
Tetraruthenium Macrocycles with Laterally Extended Bis(alkenyl)quinoxaline Ligands and Their F4TCNQ•− Salts



Abstract
:1. Introduction
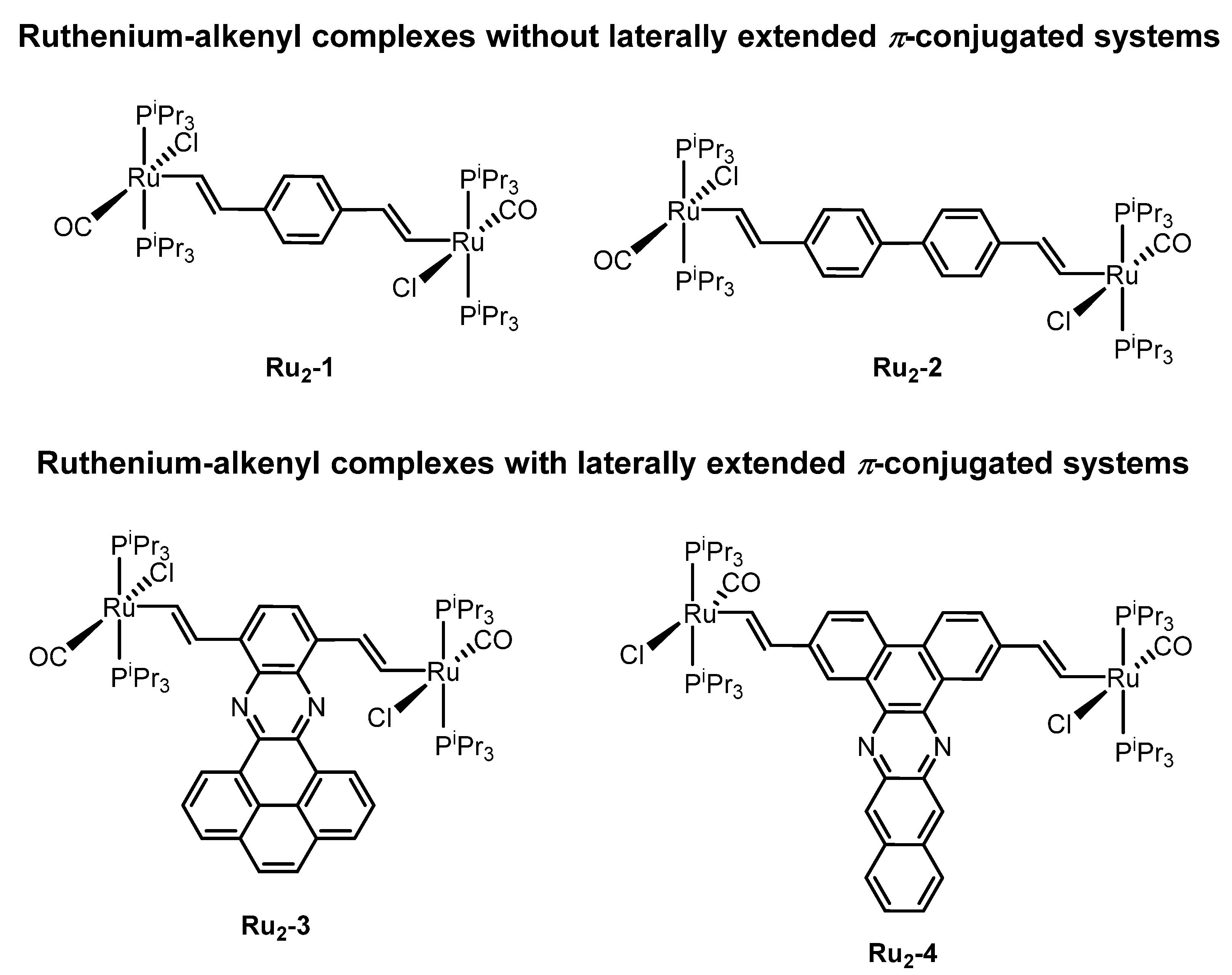
2. Results and Discussion
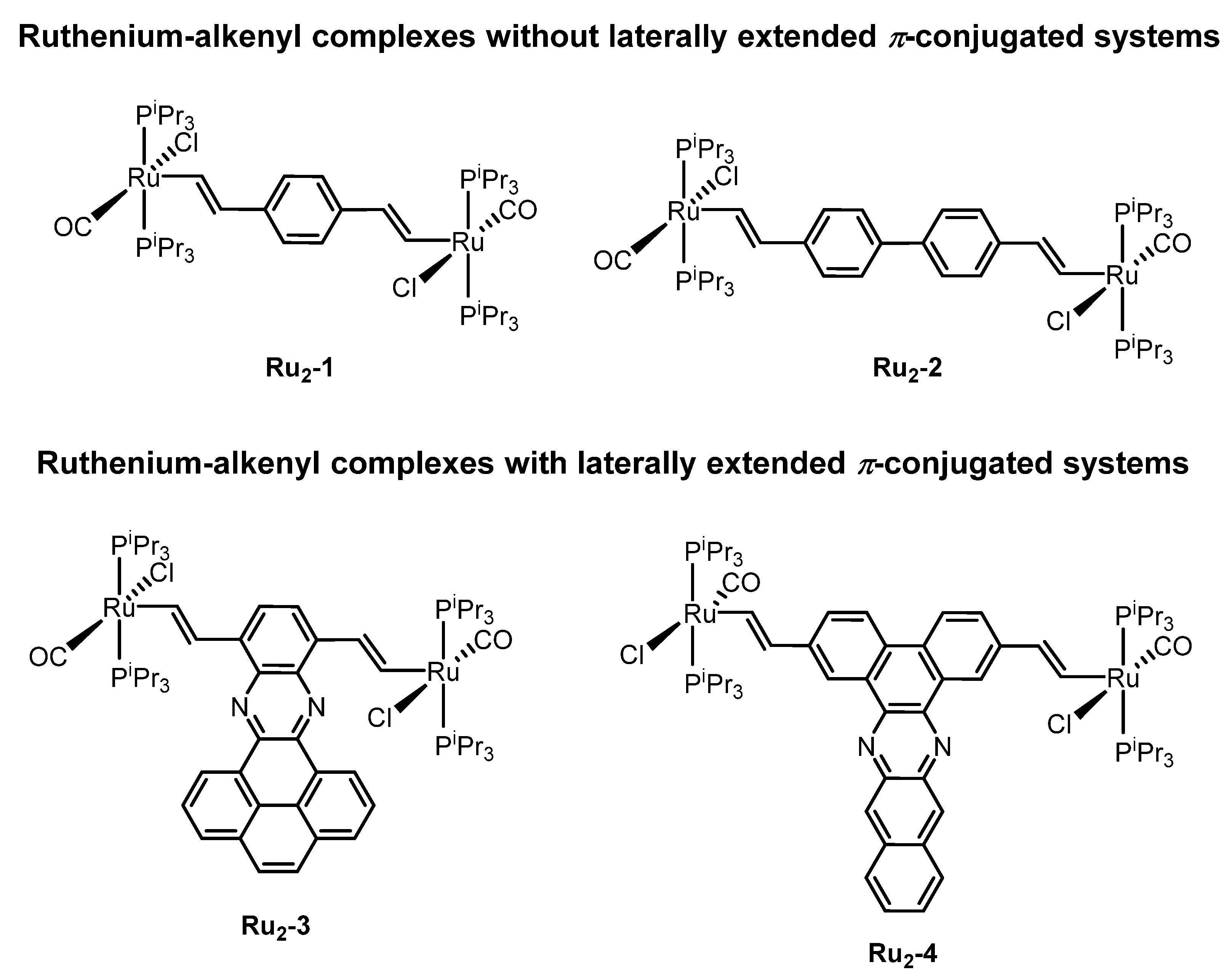
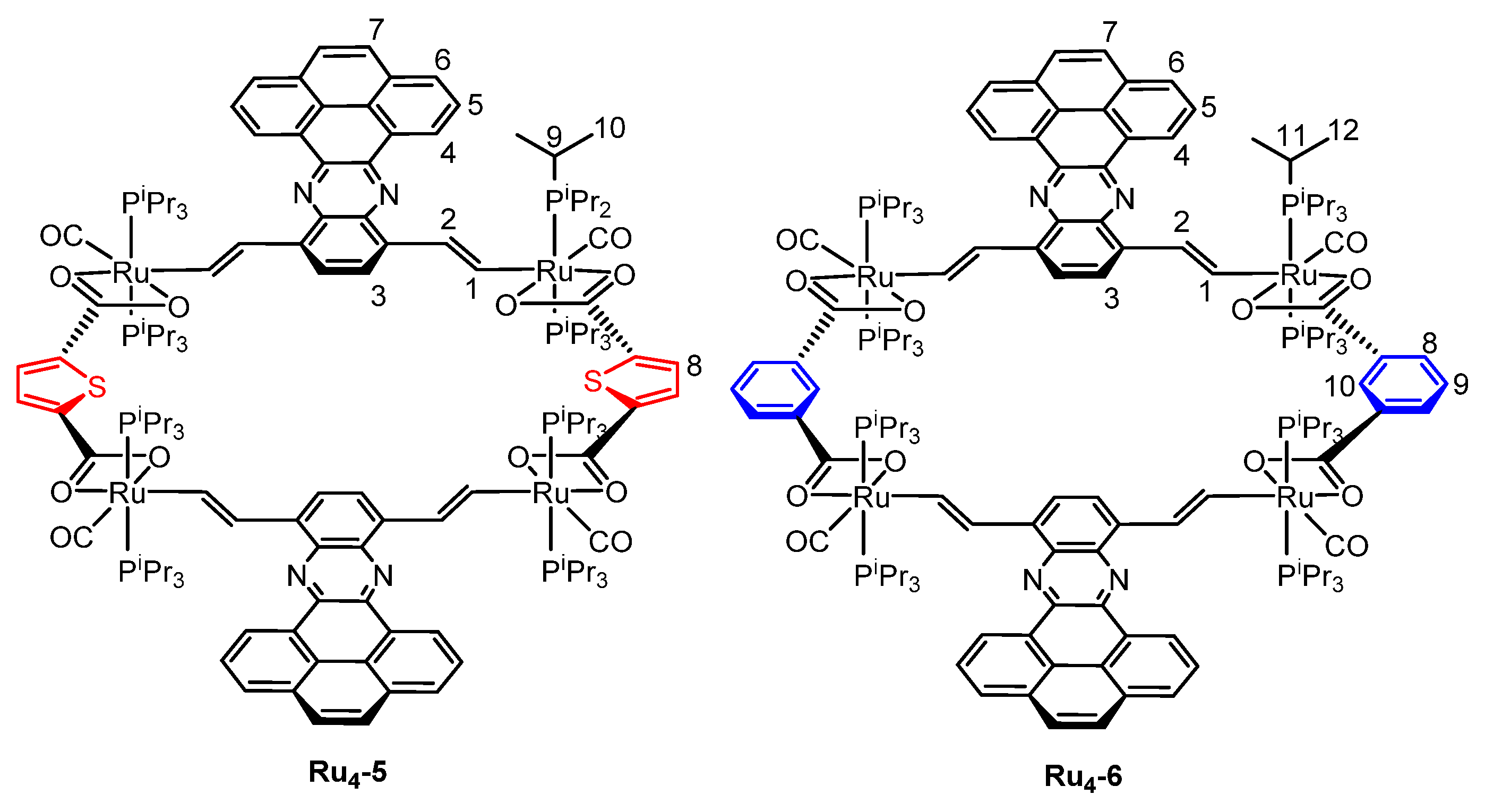
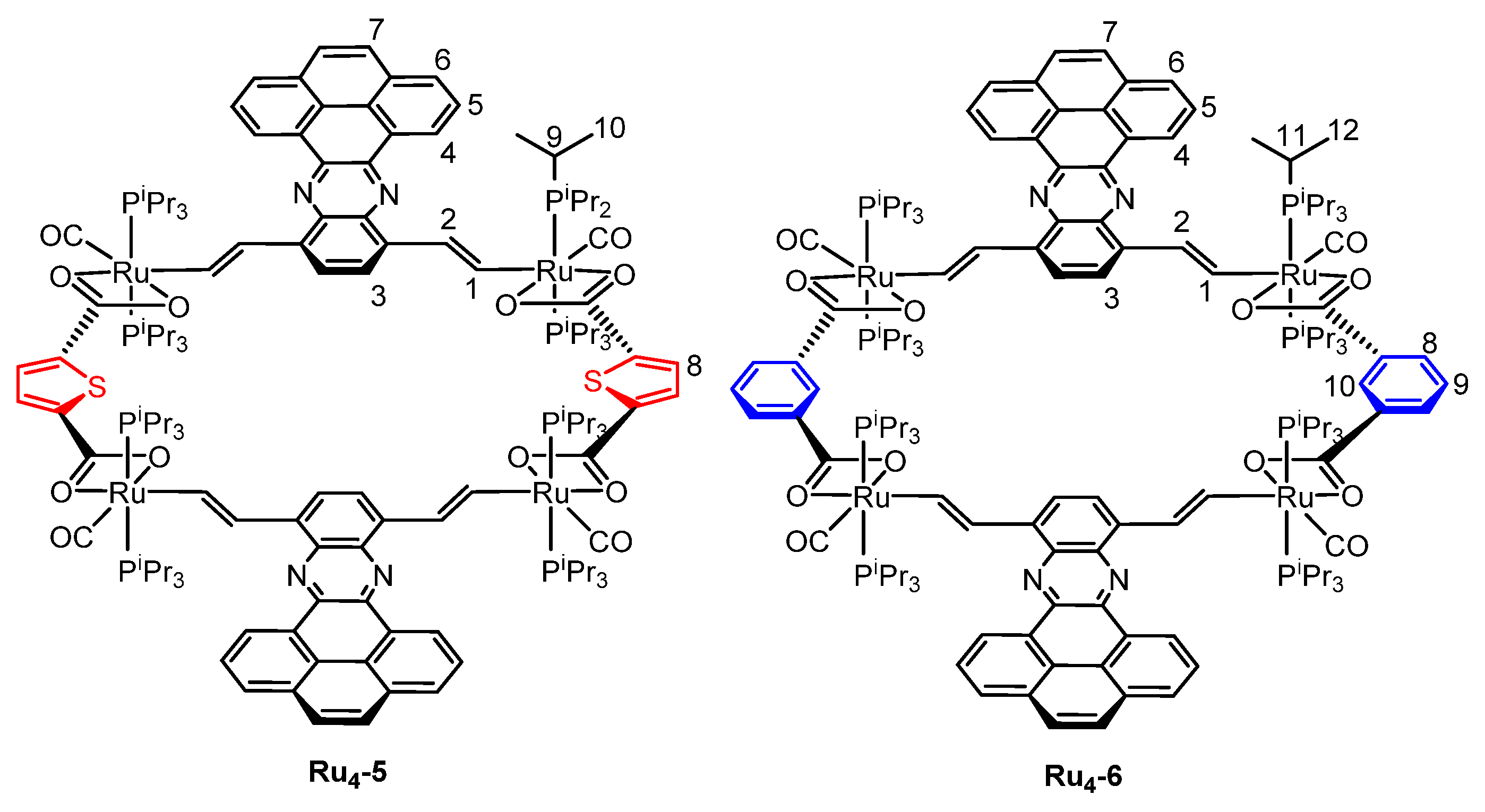
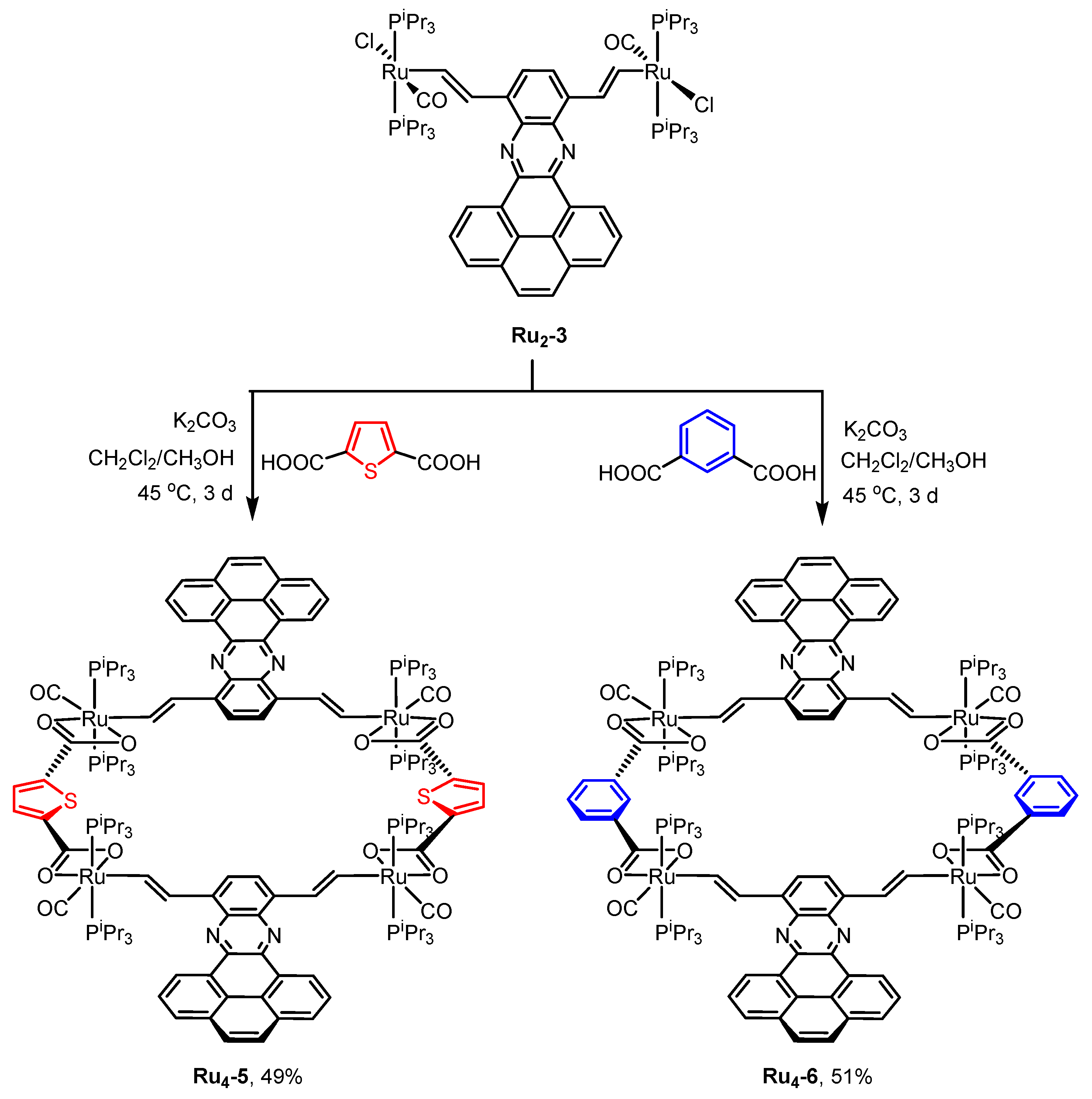
2.1. Synthesis and Spectroscopic Characterization
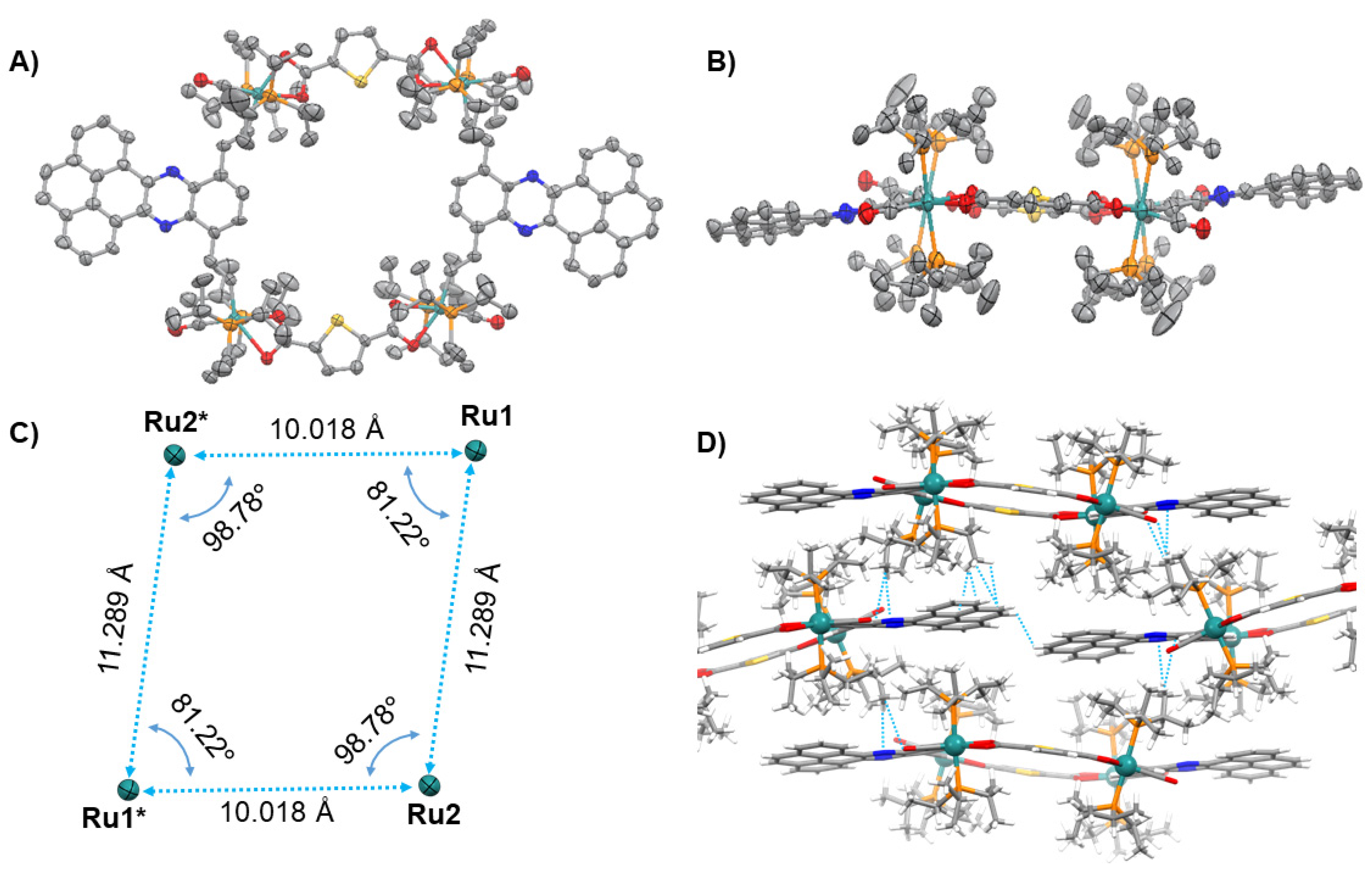
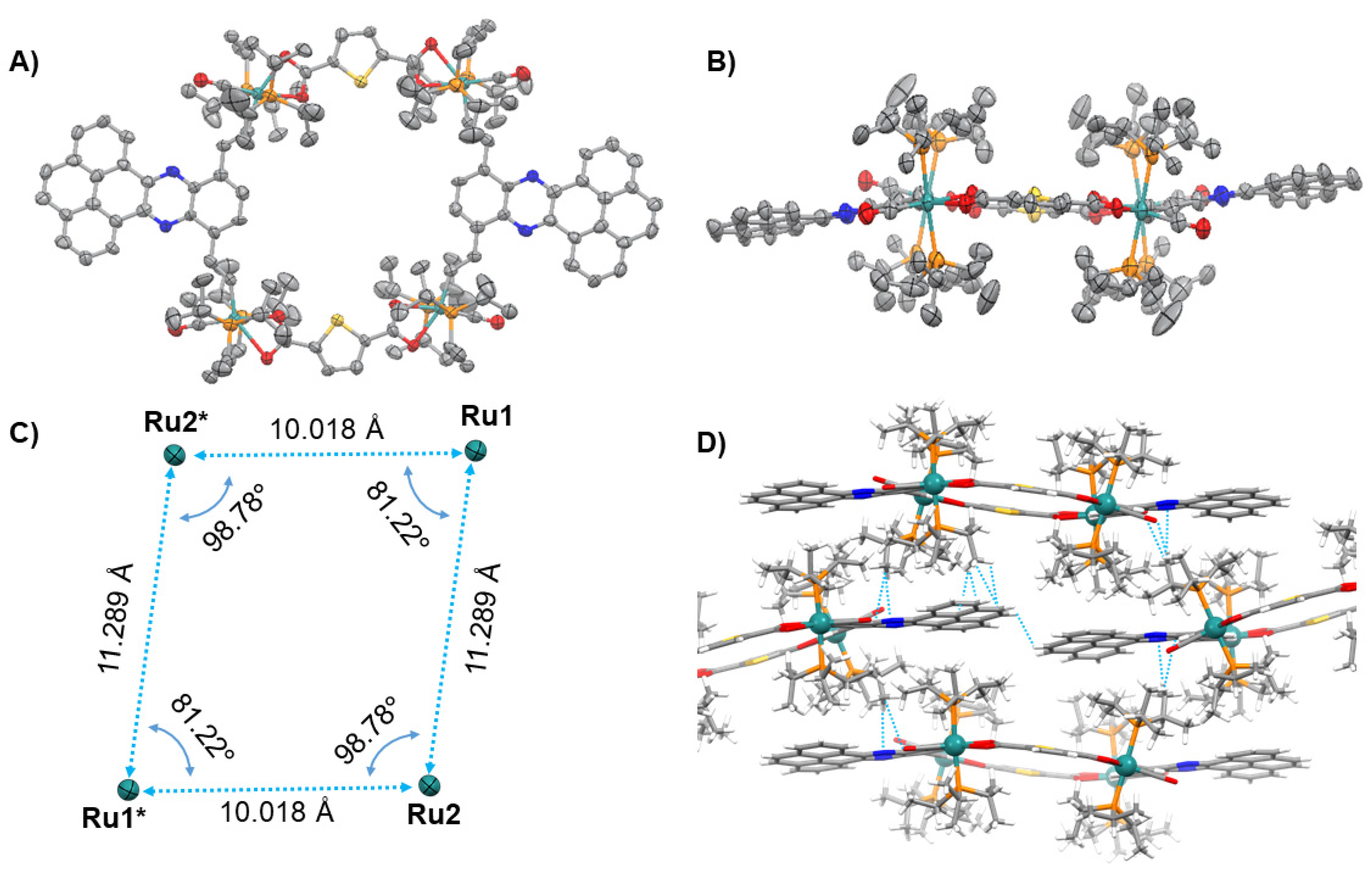
2.2. X-ray Crystallography
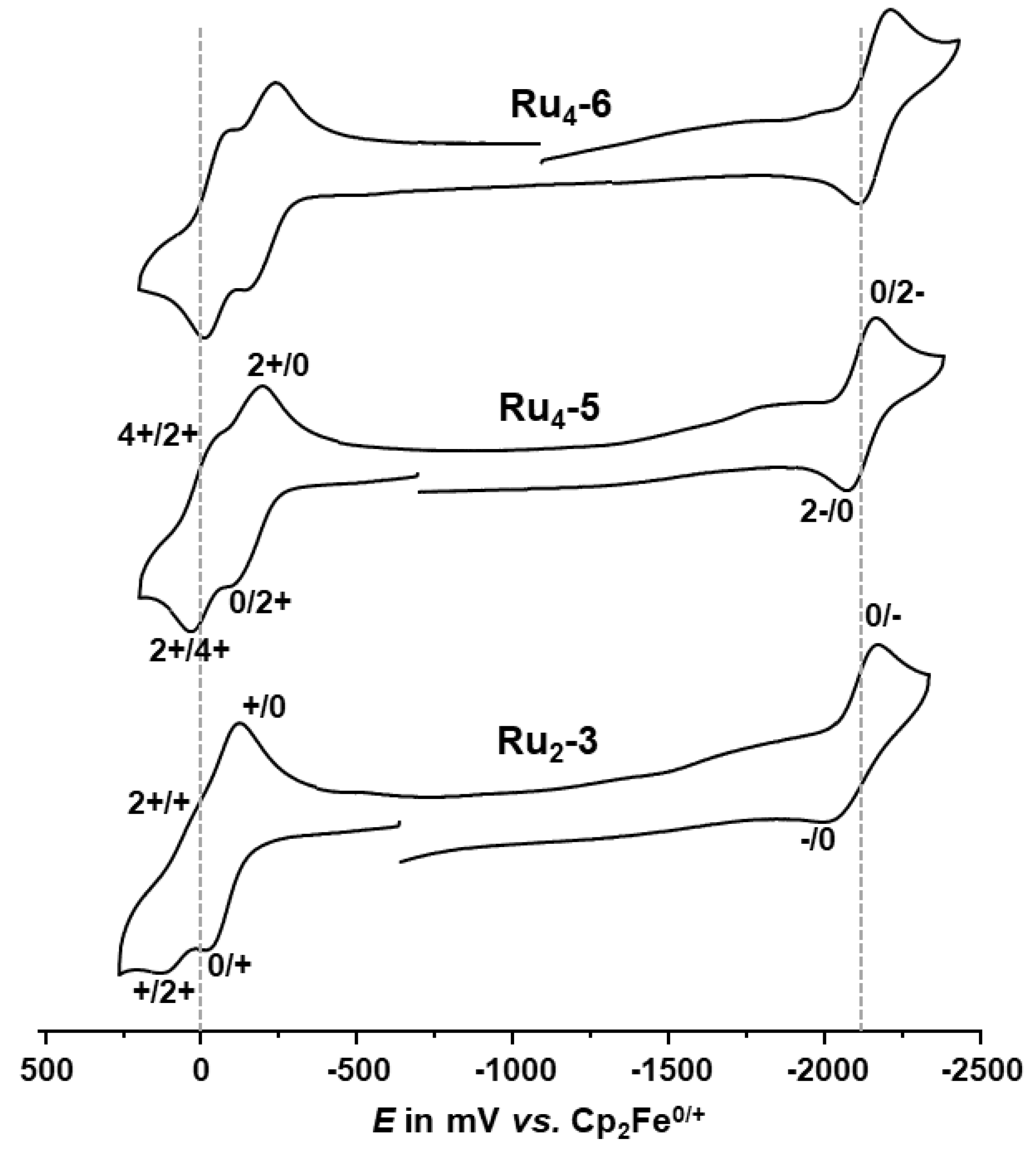
2.3. Electrochemistry
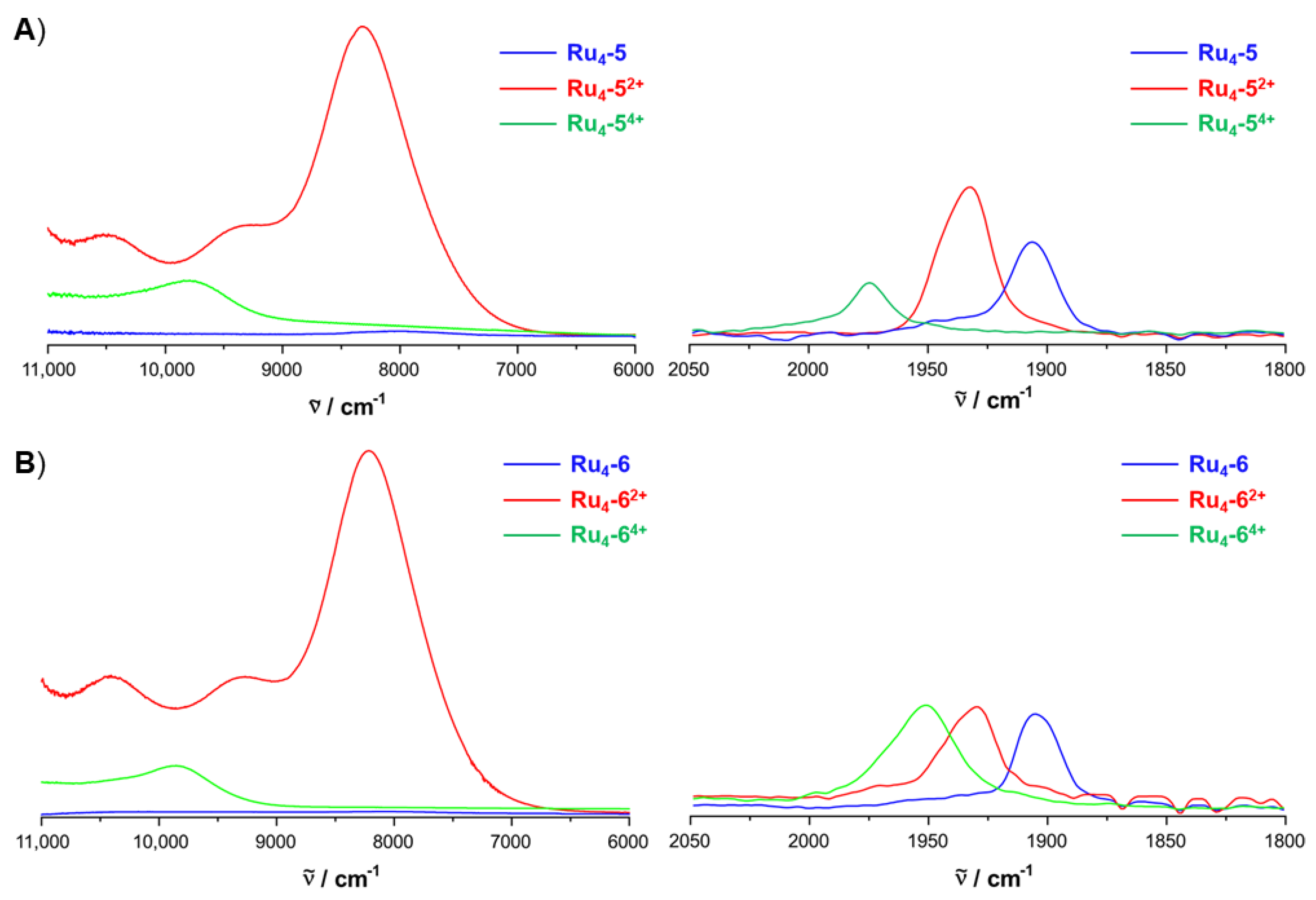
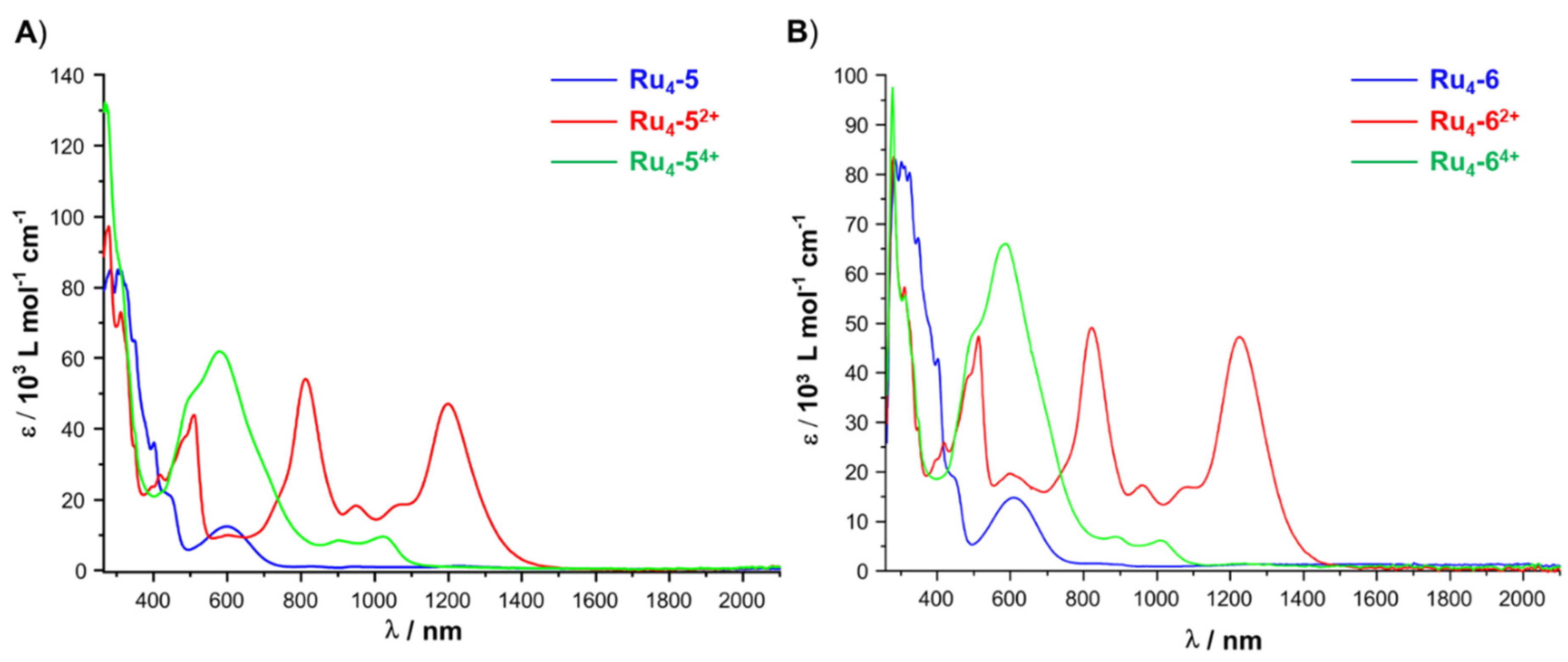
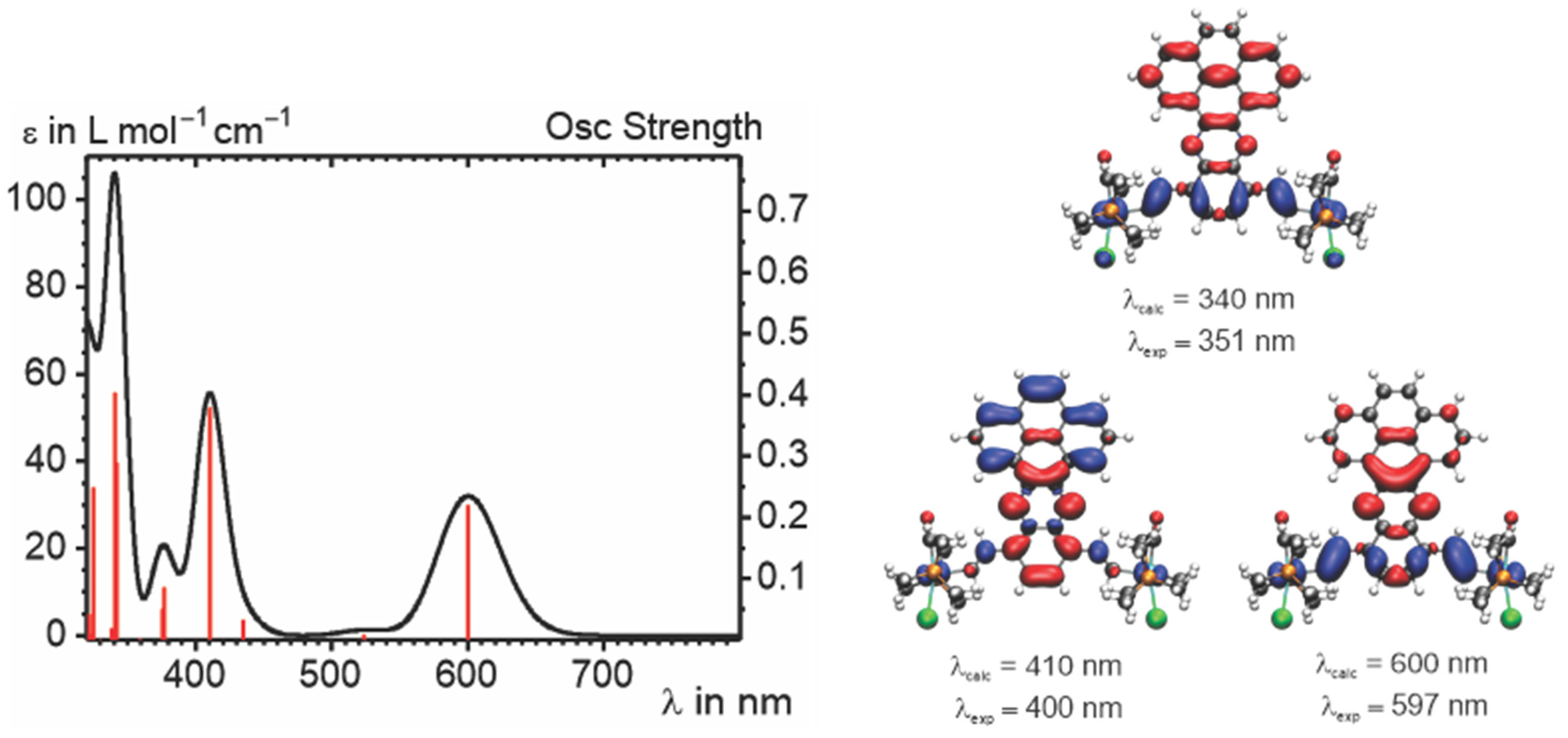
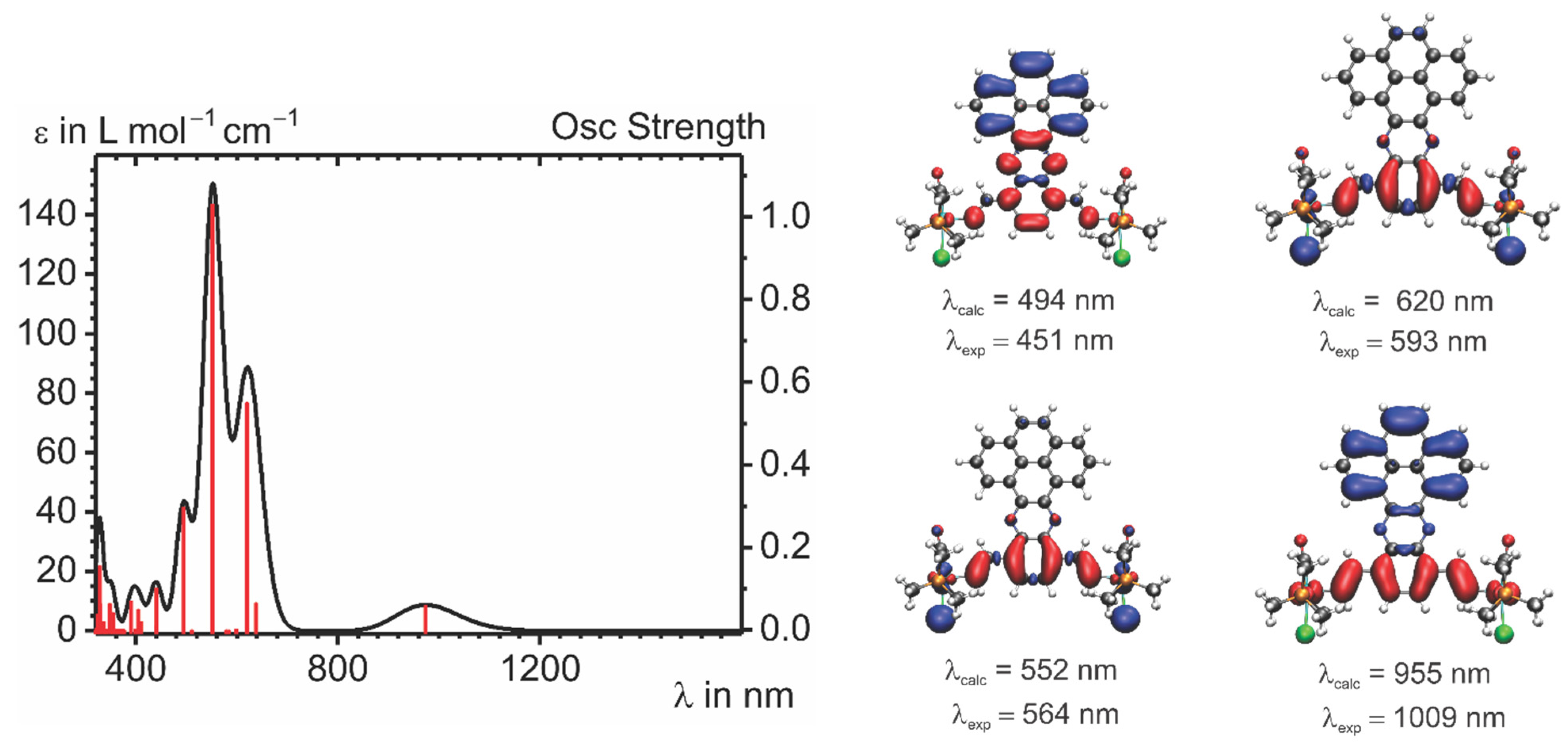
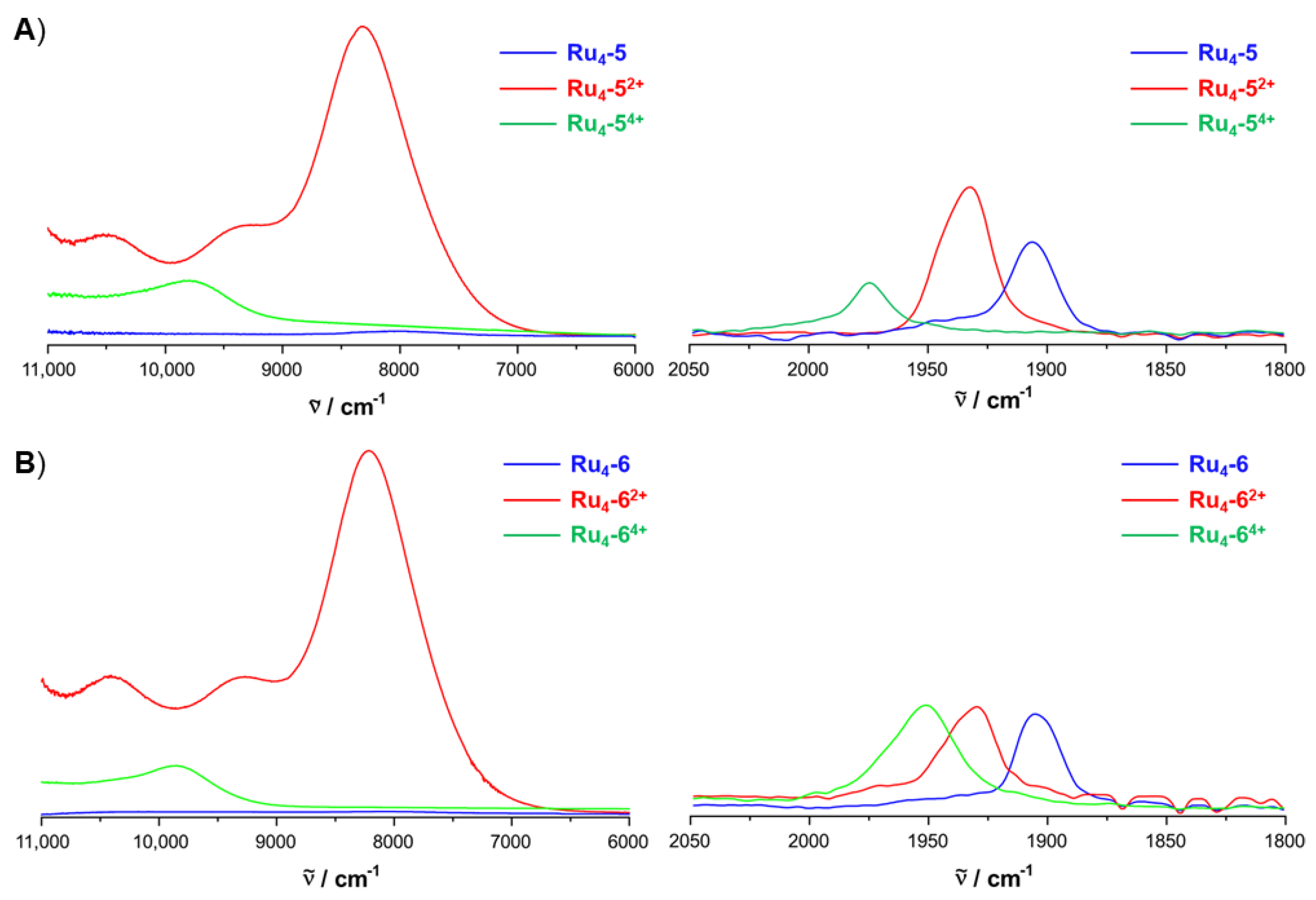
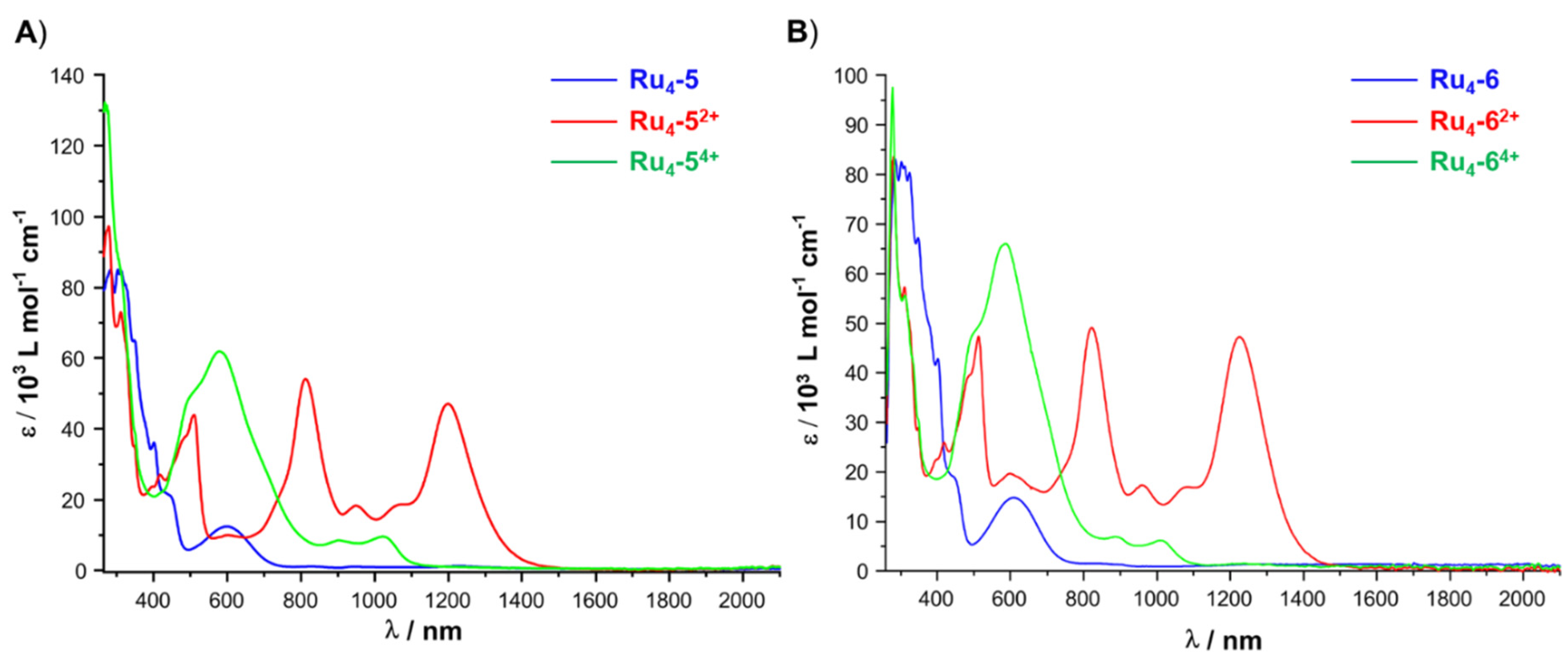
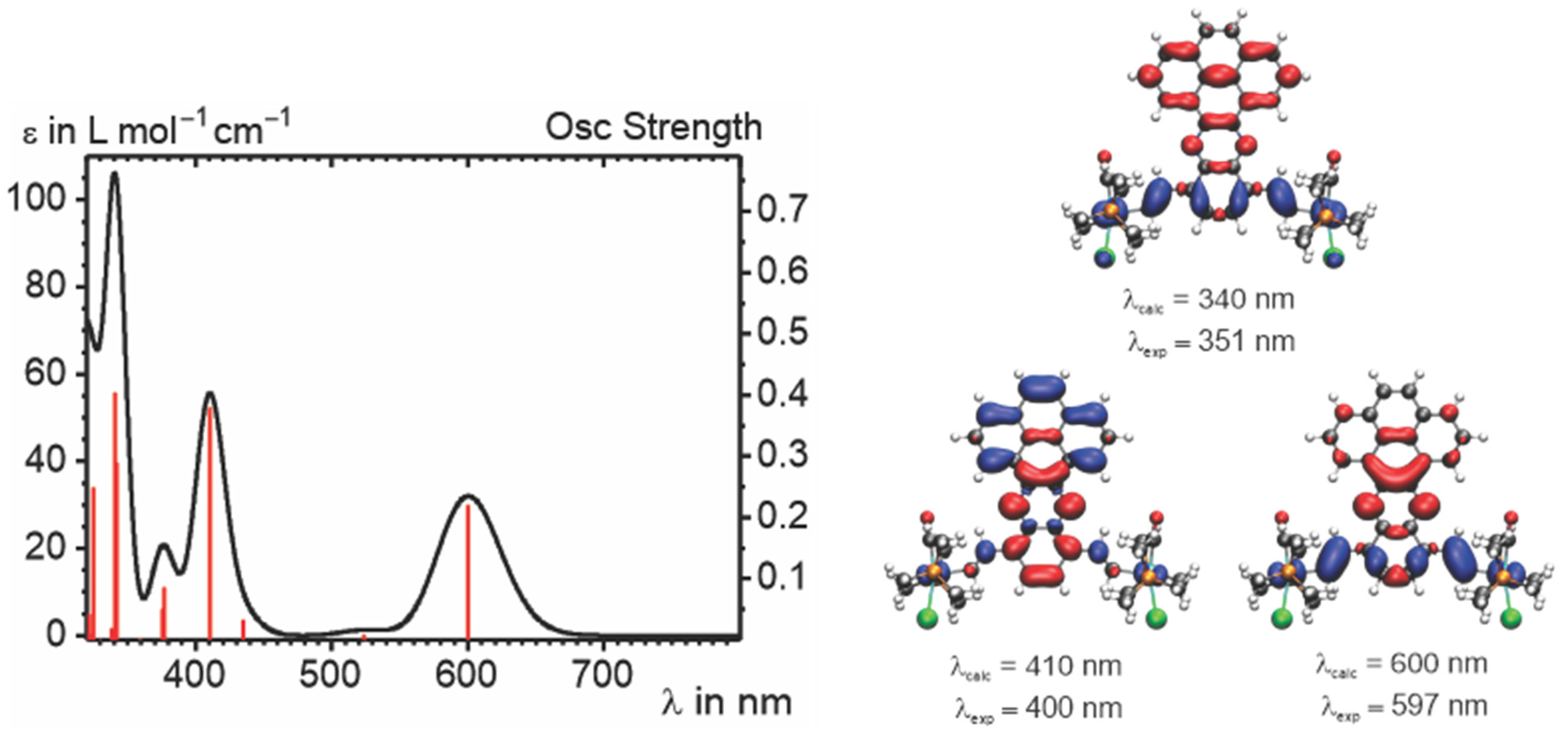
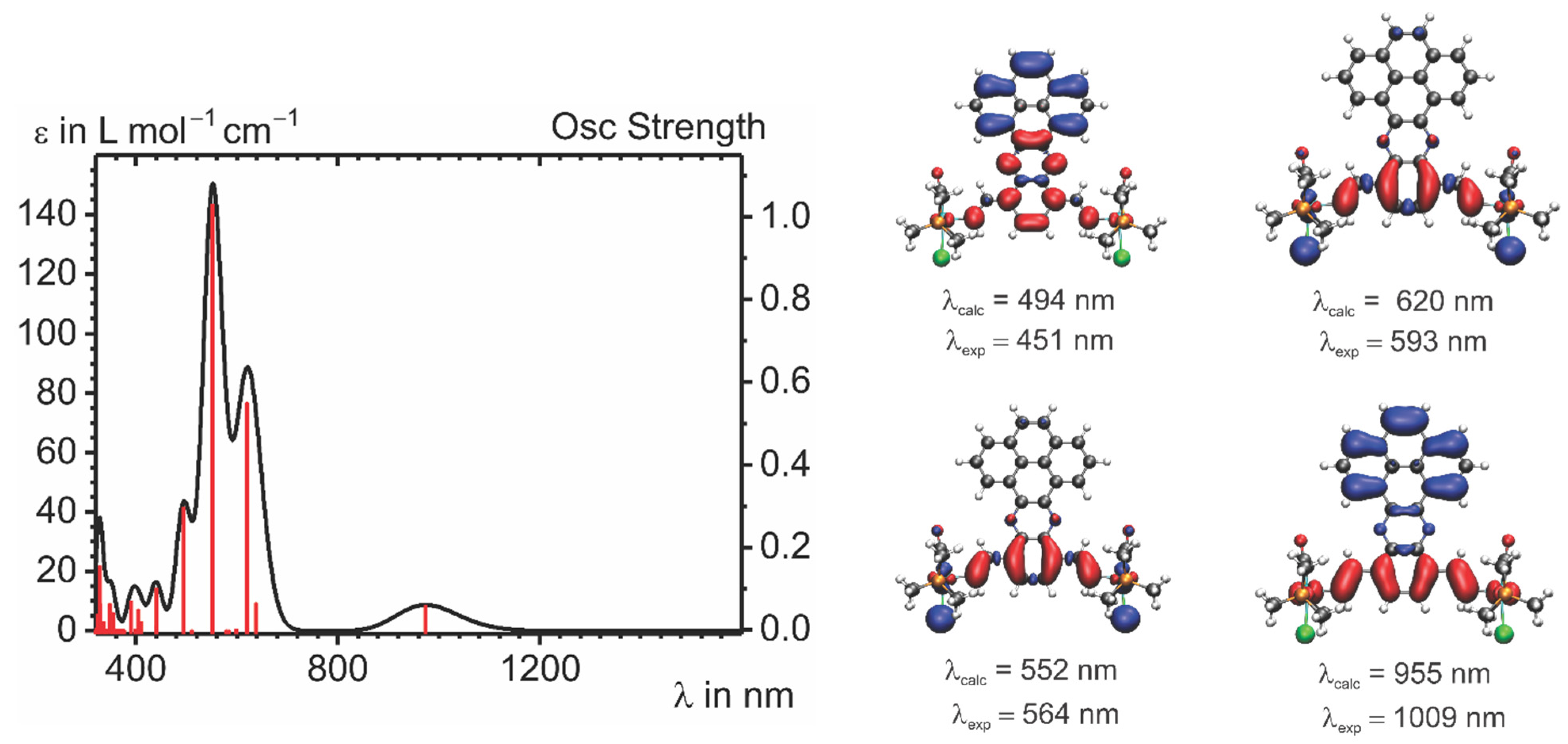
2.4. IR and UV–vis–NIR Spectroelectrochemistry
2.5. EPR Spectroscopic Studies
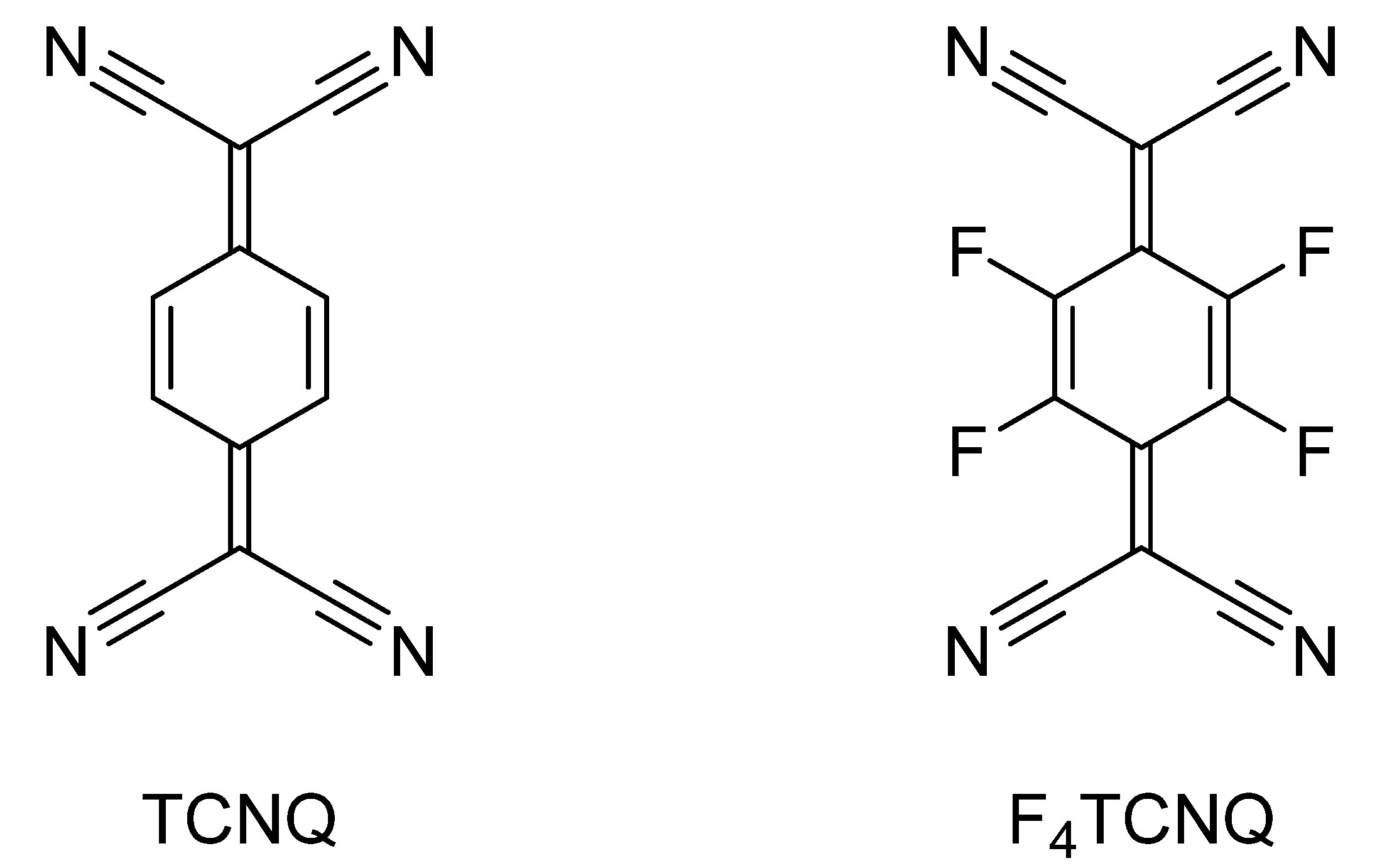
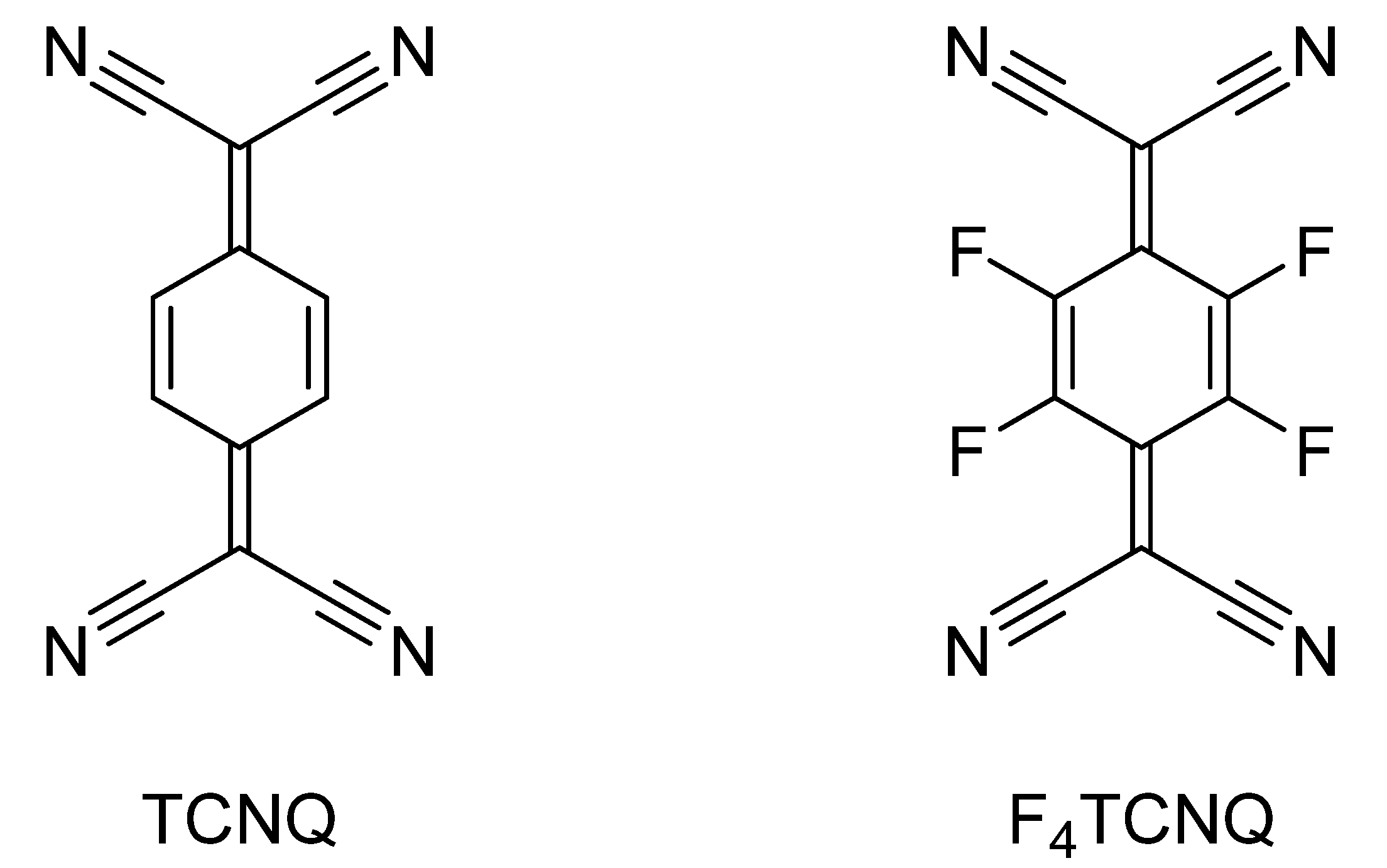
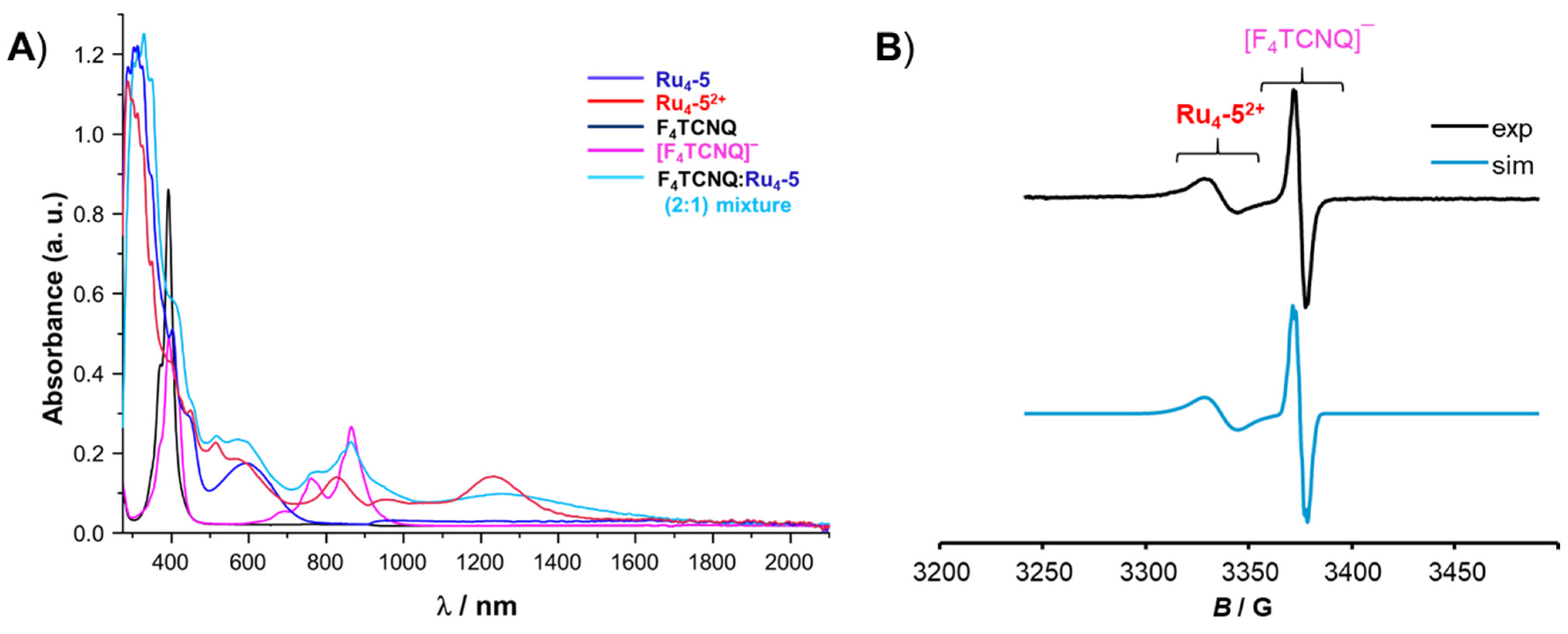
3. Charge-Transfer Salts of the Macrocycles
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cook, T.R.; Stang, P.J. Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages via Coordination. Chem. Rev. 2015, 115, 7001–7045. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, R.; Mukherjee, P.S.; Stang, P.J. Supramolecular coordination: Self-assembly of finite two- and three-dimensional ensembles. Chem. Rev. 2011, 111, 6810–6918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, M.; Tominaga, M.; Hori, A.; Therrien, B. Coordination Assemblies from a Pd(II)-Cornered Square Complex. Acc. Chem. Res. 2005, 38, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Szalóki, G.; Croué, V.; Carré, V.; Aubriet, F.; Alévêque, O.; Levillain, E.; Allain, M.; Aragó, J.; Ortí, E.; Goeb, S.; et al. Controlling the Host–Guest Interaction Mode through a Redox Stimulus. Angew. Chem. Int. Ed. 2017, 56, 16272–16276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szaloki, G.; Croue, V.; Allain, M.; Goeb, S.; Salle, M. Neutral versus polycationic coordination cages: A comparison regarding neutral guest inclusion. Chem. Commun. 2016, 52, 10012–10015. [Google Scholar] [CrossRef] [Green Version]
