
Molecular Structures and Redox Properties of Homoleptic Aluminum(III) Complexes with Azobisphenolate (azp) Ligands



Abstract
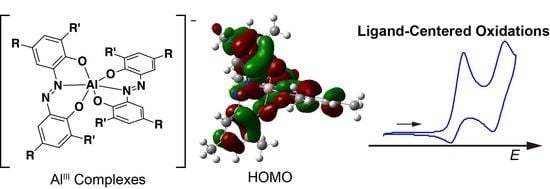
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
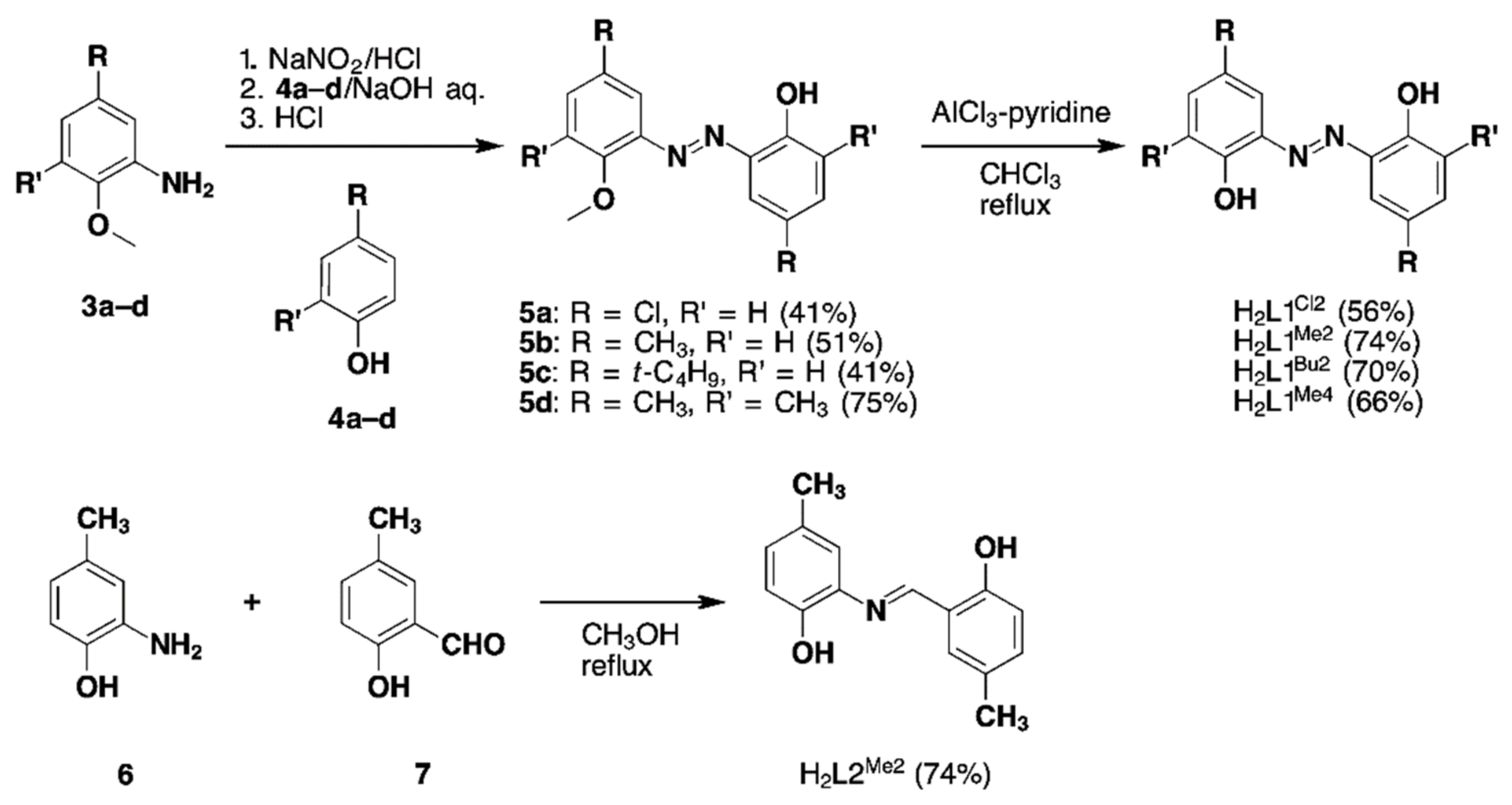
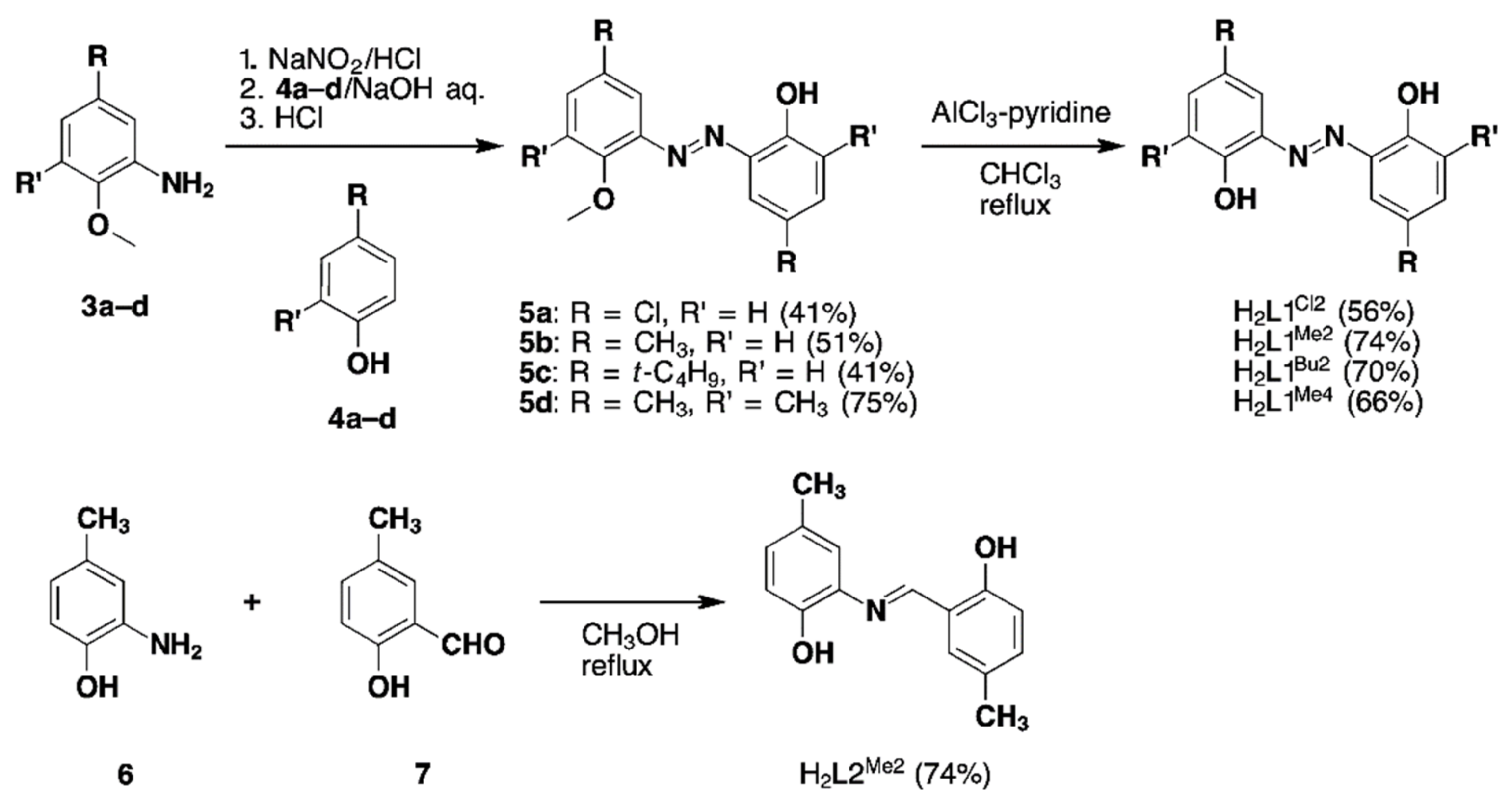
2.1.1. Synthesis of Substituted H2azp Ligands (H2L1) and Imino Derivative Ligand (H2L2)
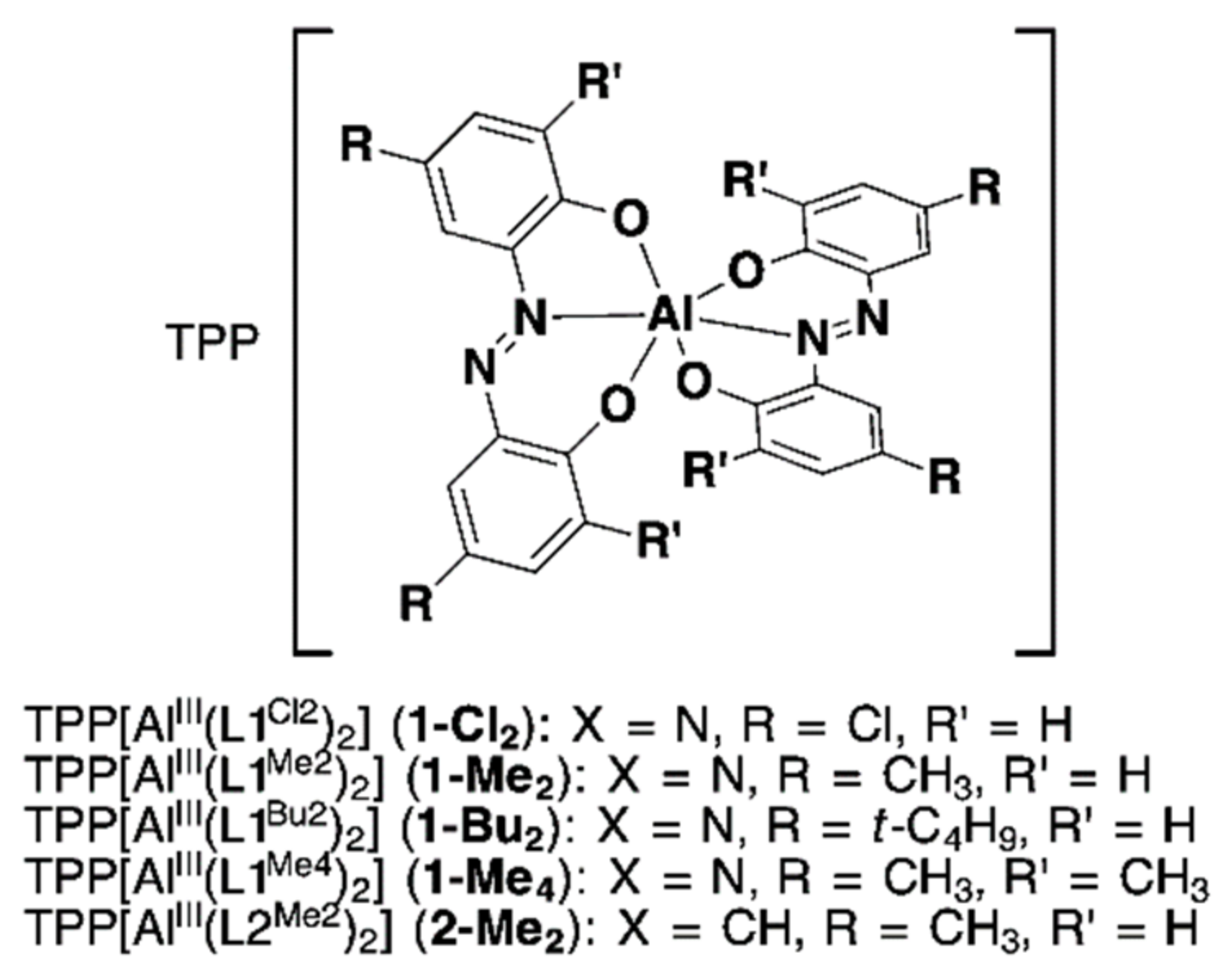
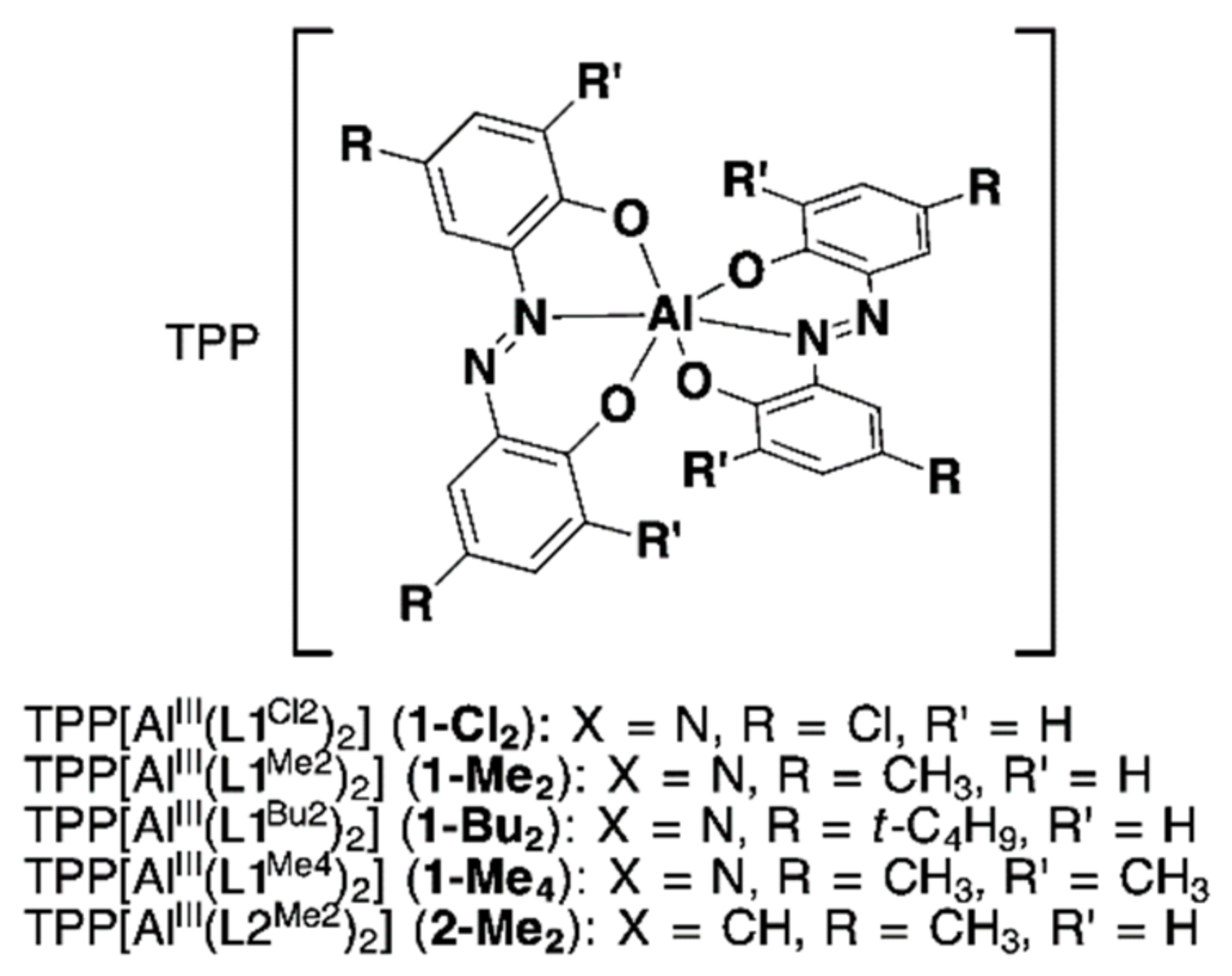
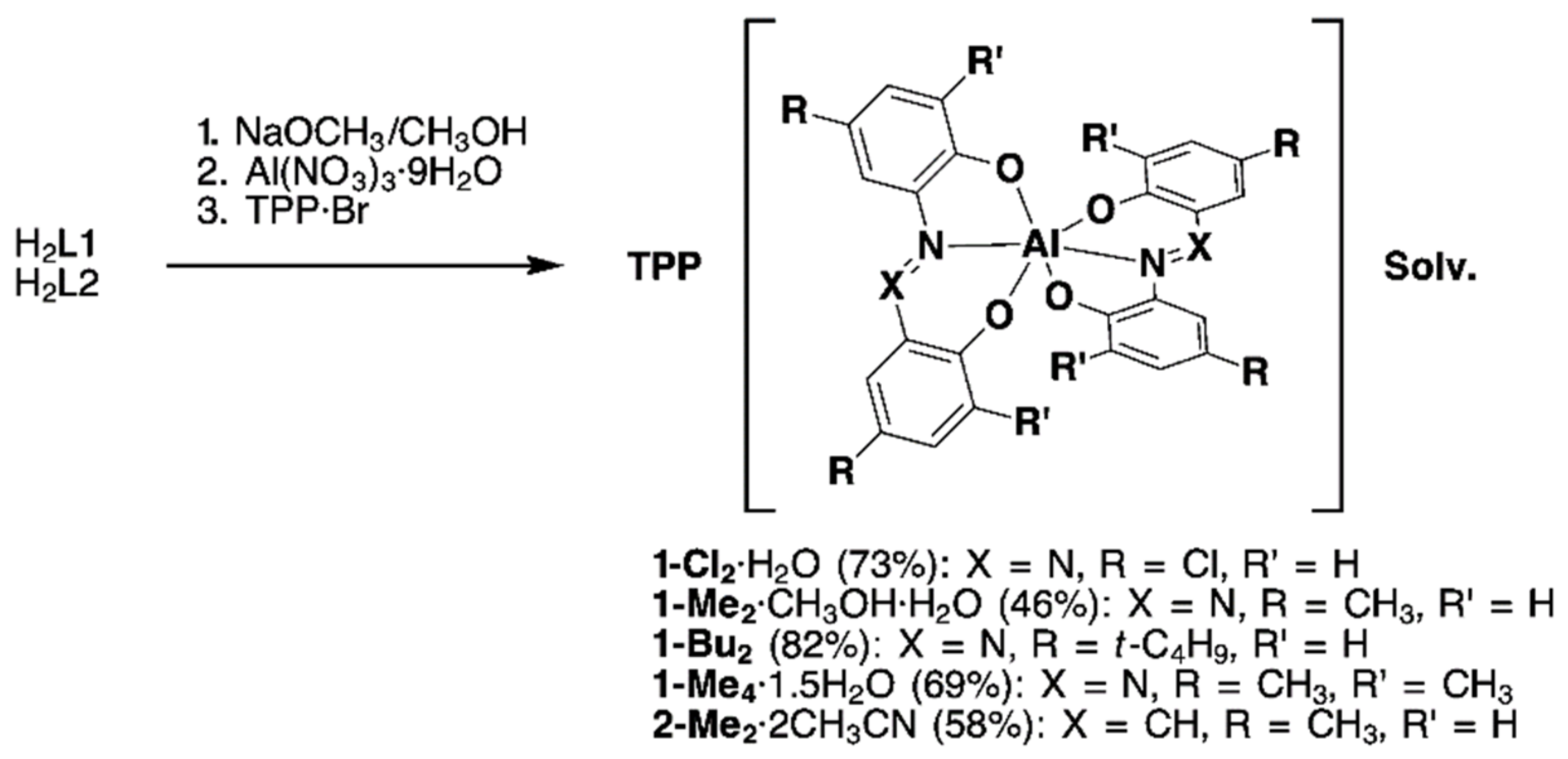
2.1.2. Synthesis of Homoleptic 1:2 AlIII Complexes
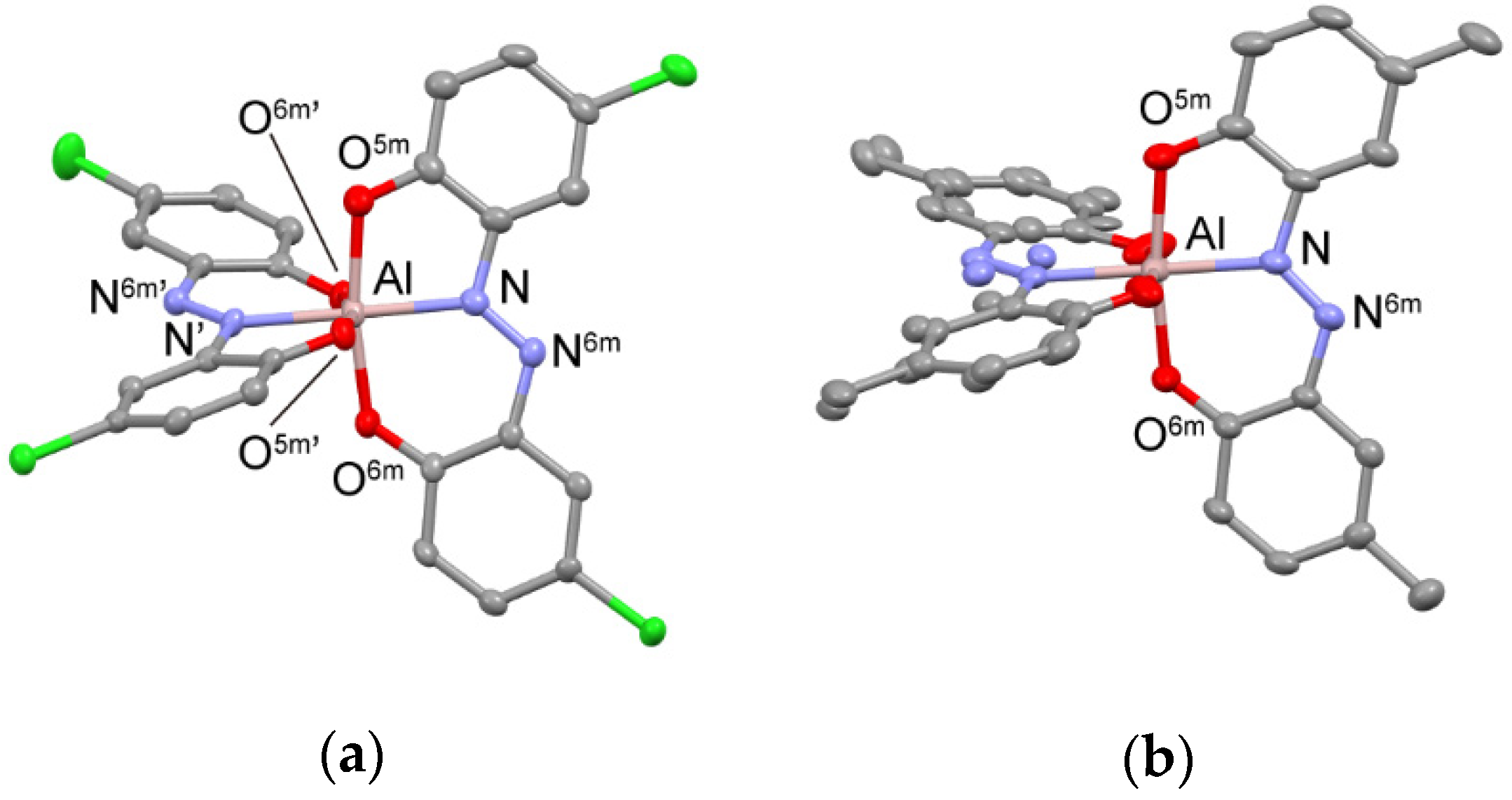
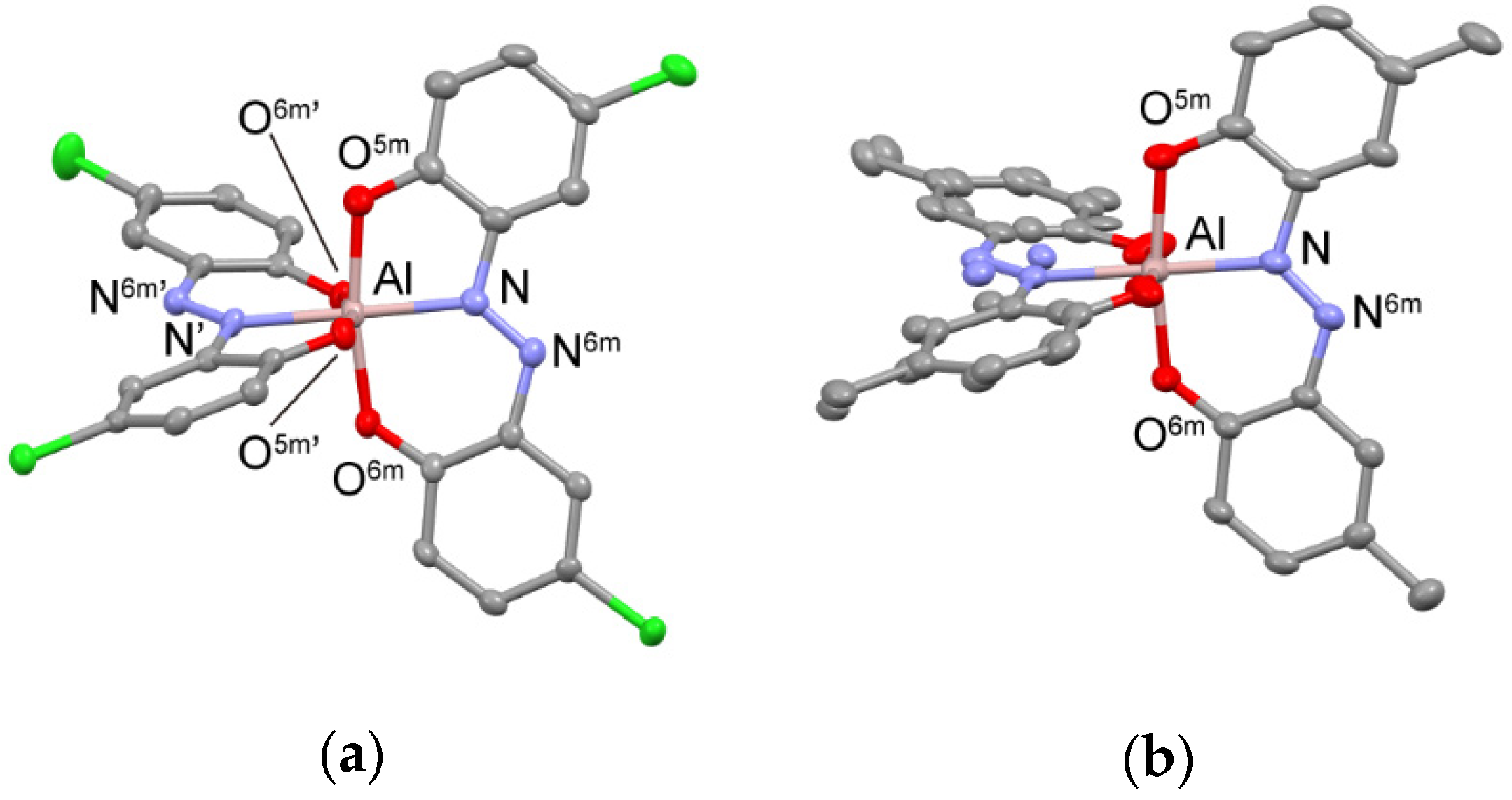
2.2. Crystal Structures of the AlIII Complexes 1 and 2
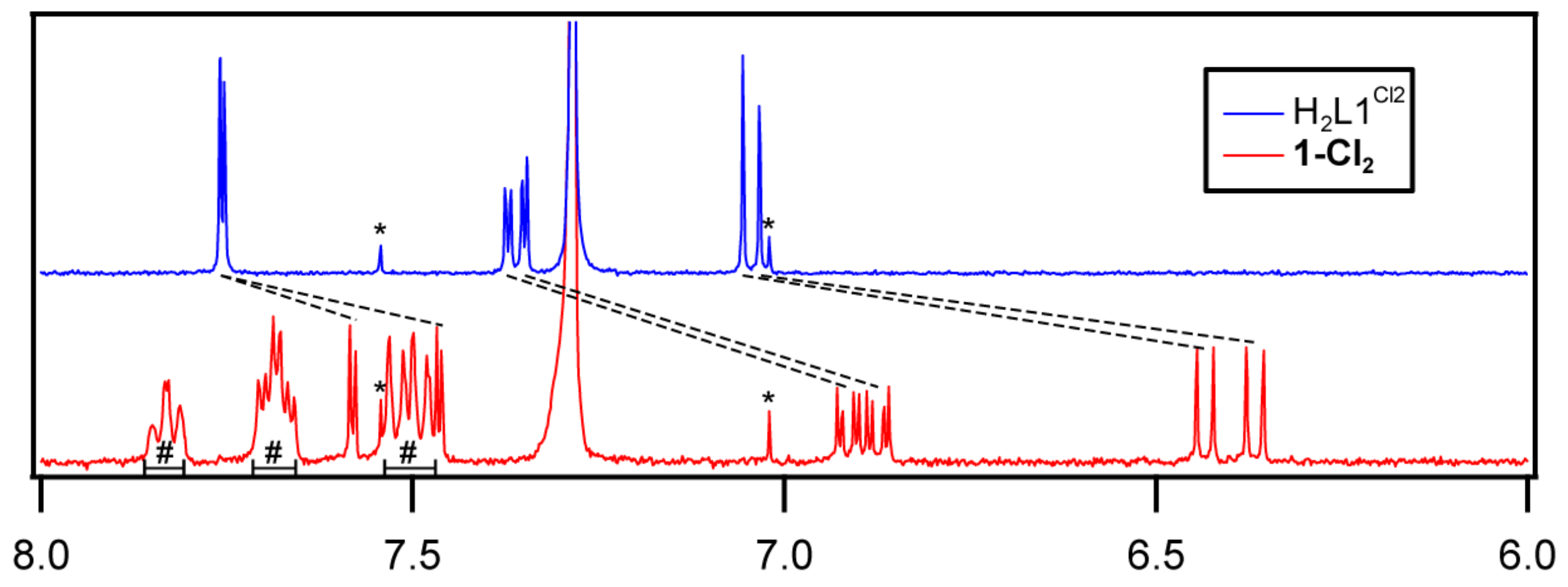
2.3. 1H NMR Spectra of the Ligands and AlIII Complexes
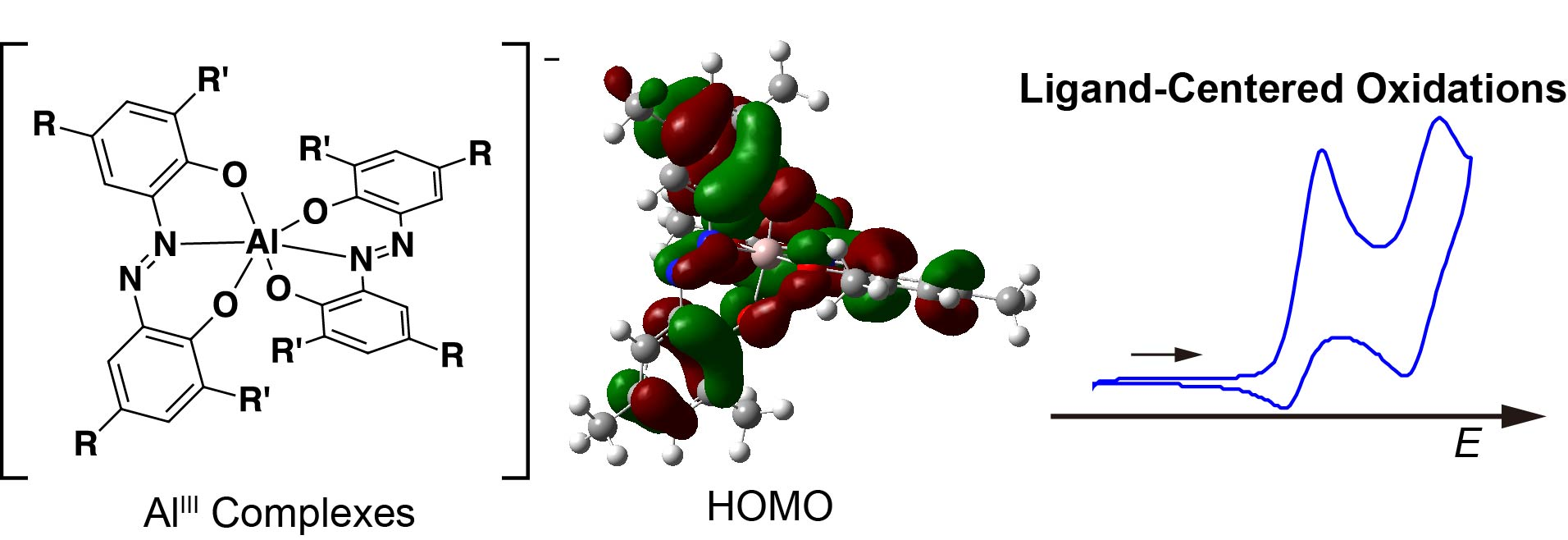
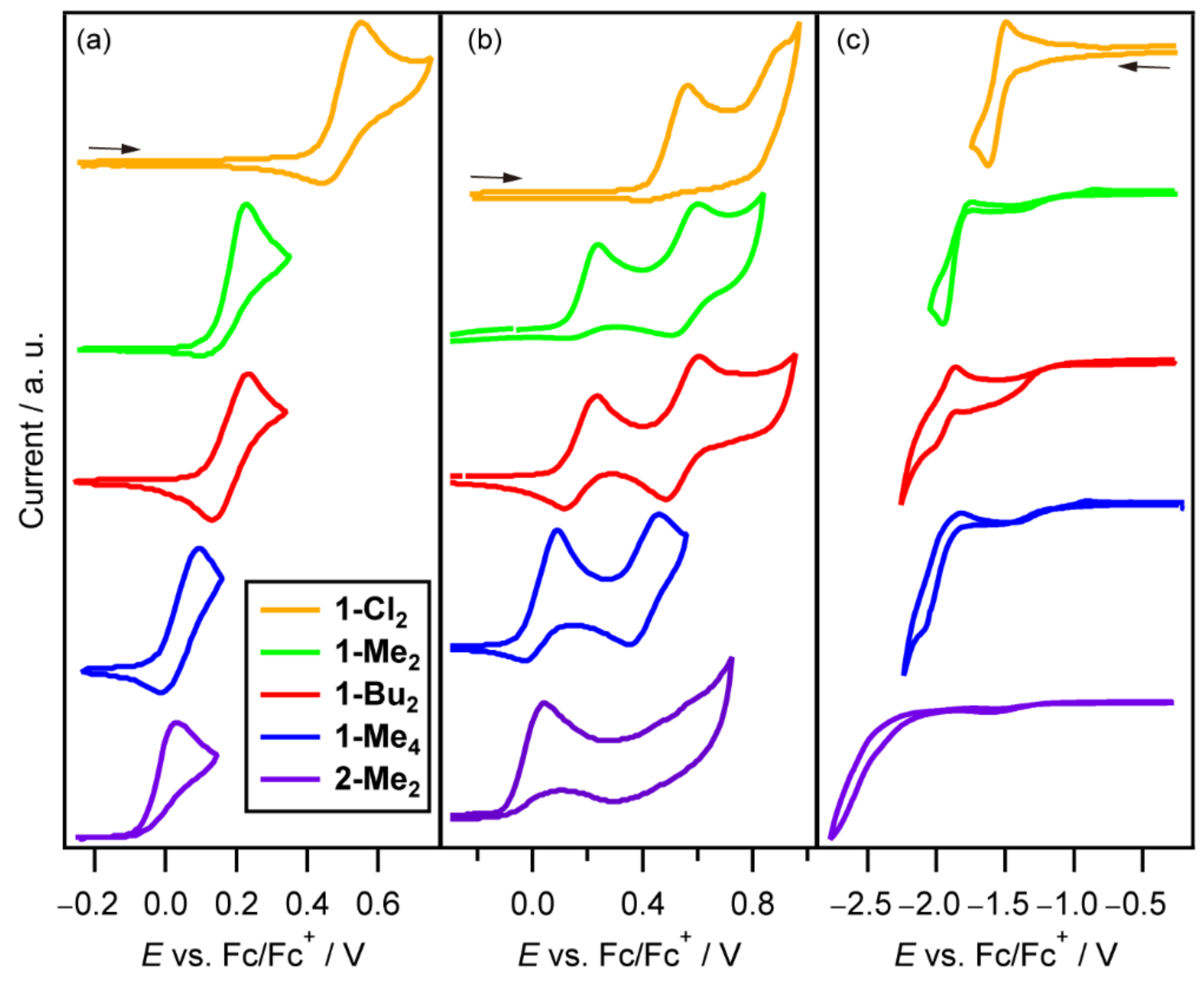
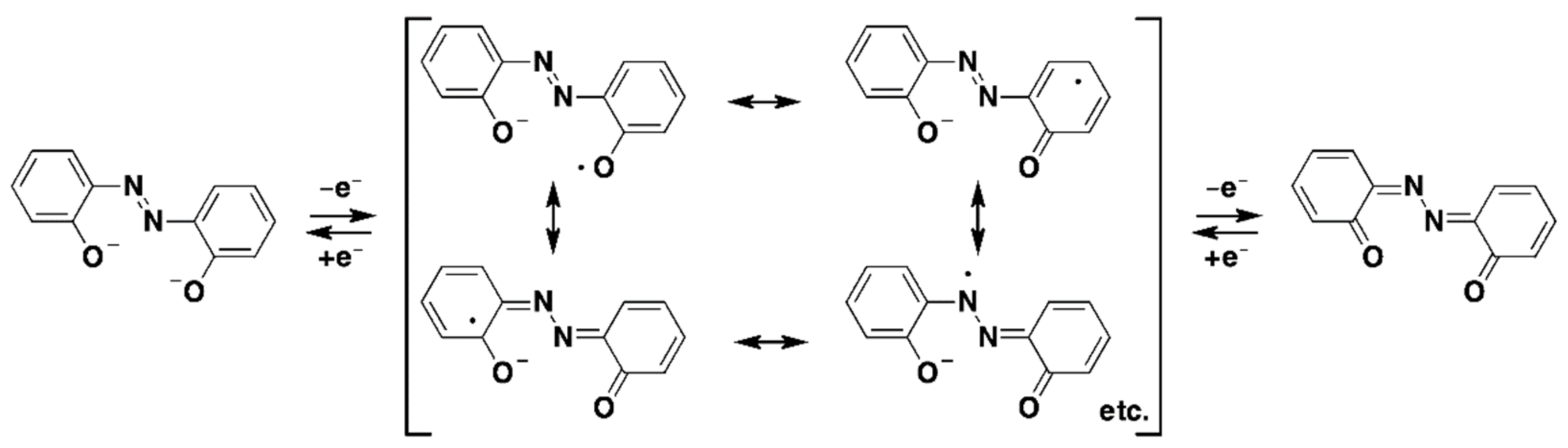
2.4. Cyclic Voltammetry (CV)
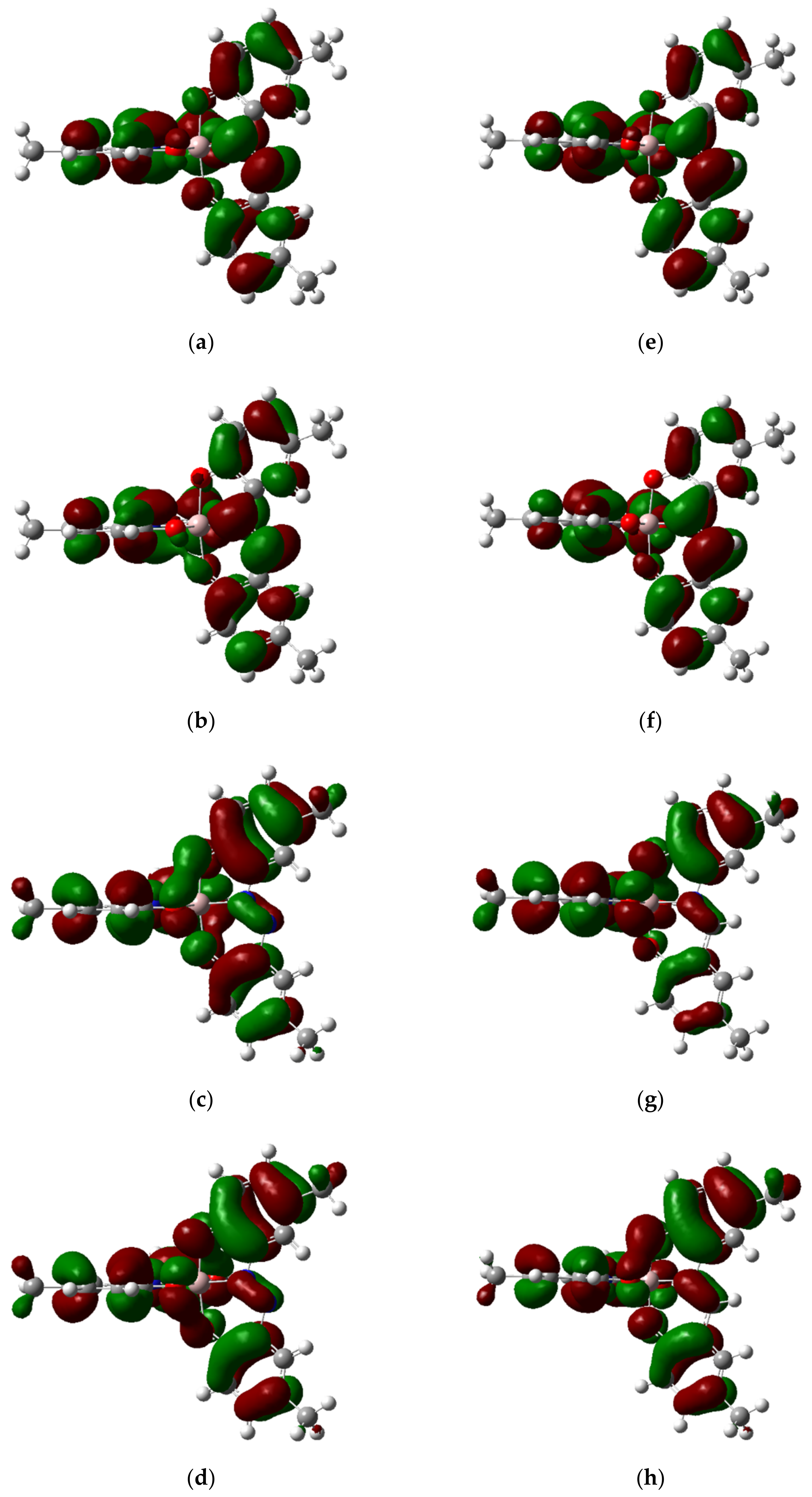
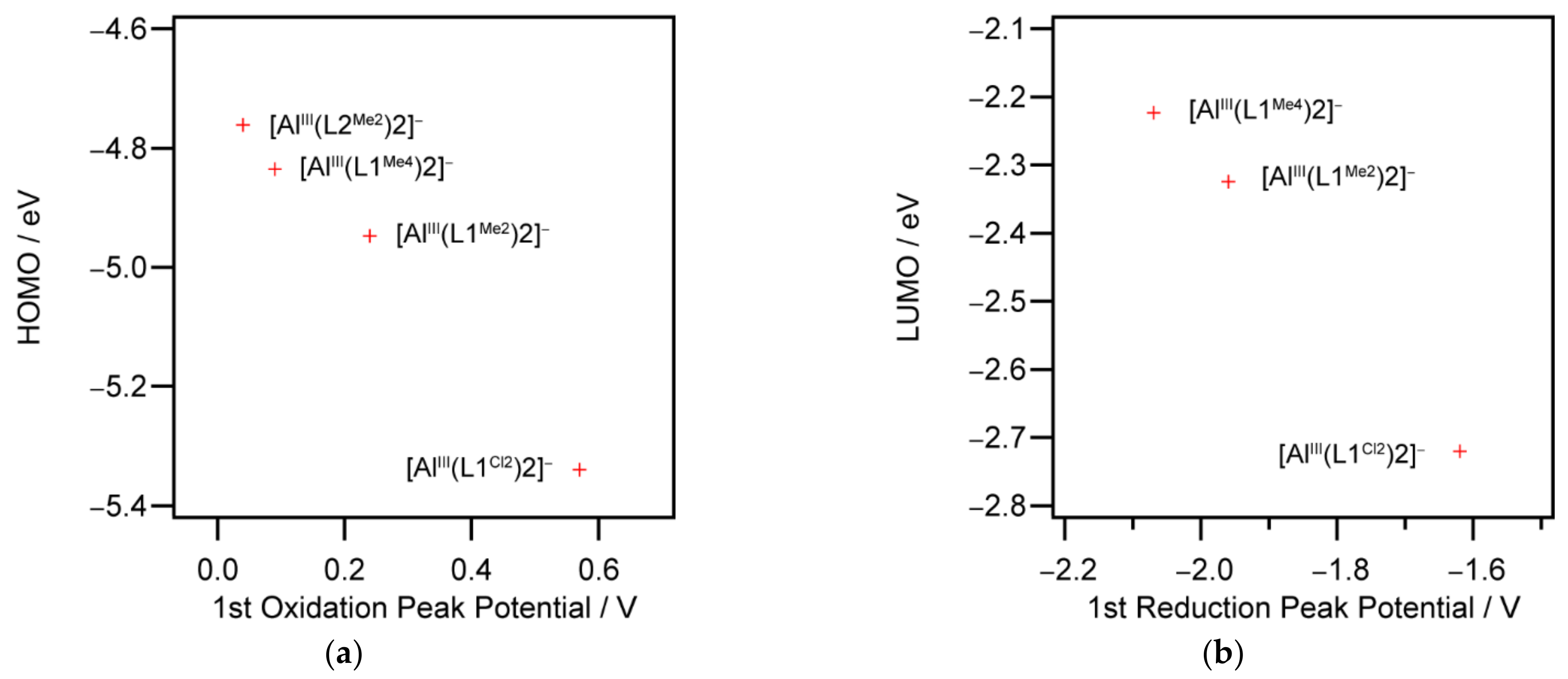
2.5. Density Functional Theory Calculations
3. Materials and Methods
3.1. Synthesis
3.1.1. General Synthetic Procedure of (2′-Methoxyphenylazo)-2-Hydroxybenzene Derivatives 5
3.1.2. General Synthetic Procedure of 2,2′-Dihydroxyazobenzene (H2azp) Derivatives H2L1
3.1.3. Synthetic Procedure of N-(2-Hydroxy-5-Methylphenyl)-5-Methylsalicylaldimine (H2L2Me2)
3.1.4. General Synthetic Procedure of the [AlIII(L1)2] (1) and [AlIII(L2)2] (2) Complexes
3.2. Physical Measurements
3.3. Crystal Structure Determinations
3.4. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tezgerevska, T.; Alley, K.G.; Boskovic, C. Valence Tautomerism in Metal Complexes: Stimulated and Reversible Intramolecular Electron Transfer between Metal Centers and Organic Ligands. Coord. Chem. Rev. 2014, 268, 23–40. [Google Scholar] [CrossRef]
- Motokawa, N.; Miyasaka, H.; Yamashita, M.; Dunbar, K.R. An Electron-Transfer Ferromagnet with Tc = 107 K Based on a Three-Dimensional [Ru2]2/TCNQ System. Angew. Chem. Int. Ed. 2008, 47, 7760–7763. [Google Scholar] [CrossRef]
