
Coupled Neural–Glial Dynamics and the Role of Astrocytes in Alzheimer’s Disease



Abstract
:1. Introduction
2. AD Network Model for Brain Connectome Data Assimilation
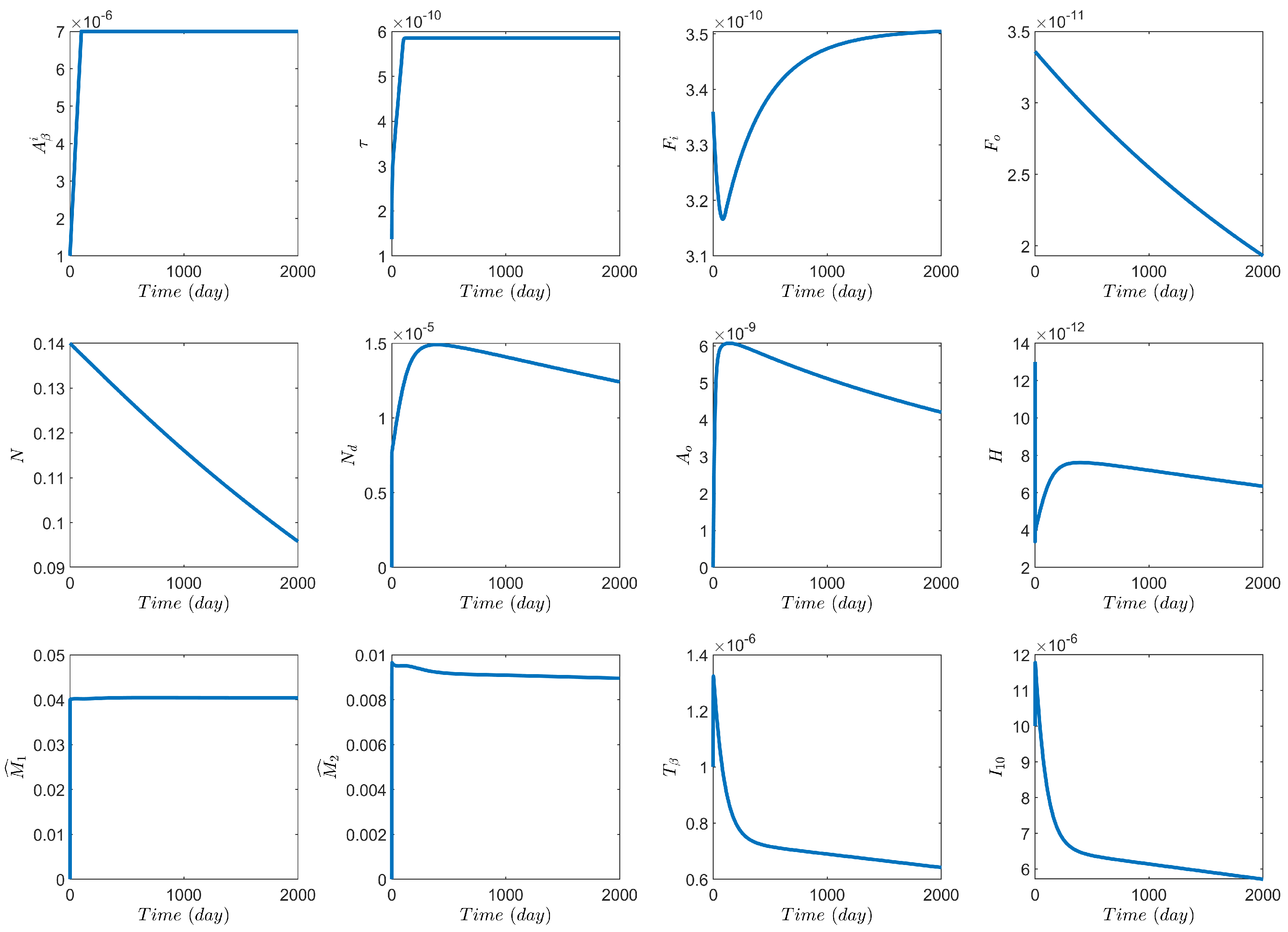
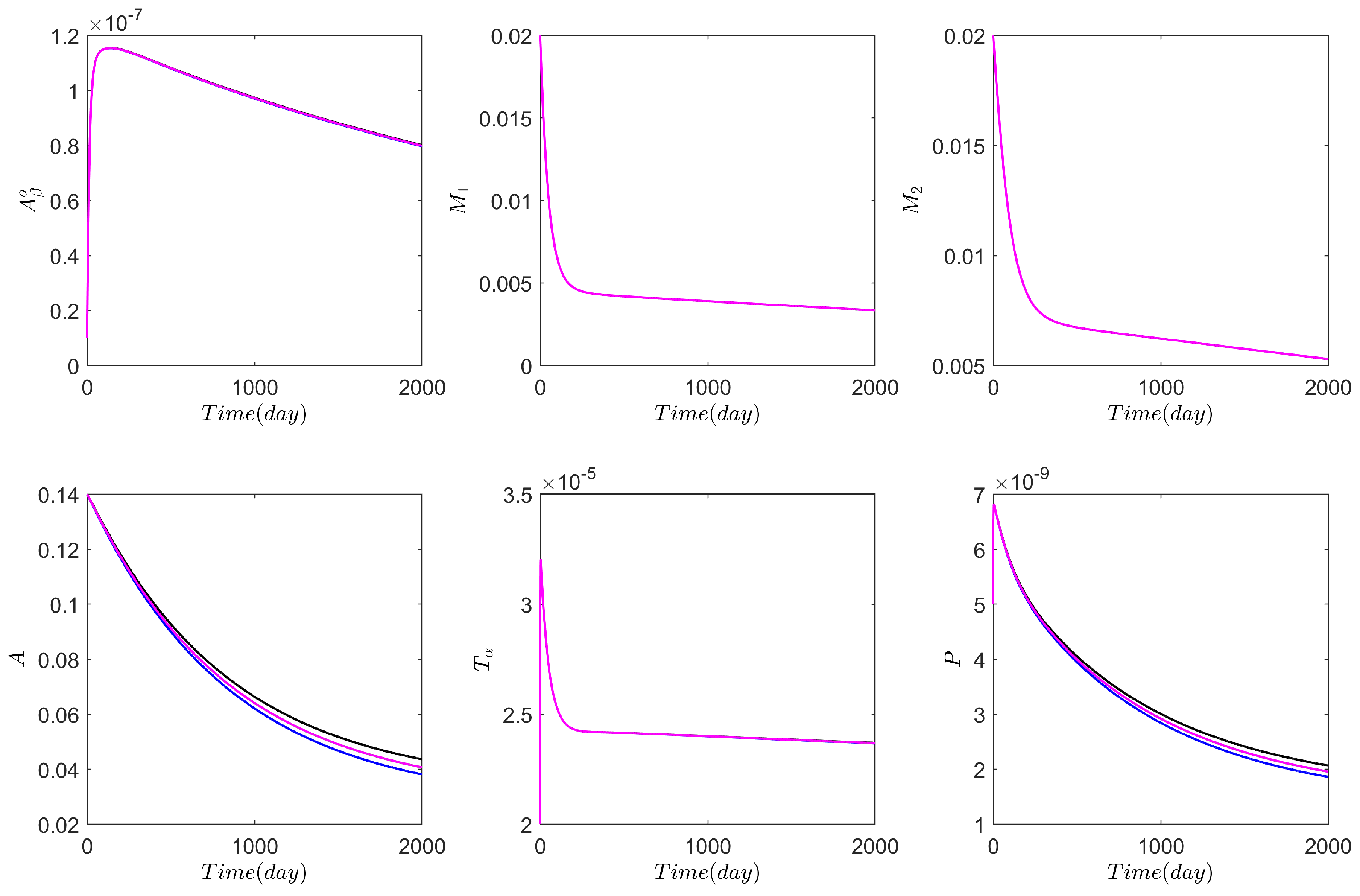
