
Ionic Conductivity of LiSiON and the Effect of Amorphization/Heterovalent Doping on Li+ Diffusion


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
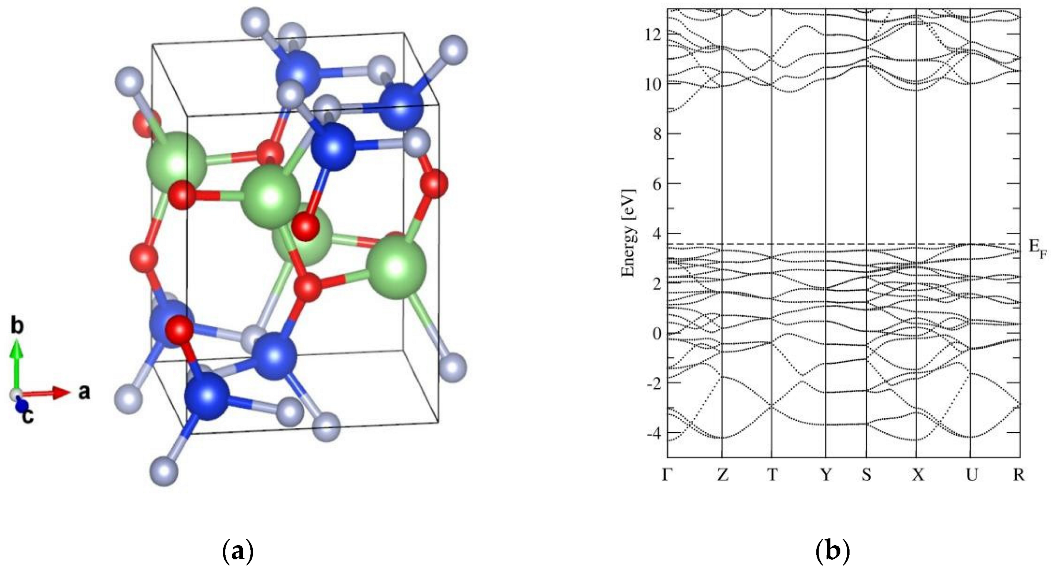
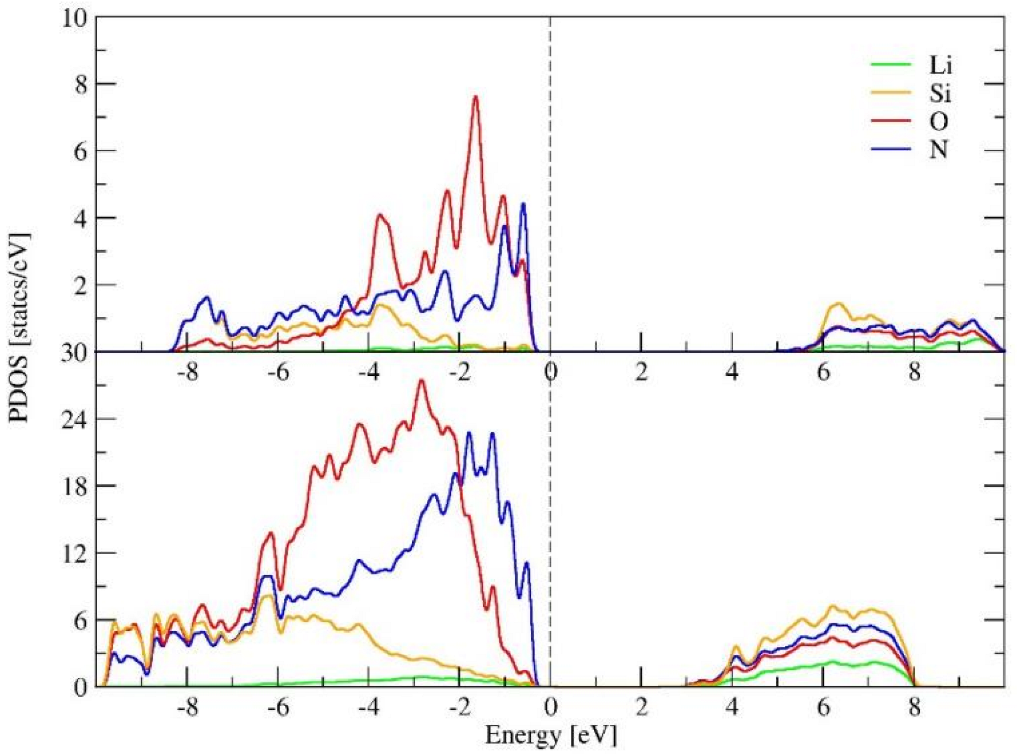
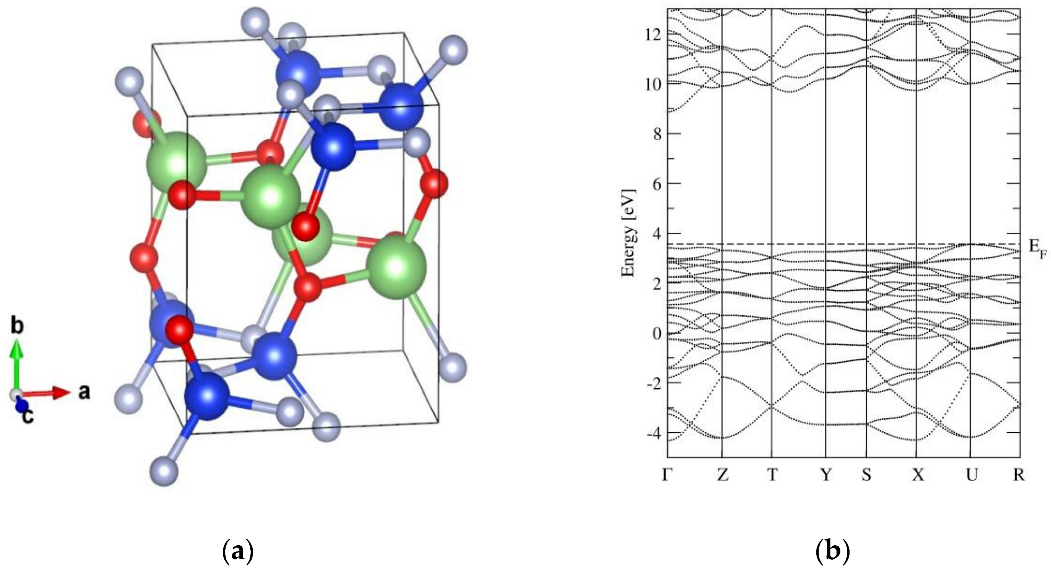
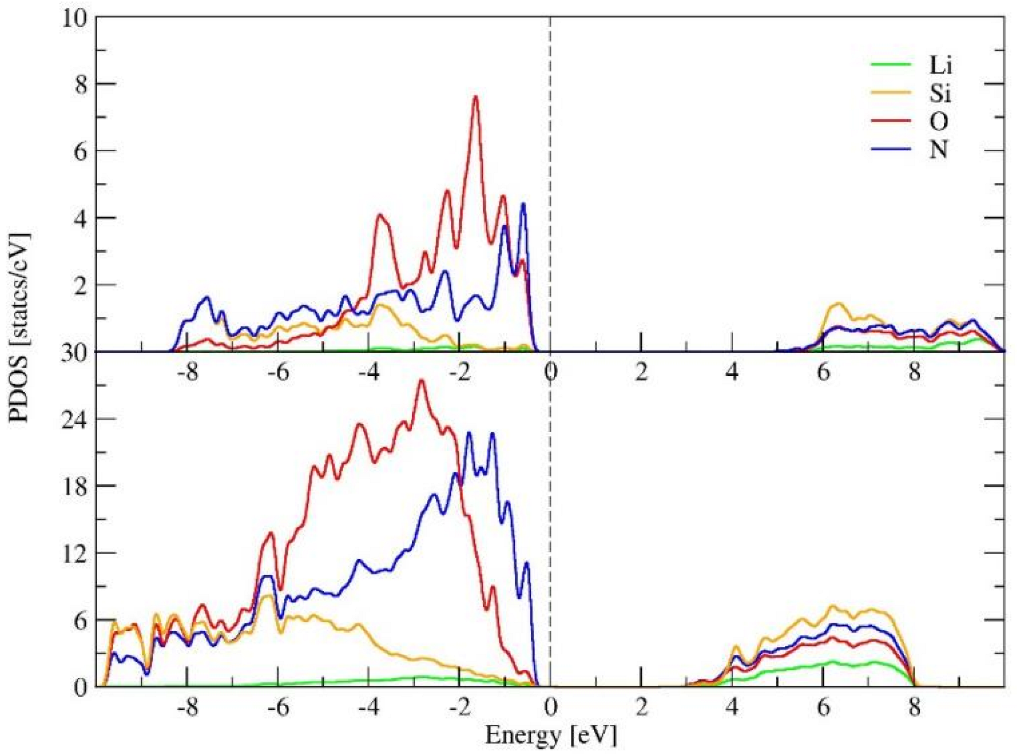
2.1. The Crystal Structure and Basic Properties of α-LiSiON
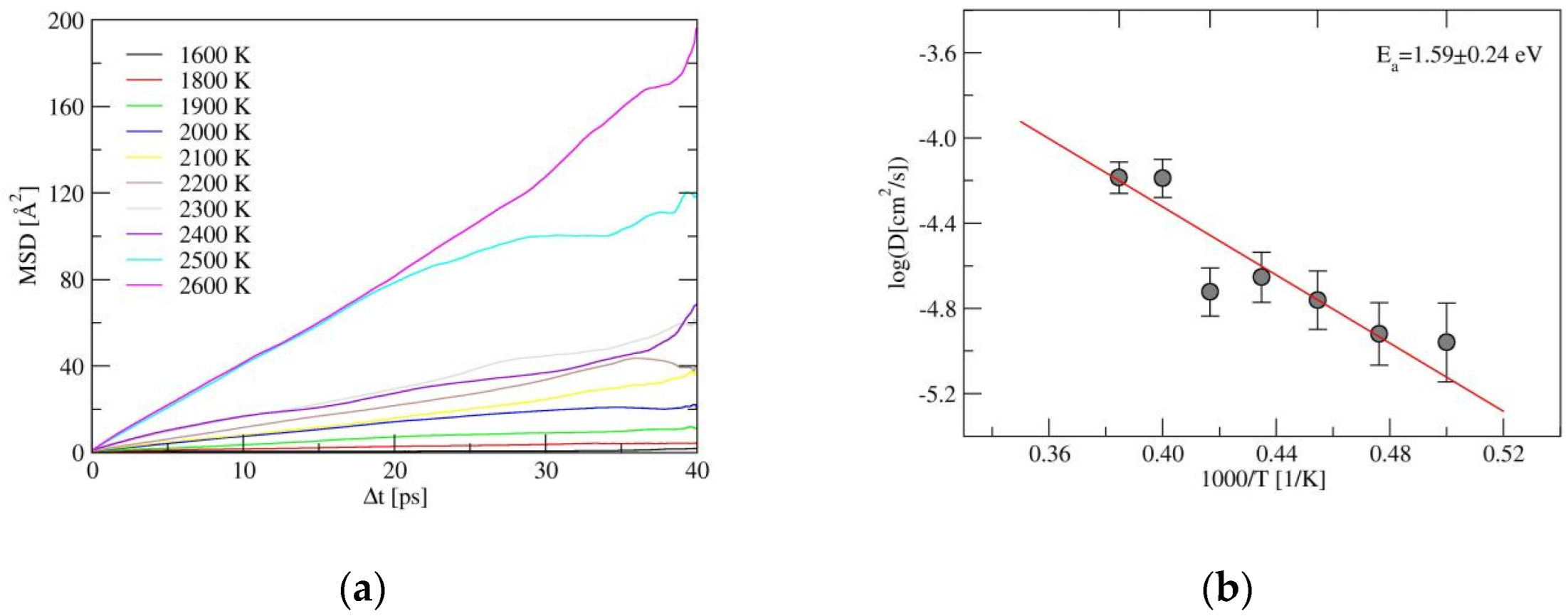
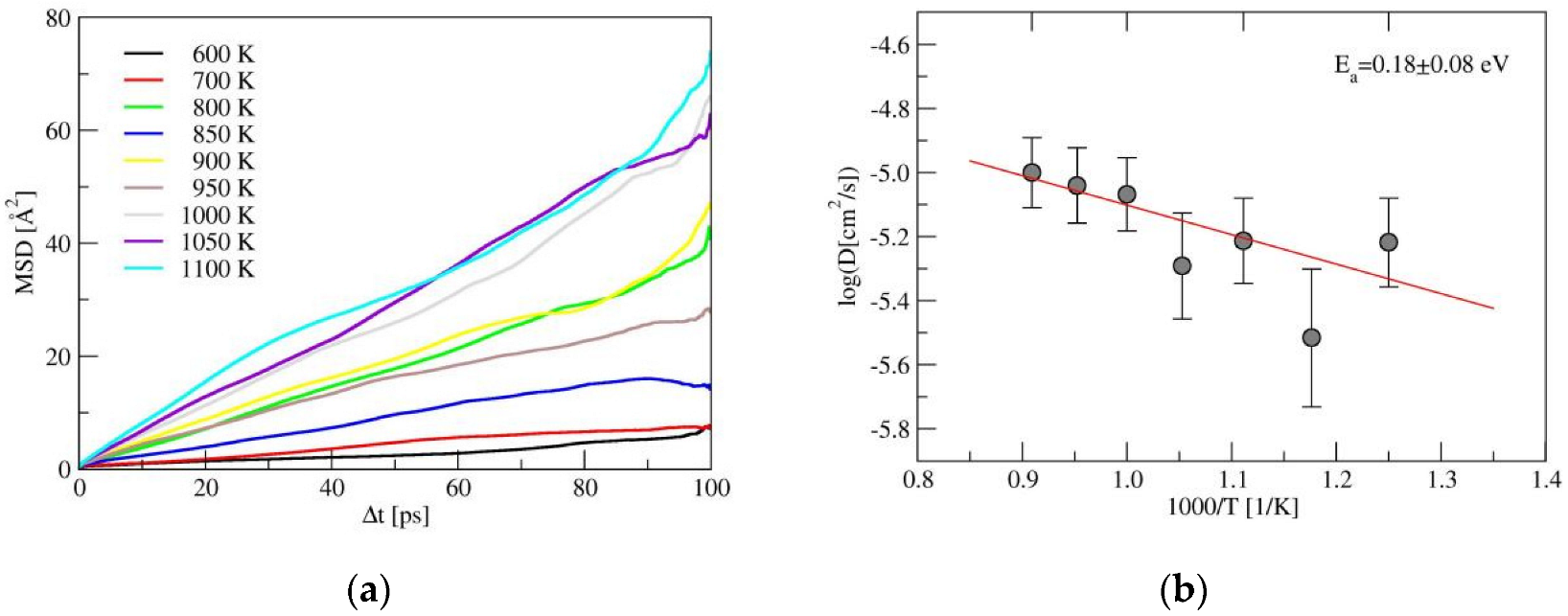
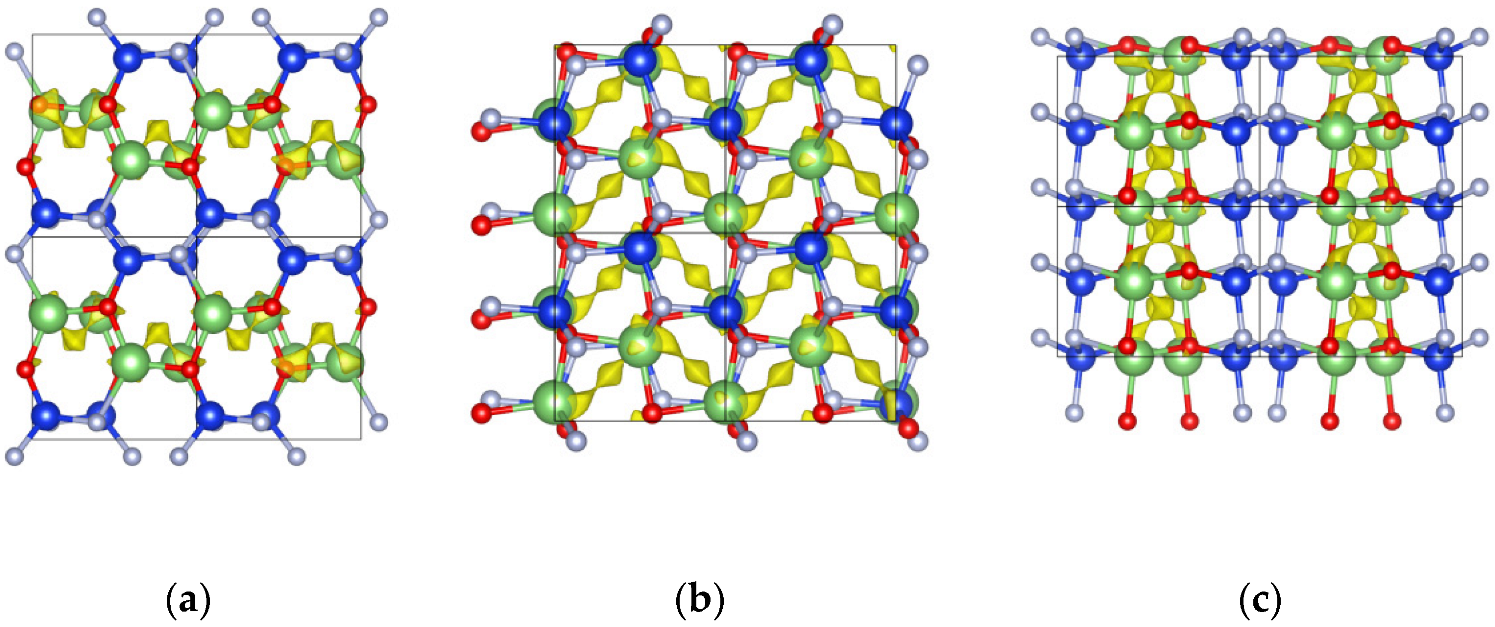
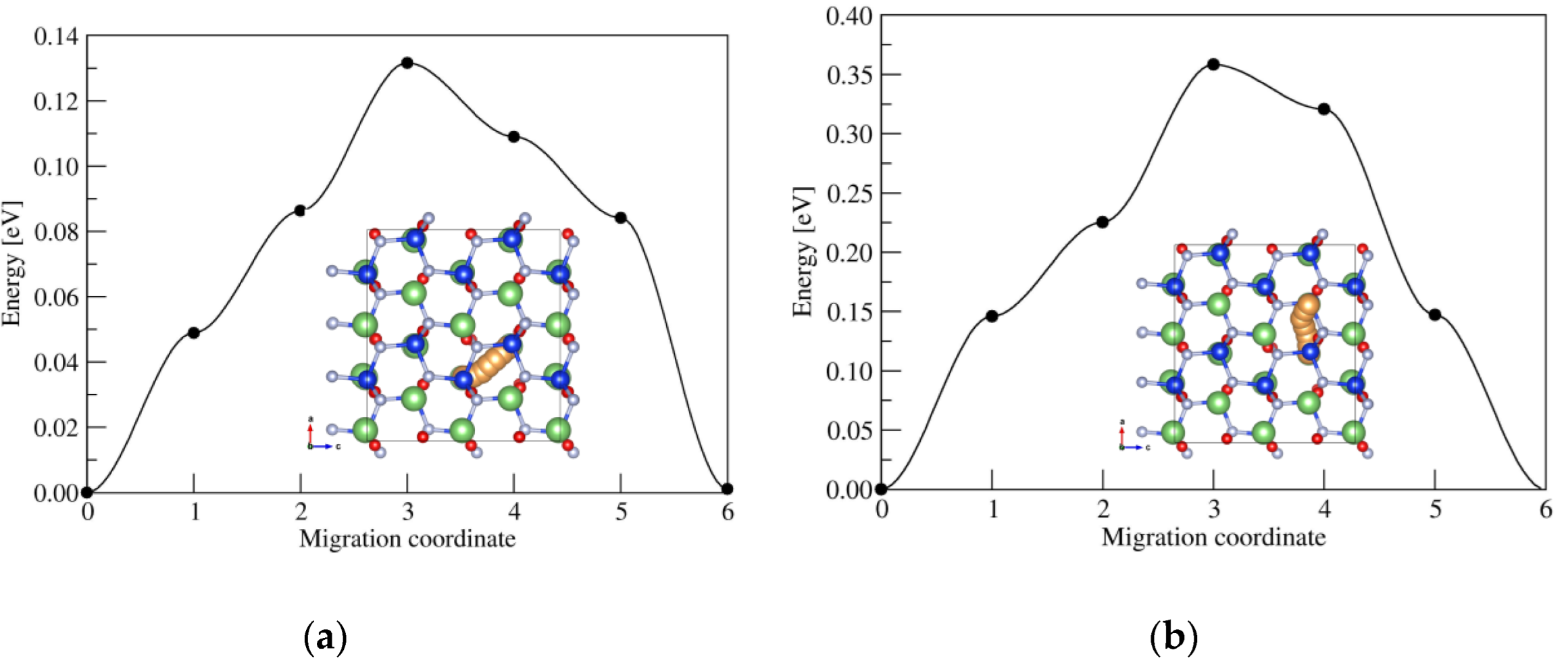
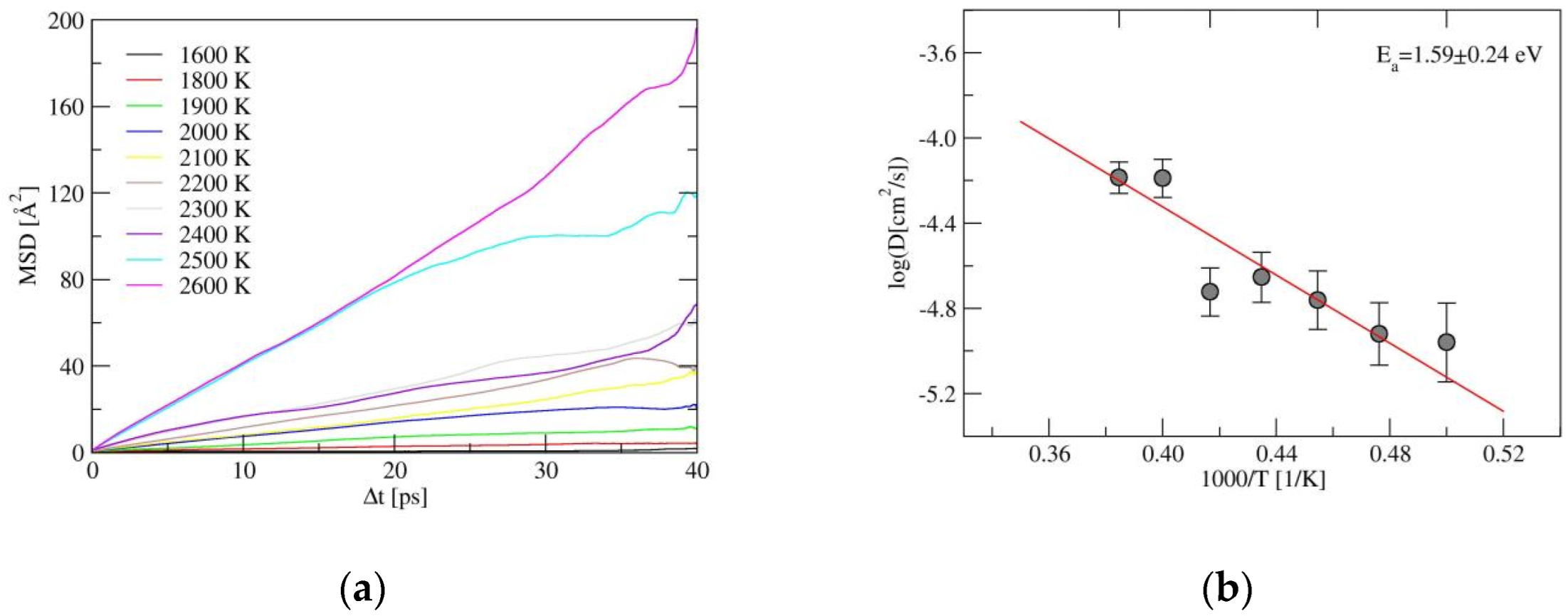
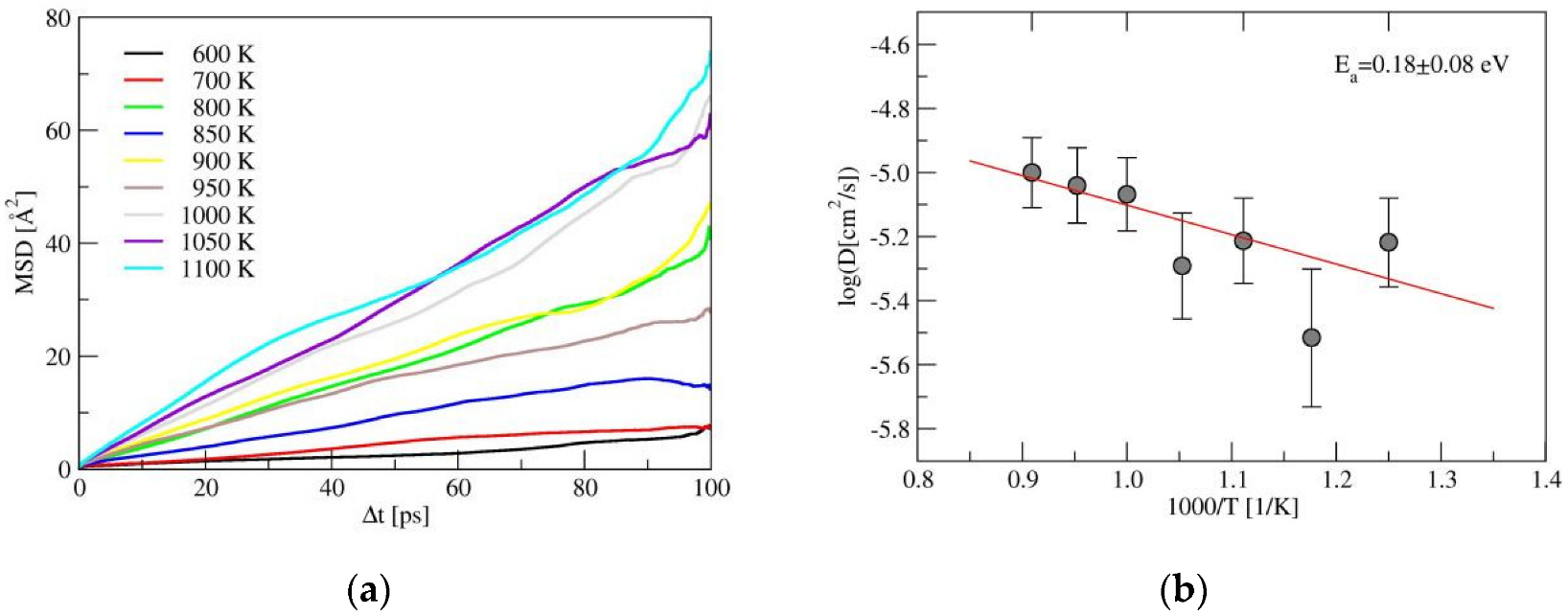
2.2. Ionic Diffusion Properties of α-LiSiON
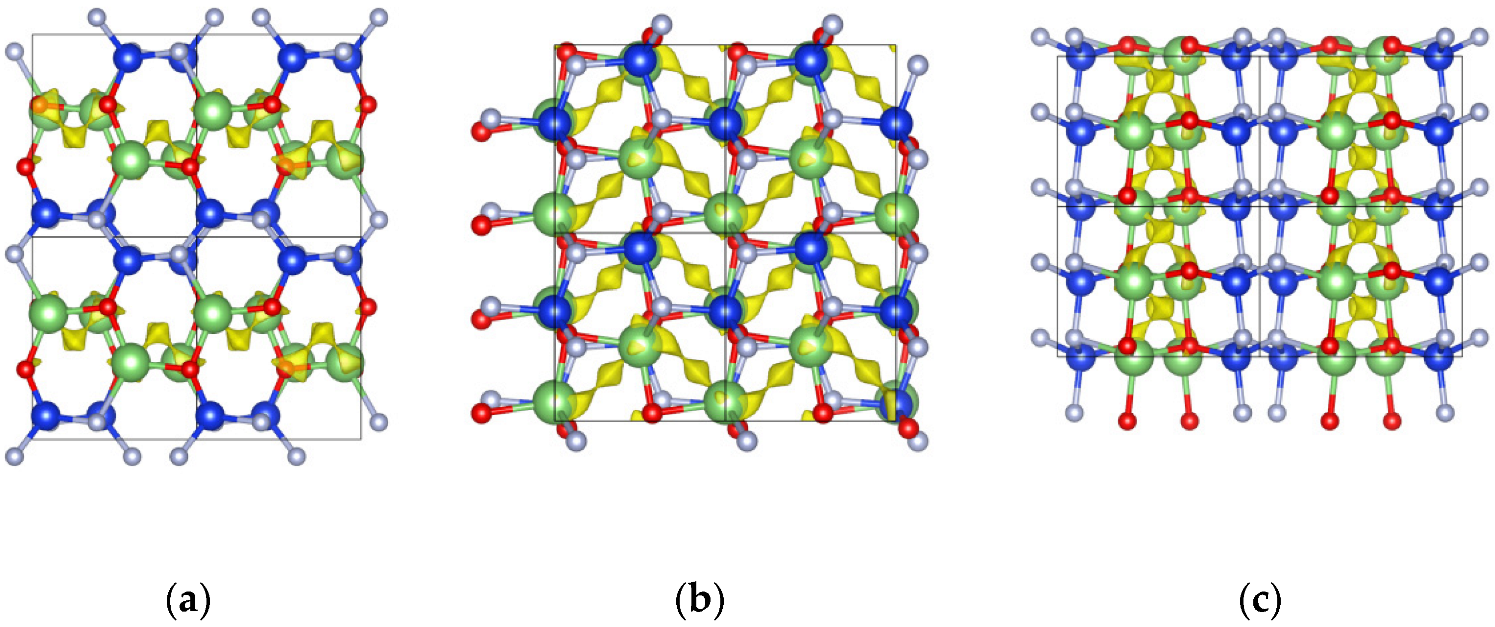
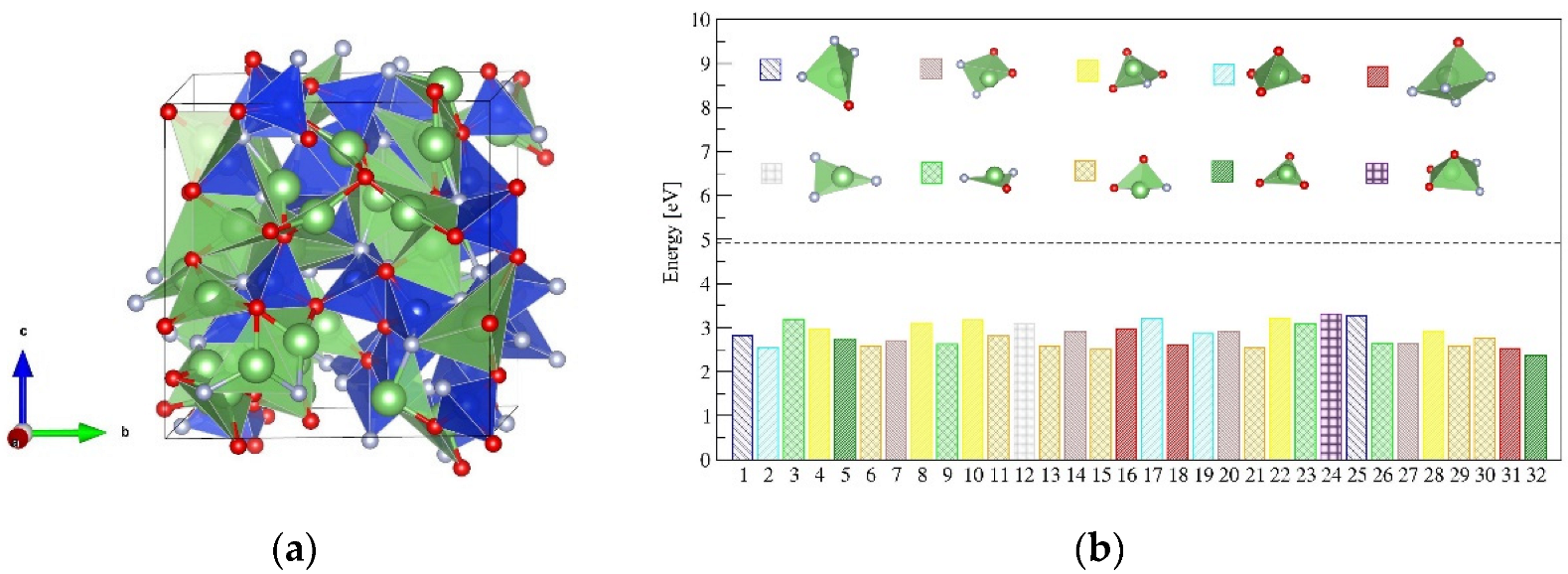
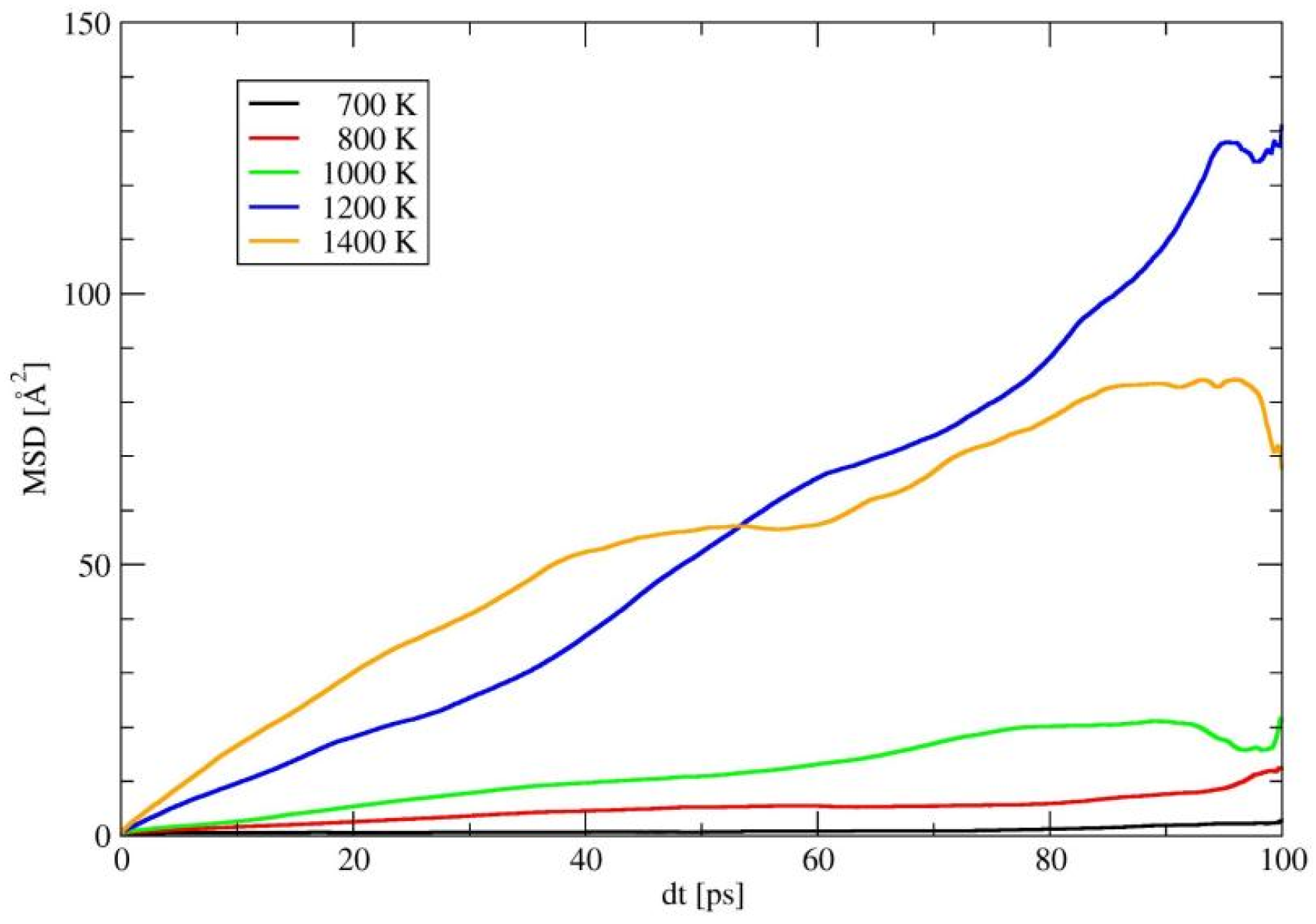
2.3. The Properties of Amorphous-LiSiON
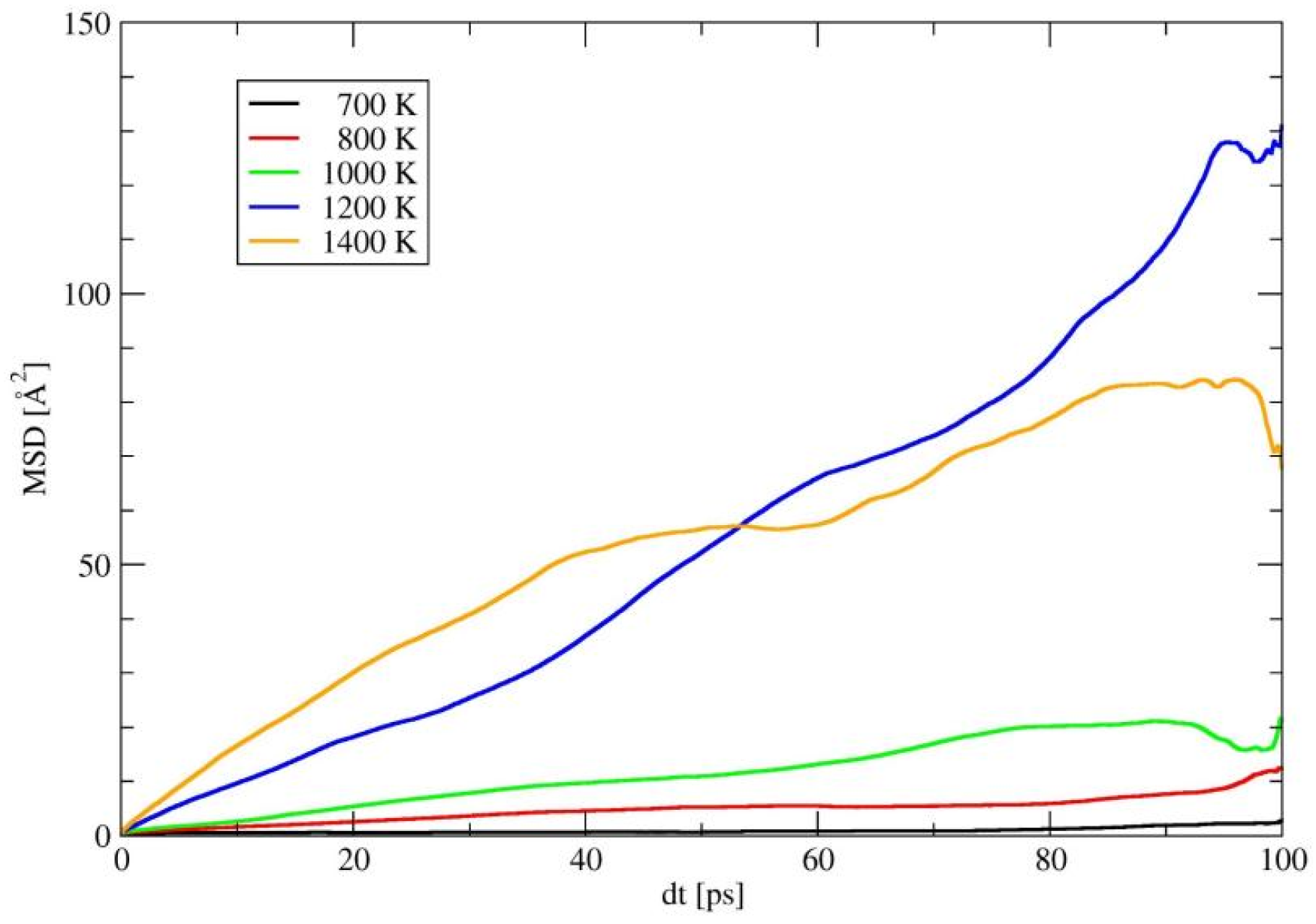
2.4. Ionic Diffusion Properties of P-Doped α-LiSiON
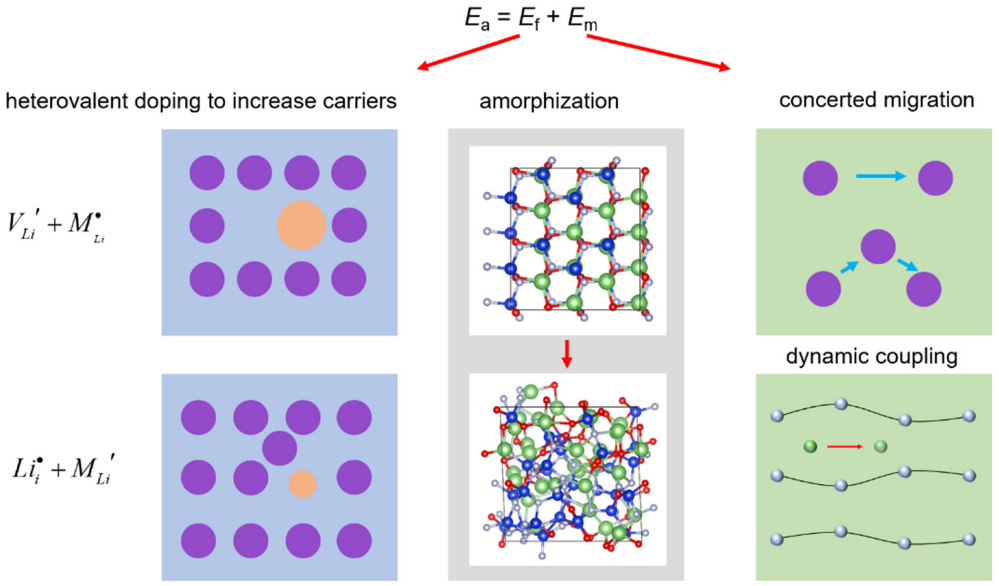
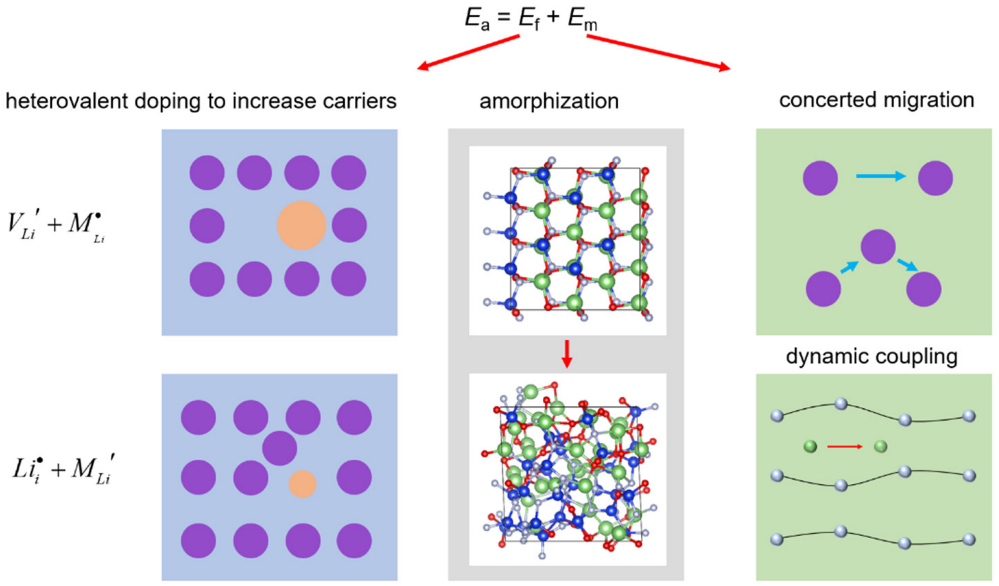
3. Discussion and Conclusions
4. Materials and Methods
4.1. DFT-Based Structural Relaxation and Band Structure Calculations
4.2. BV and NEB Simulations
4.3. The Model Building of Amorphous-LiSiON
4.4. The AIMD Simulations