- Kato, R. Conductive Copper Salts of 2,5-Disubstituted N,N′-Dicyanobenzoquinonediimines (DCNQIs): Structural and Physical Properties. Bull. Chem. Soc. Jpn. 2000, 73, 515–534. [Google Scholar] [CrossRef]
- Freeman, D.C.; White, C.E. The Structure and Characteristics of the Fluorescent Metal Chelates of o,o’-Dihydroxyazo Compounds 1. J. Am. Chem. Soc. 1956, 78, 2678–2682. [Google Scholar] [CrossRef]
- Diehl, H.; Ellingboe, J. Azo Dyes as Indicators for Calcium and Magnesium. Anal. Chem. 1960, 32, 1120–1123. [Google Scholar] [CrossRef]
- Diehl, H.; Olsen, R.; Spielholtz, G.I.; Jensen, R. Fluorometric and Spectrophotometric Determination of Magnesium with o,o′-Dihydroxyazobenzene. Anal. Chem. 1963, 35, 1144–1154. [Google Scholar] [CrossRef]
- Knoeck, J.; Buchholz, J.A. Structure, Bonding, and Fluorescence of Bivalent Metal Chelates of o,o’-Dihydroxyazobenzene. Talanta 1971, 18, 895–903. [Google Scholar] [CrossRef]
- Kirby, J.R.; Milburn, R.M.; Saylor, J.H. A Spectrophotometric Study of o,o’-Dihydroxyazobenzene and Its Chelates with Aluminum, Gallium and Indium. Anal. Chim. Acta 1962, 26, 458–469. [Google Scholar] [CrossRef]
- Hiraki, K. Metal Chelates of Aromatic o,o′-Dihydroxyazo Compounds. I. The Fluorescence Properties of the Metal Chelates of o,o′-Dihydroxyazobenzene and Their Use in Fluorometry. Bull. Chem. Soc. Jpn. 1973, 46, 2438–2443. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, H.; Yotsuyanagi, T. Highly Selective Determination of Trace Metal Ions with 2,2′-Dihydroxyazobenzene by Ion-Pair Reversed-Phase Partition HPLC-Spectrophotometric Detection System. Chem. Lett. 1984, 13, 1445–1448. [Google Scholar] [CrossRef]
- Schetty, G. Über Die Räumliche Anordnung von 1:2-Metallkomplexfarbstoffen Der Azo- Und Azomethin-Reihe. Helv. Chim. Acta 1962, 45, 1095–1102. [Google Scholar] [CrossRef]
- Schetty, G. Die Sterische Anordnung Der 1:2-Metallkomplexfarbstoffe Im Lichte Der Geometrischen Gegebenheiten. Helv. Chim. Acta 1963, 46, 1132–1143. [Google Scholar] [CrossRef]
- Schetty, G. Neuartige Isomeriefälle Bei 1:2-CrIII- Und CoIII-Komplexen von o, o’-Dihydroxyazoverbindungen: Pyramidal Gebundener Stickstoff Mit Hoher Inversionsbarriere? Helv. Chim. Acta 1970, 53, 1437–1459. [Google Scholar] [CrossRef]
