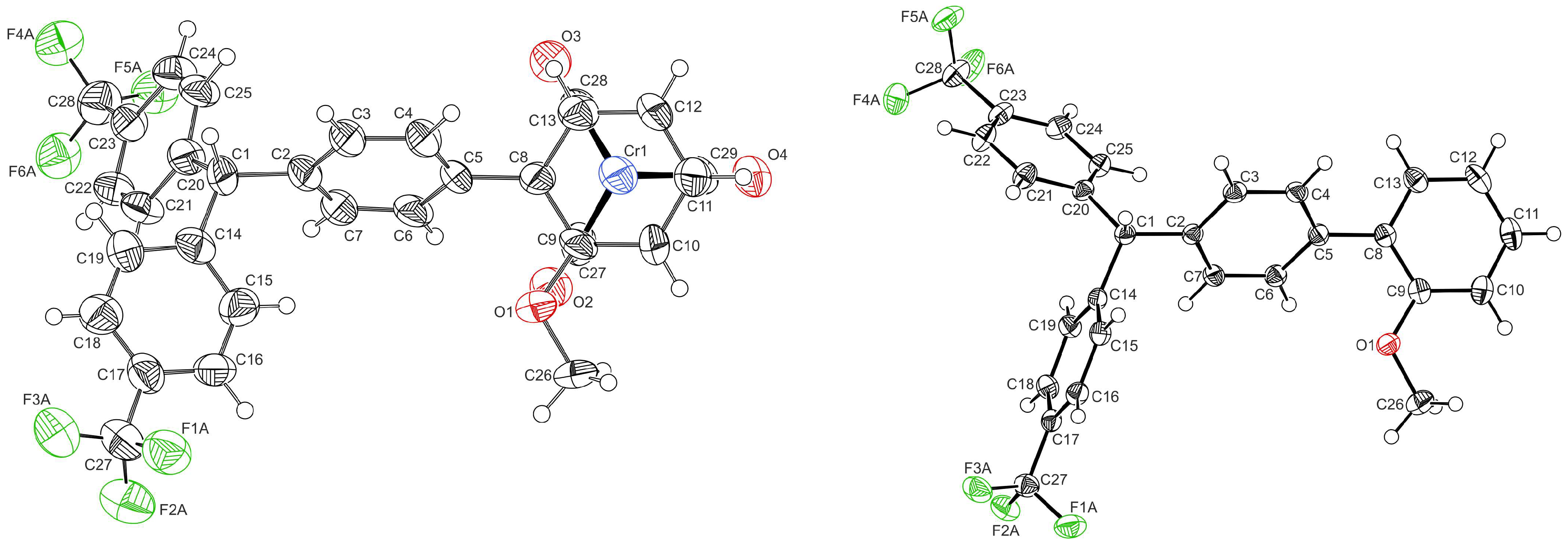
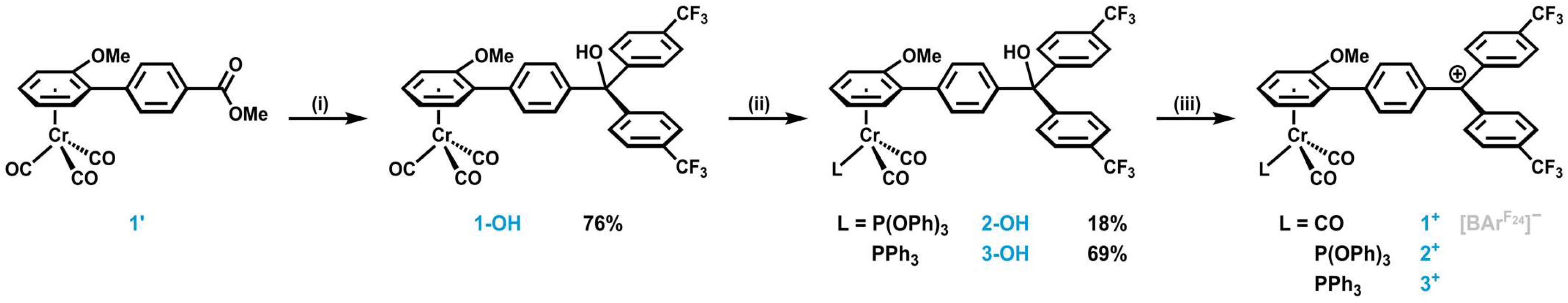
2.1. Synthesis and Characterization
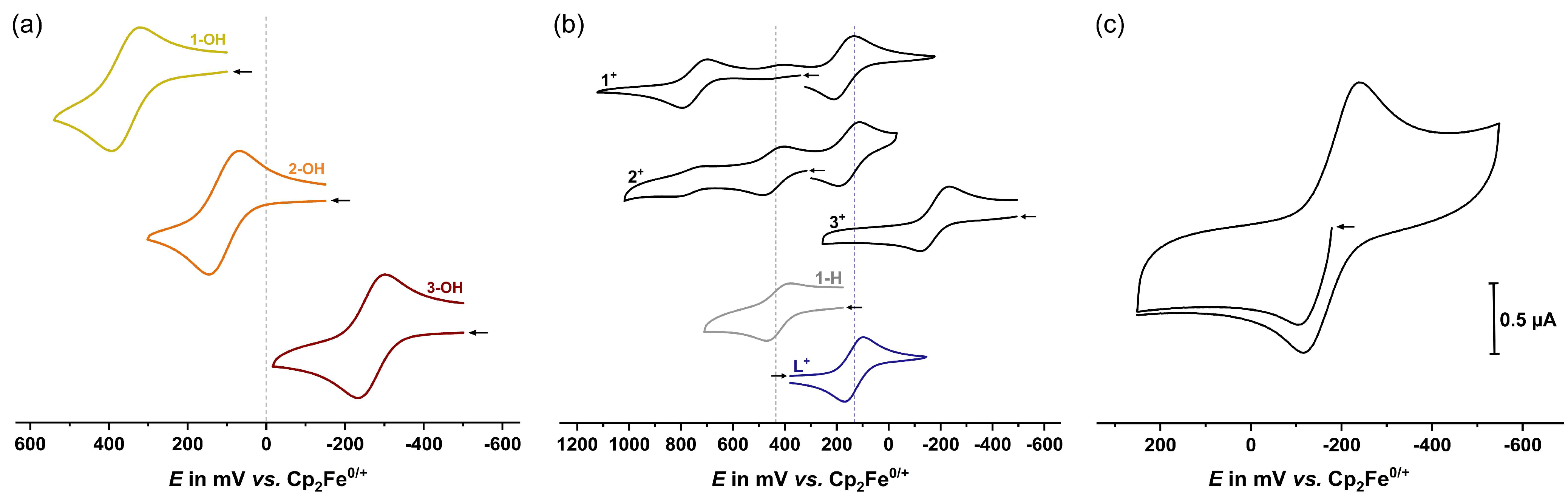
2.2. Electrochemistry
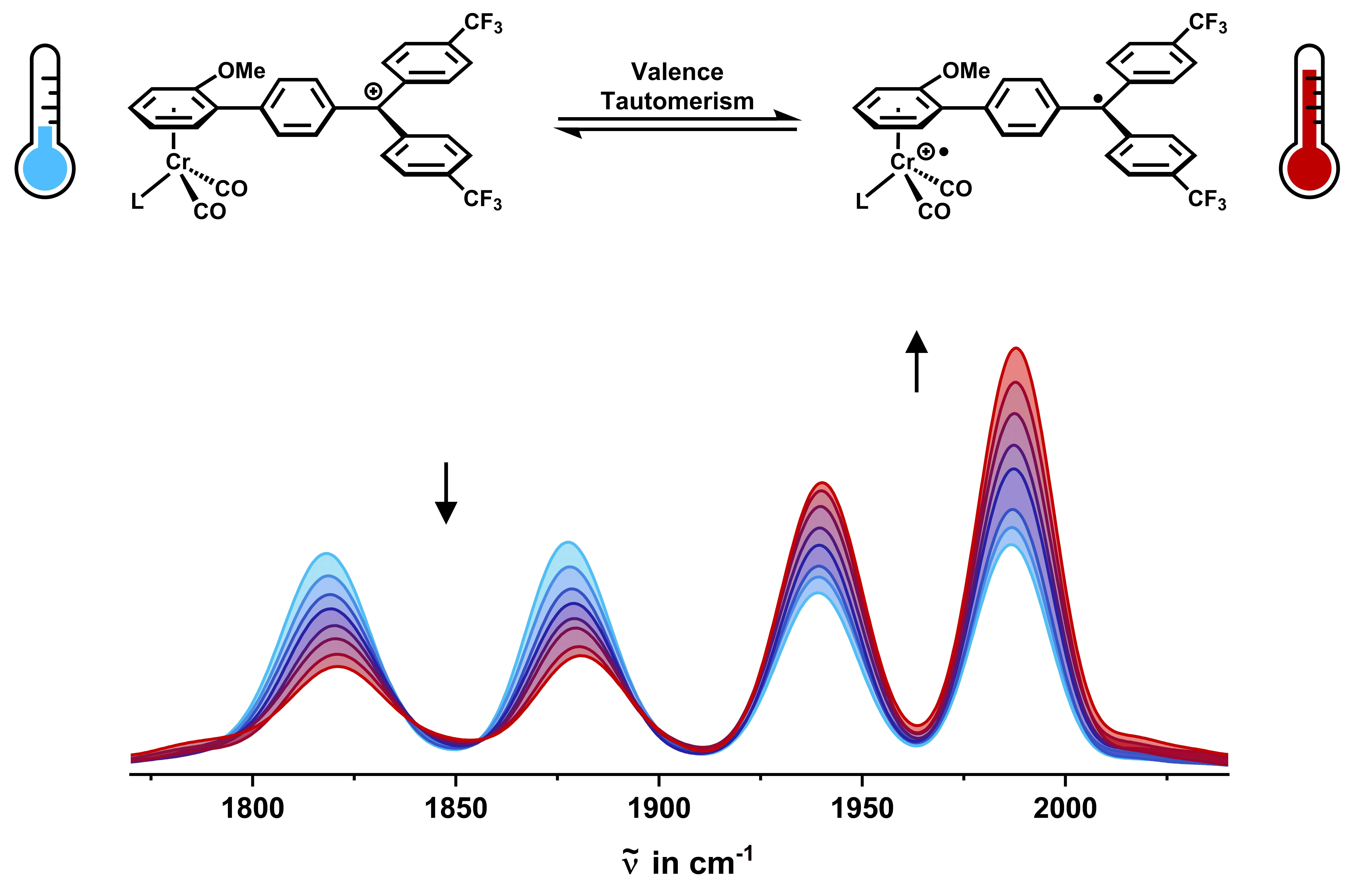
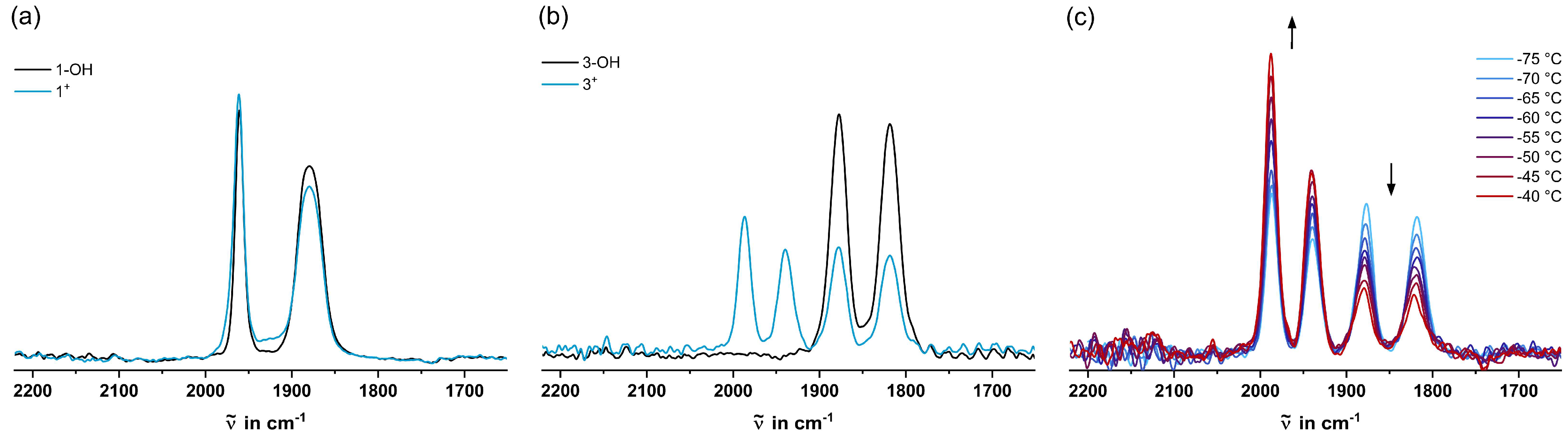
2.3. IR Spectroscopy
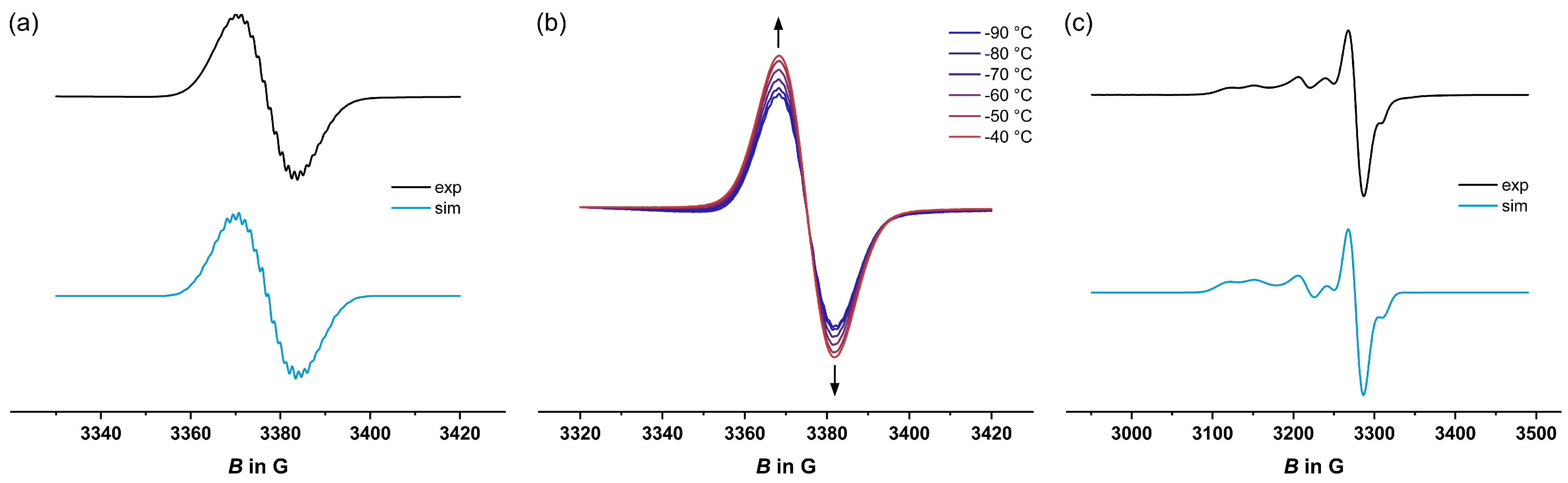
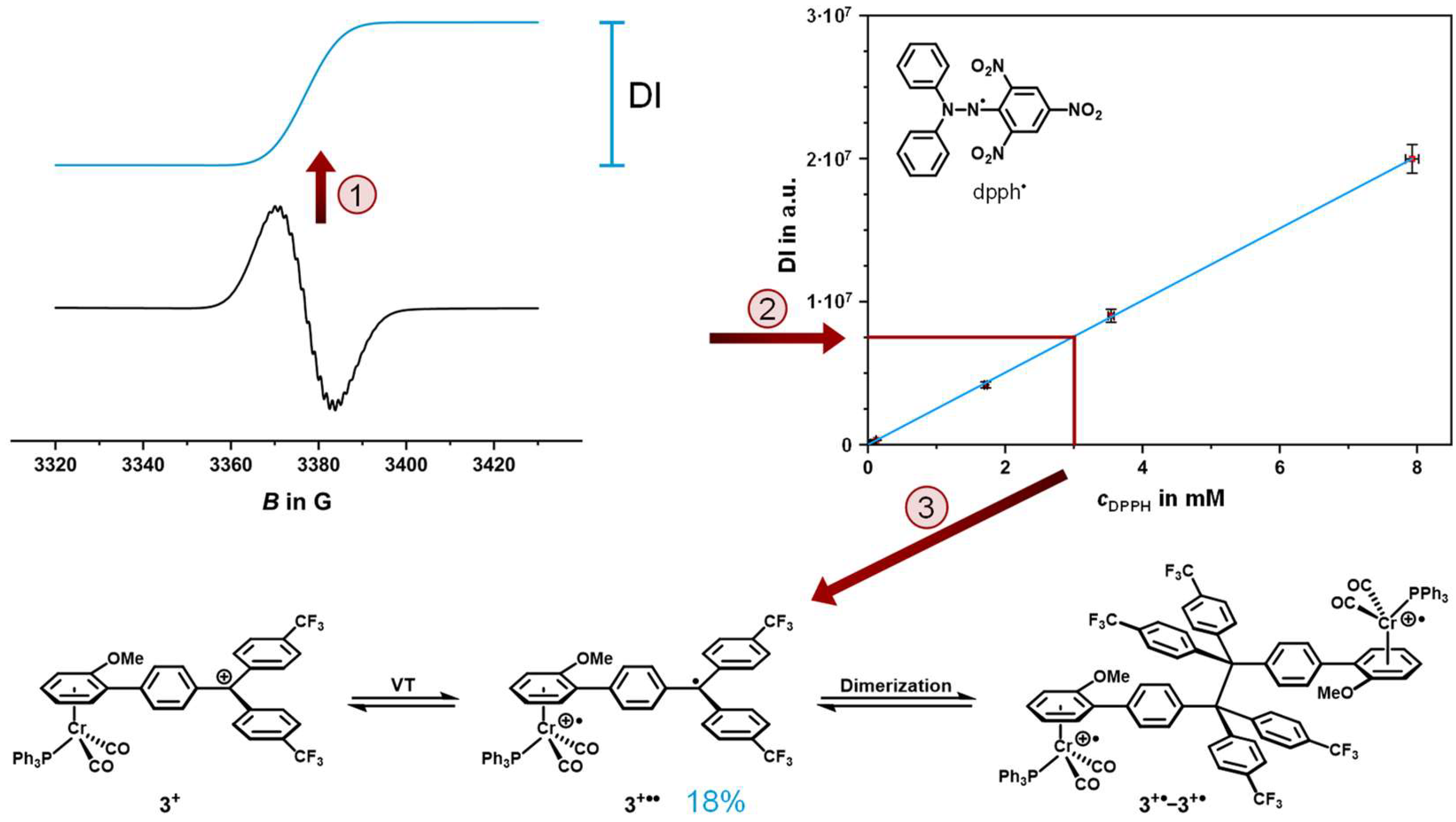
2.4. EPR Spectroscopy
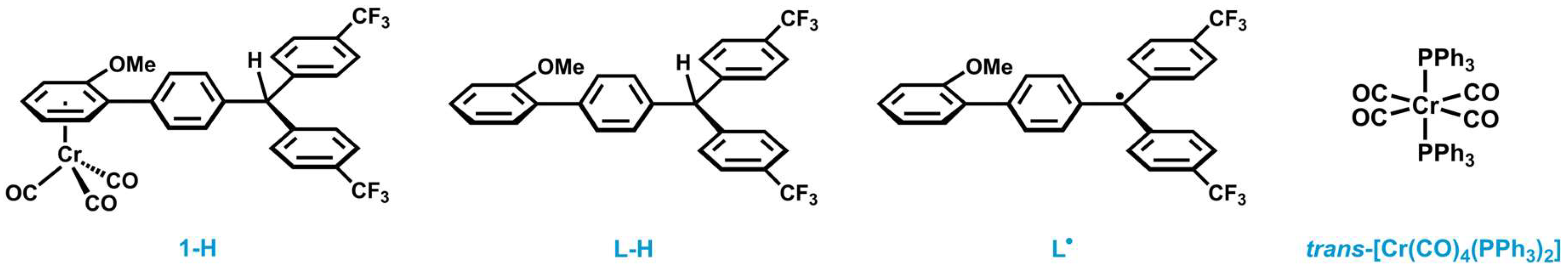
2.5. Decomposition Pathways
3. Materials and Methods
4. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pierpont, C.G. Studies on charge distribution and valence tautomerism in transition metal complexes of catecholate and semiquinonate ligands. Coord. Chem. Rev. 2001, 216, 99–125. [Google Scholar] [CrossRef]
- Adams, D.M.; Li, B.; Simon, J.D.; Hendrickson, D.N. Photoinduced Valence Tautomerism in Cobalt Complexes Containing Semiquinone Anion as Ligand: Dynamics of the High-Spin [CoII(3,5-dtbsq)2] to Low-Spin [CoIII(3,5-dtbsq)(3,5-dtbcat)] Interconversion. Angew. Chem. Int. Ed. 1995, 34, 1481–1483. [Google Scholar] [CrossRef]
- Adams, D.M.; Hendrickson, D.N. Pulsed Laser Photolysis and Thermodynamics Studies of Intramolecular Electron Transfer in Valence Tautomeric Cobalt o-Quinone Complexes. J. Am. Chem. Soc. 1996, 118, 11515–11528. [Google Scholar] [CrossRef]
- Sato, O.; Cui, A.; Matsuda, R.; Tao, J.; Hayami, S. Photo-induced Valence Tautomerism in Co Complexes. Acc. Chem. Res. 2007, 40, 361–369. [Google Scholar] [CrossRef]
- Tao, J.; Maruyama, H.; Sato, O. Valence Tautomeric Transitions with Thermal Hysteresis around Room Temperature and Photoinduced Effects Observed in a Cobalt-Tetraoxolene Complex. J. Am. Chem. Soc. 2006, 128, 1790–1791. [Google Scholar] [CrossRef]
- Francisco, T.M.; Gee, W.J.; Shepherd, H.J.; Warren, M.R.; Shultz, D.A.; Raithby, P.R.; Pinheiro, C.B. Hard X-ray-Induced Valence Tautomeric Interconversion in Cobalt-o-Dioxolene Complexes. J. Phys. Chem. Lett. 2017, 8, 4774–4778. [Google Scholar] [CrossRef]
- Poneti, G.; Mannini, M.; Sorace, L.; Sainctavit, P.; Arrio, M.-A.; Otero, E.; Criginski Cezar, J.; Dei, A. Soft-X-ray-Induced Redox Isomerism in a Cobalt Dioxolene Complex. Angew. Chem. Int. Ed. 2010, 49, 1954–1957. [Google Scholar] [CrossRef]
- Buchanan, R.M.; Pierpont, C.G. Tautomeric Catecholate-Semiquinone Interconversion via Metal-Ligand Electron Transfer. Structural, Spectral, and Magnetic Properties of (3,5-Di-tert-butylcatecholato)-(3,5-di-tert-butylsemiquinone)(bipyridyl)cobalt(III), a Complex Containing Mixed-Valence Organic Ligands. J. Am. Chem. Soc. 1980, 102, 4951–4957. [Google Scholar] [CrossRef]