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Armand, M.; Tarascon, J.M. Building better batteries. Nature 2008, 451, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, J.B.; Kim, Y. Challenges for Rechargeable Li Batteries. Chem. Mater. 2010, 22, 587–603. [Google Scholar] [CrossRef]
- Viswanathan, V.; Epstein, A.H.; Chiang, Y.; Takeuchi, E.; Bradley, M.; Langford, J.; Winter, M. The challenges and opportunities of battery-powered flight. Nature 2022, 601, 519–525. [Google Scholar] [CrossRef]
- Hu, Y. Batteries: Getting solid. Nat. Energy 2016, 1, 16042. [Google Scholar] [CrossRef]
- Janek, J.; Zeier, W. A solid future for battery development. Nat. Energy 2016, 1, 16141. [Google Scholar] [CrossRef]
- Balaish, M.; Gonzalez-Rosillo, J.; Kim, K.; Zhu, Y.; Hood, Z.; Rupp, J.M. Processing thin but robust electrolytes for solid-state batteries. Nat. Rev. Mater. 2020, 5, 229. [Google Scholar] [CrossRef]
- Xu, L.; Lu, Y.; Zhao, C.; Yuan, H.; Zhu, G.; Hou, L.; Zhang, Q.; Huang, J. Toward the Scale-Up of Solid-State Lithium Metal Batteries: The Gaps between Lab-Level Cells and Practical Large-Format Batteries. Adv. Energy Mater. 2020, 11, 2002360. [Google Scholar] [CrossRef]
- Kamaya, N.; Homma, K.; Yamakawa, Y.; Hirayama, M.; Kanno, R.; Yonemura, M.; Kamiyama, T.; Kato, Y.; Hama, S.; Kawamoto, K.; et al. A lithium superionic conductor. Nat. Mater. 2011, 10, 682–686. [Google Scholar] [CrossRef]