- Schetty, G.; Steiner, E. Untersuchungen Der Symmetrieverhältnisse in Äquatorial Koordinierten 2:1-Arylazo-CoIII-Komplexen Mit Hilfe Der Protonenresonanz. Helv. Chim. Acta 1972, 55, 1509–1532. [Google Scholar] [CrossRef]
- Schetty, G.; Steiner, E. Die Aufklärung Der Isomerie-Ursachen Bei Äquatorial Koordinierten 2:1-Arylazo-Co(III)-Komplexen Mit Hilfe Der Kernmagnetischen Protonenresonanz. Helv. Chim. Acta 1974, 57, 2149–2172. [Google Scholar] [CrossRef]
- Schetty, G. Zur Frage Der Existenz Isomerer, Äquatorial Koordinierter 1:2-Co(III)-Komplexe Aus Symmetrischen o,o’-Dihydroxyazofarbstoffen. Helv. Chim. Acta 1975, 58, 377–380. [Google Scholar] [CrossRef]
- Grieb, R.; Niggli, A. Die Strukturen Zweier 1:2-Chromkomplexe Von o,o’-Dihydroxydiaryl-Trans-Azo-Farbstoffmolekeln. Helv. Chim. Acta 1965, 48, 317–320. [Google Scholar] [CrossRef]
- Dutta, S.; Basu, P.; Chakravorty, A. Mononuclear Manganese(IV) in Tridentate ONO Coordination. Synthesis, Structure, and Redox Regulation via Oxygen Donor Variation. Inorg. Chem. 1991, 30, 4031–4037. [Google Scholar] [CrossRef]
- Hefele, H.; Ludwig, E.; Uhlemann, E.; Nöth, H. Titan(IV)-Komplexe Mit Dreizähnigen Diaciden Liganden. Kristallstruktur von Bis [2,6-Diphenacylpyridinato(2-)]Titan(IV). Z. Anorg. Allg. Chem. 1995, 621, 1431–1435. [Google Scholar] [CrossRef]
- Hefele, H.; Jancke, H.; Sawusch, S.; Schilde, U.; Uhlemann, E. 13C-CP/MAS-NMR Spectroscopy of Tridentate Diacidic Ligands and Their Titanium(IV) Chelates. Molecular Structure of Bis[Benzoylacetone Thiobenzoylhydrazonato(2−)]Titanium(IV). Struct. Chem. 1997, 8, 211–216. [Google Scholar] [CrossRef]
- Sanna, D.; Várnagy, K.; Lihi, N.; Micera, G.; Garribba, E. Formation of New Non-Oxido Vanadium(IV) Species in Aqueous Solution and in the Solid State by Tridentate (O, N, O) Ligands and Rationalization of Their EPR Behavior. Inorg. Chem. 2013, 52, 8202–8213. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, Y.; Sakurai, H.; Crans, D.C.; Micera, G.; Garribba, E. Structural and Redox Requirements for the Action of Anti-Diabetic Vanadium Compounds. Dalton Trans. 2014, 43, 6965–6972. [Google Scholar] [CrossRef] [PubMed]