- Caneschi, A.; Dei, A.; Fabrizi de Biani, F.; Gütlich, P.; Ksenofontov, V.; Levchenko, G.; Hoefer, A.; Renz, F. Pressure- and Temperature-Induced Valence Tautomeric Interconversion in a o-Dioxolene Adduct of a Cobalt-Tetraazamacrocycle Complex. Chem. Eur. J. 2001, 7, 3926–3930. [Google Scholar] [CrossRef]
- Carbonera, C.; Dei, A.; Létard, J.-F.; Sangregorio, C.; Sorace, L. Thermally and Light-Induced Valence Tautomeric Transition in a Dinuclear Cobalt-Tetraoxolene Complex. Angew. Chem. Int. Ed. 2004, 43, 3136–3138. [Google Scholar] [CrossRef]
- Leroy, L.; Francisco, T.M.; Shepherd, H.J.; Warren, M.R.; Saunders, L.K.; Shultz, D.A.; Raithby, P.R.; Pinheiro, C.B. Controlled Light and Temperature Induced Valence Tautomerism in a Cobalt-o-Dioxolene Complex. Inorg. Chem. 2021, 60, 8665–8671. [Google Scholar] [CrossRef] [PubMed]
- Ratera, I.; Ruiz-Molina, D.; Renz, F.; Ensling, J.; Wurst, K.; Rovira, C.; Gütlich, P.; Veciana, J. A New Valence Tautomerism Example in an Electroactive Ferrocene Substituted Triphenylmethyl Radical. J. Am. Chem. Soc. 2003, 125, 1462–1463. [Google Scholar] [CrossRef] [PubMed]
- Evangelio, E.; Ruiz-Molina, D. Valence Tautomerism: New Challenges for Electroactive Ligands. Eur. J. Inorg. Chem. 2005, 2005, 2957–2971. [Google Scholar] [CrossRef]
- Evangelio, E.; Ruiz-Molina, D. Valence tautomerism: More actors than just electroactive ligands and metal ions. C. R. Chim. 2008, 11, 1137–1154. [Google Scholar] [CrossRef]
- Tezgerevska, T.; Alley, K.G.; Boskovic, C. Valence tautomerism in metal complexes: Stimulated and reversible intramolecular electron transfer between metal centers and organic ligands. Coord. Chem. Rev. 2014, 268, 23–40. [Google Scholar] [CrossRef]
- Casper, L.A.; Linseis, M.; Demeshko, S.; Azarkh, M.; Drescher, M.; Winter, R.F. Tailoring Valence Tautomerism by Using Redox Potentials: Studies on Ferrocene-Based Triarylmethylium Dyes with Electron-Poor Fluorenylium and Thioxanthylium Acceptors. Chem. Eur. J. 2021, 27, 10854–10868. [Google Scholar] [CrossRef]
- Arnett, E.M.; Flowers, R.A.; Ludwig, R.T.; Meekhof, A.E.; Walek, S.A. Triarylmethanes and 9-arylxanthenes as prototypes amphihydric compounds for relating the stabilities of cations, anions and radicals by C-H bond cleavage and electron transfer. J. Phys. Org. Chem. 1997, 10, 499–513. [Google Scholar] [CrossRef]
