Electron Interference in Molecular Circular Polarization Attosecond XUV Photoionization

Abstract
:1. Introduction
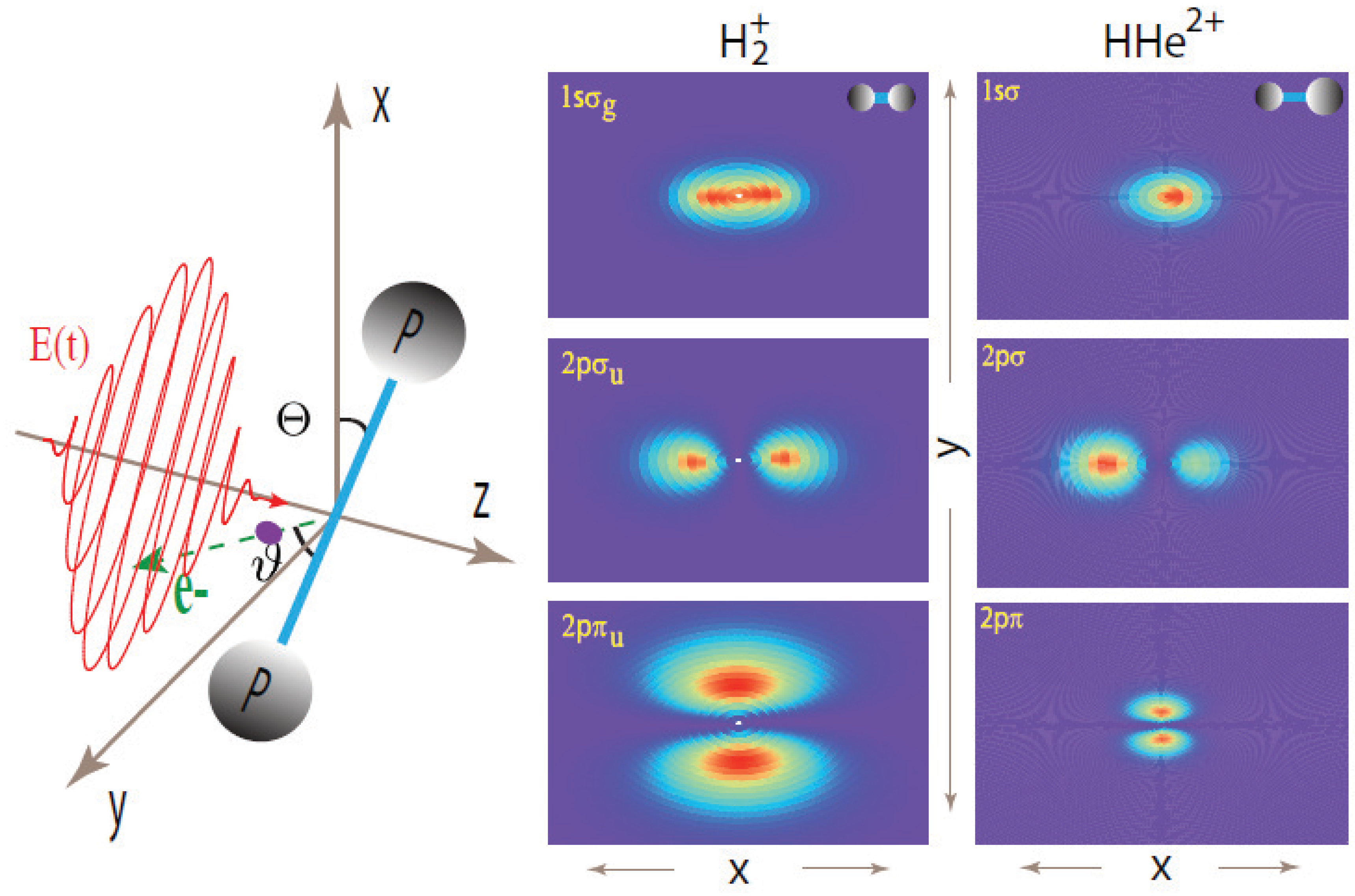
2. Two-Center Interference Models
3. Numerical methods
4. Numerical Results and Discussions
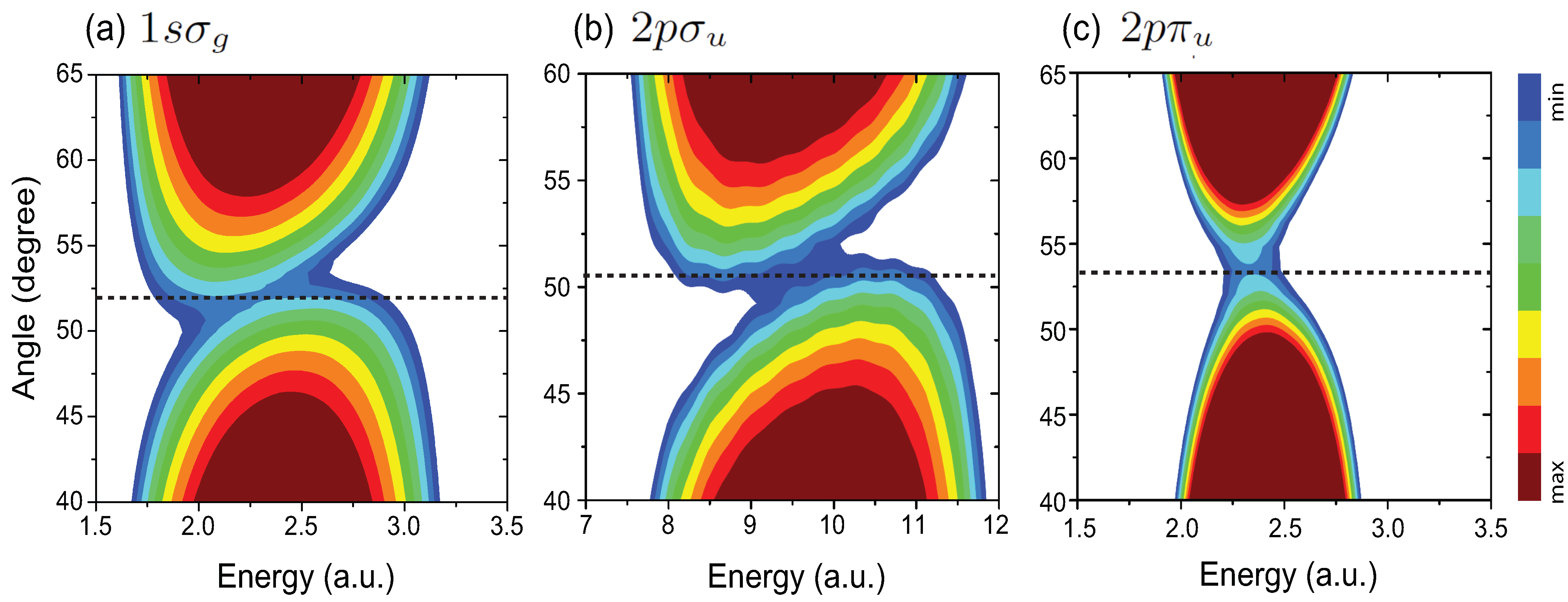
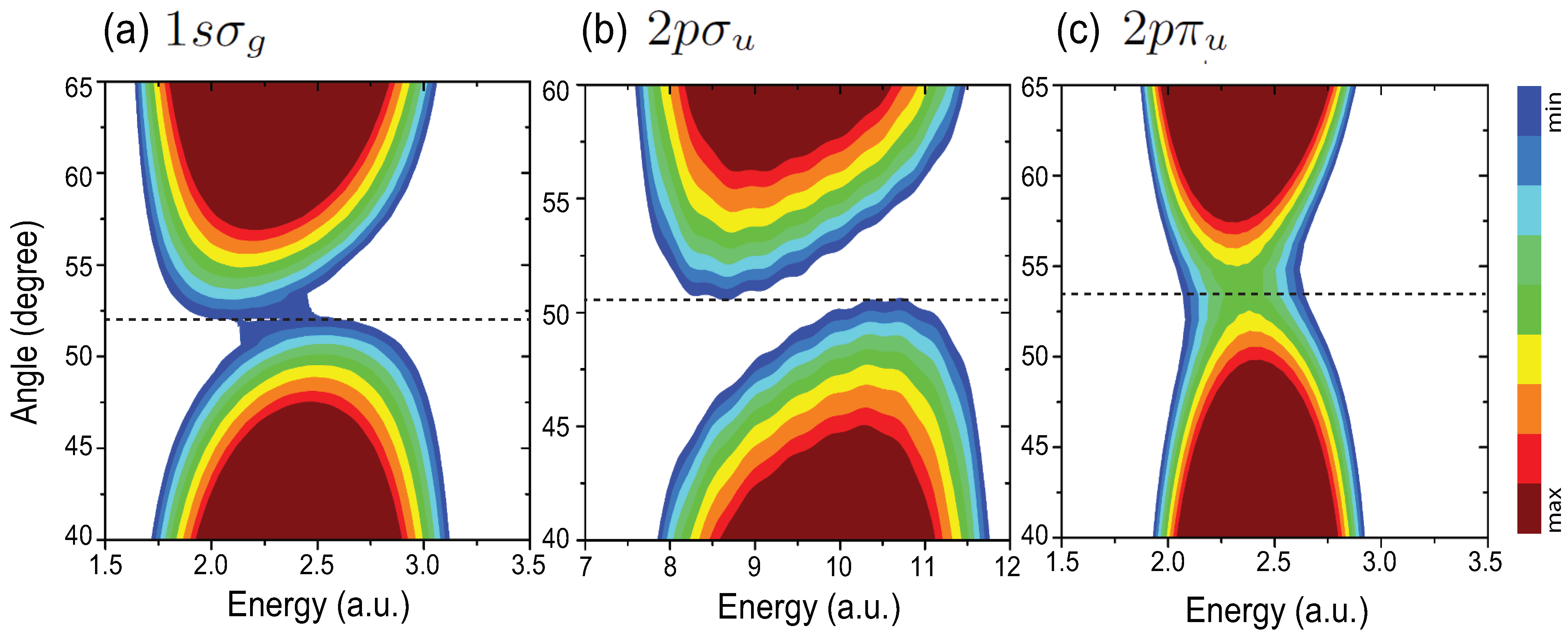
4.1. Interference Patterns in MAPES of H

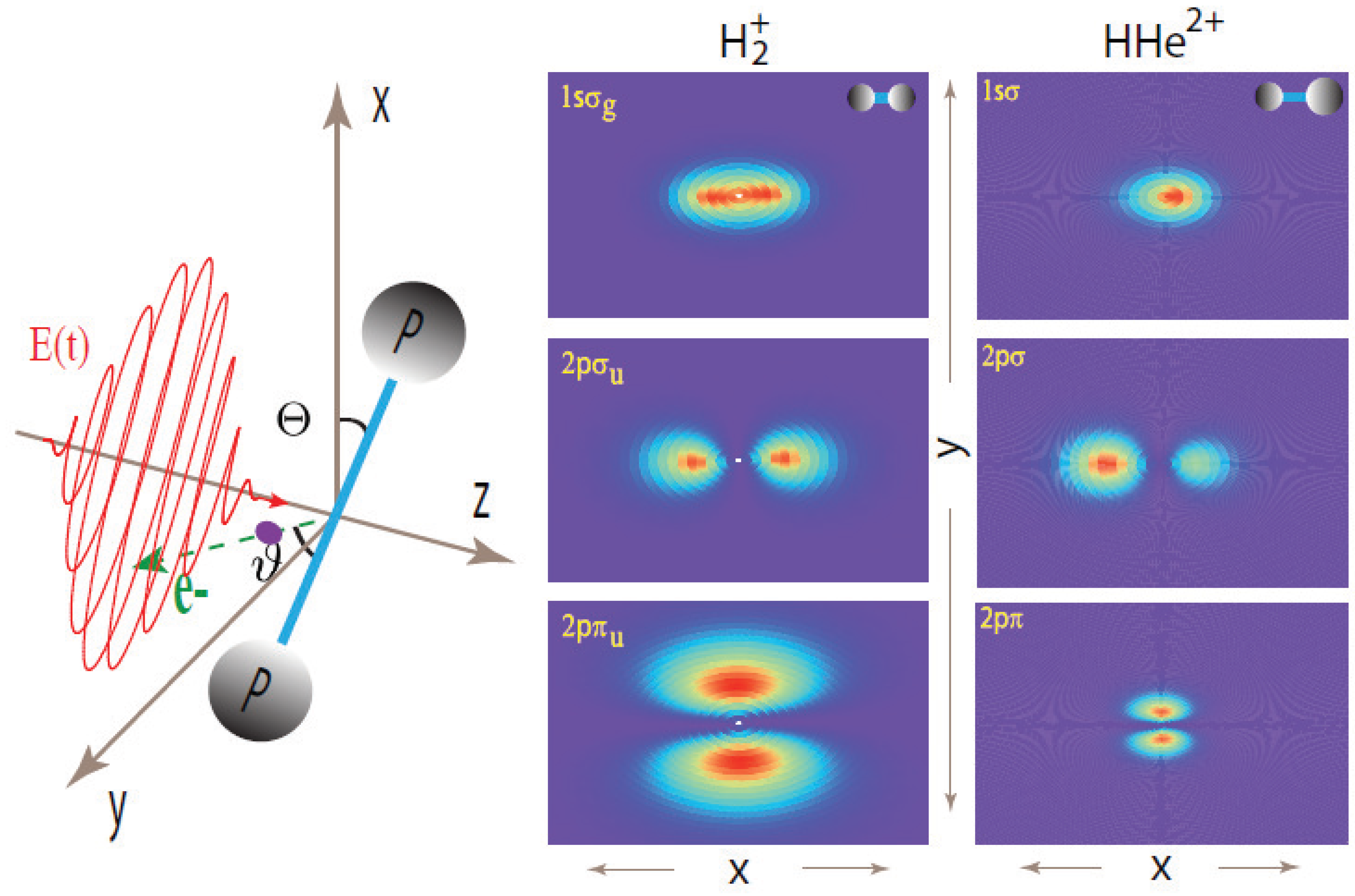{kind=link}
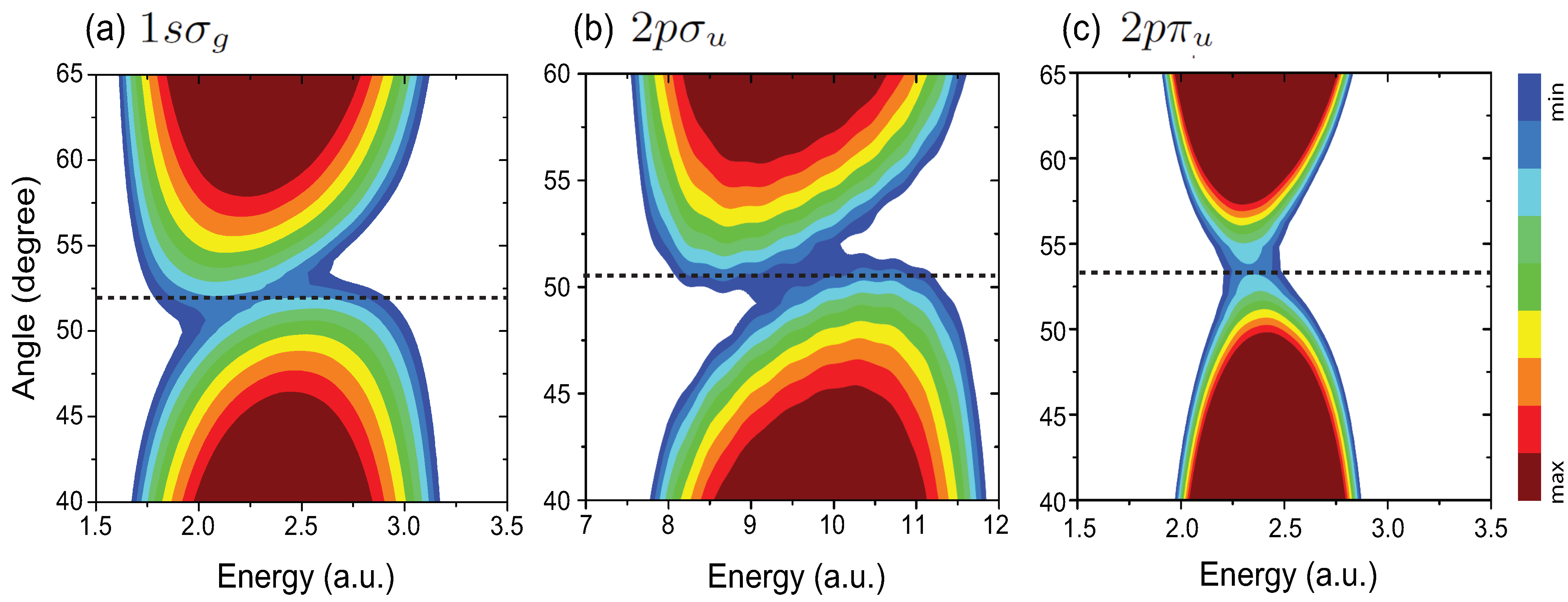{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| XUV Pulses | Photoelectron | |||
| λ | ω | p | ||
| H ( | 12.5 nm | 3.65 a.u. | 2.26 a.u. | 2.78 a.u. |
| H ( | 19.5 nm | 2.34 a.u. | 1.57 a.u. | 4.0 a.u. |
| H ( | 3.75 nm | 12.1 a.u. | 4.70 a.u. | 1.34 a.u. |
| H ( | 4.25 nm | 10.7 a.u. | 4.49 a.u. | 1.4 a.u. |
| H ( | 15.0 nm | 3.0 a.u. | 2.27 a.u. | 2.77 a.u. |
| HHe ( | 10.0 nm | 4.56 a.u. | 2.22 a.u. | 2.83 a.u. |
| HHe ( | 12.0 nm | 3.8 a.u. | 2.21 a.u. | 2.84 a.u. |
| HHe ( | 12.5 nm | 3.65 a.u. | 2.26 a.u. | 2.78 a.u. |
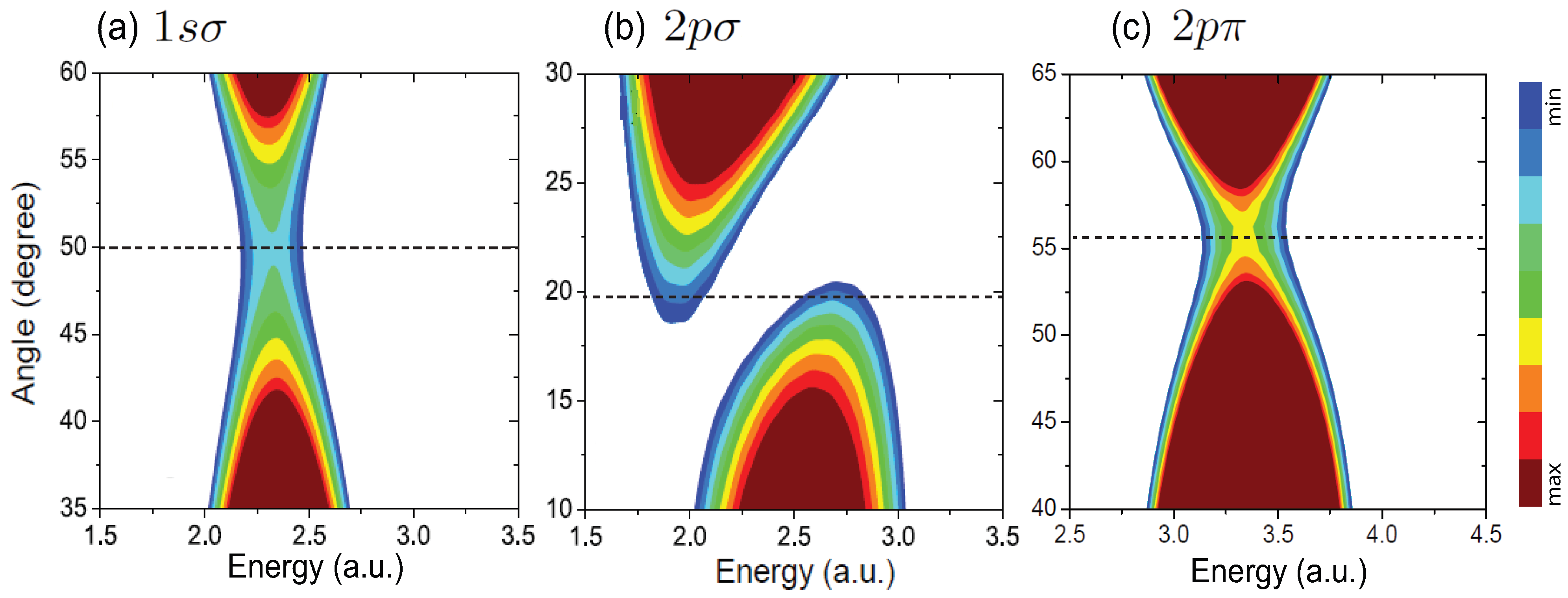
4.2. Interference in Nonsymmetric HHe

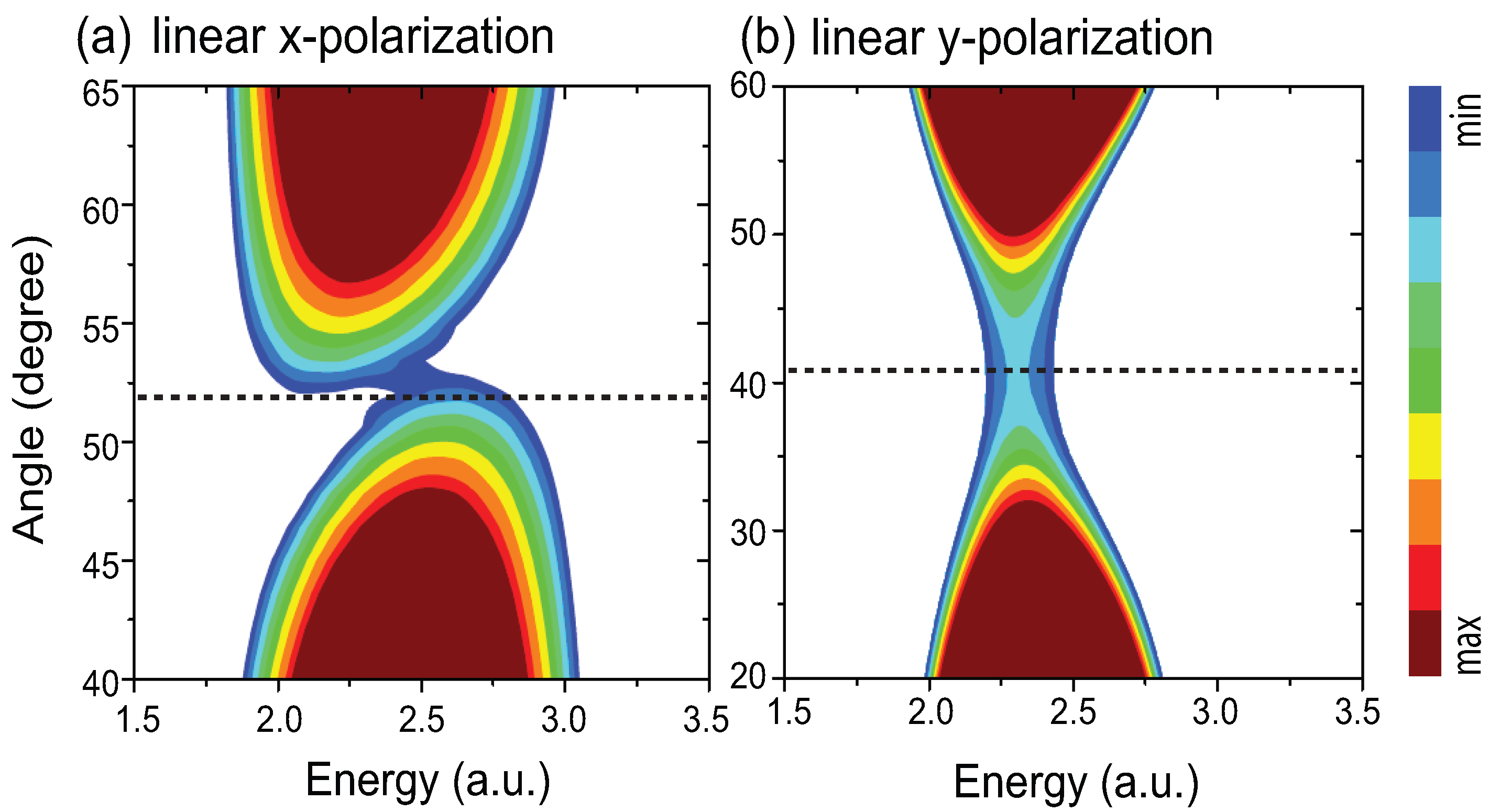
4.3. Interference patterns in elliptical polarization ionization
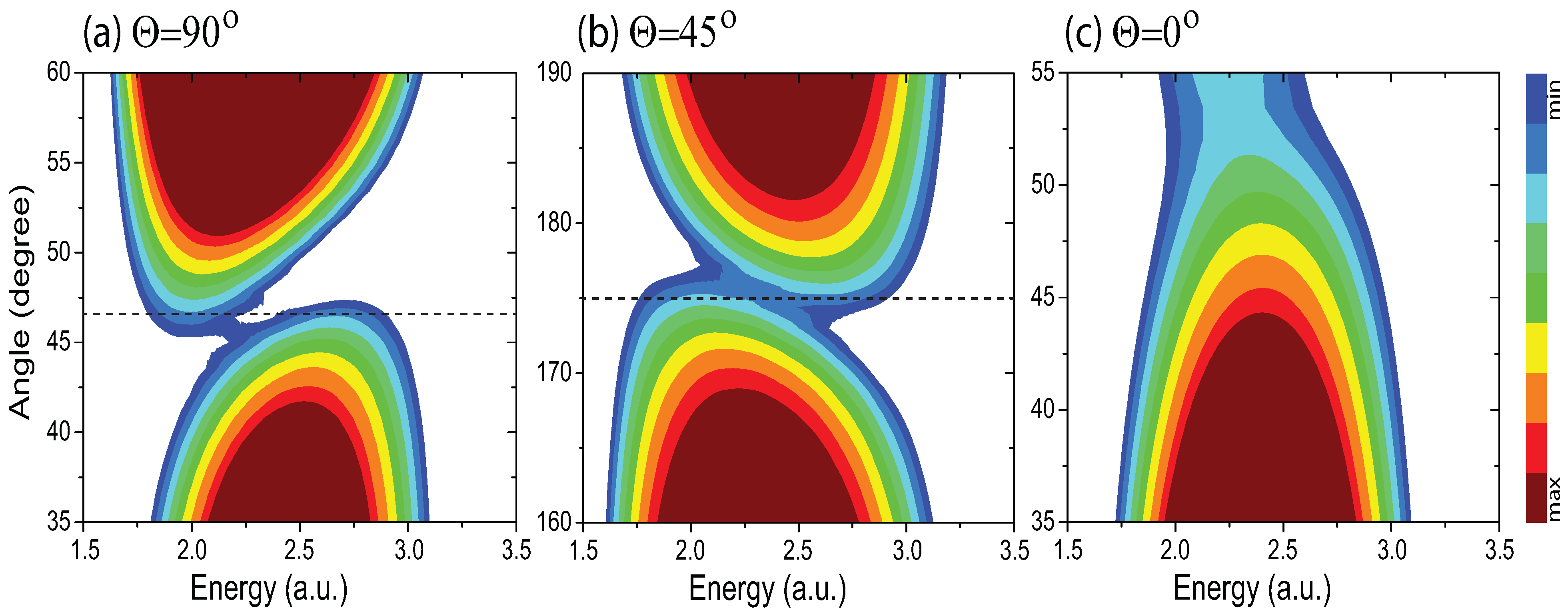
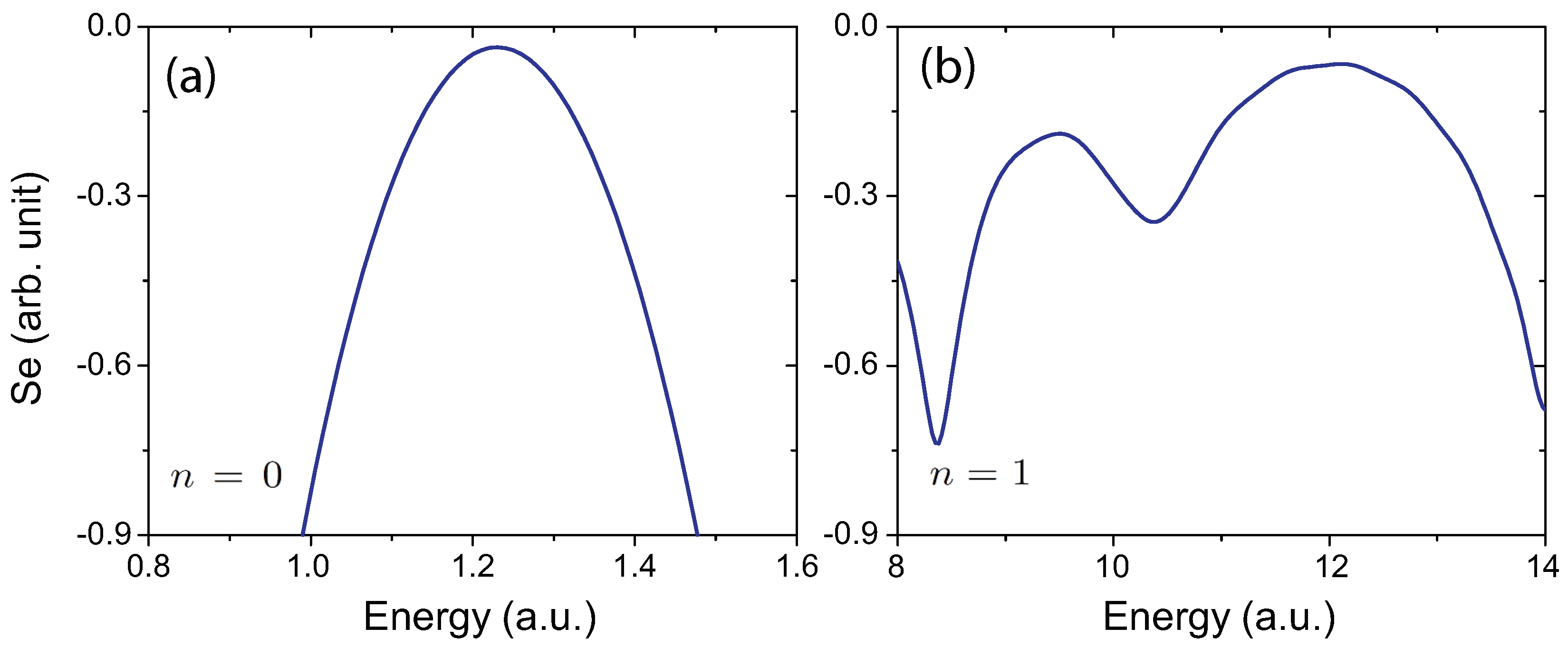
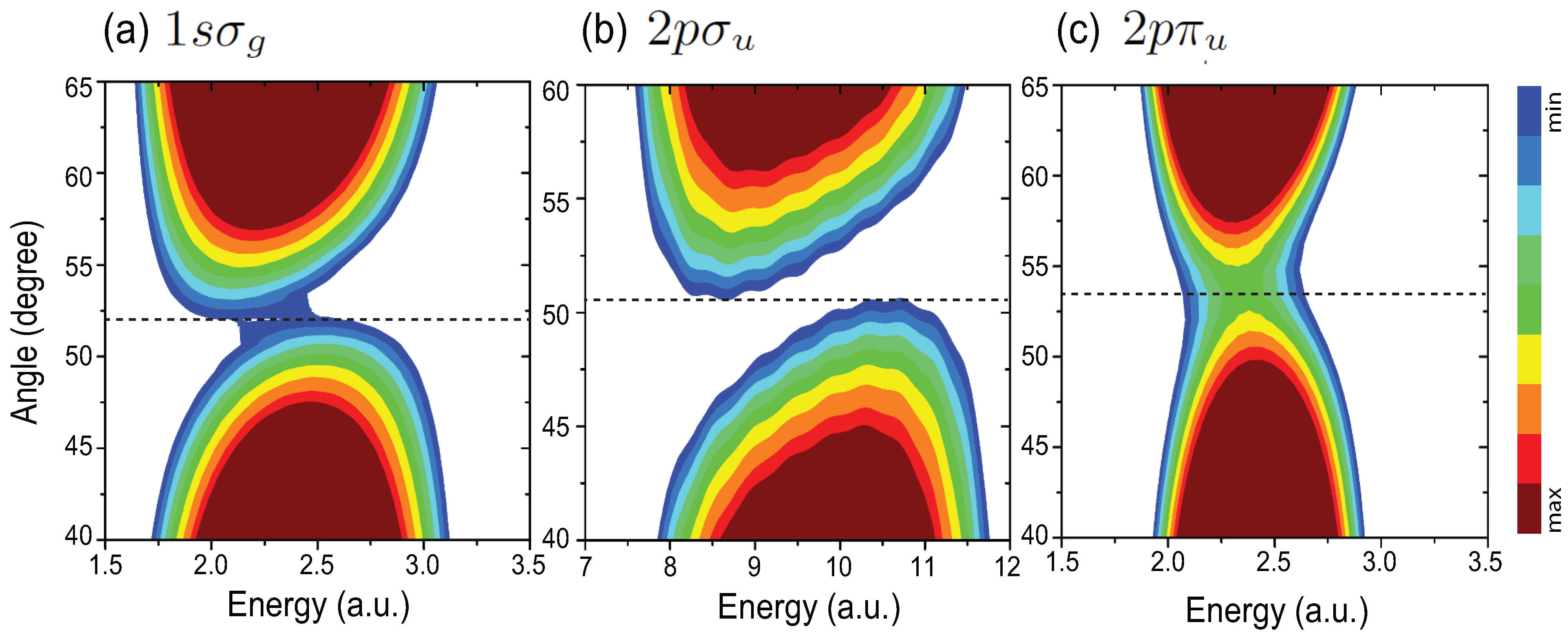
4.4. Effects of Molecular Orientation Θ on Interference Patterns

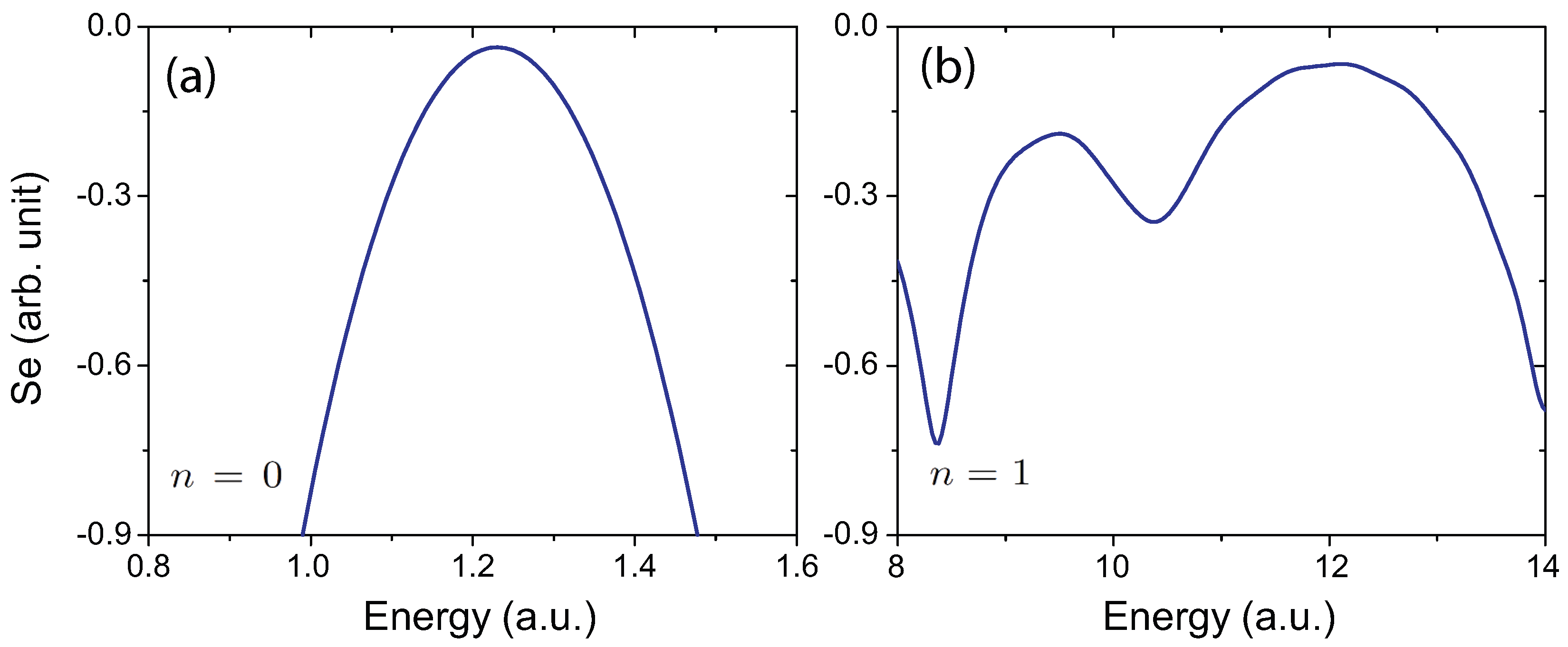
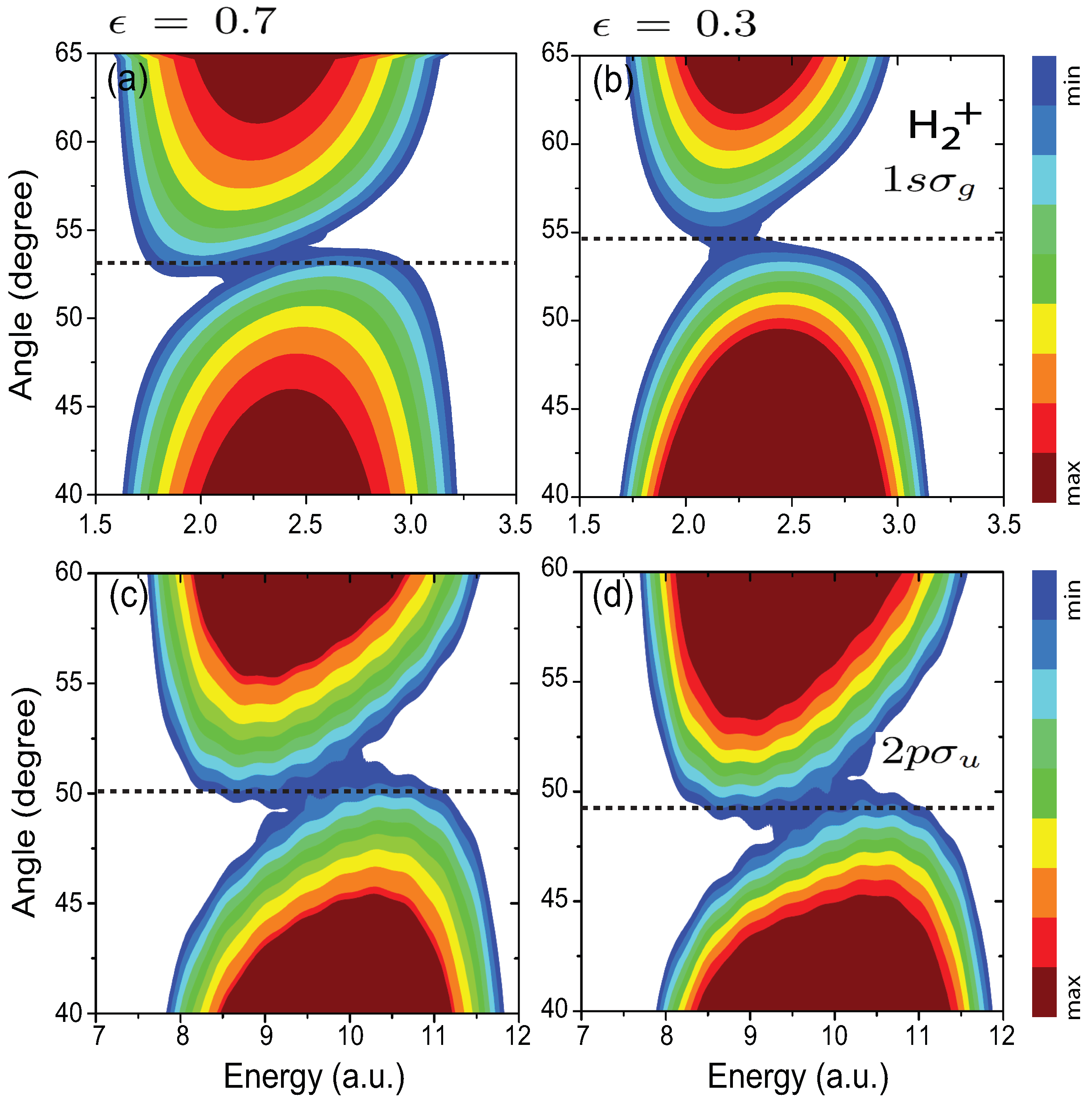
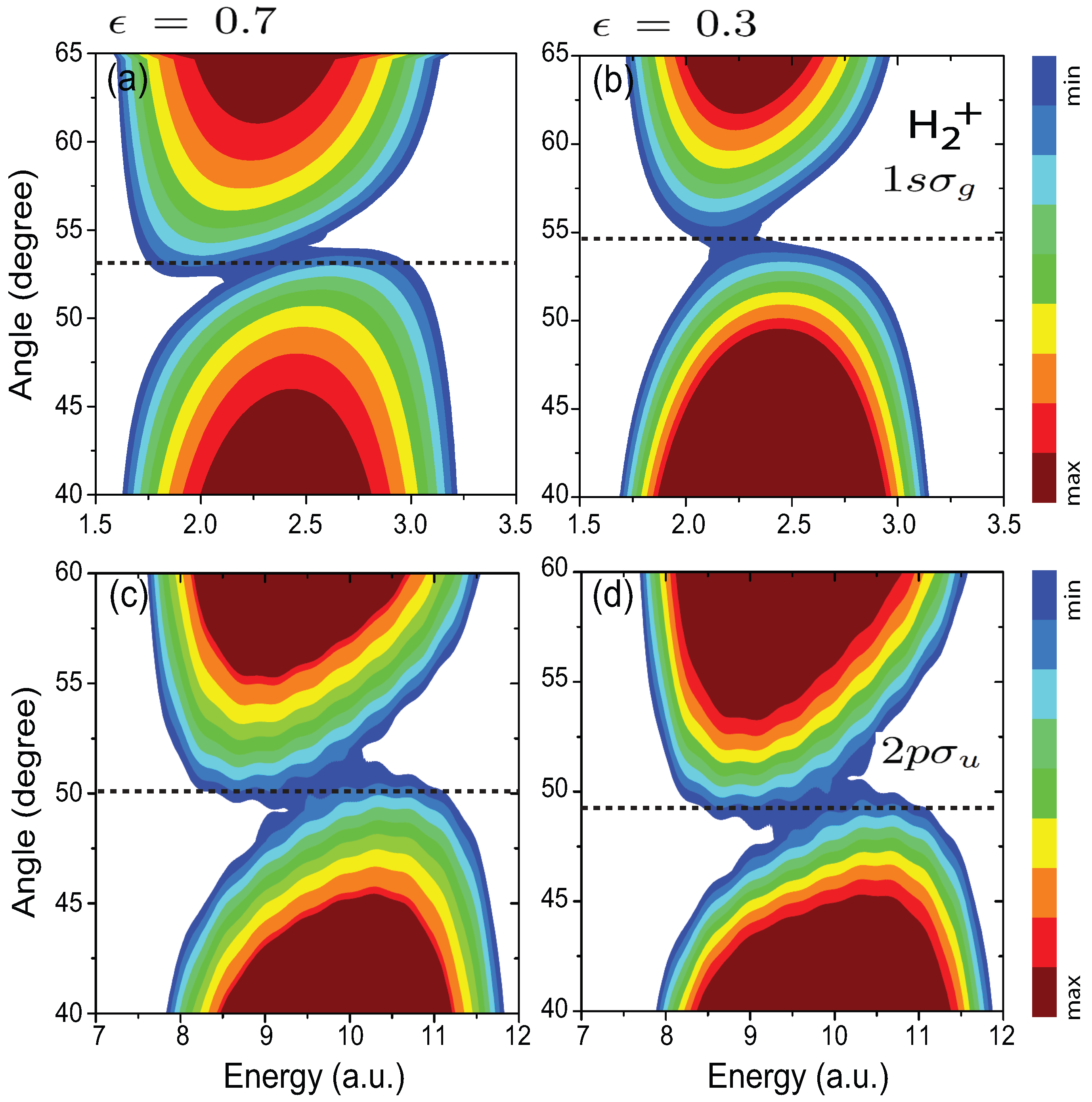
4.5. Influence of the Pulse Intensity
5. Summary
Acknowledgments
Conflicts of Interest
References
- Corkum, P.B.; Krausz, F. Attosecond science. Nature Phys. 2007, 3, 381–387. [Google Scholar] [CrossRef]
- Krausz, F.; Ivanov, M. Attosecond physics. Rev. Mod. Phys. 2009, 81, 163–234. [Google Scholar] [CrossRef]
- Chang, Z.; Corkum, P. Attosecond photon sources: the first decade and beyond. J. Opt. Soc. Am. B 2010, 27, B9–B17. [Google Scholar] [CrossRef]