- Sanna, D.; Sciortino, G.; Ugone, V.; Micera, G.; Garribba, E. Nonoxido VIV Complexes: Prediction of the EPR Spectrum and Electronic Structure of Simple Coordination Compounds and Amavadin. Inorg. Chem. 2016, 55, 7373–7387. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Chakravorty, A. Water Soluble Manganese(III) and Manganese(IV) Complexes of Tridentate Ono Ligands. Polyhedron 1994, 13, 1811–1816. [Google Scholar] [CrossRef]
- Evangelio, E.; Saiz-Poseu, J.; Maspoch, D.; Wurst, K.; Busque, F.; Ruiz-Molina, D. Synthesis, X-ray Structure and Reactivity of a Sterically Protected Azobisphenol Ligand: On the Quest for New Multifunctional Active Ligands. Eur. J. Inorg. Chem. 2008, 2008, 2278–2285. [Google Scholar] [CrossRef]
- Abildgaard, J.; Hansen, P.E.; Josephsen, J.; Lycka, A. Assignment of the Ligating Nitrogen in o,o’-Dihydroxyazoarene Complexes of Nickel-, Palladium-, and Platinum(II) by 1H and 13C NMR Spectroscopy. Inorg. Chem. 1994, 33, 5271–5277. [Google Scholar] [CrossRef]
- Abildgaard, J.; Hansen, P.E.; Josephsen, J.; Hansen, B.K.V.; Sørensen, H.O.; Larsen, S. Synthesis and Characterization of Nickel-, Palladium- and Platinum(II) Complexes of Three o,o′-Dihydroxydiarylazo Dyes: Determination of the Coordination Geometry of This Comprehensive Series of Tridentate Diaryl Dye Complexes by Combining Results from NM. Inorg. Chim. Acta 2006, 359, 4493–4502. [Google Scholar] [CrossRef]
- Speier, G.; Csihony, J.; Whalen, A.M.; Pierpont, C.G. Studies on Aerobic Reactions of Ammonia/3,5-Di-Terf-Butylcatechol Schiff-Base Condensation Products with Copper, Copper(I), and Copper(II). Strong Copper(II)-Radical Ferromagnetic Exchange and Observations on a Unique N-N Coupling Reaction. Inorg. Chem. 1996, 35, 3519–3524. [Google Scholar] [CrossRef]
- Mitra, S.; Biswas, H.; Bandyopadhyay, P. Synthesis, Spectral Properties and Redox Behaviour of Cis-Dioxo-Molybdenum(VI) Complexes with Tridentate Arylazo Ligands. Polyhedron 1995, 14, 1581–1584. [Google Scholar] [CrossRef]