- Kato, Y.; Hori, S.; Saito, T.; Suzuki, K.; Hirayama, M.; Mitsui, A.; Yonemura, M.; Iba, H.; Kanno, R. High-power all-solid-state batteries using sulfide superionic conductors. Nat. Energy 2016, 1, 16030. [Google Scholar] [CrossRef]
- Bates, J.; Engstrom, H.; Wang, J.; Larson, B.; Dudney, N.; Brundage, W. Composition, ion-ion correlations and conductivity of beta″-alumina. Solid State Ionics 1981, 5, 159–162. [Google Scholar] [CrossRef]
- Murugan, R.; Thangadurai, V.; Weppner, W. Fast Lithium Ion Conduction in Garnet-Type Li7La3Zr2O12. Angew. Chem. Int. Ed. 2007, 46, 7778–7781. [Google Scholar] [CrossRef] [PubMed]
- Inaguma, Y.; Chen, L.; Itoh, M.; Nakamura, T.; Uchida, T.; Ikuta, H.; Wakihara, M. High ionic conductivity in lithium lanthanum titanate. Solid State Commun. 1993, 86, 689–693. [Google Scholar] [CrossRef]
- Fu, J. Fast Li+ ion conducting glass-ceramics in the system Li2O–Al2O3–GeO2–P2O5. Solid State Ionics 1997, 10, 191–194. [Google Scholar] [CrossRef]
- Huang, W.; Matsui, N.; Hori, S.; Suzuki, K.; Hirayama, M.; Yonemura, M.; Saito, T.; Kamiyama, T.; Sasaki, Y.; Yoon, Y.; et al. Anomalously High Ionic Conductivity of Li2SiS3-Type Conductors. J. Am. Chem. Soc. 2022, 144, 4989–4994. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Li, H.; Chen, L. High-throughput design and optimization of fast lithium ion conductors by the combination of bond-valence method and density functional theory. Sci. Rep. 2015, 5, 14227. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xiao, R.; Li, H.; Chen, L. Oxygen-driven transition from two-dimensional to three-dimensional transport behaviour in β-Li3PS4 electrolyte. Phys. Chem. Chem. Phys. 2016, 18, 21269–21277. [Google Scholar] [CrossRef]
- Wang, X.; Xiao, R.; Li, H.; Chen, L. Oxysulfide LiAlSO: A Lithium Superionic Conductor from First Principles. Phys. Rev. Lett. 2017, 118, 195901. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Yao, Z.; Hegde, V.; Naghavi, S.; Shen, J.; Bushick, K.; Wolverton, C. Computational Discovery of Stable Heteroanionic Oxychalcogenides ABXO (A, B = Metals; X = S, Se, and Te) and Their Potential Applications. Chem. Mater. 2020, 32, 8229–8242. [Google Scholar] [CrossRef]
- Angell, C.A. Mobile Ions in Amorphous Solids. Annu. Rev. Phys. Chem. 1992, 43, 693–717. [Google Scholar] [CrossRef]
- Bates, J.B.; Dudney, N.J.; Gruzalski, G.R.; Zuhr, R.A.; Choudhury, A.; Luck, C.F.; Robertson, J.D. Fabrication and characterization of amorphous lithium electrolyte thin films and rechargeable thin-film batteries. J. Power Source 1993, 43, 103–110. [Google Scholar] [CrossRef]
- Takuya, K.; Chie, H.; Atsushi, S.; Akitoshi, H.; Masahiro, T. Mechanochemical synthesis and characterization of amorphous Li2CN2 as a lithium ion conductor. J. Ceram. Soc. Jpn. 2019, 127, 518–520. [Google Scholar]