- Cheng, J.; Handoo, K.L.; Parker, V.D. Hydride Affinities of Carbenium Ions in Acetonitrile and Dimethyl Sulfoxide Solution. J. Am. Chem. Soc. 1993, 115, 2655–2660. [Google Scholar] [CrossRef]
- Breslow, R.; Mazur, S. Electrochemical Determination of pKR+ for Some Antiaromatic Cyclopentadienyl Cations. J. Am. Chem. Soc. 1973, 95, 584–585. [Google Scholar] [CrossRef]
- Strohbusch, F. Polarographische Untersuchungen der Substituenteneffekte in Triarylmethylkationen. Ber. Bunsenges. Phys. Chem. 1972, 76, 622–628. [Google Scholar] [CrossRef]
- Nau, M.; Casper, L.A.; Haug, G.; Linseis, M.; Demeshko, S.; Winter, R.F. Linker permethylation as a means to foster valence tautomerism and thwart dimerization in ferrocenyl-triarylmethylium cations. Dalton Trans. 2023, 52, 4674–4677. [Google Scholar] [CrossRef] [PubMed]
- Prins, R. Electronic structure of the ferricenium cation. Mol. Phys. 1970, 19, 603–620. [Google Scholar] [CrossRef]
- Prins, R.; Reinders, F.J. Electron Spin Resonance of the Cation of Ferrocene. J. Am. Chem. Soc. 1969, 91, 4929–4931. [Google Scholar] [CrossRef]
- Fischer, E.O.; Öfele, K. Über Aromatenkomplexe von Metallen, XIII Benzol-Chrom-Tricarbonyl. Chem. Ber. 1957, 90, 2532–2535. [Google Scholar] [CrossRef]
- Berger, A.; Djukic, J.-P.; Michon, C. Metalated (η6-arene)tricarbonylchromium complexes in organometallic chemistry. Coord. Chem. Rev. 2002, 225, 215–238. [Google Scholar] [CrossRef]
- Rosillo, M.; Domínguez, G.; Pérez-Castells, J. Chromium arene complexes in organic synthesis. Chem. Soc. Rev. 2007, 36, 1589–1604. [Google Scholar] [CrossRef]
- Semmelhack, M.F. Transition Metal Arene Complexes: Nucleophilic Addition. In Comprehensive Organometallic Chemistry II; Abel, E.W., Stone, F.G.A., Wilkinson, G., Eds.; Elsevier: Oxford, UK, 1995; pp. 979–1015. [Google Scholar]
- Nicholls, B.; Whiting, M.C. The Organic Chemistry of the Transition Elements. Part I. Tricarbonylchromium Derivatives of Aromatic Compounds. J. Chem. Soc. 1959, 551–556. [Google Scholar] [CrossRef]
- Strohmeier, W. Eine verbesserte Darstellung von Aromaten- und Cycloheptatrien-Chromtricarbonylen. Chem. Ber. 1961, 94, 2490–2493. [Google Scholar] [CrossRef]
- Mahaffy, C.A.L.; Pauson, P.L.; Rausch, M.D.; Lee, W. (η6-Arene)Tricarbonylchromium Complexes. In Inorganic Syntheses; Shriver, D.F., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1979; pp. 154–158. [Google Scholar]