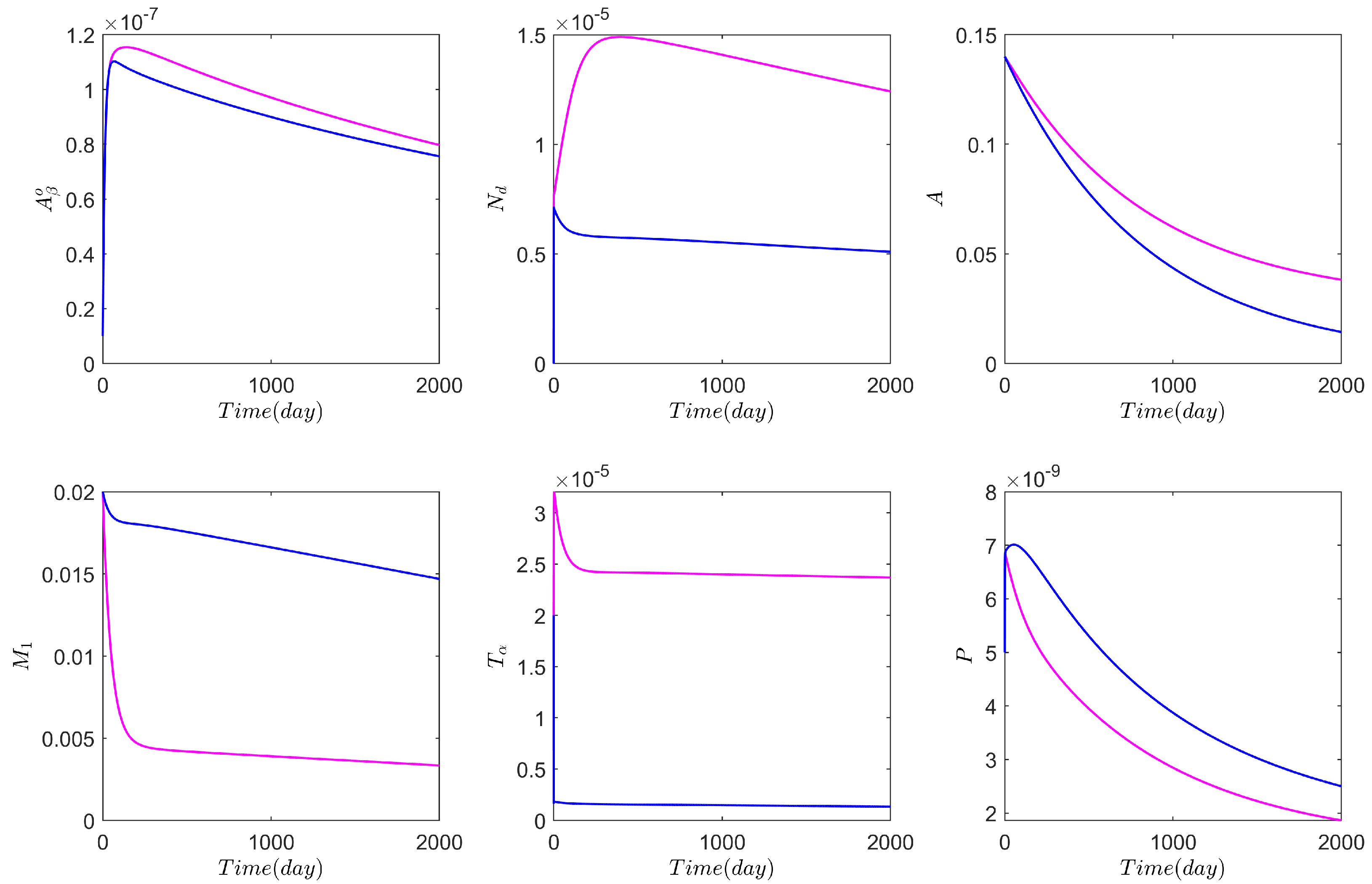
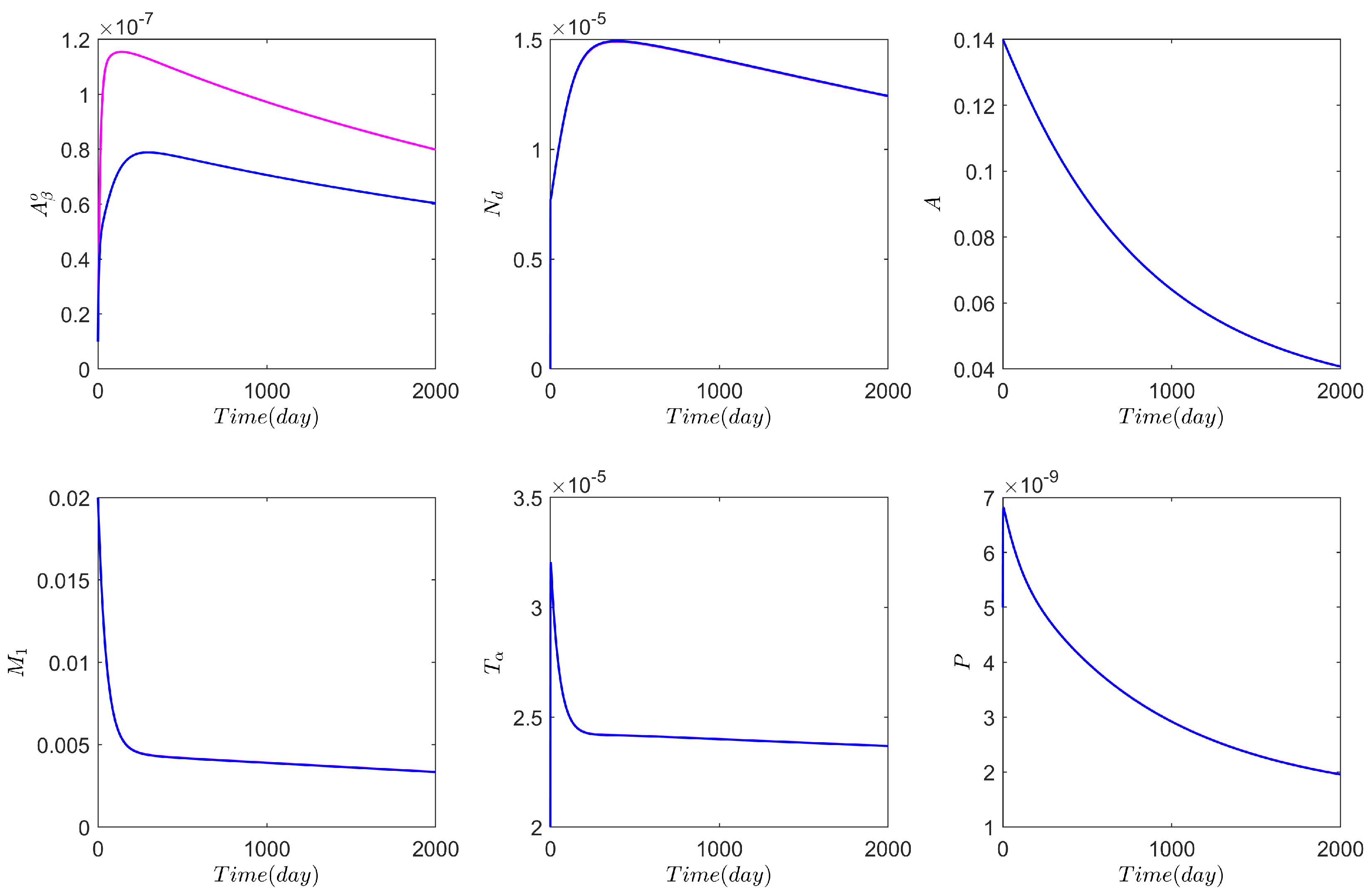