- Zhao, K.; Zhang, Q.; Chini, M.; Wu, Y.; Wang, X.; Chang, Z. Tailoring a 67 attosecond pulse through advantageous phase-mismatch. Opt. Lett. 2012, 37, 3891–3893. [Google Scholar] [CrossRef] [PubMed]
- Popmintchev, T.; Chen, M.-C.; Popmintchev, D.; Arpin, P.; Brown, S.; Ališauskas, S.; Andriukaitis, G.; Balčiunas, T.; Mücke, O.D.; Pugzlys, A.; Baltuška, A.; Shim, B.; Schrauth, S.E.; Gaeta, A.; Hernández-García, C.; Plaja, L.; Becker, A.; Jaron-Becker, A.; Murnane, M.M.; Kapteyn, H.C. Bright coherent ultrahigh harmonics in the keV x-ray regime from mid-infrared femtosecond lasers. Science 2012, 336, 1287–1291. [Google Scholar]
- Hernández-García, C.; Pérez-Hernández, J.A.; Popmintchev, T.; Murnane, M.M.; Kapteyn, H.C.; Jaron-Becker, A.; Becker, A.; Plaja, L. Zeptosecond high harmonic keV X-ray waveforms driven by mid-infrared laser pulses. Phys. Rev. Lett. 2013, 111, 033002. [Google Scholar] [CrossRef] [PubMed]
- Bandrauk, A.D.; Chelkowski, S.; Diestler, D.J.; Manz, J.; Yuan, K.-J. Quantum-mechanical models for photo-ionization: Uni-directional electron re-scattering by a laser pulse. Int. J. Mass Spectrom. 2008, 277, 189–196. [Google Scholar] [CrossRef]
- Vrakking, M. Chemical physics: Electronic movies. Nature 2009, 460, 960. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.D.; Fano, U. Interference in the photo-ionization of molecules. Phys. Rev. 1966, 150, 30. [Google Scholar] [CrossRef]
- Kaplan, I.G.; Markin, A.P. Interference phenomena in photoionization of molecules. Sov. Phys. Dokl. 1969, 14, 36. [Google Scholar]
- Zuo, T.; Bandrauk, A.D.; Corkum, P.B. Laser-induced electron diffraction: A new tool for probing ultrafast molecular dynamics. Chem. Phys. Lett. 1996, 259, 313. [Google Scholar] [CrossRef]
- Hu, S.X.; Collins, L.A. Imaging molecular structures by electron diffraction using an intense few-cycle pulse. Phys. Rev. Lett. 2005, 94, 073004. [Google Scholar] [PubMed]