- Freudenreich, J.; Dalvit, C.; Süss-Fink, G.; Therrien, B. Encapsulation of Photosensitizers in Hexa- and Octanuclear Organometallic Cages: Synthesis and Characterization of Carceplex and Host–Guest Systems in Solution. Organometallics 2013, 32, 3018–3033. [Google Scholar] [CrossRef]
- Barry, N.P.E.; Zava, O.; Dyson, P.J.; Therrien, B. Encapsulation of inorganic and organic guest molecules into an organometallic hexacationic arene osmium metalla-prism: Synthesis, characterisation and anticancer activity. J. Organomet. Chem. 2012, 705, 1–6. [Google Scholar] [CrossRef]
- Ward, M.D.; Hunter, C.A.; Williams, N.H. Coordination Cages Based on Bis(pyrazolylpyridine) Ligands: Structures, Dynamic Behavior, Guest Binding, and Catalysis. Acc. Chem. Res. 2018, 51, 2073–2082. [Google Scholar] [CrossRef] [Green Version]
- Pluth, M.D.; Raymond, K.N. Reversible guest exchange mechanisms in supramolecular host-guest assemblies. Chem. Soc. Rev. 2007, 36, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Fujii, S.; Tada, T.; Komoto, Y.; Osuga, T.; Murase, T.; Fujita, M.; Kiguchi, M. Rectifying Electron-Transport Properties through Stacks of Aromatic Molecules Inserted into a Self-Assembled Cage. J. Am. Chem. Soc. 2015, 137, 5939–5947. [Google Scholar] [CrossRef]
- Kiguchi, M.; Ohto, T.; Fujii, S.; Sugiyasu, K.; Nakajima, S.; Takeuchi, M.; Nakamura, H. Single Molecular Resistive Switch Obtained via Sliding Multiple Anchoring Points and Varying Effective Wire Length. J. Am. Chem. Soc. 2014, 136, 7327–7332. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, M.; Takahashi, T.; Takahashi, Y.; Yamauchi, Y.; Murase, T.; Fujita, M.; Tada, T.; Watanabe, S. Electron Transport through Single Molecules Comprising Aromatic Stacks Enclosed in Self-Assembled Cages. Angew. Chem. Int. Ed. 2011, 50, 5708–5711. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Fraser Stoddart, J. From molecular to supramolecular electronics. Nat. Rev. Mater. 2021, 6, 804–828. [Google Scholar] [CrossRef]
- Das, R.; Linseis, M.; Schupp, S.M.; Schmidt-Mende, L.; Winter, R.F. Electron-Rich Diruthenium Complexes with π-Extended Alkenyl Ligands and Their F4TCNQ Charge-Transfer Salts. Chem. Eur. J. 2022, 28, e202104403. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.; Weibert, B.; Winter, R.F. Redox-active tetraruthenium metallacycles: Reversible release of up to eight electrons resulting in strong electrochromism. Chem. Commun. 2016, 52, 6103–6106. [Google Scholar] [CrossRef] [Green Version]