- Minami, T.; Hayashi, A.; Tatsumisago, M. Recent progress of glass and glass-ceramics as solid electrolytes for lithium secondary batteries. Solid State Ionics 2006, 177, 2715–2720. [Google Scholar] [CrossRef]
- Laurent, Y.; Guyader, J.; Roult, G. Etude par diffraction de neutrons selon la method du temps de vol de LiSiONα. Acta Cryst. 1981, 37, 911–913. [Google Scholar] [CrossRef]
- Podsiadlo, S. Formation and Thermal Decomposition of Silicon Oxynitride Compounds II. J. Therm. Anal. 1987, 32, 445–449. [Google Scholar] [CrossRef]
- Du, Y.A.; Holzwarth, N.A.W. First-principles study of LiPON and related solid electrolytes. Phys. Rev. B 2010, 81, 184106. [Google Scholar]
- Adams, S.; Rao, R.P. High power lithium ion battery materials by computational design. Phys. Status Solidi A 2011, 208, 1746–1753. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Zhu, Y.; Epstein, A.; Mo, Y. Statistical variances of diffusional properties from ab initio molecular dynamics simulations. npj Comput. Mater. 2018, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Stefano, D.D.; Miglio, A.; Robeyns, K.; Filinchuk, Y.; Lechartier, M.; Senyshyn, A.; Ishida, H.; Spannenberger, S.; Prutsch, D.; Lunghammer, S.; et al. Superionic diffusion through frustrated energy landscape. Chem 2019, 5, 2450–2460. [Google Scholar] [CrossRef] [Green Version]