- Connelly, N.G.; Demidowicz, Z.; Kelly, R.L. Reactions of Tricarbonyl(η-hexamethylbenzene)chromium Derivatives with Nitrosonium and Benzenediazonium Ions: Reversible Oxidation versus Nitrosyl- or Areneazo-complex Formation. J. Chem. Soc. Dalton Trans. 1975, 22, 2335–2340. [Google Scholar] [CrossRef]
- Ohrenberg, N.C.; Paradee, L.M.; DeWitte, R.J.; Chong, D.; Geiger, W.E. Spectra and Synthetic-Time-Scale Substitution Reactions of Electrochemically Produced [Cr(CO)3(η6-arene)]+ Complexes. Organometallics 2010, 29, 3179–3186. [Google Scholar] [CrossRef]
- Van Order, N., Jr.; Geiger, W.E.; Bitterwolf, T.E.; Rheingold, A.L. Mixed-Valent Cations of Dinuclear Chromium Arene Complexes: Electrochemical, Spectroscopic, and Structural Considerations. J. Am. Chem. Soc. 1987, 109, 5680–5690. [Google Scholar] [CrossRef]
- Pierce, D.T.; Geiger, W.E. Mixed-Valent Interactions in Rigid Dinuclear Systems: Electrochemical and Spectroscopic Studies of CrICr0 Ions with Controlled Torsion of the Biphenyl Bridge. Inorg. Chem. 1994, 33, 373–381. [Google Scholar] [CrossRef]
- Castellani, M.P.; Connelly, N.G.; Pike, R.D.; Rieger, A.L.; Rieger, P.H. EPR Spectra of [Cr(CO)2L(η-C6Me6)]+ (L= PEt3, PPh3, P(OEt)3, P(OPh)3): Analysis of Line Widths and Determination of Ground State Configuration from Interpretation of 31P Couplings. Organometallics 1997, 16, 4369–4376. [Google Scholar] [CrossRef]
- Momoi, Y.; Okano, K.; Tokuyama, H. Generation of Aryl Grignard Reagents from Arene Chromium Tricarbonyl Complexes by Mg(TMP)2·2LiCl and Their Application to Murahashi Coupling. Synlett 2014, 25, 2503–2507. [Google Scholar] [CrossRef]
- Strohmeier, W.; Hellmann, H. Photochemisch hergestellte Derivate von Aromatenchromtricarbonylen und ihre Stabilität als Funktion der Substituenten am Benzolring. Chem. Ber. 1963, 96, 2859–2866. [Google Scholar] [CrossRef]
- Cais, M.; Kaftory, M.; Kohn, D.H.; Tatarsky, D. Structural and catalytic activity studies on phosphine- and phosphite-dicarbonylchromium complexes of phenanthrene and naphthalene. J. Organomet. Chem. 1980, 184, 103–112. [Google Scholar] [CrossRef]
- Brookhart, M.; Grant, B.; Volpe, A.F., Jr. [(3,5-(CF3)2C6H3)4B]-[H(OEt2)2]+: A Convenient Reagent for Generation and Stabilization of Cationic, Highly Electrophilic Organometallic Complexes. Organometallics 1992, 11, 3920–3922. [Google Scholar] [CrossRef]