- Fink, D.; Bodensteiner, M.; Linseis, M.; Winter, R.F. Macrocyclic Triruthenium Complexes Having Electronically Coupled Mixed-Valent States. Chem. Eur. J. 2018, 24, 992–996. [Google Scholar] [CrossRef]
- Scheerer, S.; Linseis, M.; Wuttke, E.; Weickert, S.; Drescher, M.; Troppner, O.; Ivanovic-Burmazovic, I.; Irmler, A.; Pauly, F.; Winter, R.F. Redox-Active Tetraruthenium Macrocycles Built from 1,4-Divinylphenylene-Bridged Diruthenium Complexes. Chem. Eur. J. 2016, 22, 9574–9590. [Google Scholar] [CrossRef]
- Fink, D.; Orth, N.; Linseis, M.; Ivanovic-Burmazovic, I.; Winter, R.F. Ring size matters: Supramolecular isomerism in self-assembled redox-active tetra- and hexaruthenium macrocycles. Chem. Commun. 2020, 56, 1062–1065. [Google Scholar] [CrossRef]
- Fink, D.; Orth, N.; Linseis, M.; Ivanović-Burmazović, I.; Winter, R.F. Structural Versatility and Supramolecular Isomerism in Redox-Active Tetra- and Hexaruthenium Macrocycles. Eur. J. Inorg. Chem. 2020, 2020, 2816–2829. [Google Scholar] [CrossRef]
- Rotthowe, N.; Linseis, M.; Vogelsang, L.; Orth, N.; Ivanović-Burmazović, I.; Winter, R.F. A “Pretender” Croconate-Bridged Macrocyclic Tetraruthenium Complex: Sizable Redox Potential Splittings despite Electronically Insulated Divinylphenylene Diruthenium Entities. Molecules 2021, 26, 5232. [Google Scholar] [CrossRef]
- Fujita, M.; Sasaki, O.; Mitsuhashi, T.; Fujita, T.; Yazaki, J.; Yamaguchi, K.; Ogura, K. On the structure of transition-metal-linked molecular squares. Chem. Commun. 1996, 13, 1535–1536. [Google Scholar] [CrossRef]
- Suzuki, K.; Kawano, M.; Fujita, M. Solvato-Controlled Assembly of Pd3L6 and Pd4L8 Coordination “Boxes”. Angew. Chem. Int. Ed. 2007, 46, 2819–2822. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Beves, J.E.; Blight, B.A.; Campbell, C.J.; Leigh, D.A.; McBurney, R.T. Strategies and tactics for the metal-directed synthesis of rotaxanes, knots, catenanes, and higher order links. Angew. Chem. Int. Ed. 2011, 50, 9260–9327. [Google Scholar] [CrossRef] [PubMed]
- Neogi, S.; Lorenz, Y.; Engeser, M.; Samanta, D.; Schmittel, M. Heteroleptic Metallosupramolecular Racks, Rectangles, and Trigonal Prisms: Stoichiometry-Controlled Reversible Interconversion. Inorg. Chem. 2013, 52, 6975–6984. [Google Scholar] [CrossRef] [PubMed]