- Meckel, M.; Comtois, D.; Zeidler, D.; Staudte, A.; Pavicic, D.; Bandulet, H.C.; Pépin, H.; Kieffer, J.C.; Dörner, R.; Villeneuve, D.M.; Corkum, P.B. Laser-induced electron tunneling and diffraction. Science 2008, 320, 1478. [Google Scholar] [CrossRef] [PubMed]
- Matta, C.F.; Boyd, R.J. The Quantum Theory of Atoms in Molecules; Wiley-VCH: New York, NY, USA, 2009. [Google Scholar]
- Lein, M.; Marangos, J.P.; Knight, P.L. Electron diffraction in above-threshold ionization of molecules. Phys. Rev. A 2002, 66, 051404(R). [Google Scholar] [CrossRef]
- Spanner, M.; Smirnova, O.; Corkum, P.B.; Ivanov, M.Y. Reading diffraction images in strong field ionization of diatomic molecules. J. Phys. B 2004, 37, L243. [Google Scholar] [CrossRef]
- Kamta, G.L.; Bandrauk, A.D. Orbital symmetry and interference effects in molecular high-order harmonic generation. Phys. Rev. A 2009, 80, 041403(R). [Google Scholar]
- Picón, A.; Bahabad, A.; Kapteyn, H.C.; Murnane, M.M.; Becker, A. Two-center interferences in photoionization of a dissociating H molecule. Phys. Rev. A 2009, 83, 013414. [Google Scholar] [CrossRef]
- Peters, M.; Nguyen-Dang, T.T.; Cornaggia, C.; Saugout, S.; Charron, E.; Keller, A.; Atabek, O. Ultrafast molecular imaging by laser-induced electron diffraction. Phys. Rev. A 2011, 83, 051403(R). [Google Scholar] [CrossRef]
- Canton, S.E.; Plésiat, E.; Bozek, J.D.; Rude, B.S.; Decleva, P.; Martín, F. Direct observation of Young’s double-slit interferences in vibrationally resolved photoionization of diatomic molecules. Proc. Natl. Acad. Sci. USA 2011, 108, 7302. [Google Scholar] [CrossRef]
- Blaga, C.I.; Xu, J.; DiChiara, A.D.; Sistrunk, E.; Zhang, K.; Miller, T.; Agostini, P.; DiMauro, L.F.; Lin, C.D. Imaging ultrafast molecular dynamics with laser-induced electron diffraction. Nature 2012, 483, 194. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.B.; Bandrauk, A.D. Attosecond time-resolved imaging of molecular structure by photoelectron holography. Phys. Rev. Lett. 2012, 108, 263003. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; DuToit, R.C.; Bartschat, K. Photoionization of the H ion by ultrashort elliptically polarized laser pulses. Phys. Rev. A 2013, 87, 053410. [Google Scholar] [CrossRef]