- Oßwald, S.; Casper, L.A.; Anders, P.; Schiebel, E.; Demeshko, S.; Winter, R.F. Electrochemical, Spectroelectrochemical, Mößbauer, and EPR Spectroscopic Studies on Ferrocenyl-Substituted Tritylium Dyes. Chem. Eur. J. 2018, 24, 12524–12538. [Google Scholar] [CrossRef]
- Casper, L.A.; Oßwald, S.; Anders, P.; Rosenbaum, L.-C.; Winter, R.F. Extremely Electron-Poor Bis(diarylmethylium)-Substituted Ferrocenes and the First Peroxoferrocenophane. Z. Anorg. Allg. Chem. 2020, 646, 712–725. [Google Scholar] [CrossRef]
- Casper, L.A.; Wursthorn, L.; Geppert, M.; Roser, P.; Linseis, M.; Drescher, M.; Winter, R.F. 4-Ferrocenylphenyl-Substituted Tritylium Dyes with Open and Interlinked C+Ar2 Entities: Redox Behavior, Electrochromism, and a Quantitative Study of the Dimerization of Their Neutral Radicals. Organometallics 2020, 39, 3275–3289. [Google Scholar] [CrossRef]
- Tan, Y.-L.; White, A.J.P.; Widdowson, D.A.; Wilhelm, R.; Williams, D.J. Dilithiation of arenetricarbonylchromium(0) complexes with enantioselective quench: Application to chiral biaryl synthesis. J. Chem. Soc. Perkin Trans. 2001, 24, 3269–3280. [Google Scholar] [CrossRef]
- Uemura, M.; Nishikawa, N.; Take, K.; Ohnishi, M.; Hirotsu, K.; Higuchi, T.; Hayashi, Y. Arene-Metal Complex in Organic Synthesis: Directed Regioselective Lithiation of (π-Substituted benzene)chromium Tricarbonyl Complexes. J. Org. Chem. 1983, 48, 2349–2356. [Google Scholar] [CrossRef]
- Carter, O.L.; McPhail, A.T.; Sim, G.A. Metal-Carbonyl and Metal-Nitrosyl Complexes. Part II. Crystal and Molecular Structure of the Tricarbonylchromiumanisole-1,3,5-Trinitrobenzene Complex. J. Chem. Soc. A 1966, 822–838. [Google Scholar] [CrossRef]
- Alberico, E.; Braun, W.; Calmuschi-Cula, B.; Englert, U.; Salzer, A.; Totev, D. Expanding the Range of “Daniphos”-Type P∩P- and P∩N-Ligands: Synthesis and Structural Characterisation of New [(η6-arene)Cr(CO)3] Complexes. Eur. J. Inorg. Chem. 2007, 2007, 4923–4945. [Google Scholar] [CrossRef]
- Cambie, R.C.; Clark, G.R.; Gourdie, A.C.; Rutledge, P.S.; Woodgate, P.D. Synthesis and crystal structures of the α and β stereoisomers of tricarbonyl[(8,9,11,12,13,14-η)-methyl 12-methoxypodocarpa-8,11,13-trien-19-oate]chromium. J. Organomet. Chem. 1985, 297, 177–184. [Google Scholar] [CrossRef]
- Gilday, J.P.; Widdowson, D.A. Lithiation of 2-, 3-, and 4-Fluoroanisole(tricarbonyl)chromium(0) Complexes: A Reversal of Normal Regiocontrol. J. Chem. Soc. Chem. Commun. 1986, 16, 1235–1237. [Google Scholar] [CrossRef]