- Rancan, M.; Dolmella, A.; Seraglia, R.; Orlandi, S.; Quici, S.; Armelao, L. A templating guest sorts out a molecular triangle from a dimer–trimer constitutional dynamic library. Chem. Commun. 2012, 48, 3115–3117. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.-X.; Yang, H.-B. Supramolecular transformations within discrete coordination-driven supramolecular architectures. Chem. Soc. Rev. 2016, 45, 2656–2693. [Google Scholar] [CrossRef]
- Garci, A.; Marti, S.; Schurch, S.; Therrien, B. Insight into the dynamic ligand exchange process involved in bipyridyl linked arene ruthenium metalla-rectangles. RSC Adv. 2014, 4, 8597–8604. [Google Scholar] [CrossRef]
- Goswami, A.; Saha, S.; Biswas, P.K.; Schmittel, M. (Nano)mechanical Motion Triggered by Metal Coordination: From Functional Devices to Networked Multicomponent Catalytic Machinery. Chem. Rev. 2020, 120, 125–199. [Google Scholar] [CrossRef]
- Anyfanti, G.; Bauza, A.; Gentiluomo, L.; Rodrigues, J.; Portalone, G.; Frontera, A.; Rissanen, K.; Puttreddy, R. Short X…N Halogen Bonds With Hexamethylenetetraamine as the Acceptor. Front. Chem. 2021, 9, 623595. [Google Scholar] [CrossRef]
- Dang, L.-L.; Feng, H.-J.; Lin, Y.-J.; Jin, G.-X. Self-Assembly of Molecular Figure-Eight Knots Induced by Quadruple Stacking Interactions. J. Am. Chem. Soc. 2020, 142, 18946–18954. [Google Scholar] [CrossRef]
- Gao, X.; Cui, Z.; Shen, Y.-R.; Liu, D.; Lin, Y.-J.; Jin, G.-X. Synthesis and Near-Infrared Photothermal Conversion of Discrete Supramolecular Topologies Featuring Half-Sandwich [Cp*Rh] Units. J. Am. Chem. Soc. 2021, 143, 17833–17842. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, W.; Sheng, P.; Zhao, G.; Zhu, D. Organic Donor-Acceptor Complexes as Novel Organic Semiconductors. Acc. Chem. Res. 2017, 50, 1654–1662. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Luo, L.; Sheng, P.; Zhang, J.; Zhang, Q. Multifunctional Features of Organic Charge-Transfer Complexes: Advances and Perspectives. Chem. Eur. J. 2021, 27, 464–490. [Google Scholar] [CrossRef] [PubMed]
- Goetz, K.P.; Vermeulen, D.; Payne, M.E.; Kloc, C.; McNeil, L.E.; Jurchescu, O.D. Charge-transfer complexes: New perspectives on an old class of compounds. J. Mater. Chem. C 2014, 2, 3065–3076. [Google Scholar] [CrossRef]
- Bassler, H.; Kohler, A. Charge transport in organic semiconductors. Top. Curr. Chem. 2012, 312, 1–65. [Google Scholar] [CrossRef]