- Yuan, K.J.; Bian, X.B.; Bandrauk, A.D. Two-center interference in molecular photoelectron energy spectra with intense attosecond circularly polarized XUV laser pulses. Phys. Rev. A 2014, 90, 023407. [Google Scholar] [CrossRef]
- Corkum, P.B. Plasma perspective on strong field multiphoton ionization. Phys. Rev. Lett. 1993, 71, 1994–1997. [Google Scholar] [CrossRef] [PubMed]
- Lewenstein, M.; Balcou, Ph.; Ivanov, M.Y.; L’Huillier, A.; Corkum, P.B. Theory of high-harmonic generation by low-frequency laser fields. Phys. Rev. A 1994, 49, 2117–2132. [Google Scholar] [CrossRef] [PubMed]
- Bandrauk, A.D.; Chelkowski, S.; Goudreau, S. Control of harmonic generation using two-colour femtosecond-attosecond laser fields: quantum and classical perspectives. J. Mod. Opt. 2005, 52, 411–428. [Google Scholar] [CrossRef]
- Bandrauk, A.D.; Chelkowski, S.; Yu, H.; Constant, E. Enhanced harmonic generation in extended molecular systems by two-color excitation. Phys. Rev. A 1997, 56, R2537–R2540. [Google Scholar] [CrossRef]
- Moreno, P.; Plaja, L.; Roso, L. Ultrahigh harmonic generation from diatomic molecular ions in highly excited vibrational states. Phys. Rev. A 1997, 55, R1593–R1596. [Google Scholar] [CrossRef]
- Hetzheim, H.; Figueira de Morisson Faria, C.; Becker, W. Interference effects in above-threshold ionization from diatomic molecules: Determining the internuclear separation. Phys. Rev. A 2007, 76, 023418. [Google Scholar] [CrossRef]
- Okunishi, M.; Itaya, R.; Shimada, K.; Prümper, G.; Ueda, K.; Busuladžić, M.; Gazibegović-Busuladžić, A.; Milošević, D.B.; Becker, W. Two-source double-slit interference in angle-resolved high-energy above-threshold ionization spectra of diatoms. Phys. Rev. Lett. 2009, 103, 043001. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zhang, X.; Xu, Y.; Ma, H.; Yang, J. Two-center interference in photoionization of H in circularly polarized laser fields. J. Opt. Sco. Am. B 2012, 29, 2124. [Google Scholar] [CrossRef]