- Batuecas, M.; Luo, J.; Gergelitsová, I.; Krämer, K.; Whitaker, D.; Vitorica-Yrezabal, I.J.; Larrosa, I. Catalytic Asymmetric C-H Arylation of (η6-Arene)Chromium Complexes: Facile Access to Planar-Chiral Phosphines. ACS Catal. 2019, 9, 5268–5278. [Google Scholar] [CrossRef]
- Camire, N.; Nafady, A.; Geiger, W.E. Characterization and Reactions of Previously Elusive 17-Electron Cations: Electrochemical Oxidations of (C6H6)Cr(CO)3 and (C5H5)Co(CO)2 in the Presence of [B(C6F5)4]−. J. Am. Chem. Soc. 2002, 124, 7260–7261. [Google Scholar] [CrossRef]
- Hunter, A.D.; Mozol, V.; Tsai, S.D. Nonlinear Substituent Interactions and the Electron Richness of Substituted (η6-Arene)Cr(CO)3 Complexes as Measured by IR and 13C NMR Spectroscopy and Cyclic Voltammetry: Role of π-Donor and π-Acceptor Interactions. Organometallics 1992, 11, 2251–2262. [Google Scholar] [CrossRef]
- Connelly, N.G.; Geiger, W.E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef]
- Lever, A.B.P. Electrochemical Parametrization of Metal Complex Redox Potentials, Using the Ruthenium(III)/Ruthenium(II) Couple to Generate a Ligand Electrochemical Series. Inorg. Chem. 1990, 29, 1271–1285. [Google Scholar] [CrossRef]
- Richardson, D.E.; Taube, H. Determination of E20-E10 in Multistep Charge Transfer by Stationary-Electrode Pulse and Cyclic Voltammetry: Application to Binuclear Ruthenium Ammines. Inorg. Chem. 1981, 20, 1278–1285. [Google Scholar] [CrossRef]
- Mahaffy, C.A.L.; Pauson, P.L.; Rausch, M.D.; Lee, W. (η6-Arene)Tricarbonylchromium Complexes. In Reagents for Transition Metal Complex and Organometallic Syntheses; Angelici, R.J., Basolo, F., Eds.; Wiley: New York, NY, USA, 1990; pp. 136–140. [Google Scholar]
- Trapp, C.; Wang, C.-S.; Filler, R. Electron Spin Resonance Absorption in the Tris(pentafluorophenyl)methyl Radical. J. Chem. Phys. 2004, 45, 3472–3474. [Google Scholar] [CrossRef]
- Hoffmann, K.F.; Battke, D.; Golz, P.; Rupf, S.M.; Malischewski, M.; Riedel, S. The Tris(pentafluorophenyl)methylium Cation: Isolation and Reactivity. Angew. Chem. Int. Ed. 2022, 61, e202203777. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Eaton, S.S.; Eaton, G.R. Signal Area Measurements in EPR. Bull. Magn. Reson. 1979, 1, 130–138. [Google Scholar]