- Takahashi, K.; Kawamukai, K.; Okai, M.; Mochida, T.; Sakurai, T.; Ohta, H.; Yamamoto, T.; Einaga, Y.; Shiota, Y.; Yoshizawa, K. A New Family of Anionic FeIII Spin Crossover Complexes Featuring a Weak-Field N2O4 Coordination Octahedron. Chem. Eur. J. 2016, 22, 1253–1257. [Google Scholar] [CrossRef]
- Murata, S.; Takahashi, K.; Sakurai, T.; Ohta, H.; Yamamoto, T.; Einaga, Y.; Shiota, Y.; Yoshizawa, K. The Role of Coulomb Interactions for Spin Crossover Behaviors and Crystal Structural Transformation in Novel Anionic Fe(III) Complexes from a π-Extended ONO Ligand. Crystals 2016, 6, 49. [Google Scholar] [CrossRef] [Green Version]
- Murata, S.; Takahashi, K.; Mochida, T.; Sakurai, T.; Ohta, H.; Yamamoto, T.; Einaga, Y. Cooperative Spin-Crossover Transition from Three-Dimensional Purely π-Stacking Interactions in a Neutral Heteroleptic Azobisphenolate FeIII Complex with a N3O3 Coordination Sphere. Dalton Trans. 2017, 46, 5786–5789. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A.; Mochida, T.; Sakurai, T.; Ohta, H.; Takahashi, K. The Impact of the Next-Nearest Neighbor Dispersion Interactions on Spin Crossover Transition Enthalpy Evidenced by Experimental and Computational Analyses of Neutral π-Extended Heteroleptic Fe(III) Complexes. Inorg. Chem. 2020, 59, 12295–12303. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Takahashi, K.; Sakurai, T.; Ohta, H. Single-Crystal-to-Single-Crystal Transformation in Hydrogen-Bond-Induced High-Spin Pseudopolymorphs from Protonated Cation Salts with a π-Extended Spin Crossover Fe(III) Complex Anion. Polyhedron 2017, 136, 170–175. [Google Scholar] [CrossRef]
- Miyawaki, A.; Eda, K.; Mochida, T.; Sakurai, T.; Ohta, H.; Nakajima, T.; Takahashi, K. Spin-Crossover-Triggered Linkage Isomerization by the Pedal-like Motion of the Azobenzene Ligand in a Neutral Heteroleptic Iron(III) Complex. Inorg. Chem. 2021, 60, 12735–12739. [Google Scholar] [CrossRef] [PubMed]
- Phonsri, W.; Lewis, B.A.I.; Jameson, G.N.L.; Murray, K.S. Double Spin Crossovers: A New Double Salt Strategy to Improve Magnetic and Memory Properties. Chem. Commun. 2019, 55, 14031–14034. [Google Scholar] [CrossRef]