- Busuladžić, M.; Gazibegović-Busuladžić, A.; Milošević, D.B.; Becker, W. Molecular above-threshold ionization with a circularly polarized laser field. Eurp. Phys. J. D 2013, 67, 61. [Google Scholar] [CrossRef]
- Korneev, P.A.; Popruzhenko, S.V.; Goreslavski, S.P.; Becker, W.; Paulus, G.G.; Fetić, B.; Milošević, D.B. Interference structure of above-threshold ionization versus above-threshold detachment. New J. Phys. 2012, 14, 055019. [Google Scholar] [CrossRef]
- Yuan, K.J.; Bandrauk, A.D. Angle-dependent molecular above-threshold ionization with ultrashort intense linearly and circularly polarized laser pulses. Phys. Rev. A 2011, 84, 013426. [Google Scholar] [CrossRef]
- Yuan, K.J.; Lu, H.Z.; Bandrauk, A.D. Electron interference in molecular photoionization by attosecond laser pulses. Chem. Phys. Chem. 2013, 14, 1496. [Google Scholar] [CrossRef] [PubMed]
- Milošević, D.B.; Sandner, W. Extreme-ultraviolet harmonic generation near 13 nm with a two-color elliptically polarized laser field. Opt. Lett. 2000, 25, 1532. [Google Scholar] [CrossRef] [PubMed]
- Milošević, D.B.; Becker, W. Attosecond pulse trains with unusual nonlinear polarization. Phys. Rev. A 2000, 62, 011403(R). [Google Scholar] [CrossRef]
- Fleischer, A.; Kfir, O.; Diskin, T.; Sidorenko, P.; Cohen, O. Spin angular momentum and tunable polarization in high-harmonic generation. Nature Photonics 2014, 8, 543. [Google Scholar] [CrossRef]
- Yuan, K.-J.; Bandrauk, A.D. Circularly polarized attosecond pulses from molecular high-order harmonic generation by ultrashort intense bichromatic circularly and linearly polarized laser pulses. J. Phys. B 2012, 45, 074001. [Google Scholar] [CrossRef]