- Wosnitza, J. Superconductivity in Layered Organic Metals. Crystals 2012, 2, 248–265. [Google Scholar] [CrossRef]
- Fink, D.; Orth, N.; Ebel, V.; Gogesch, F.S.; Staiger, A.; Linseis, M.; Ivanović-Burmazović, I.; Winter, R.F. Self-Assembled Redox-Active Tetraruthenium Macrocycles with Large Intracyclic Cavities. Organometallics 2020, 39, 1861–1880. [Google Scholar] [CrossRef]
- Rudolph, M.; Feldberg, S. DigiSim3; Version 3.03; Bioanalytical Systems, Inc.: West Lafayette, IN, USA, 1994. [Google Scholar]
- Fink, D.; Linseis, M.; Winter, R.F. Constitutional Isomers of Macrocyclic Tetraruthenium Complexes with Vastly Different Spectroscopic and Electrochemical Properties. Organometallics 2018, 37, 1817–1820. [Google Scholar] [CrossRef]
- Fink, D.; Staiger, A.; Orth, N.; Linseis, M.; Ivanovic-Burmazovic, I.; Winter, R.F. Redox-Induced Hydrogen Bond Reorientation Mimicking Electronic Coupling in Mixed-Valent Diruthenium and Macrocyclic Tetraruthenium Complexes. Inorg. Chem. 2020, 59, 16703–16715. [Google Scholar] [CrossRef]
- Sherlock, S.J.; Boyd, D.C.; Moasser, B.; Gladfelter, W.L. Homogeneous catalytic carbonylation of nitroaromatics. 4. Preparation and characterization of ruthenium radical cations. Inorg. Chem. 1991, 30, 3626–3632. [Google Scholar] [CrossRef]
- Maurer, J.; Winter, R.F.; Sarkar, B.; Fiedler, J.; Záliš, S. Bridge dominated oxidation of a diruthenium 1,3-divinylphenylene complex. Chem. Commun. 2004, 17, 1900–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, J.; Sarkar, B.; Schwederski, B.; Kaim, W.; Winter, R.F.; Záliš, S. Divinylphenylene-Bridged Diruthenium Complexes Bearing Ru(CO)Cl(PiPr3)2 Entities. Organometallics 2006, 25, 3701–3712. [Google Scholar] [CrossRef] [Green Version]
- Mücke, P.; Linseis, M.; Záliš, S.; Winter, R.F. Vinyl-ruthenium entities as markers for intramolecular electron transfer processes. Inorg. Chim. Acta 2011, 374, 36–50. [Google Scholar] [CrossRef] [Green Version]
- Scheerer, S.; Rotthowe, N.; Abdel-Rahman, O.S.; He, X.; Rigaut, S.; Kvapilova, H.; Zalis, S.; Winter, R.F. Vinyl ruthenium-modified biphenyl and 2,2′-bipyridines. Inorg. Chem. 2015, 54, 3387–3402. [Google Scholar] [CrossRef] [PubMed]
- Linseis, M.; Zalis, S.; Zabel, M.; Winter, R.F. Ruthenium stilbenyl and diruthenium distyrylethene complexes: Aspects of electron delocalization and electrocatalyzed isomerization of the Z-isomer. J. Am. Chem. Soc. 2012, 134, 16671–16692. [Google Scholar] [CrossRef]