- Smékal, Z.; Novák, P.; Zeller, M.; Antal, P.; Čižmár, E.; Herchel, R. Synthesis, Crystal Structure, 57Fe Mössbauer Spectroscopy and Magnetic Properties of High-Spin Iron(III) Anionic Complexes [Fe(Azp)2]− (H2azp = 2,2′-Dihydroxyazobenzene) with Organic Cations. Polyhedron 2022, 212, 115586. [Google Scholar] [CrossRef]
- Willard, H.H.; Dean, J.A. Polarographic Determination of Aluminum: Use of an Organic Reagent. Anal. Chem. 1950, 22, 1264–1267. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Y.; Deng, L. Three-Coordinate Cobalt(IV) and Cobalt(V) Imido Complexes with N-Heterocyclic Carbene Ligation: Synthesis, Structure, and Their Distinct Reactivity in C–H Bond Amination. J. Am. Chem. Soc. 2014, 136, 15525–15528. [Google Scholar] [CrossRef]
- Kwon, Y.M.; Lee, Y.; Evenson, G.E.; Jackson, T.A.; Wang, D. Crystal Structure and C–H Bond-Cleaving Reactivity of a Mononuclear CoIV–Dinitrate Complex. J. Am. Chem. Soc. 2020, 142, 13435–13441. [Google Scholar] [CrossRef]
- Cui, C.; Roesky, H.W.; Schmidt, H.G.; Noltemeyer, M.; Hao, H.; Cimpoesu, F. Synthesis and Structure of a Monomeric Aluminum(I) Compound [{HC(CMeNAr)2}Al] (Ar = 2,6-IPr2C6H3): A Stable Aluminum Analogue of a Carbene. Angew. Chem. Int. Ed. 2000, 39, 4274–4276. [Google Scholar] [CrossRef]
- Lyčka, A.; Rys, P.; Skrabal, P. 27Al, 15N, 13C and 1H NMR Spectra of the 2:1 Aluminium(III) Complexes of Some Azo Dyes. Magn. Reson. Chem. 1998, 36, 279–284. [Google Scholar] [CrossRef]
- Freeman, D.C., Jr.; White, C.E. Notes-Preparation of o,o’-Dihydroxyazo Compounds. J. Org. Chem. 1956, 21, 379. [Google Scholar] [CrossRef]
- Hunter, L.; Barnes, R.S. CCLXVII.—Halogen Derivatives of o- and p-Azophenol. J. Chem. Soc. 1928, 2051–2058. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Cryst. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Witwicki, M.; Jezierska, J. Effects of solvents, ligand aromaticity, and coordination sphere on the g tensor of anionic o-semiquinone radicals complexed by Mg2+ ions: DFT studies. J. Phys. Chem. B 2011, 115, 3172–3184. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A Tool for the Calculation of the Disordered Solvent Contribution to the Calculated Structure Factors. Acta Cryst. C 2015, 71, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. A New Mixing of Hartree-Fock and Local Density-Functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3-21+G Basis Set for First-Row Elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian Basis Sets for Molecular Calculations. I. Second Row Atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent Molecular Orbital Methods. XXIII. A Polarization-type Basis Set for Second-row Elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef] [Green Version]
- Spitznagel, G.W.; Clark, T.; von Ragué Schleyer, P.; Hehre, W.J. An Evaluation of the Performance of Diffuse Function-Augmented Basis Sets for Second Row Elements, Na-Cl. J. Comput. Chem. 1987, 8, 1109–1111. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal Ion | AlIII | CoIII | CuII | FeIII | |||||
|---|---|---|---|---|---|---|---|---|---|
| Spin-state a | — | LS | — | LS | HS | ||||
| Geometry b | Oh | Oh | Sq | Oh | Oh | ||||
| Reffc/Å | 0.535 | 0.545 | 0.570 | 0.550 | 0.645 | ||||
| Complex | 1-Cl2 | 1-Me2 | Co(L) | Cu(L) | TMA[Fe(azp)2] | ||||
| Temp. | 90 K | 90 K | 233 K | 296 K | 90 K | 293 K | |||
| Bond Length/Å | |||||||||
| M-O5m | 1.882(4) | 1.870(4) | 1.879(3) | 1.888(3) | 1.890(2) | 1.868(4) | 1.905(4) | 1.9217(14) | 1.9842(18) |
| M-O6m | 1.855(4) | 1.851(3) | 1.845(4) | 1.856(3) | 1.843(2) | 1.856(4) | 1.867(4) | 1.8898(14) | 1.9425(19) |
| M-N | 2.020(4) | 2.030(4) | 2.030(4) | 2.030(4) | 2.008(2) | 1.849(6) | 1.923(6) | 1.9220(15) | 2.1534(18) |
| C-O5m | 1.325(5) | 1.330(6) | 1.319(5) | 1.325(6) | 1.328(4) | 1.334(8) | 1.331(9) | 1.312(2) | 1.311(3) |
| C-N | 1.414(6) | 1.400(6) | 1.417(6) | 1.415(6) | 1.416(3) | 1.422(8) | 1.405(8) | 1.416(2) | 1.415(3) |
| C-O6m | 1.316(6) | 1.320(5) | 1.310(6) | 1.314(5) | 1.302(3) | 1.326(8) | 1.309(8) | 1.319(2) | 1.299(3) |
| C-N6m | 1.384(6) | 1.406(6) | 1.403(6) | 1.407(5) | 1.391(3) | 1.386(9) | 1.390(9) | 1.388(2) | 1.391(3) |