- Xiao, R.; Li, H.; Chen, L.Q. High-throughput computational discovery of K2CdO2 as an ion conductor for solid-state potassium-ion batteries. J. Mater. Chem. A 2020, 8, 5157. [Google Scholar] [CrossRef]
- Singh, S.P.; Schneider, J.F.; Kundu, S.; Rodrigues, A.C.M.; de Mattos, P.P.G.; Zanotto, E.D.; Rocherullé, J.; Bénard-Rocherullé, P.; Lebullenger, R. Structure and ionic conductivity of nitrated lithium disilicate (LiSiON) glasses. Mater. Chem. Phys. 2018, 211, 438–444. [Google Scholar] [CrossRef]
- Famprikis, T.; Galipaud, J.; Clemens, O.; Pecquenard, B.; Le Cras, F. Composition Dependence of Ionic Conductivity in LiSiPO(N) Thin-Film Electrolytes for Solid-State Batteries. ACS Appl. Energy Mater. 2019, 2, 4782–4791. [Google Scholar] [CrossRef]
- Lee, S.J.; Bae, J.H.; Lee, H.W.; Baik, H.K.; Lee, S.M. Electrical conductivity in Li–Si–P–O–N oxynitride thin-films. J. Power Sources 2003, 123, 61–64. [Google Scholar] [CrossRef]
- Xia, Q.; Sun, S.; Zan, F.; Xu, J.; Xia, H. Amorphous LiSiON Thin Film Electrolyte for All-solid-state Thin Film Lithium Battery. J. Inorg. Mater. 2022, 37, 230–236. [Google Scholar] [CrossRef]
- He, X.; Zhu, Y.; Mo, Y. Origin of fast ion diffusion in super-ionic conductors. Nat. Comm. 2017, 8, 15893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Roy, P.N.; Li, H.; Avdeev, M.; Nazar, L.F. Coupled