- Yuan, K.-J.; Bandrauk, A.D. Single circularly polarized attosecond pulse generation by intense few cycle elliptically polarized laser pulses and terahertz fields from molecular media. Phys. Rev. Lett. 2013, 110, 023003. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.-J.; Bandrauk, A.D. Generation of circularly polarized attosecond pulses by intense ultrashort laser pulses from extended asymmetric molecular ions. Phys. Rev. A 2011, 84, 023410. [Google Scholar] [CrossRef]
- Horner, D.A.; Miyabe, S.; Rescigno, T.N.; McCurdy, C.W.; Morales, F.; Martín, F. Classical two-slit interference effects in double photoionization of molecular hydrogen at high energies. Phys. Rev. Lett. 2008, 101, 183002. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.-J.; Lu, H.Z.; Bandrauk, A.D. Laser-induced electron diffraction in H2 with linear and circular polarization ultrashort XUV laser pulses. Phys. Rev. A 2009, 80, 061403(R). [Google Scholar] [CrossRef]
- Yuan, K.-J.; Lu, H.Z.; Bandrauk, A.D. Linear- and circular-polarization photoionization angular distributions in H2 and H by attosecond xuv laser pulses. Phys. Rev. A 2011, 83, 043418. [Google Scholar] [CrossRef]
- Huang, C.; Zhou, Y.; Liao, Q.; Lu, P. Imaging molecular structures with high-energy photoelectrons produced by extreme ultraviolet pulses. J. Opt. Soc. Am. B 2012, 29, 734. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, Q.; Hong, W.; Lu, P.; Xu, Z. Molecular orbital imaging via above-threshold ionization with circularly polarized pulses. Opt. Express 2011, 19, 13722. [Google Scholar] [CrossRef] [PubMed]
- Akoury, D.; Kreidi, K.; Jahnke, T.; Weber, T.; Staudte, A.; Schöffler, M.; Neumann, N.; Titze, J.; Schmidt, L.P.H.; Czasch, A.; et al. The Simplest Double Slit: Interference and Entanglement in Double Photoionization of H2. Science 2007, 318, 949–952. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.J.; Bandrauk, A.D. Molecular above-threshold-ionization angular distributions with intense circularly polarized attosecond XUV laser pulses. Phys. Rev. A 2012, 85, 053419. [Google Scholar] [CrossRef]