- Rotthowe, N.; Zwicker, J.; Winter, R.F. Influence of Quinoidal Distortion on the Electronic Properties of Oxidized Divinylarylene-Bridged Diruthenium Complexes. Organometallics 2019, 38, 2782–2799. [Google Scholar] [CrossRef]
- Krejčik, M.; Daněk, M.; Hartl, F. Simple construction of an infrared optically transparent thin-layer cell: Applications to the redox reactions of ferrocene, Mn2(CO)10 and Mn(CO)3(3,5-di-t-butyl-catecholate). J. Electroanal. Chem. Interfac. Electrochem. 1991, 317, 179–187. [Google Scholar] [CrossRef]
- Connelly, N.G.; Geiger, W.E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, G.; Zhou, Y.; Long, G.; Gu, P.; Zhang, Q. Solvent Accommodation: Functionalities Can Be Tailored through Co-Crystallization Based on 1:1 Coronene-F4TCNQ Charge-Transfer Complex. ACS Appl. Mater. Interfaces 2017, 9, 1183–1188. [Google Scholar] [CrossRef]
- Hu, P.; Li, H.; Li, Y.; Jiang, H.; Kloc, C. Single-crystal growth, structures, charge transfer and transport properties of anthracene-F4TCNQ and tetracene-F4TCNQ charge-transfer compounds. CrystEngComm 2017, 19, 618–624. [Google Scholar] [CrossRef]
- Cochran, J.E.; Junk, M.J.N.; Glaudell, A.M.; Miller, P.L.; Cowart, J.S.; Toney, M.F.; Hawker, C.J.; Chmelka, B.F.; Chabinyc, M.L. Molecular Interactions and Ordering in Electrically Doped Polymers: Blends of PBTTT and F4TCNQ. Macromolecules 2014, 47, 6836–6846. [Google Scholar] [CrossRef]
- Le, T.H.; Nafady, A.; Vo, N.T.; Elliott, R.W.; Hudson, T.A.; Robson, R.; Abrahams, B.F.; Martin, L.L.; Bond, A.M. Electrochemically directed synthesis of Cu2(I)(TCNQF4(II−))(MeCN)2 (TCNQF4 = 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane): Voltammetry, simulations, bulk electrolysis, spectroscopy, photoactivity, and X-ray crystal structure of the Cu2(I)(TCNQF4(II))(EtCN)2 analogue. Inorg. Chem. 2014, 53, 3230–3242. [Google Scholar] [CrossRef] [PubMed]
- Pingel, P.; Neher, D. Comprehensive picture of p-type doping of P3HT with the molecular acceptor F4TCNQ. Phys. Rev. B 2013, 87, 115209. [Google Scholar] [CrossRef]
- O’Kane, S.A.; Clérac, R.; Zhao, H.; Ouyang, X.; Galán-Mascarós, J.R.; Heintz, R.; Dunbar, K.R. New Crystalline Polymers of Ag(TCNQ) and Ag(TCNQF4): Structures and Magnetic Properties. J. Solid State Chem. 2000, 152, 159–173. [Google Scholar] [CrossRef]