Cation–Anion Dynamics Enhances Cation Mobility in Room-Temperature Superionic Solid-State Electrolytes. J. Am. Chem. Soc. 2019, 141, 19360–19372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, H.; Kaup, K.; Zhou, L.; Roy, P.N.; Nazar, L.F. Targeting Superionic Conductivity by Turning on Anion Rotation at Room Temperature in Fast Ion Conductors. Matter 2020, 2, 1667–1684. [Google Scholar] [CrossRef]
- Zhang, Z.; Nazar, L. Exploiting the paddle-wheel mechanism for the design of fast ion conductors. Nat. Rev. Mater. 2022, 1–17. [Google Scholar] [CrossRef]
- Wu, S.; Xiao, R.; Li, H.; Chen, L. New insights into the mechanism of cation migration induced by cation–anion dynamic coupling in superionic conductors. J. Mater. Chem. A 2022, 10, 3093–3101. [Google Scholar] [CrossRef]
- Morgan, B.J.; Madden, P.A. Effects of Lattice Polarity on Interfacial Space Charges and Defect Disorder in Ionically Conducting AgI Heterostructures. Phys. Rev. Lett. 2011, 107, 206102. [Google Scholar] [CrossRef]
- Fu, L.; Chen, C.C.; Samuelis, D.; Maier, J. Thermodynamics of Lithium Storage at Abrupt Junctions: Modeling and Experimental Evidence. Phys. Rev. Lett. 2014, 112, 208301. [Google Scholar] [CrossRef]