- Yuan, K.J.; Chelkowski, S.; Bandrauk, A.D. Rotations of molecular photoelectron angular distributions with intense ultrashort circularly polarized attosecond laser pulses. J. Chem. Phys. 2013, 138, 134316. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.J.; Chelkowski, S.; Bandrauk, A.D. Molecular photoelectron angular distributions with intense attosecond circularly polarized UV laser pulses. Chem. Phys. Lett. 2014, 592, 334. [Google Scholar] [CrossRef]
- Yuan, K.J.; Chelkowski, S.; Bandrauk, A.D. Rotations of molecular photoelectron angular distributions in above threshold ionization of H by intense circularly polarized attosecond UV laser pulses. J. Phys. B 2014, 47, 204009. [Google Scholar] [CrossRef]
- Lagmago Kamta, G.; Bandrauk, A.D. Three-dimensional time-profile analysis of high-order harmonic generation in molecules: Nuclear interferences in H. Phys. Rev. A 2005, 71, 053407. [Google Scholar] [CrossRef]
- Pronin, E.A.; Starace, A.F.; Frolov, M.V.; Manakov, N.L. Perturbation theory analysis of attosecond photoionization. Phys. Rev. A 2009, 80, 063403. [Google Scholar] [CrossRef]
- Odžak, S.; Milošević, D.B. Interference effects in high-order harmonic generation by homonuclear diatomic molecules. Phys. Rev. A 2009, 79, 023414. [Google Scholar] [CrossRef]
- Marques, M.A.L.; Maitra, N.T.; Nogueira, F.M.S.; Gross, E.K.U.; Rubio, A. Fundamentals of Time-Dependent Density Functional Theory; Springer: Berlin, Germany, 2012. [Google Scholar]
- Girardeau, M.D.; Kim, K.G.; Widmayer, C.C. Theory of atomic excitation and ionization by ultrashort laser pulses. Phys. Rev. A 1992, 46, 5932. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.J.; Bandrauk, A.D. Photoelectron angular distributions of H and HHe by intense circularly polarized extreme ultraviolet laser pulses. J. Phys. B 2012, 45, 105601. [Google Scholar] [CrossRef]
- Bandrauk, A.D.; Shen, H. Exponential split operator methods for solving coupled time-dependent Schrödinger equations. J. Chem. Phys. 1993, 99, 1185. [Google Scholar] [CrossRef]