- Le, T.H.; Nafady, A.; Lu, J.; Peleckis, G.; Bond, A.M.; Martin, L.L. Electrochemical Synthesis and Characterization of Semiconducting Ni(TCNQF4)2(H2O)2 (TCNQF4 = 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane). Eur. J. Inorg. Chem. 2012, 2012, 2889–2897. [Google Scholar] [CrossRef]
- Murata, T.; Saito, G.; Nakamura, K.; Maesato, M.; Hiramatsu, T.; Yoshida, Y. Exploration of Charge-Transfer Solids Utilizing Nucleobases: Nanoarchitectures by Hydrogen-Bonds in the Ionic Assemblies of Guanine and TCNQ Derivatives. Cryst. Growth Des. 2013, 13, 2778–2792. [Google Scholar] [CrossRef]
- Gerson, F.; Heckendorn, R.; Cowan, D.O.; Kini, A.M.; Maxfield, M. Radical anions and radical trianions of tetracyanoarenoquinodimethanes. An ESR and ENDOR study. J. Am. Chem. Soc. 1983, 105, 7017–7023. [Google Scholar] [CrossRef]
- Jones, M.T.; Maruo, T.; Jansen, S.; Roble, J.; Rataiczak, R.D. Comparative ESR Studies of Solid Polycrystalline Alkali Metal Salts of TCNQ and TCNQF4. Mol. Cryst. Liq. Cryst. 1986, 134, 21–30. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 7, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGXandORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the rare earth elements. J. Chem. Phys. 1989, 90, 1730–1734. [Google Scholar] [CrossRef]
- Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted pseudopotentials for the actinides. Parameter sets and test calculations for thorium and thorium monoxide. J. Chem. Phys. 1994, 100, 7535–7542. [Google Scholar] [CrossRef]
- Andrae, D.; Haeussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Hariharan, P.H.; Pople, J.A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 6158–6170. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tange, O. GNU Parallel—The Command-Line Power Tool. USENIX Mag. 2011, 36, 42–47. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Persistence of Vision Raytracer; Version 3.7; Persistence of Vision Raytracer Pty. Ltd.: Williamstown, Australia, 2004.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E1/20/+ (ΔEp) | E1/2+/2+ (ΔEp) | E1/22+/3+ (ΔEp) | E1/23+/4+ (ΔEp) | ΔE1/22+/4+ | E1/20/− or E1/20/2− (ΔEp) | |
|---|---|---|---|---|---|---|
| Ru2-3 | −68 (111) [−154 (64)] | 77 (104) [78 (67)] | − | − | 145 [232] | −2085 (175) [−1510 (-)] 2 |
| Ru4-5 | −134 (101) [−280 (-)] 3 | −134 (101) [−200 (-)] 3 | −6 (106) [−28 (-)] 3 | −6 (106) [30 (-)] 3 | 128 [240] | −2120 (96) [−2074 (128)] 2 |