- Holden, A.N.; Kittel, C.; Merritt, F.R.; Yager, W.A. Determination of g-Values in Paramagnetic Organic Compounds by Microwave Resonance. Phys. Rev. 1950, 77, 147–148. [Google Scholar] [CrossRef]
- Bagchi, R.N.; Bond, A.M.; Brain, G.; Colton, R.; Henderson, T.L.E.; Kevekordes, J.E. Electrochemical, Chemical and Spectroscopic Characterization of the trans-[Tetracarbonylbis(triphenylphosphine)chromium]+/0 Redox Couple. Organometallics 1984, 3, 4–9. [Google Scholar] [CrossRef]
- Bond, A.M.; Colton, R.; Kevekordes, J.E. Redox Reactions of Chromium Tetracarbonyl and Tricarbonyl Complexes: Thermodynamic, Kinetic, and Catalytic Aspects of Isomerization in the fac/mer-Tricarbonyltris(trimethyl phosphite)chromium(1+/0) System. Inorg. Chem. 1986, 25, 749–756. [Google Scholar] [CrossRef]
- Hunter, A.D.; Shilliday, L.; Furey, W.S.; Zaworotko, M.J. Systematic X-ray Crystallographic Study of the Structural Manifestations of π-Donor and π-Acceptor Substituent Effects in Substituted (η6-Arene)Cr(CO)3 Complexes. Organometallics 1992, 11, 1550–1560. [Google Scholar] [CrossRef]
- Reger, D.L.; Wright, T.D.; Little, C.A.; Lamba, J.J.S.; Smith, M.D. Control of the Stereochemical Impact of the Lone Pair in Lead(II) Tris(pyrazolyl)methane Complexes. Improved Preparation of Na{B[3,5-(CF3)2C6H3]4}. Inorg. Chem. 2001, 40, 3810–3814. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Josowicz, M.; Tolbert, L.M. Diferrocenyl Molecular Wires. The Role of Heteroatom Linkers. J. Am. Chem. Soc. 2010, 132, 10374–10382. [Google Scholar] [CrossRef] [PubMed]
- Hunter, G.; Weakley, T.J.R.; Weissensteiner, W. The Triphenylphosphine Cone Angle and Restricted Rotation about the Chromium-Phosphorus Bond in Dicarbonyl(η6-hexa-alkylbenzene)(triphenylphosphine)chromium(0) Complexes. Crystal and Molecular Structure of Dicarbonyl(η6-hexa-n-propylbenzene)(triphenylphosphine)chromium(0). J. Chem. Soc. Dalton Trans. 1987, 6, 1545–1550. [Google Scholar] [CrossRef]
- Kratzert, D.; Holstein, J.J.; Krossing, I. DSR: Enhanced modelling and refinement of disordered structures with SHELXL. J. Appl. Crystallogr. 2015, 48, 933–938. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1′ | 3-OH | 1-H | L-H | |
|---|---|---|---|---|
| Crystal system | triclinic | monoclinic | monoclinic | orthorhombic |
| Space group | P | P21/c | P21/n | Pbca |
| ∠PhCr-Ph1 | 53.63(17)° | 52.55(14)° | 55.3(5)° | 47.09(8)° |
| ∠Ph1-Ph2 | n.a. | 75.00(16)° | 78.1(5)° | 77.00(8)° |
| ∠Ph1-Ph3 | n.a. | 76.50(18)° | 84.1(5)° | 82.95(8)° |