- Swift, M.; Qi, Y. First-Principles Prediction of Potentials and Space-Charge Layers in All-Solid-State Batteries. Phys. Rev. Lett. 2019, 122, 167701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwietert, T.K.; Vasileiadis, A.; Wagemaker, M. First-Principles Prediction of the Electrochemical Stability and Reaction Mechanisms of Solid-State Electrolytes. J. Am. Chem. Soc. Au 2021, 1, 1488–1496. [Google Scholar] [CrossRef] [PubMed]
- Richards, W.D.; Miara, L.J.; Wang, Y.; Kim, J.C.; Ceder, G. Interface Stability in Solid-State Batteries. Chem. Mater. 2016, 28, 266–273. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, S.; Xiao, R.; Li, H.; Chen, L. Ionic Conductivity of LiSiON and the Effect of Amorphization/Heterovalent Doping on Li+ Diffusion. Inorganics 2022, 10, 45. https://doi.org/10.3390/inorganics10040045
Wu S, Xiao R, Li H, Chen L. Ionic Conductivity of LiSiON and the Effect of Amorphization/Heterovalent Doping on Li+ Diffusion. Inorganics. 2022; 10(4):45. https://doi.org/10.3390/inorganics10040045
Chicago/Turabian StyleWu, Siyuan, Ruijuan Xiao, Hong Li, and Liquan Chen. 2022. "Ionic Conductivity of LiSiON and the Effect of Amorphization/Heterovalent Doping on Li+ Diffusion" Inorganics 10, no. 4: 45. https://doi.org/10.3390/inorganics10040045
APA StyleWu, S., Xiao, R., Li, H., & Chen, L. (2022). Ionic Conductivity of LiSiON and the Effect of Amorphization/Heterovalent Doping on Li+ Diffusion. Inorganics, 10(4), 45. https://doi.org/10.3390/inorganics10040045