| Ru4-6 | −174 (102) [−320 (-)] 3 | −174 (102) [−240(-)] 3 | −36 (89) [−66(-)] 3 | −36 (89) [−10(-)] 3 | 138 [240] | −2159 (102) [−2118(157)] 2 |
| νCO (cm−1) | λ (nm) (εmax [M−1cm−1]) | |
|---|---|---|
| Ru2-3 | 1912 | 351 (48,000), 400 (26,300), 597 (11,200) |
| [Ru2-3]+ | 1934, 1945 (sh) | 350 (33,700), 452 (17,000), 512 (17,700), 602 (20,500), 814 (8800), 947 (5500), 1220 (7900) |
| [Ru2-3]2+ | 1955 | 349 (35,000), 451 (14,400), 564 (1,3000), 1009 (9800) |
| Ru4-5 | 1906 | 312 (84,500), 347 (65,000), 403 (36,000), 444 (22,200), 599 (12,400) |
| [Ru4-5]2+ | 1932, 1946 | 312 (73,000), 349 (35,200), 417 (27,100), 482 (37,500), 509 (44,000), 811 (54,000), 950 (18,300), 1065 (18,700), 1200 (47,000) |
| [Ru4-5]4+ | 1973 | 306 (87,000), 347 (40,000), 505 (50,000), 576 (62,000), 900 (8450), 1020 (9700) |
| Ru4-6 | 1905 | 285 (83,000), 302 (82,500), 311 (82,000), 324 (80,000), 348 (67,200), 402 (42,800), 445 (19,000), 606 (14,800) |
| [Ru4-6]2+ | 1930, 1940 | 311 (55,840), 418 (26,000), 488 (39,500), 512 (47,100), 598 (19,700), 818 (50,000), 960 (17,300), 1084 (17,000), 1222 (47,200) |
| [Ru4-6]4+ | 1952 | 278 (97,500), 315 (55,000), 494 (47,400), 585 (66,000), 890 (6900), 1010 (6200) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, R.; Linseis, M.; Senft, L.; Ivanović-Burmazović, I.; Winter, R.F. Tetraruthenium Macrocycles with Laterally Extended Bis(alkenyl)quinoxaline Ligands and Their F4TCNQ•− Salts. Inorganics 2022, 10, 82. https://doi.org/10.3390/inorganics10060082
Das R, Linseis M, Senft L, Ivanović-Burmazović I, Winter RF. Tetraruthenium Macrocycles with Laterally Extended Bis(alkenyl)quinoxaline Ligands and Their F4TCNQ•− Salts. Inorganics. 2022; 10(6):82. https://doi.org/10.3390/inorganics10060082
Chicago/Turabian StyleDas, Rajorshi, Michael Linseis, Laura Senft, Ivana Ivanović-Burmazović, and Rainer F. Winter. 2022. "Tetraruthenium Macrocycles with Laterally Extended Bis(alkenyl)quinoxaline Ligands and Their F4TCNQ•− Salts" Inorganics 10, no. 6: 82. https://doi.org/10.3390/inorganics10060082
APA StyleDas, R., Linseis, M., Senft, L., Ivanović-Burmazović, I., & Winter, R. F. (2022). Tetraruthenium Macrocycles with Laterally Extended Bis(alkenyl)quinoxaline Ligands and Their F4TCNQ•− Salts. Inorganics, 10(6), 82. https://doi.org/10.3390/inorganics10060082