- Keldysh, L.V. Ionization in the field of a strong electromagnetic wave. Sov. Phys. JETP 1965, 20, 1307. [Google Scholar]
- Yuan, K.J.; Bandrauk, A.D. Molecular above threshold ionization angular distributions with attosecond bichrimatic intense xuv laser pulses. Phys. Rev. A 2012, 85, 013413. [Google Scholar] [CrossRef]
- Bian, X.B.; Bandrauk, A.D. Multichannel molecular high-order harmonic generation from asymmetric diatomic molecules. Phys. Rev. Lett. 2010, 105, 093903. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.B.; Trevisan, C.S.; Schöffler, M.S.; Jahnke, T.; Bocharova, I.; Kim, H.; Ulrich, B.; Wallauer, R.; Sturm, F.; Rescigno, T.N.; et al. Imaging polyatomic molecules in three dimensions using molecular frame photoelectron angular distributions. Phys. Rev. Lett. 2012, 108, 233002. [Google Scholar] [CrossRef] [PubMed]
- Etches, A.; Gaarde, M.B.; Madsen, L.B. Two-center minima in harmonic spectra from aligned polar molecules. Phys. Rev. A 2011, 84, 023418. [Google Scholar] [CrossRef]
- Bandrauk, A.D.; Chelkowski, S.; Yuan, K.J. Controlling electron collisions-recollisions with ultrashort intense laser pulses-from femto to attosecond science. Int. Rev. At. Mol. Phys. 2011, 2, 1–23. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, K.-J.; Bandrauk, A.D. Electron Interference in Molecular Circular Polarization Attosecond XUV Photoionization. Photonics 2015, 2, 71-92. https://doi.org/10.3390/photonics2010071
Yuan K-J, Bandrauk AD. Electron Interference in Molecular Circular Polarization Attosecond XUV Photoionization. Photonics. 2015; 2(1):71-92. https://doi.org/10.3390/photonics2010071
Chicago/Turabian StyleYuan, Kai-Jun, and André D. Bandrauk. 2015. "Electron Interference in Molecular Circular Polarization Attosecond XUV Photoionization" Photonics 2, no. 1: 71-92. https://doi.org/10.3390/photonics2010071
APA StyleYuan, K.-J., & Bandrauk, A. D. (2015). Electron Interference in Molecular Circular Polarization Attosecond XUV Photoionization. Photonics, 2(1), 71-92. https://doi.org/10.3390/photonics2010071