| ∠Ph1-C-OH | n.a. | 110.1(3)° | n.a. | n.a. |
| ∠Ph2-C-OH | n.a. | 105.9(3)° | n.a. | n.a. |
| ∠Ph3-C-OH | n.a. | 106.8(3)° | n.a. | n.a. |
| d C-OH | n.a. | 1.433(4) | n.a. | n.a. |
| d Cr-PhCr | 1.7151(5) | 1.703(5) | 1.720(5) | n.a. |
| d Cr-CO1 | 1.837(4) | 1.821(3) | 1.819(12) | n.a. |
| d Cr-CO2 | 1.837(3) | 1.810(3) | 1.786(12) | n.a. |
| d Cr-CO3/P | 1.844(5) | 2.303(1) | 1.801(12) | n.a. |
| Oxidation | Reduction | Potential Difference | |||
|---|---|---|---|---|---|
| E1/2 | ΔEp | E1/2 | ΔEp | ΔE1/2 | |
| 1-OH | 360 | 71 | - | - | - |
| 2-OH | 110 | 81 | - | - | - |
| 3-OH | −265 | 68 | - | - | - |
| 1+[a] | 750 | 96 | 175 | 78 | 575 |
| 2+[b] | 445 | 77 | 155 | 79 | 290 |
| 3+[c] | −180 | 110 | −180 | 110 | < 50 |
| 1-H | 435 | 83 | - | - | - |
| L+ | - | - | 135 | 72 | - |
| (cm−1) | (cm−1) [a] | |
|---|---|---|
| 1-OH | 1965, 1886 | 1912 |
| 2-OH | 1910, 1853 | 1872 |
| 3-OH | 1880, 1821 | 1840 |
| 1+ | 1965, 1886 | 1912 |
| 2+ | 2012, 1964 [b] 1904, 1848 | 1980 [b] 1867 |
| 3+ | 1988, 1940 [b] 1878, 1819 | 1956 [b] 1839 |
| 1-H | 1965, 1888 | 1914 |
| T in K | Metal-Centered Spin | Organic Spin | |||||||
|---|---|---|---|---|---|---|---|---|---|
| g1 | g2 | g3 | A1 | A2 | A3 | giso | A | ||
| 1+•• | 77 | -[a] | 2.003 | -[b] | |||||
| 203 | - | 2.005 | 5.4 (4H), 1.4 (2H), 2.7 (6F), 1.4 (1H) | ||||||
| 3+•• | 77 | 2.095 | 2.031 | 1.991 | 34.6 (31P) | 33.9 (31P) | 27.8 (31P) | 2.003 | -[b] |
| 203 | - | 2.003 | 5.1 (4H), 1.4 (2H), 2.7 (6F), 1.4 (1H) | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehse, A.; Linseis, M.; Azarkh, M.; Drescher, M.; Winter, R.F. Valence Tautomerism in Chromium Half-Sandwich Triarylmethylium Dyads. Inorganics 2023, 11, 448. https://doi.org/10.3390/inorganics11110448
Rehse A, Linseis M, Azarkh M, Drescher M, Winter RF. Valence Tautomerism in Chromium Half-Sandwich Triarylmethylium Dyads. Inorganics. 2023; 11(11):448. https://doi.org/10.3390/inorganics11110448
Chicago/Turabian StyleRehse, Anja, Michael Linseis, Mykhailo Azarkh, Malte Drescher, and Rainer F. Winter. 2023. "Valence Tautomerism in Chromium Half-Sandwich Triarylmethylium Dyads" Inorganics 11, no. 11: 448. https://doi.org/10.3390/inorganics11110448
APA StyleRehse, A., Linseis, M., Azarkh, M., Drescher, M., & Winter, R. F. (2023). Valence Tautomerism in Chromium Half-Sandwich Triarylmethylium Dyads. Inorganics, 11(11), 448. https://doi.org/10.3390/inorganics11110448