| N-N6m | 1.278(5) | 1.276(5) | 1.271(5) | 1.268(5) | 1.277(3) | 1.266(8) | 1.263(8) | 1.267(2) | 1.274(3) |
| reference | this work | ref. [25] | ref. [28] | ref. [30] | |||||
| Complex | Ered1 | Eox1 | Eox2 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Epa | Epc | E1/2b | Epa | Epc | E1/2b | Epa | Epc | E1/2b | |
| 1-Cl2 | −1.50 | −1.62 | −1.56 | 0.57 | 0.40 c | 0.49 | 0.90 d | — | — |
| 1-Me2 | −1.73 c | −1.96 | — | 0.24 | 0.11 | 0.18 | 0.60 | 0.50 | 0.55 |
| 1-Bu2 | −1.86 | −1.98 d | — | 0.24 | 0.12 | 0.18 | 0.61 | 0.49 | 0.55 |
| 1-Me4 | −1.82 | −2.07 d | — | 0.09 | −0.03 | 0.03 | 0.46 | 0.35 | 0.41 |
| 2-Me2 | — | — | — | 0.04 | — | — | — | — | — |
| Anion | LUMO+1 | LUMO | HOMO | HOMO−1 |
|---|---|---|---|---|
| [AlIII(L1Cl2)2] | −2.527 | −2.720 | −5.339 | −5.343 |
| [AlIII(L1Me2)2] | −2.132 | −2.324 | −4.946 | −4.953 |
| [AlIII(L1Me4)2] | −2.025 | −2.223 | −4.836 | −4.839 |
| [AlIII(L2Me2)2] | −1.562 | −1.742 | −4.762 | −4.763 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahashi, K.; Noguchi, T.; Ueda, K.; Miyawaki, A.; Murata, S. Molecular Structures and Redox Properties of Homoleptic Aluminum(III) Complexes with Azobisphenolate (azp) Ligands. Inorganics 2022, 10, 84. https://doi.org/10.3390/inorganics10060084
Takahashi K, Noguchi T, Ueda K, Miyawaki A, Murata S. Molecular Structures and Redox Properties of Homoleptic Aluminum(III) Complexes with Azobisphenolate (azp) Ligands. Inorganics. 2022; 10(6):84. https://doi.org/10.3390/inorganics10060084
Chicago/Turabian StyleTakahashi, Kazuyuki, Takumi Noguchi, Keiji Ueda, Atsuhiro Miyawaki, and Suguru Murata. 2022. "Molecular Structures and Redox Properties of Homoleptic Aluminum(III) Complexes with Azobisphenolate (azp) Ligands" Inorganics 10, no. 6: 84. https://doi.org/10.3390/inorganics10060084
APA StyleTakahashi, K., Noguchi, T., Ueda, K., Miyawaki, A., & Murata, S. (2022). Molecular Structures and Redox Properties of Homoleptic Aluminum(III) Complexes with Azobisphenolate (azp) Ligands. Inorganics, 10(6), 84. https://doi.org/10.3390/inorganics10060084