3. Numerical Results Based on the Network Model
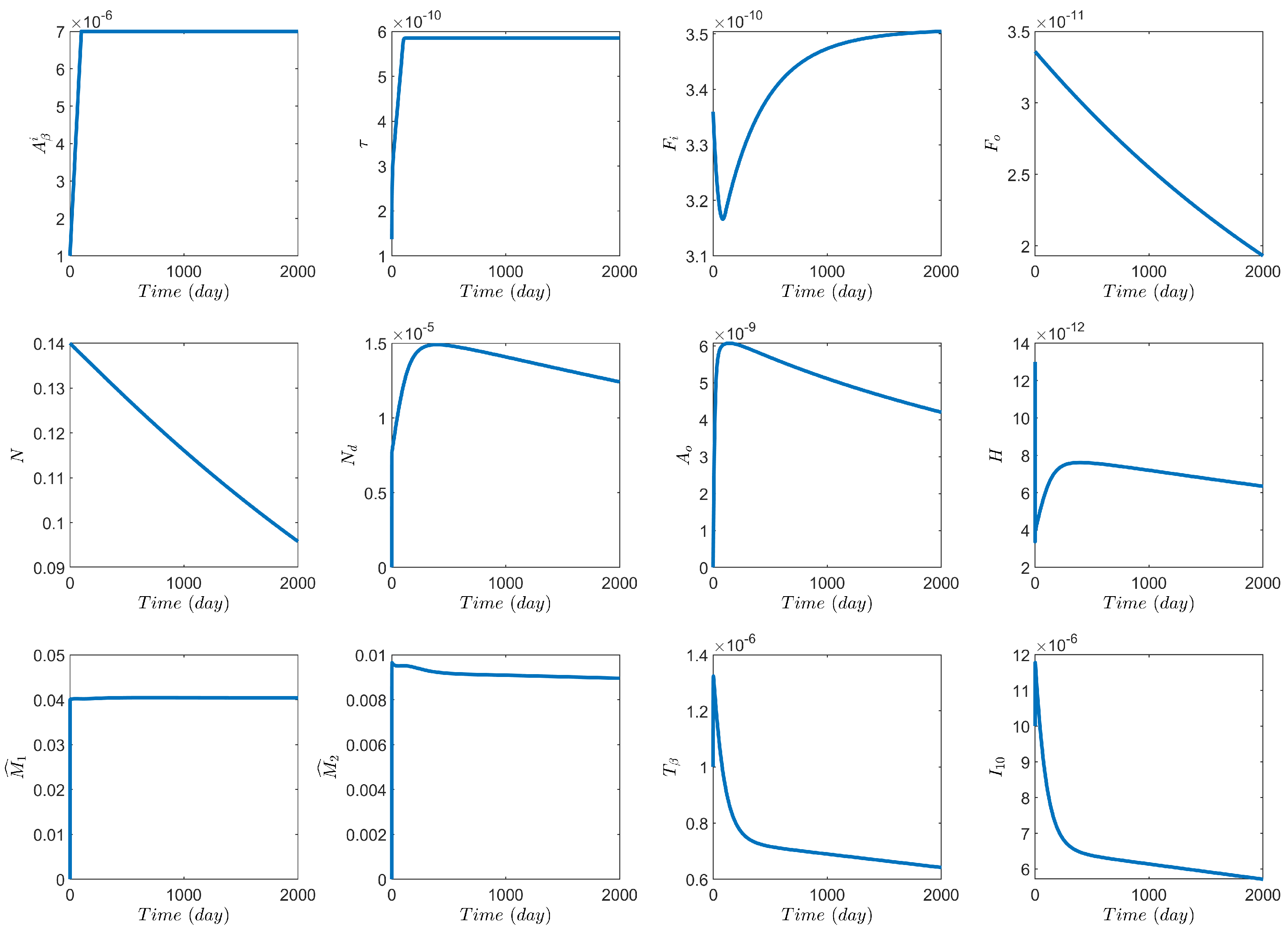
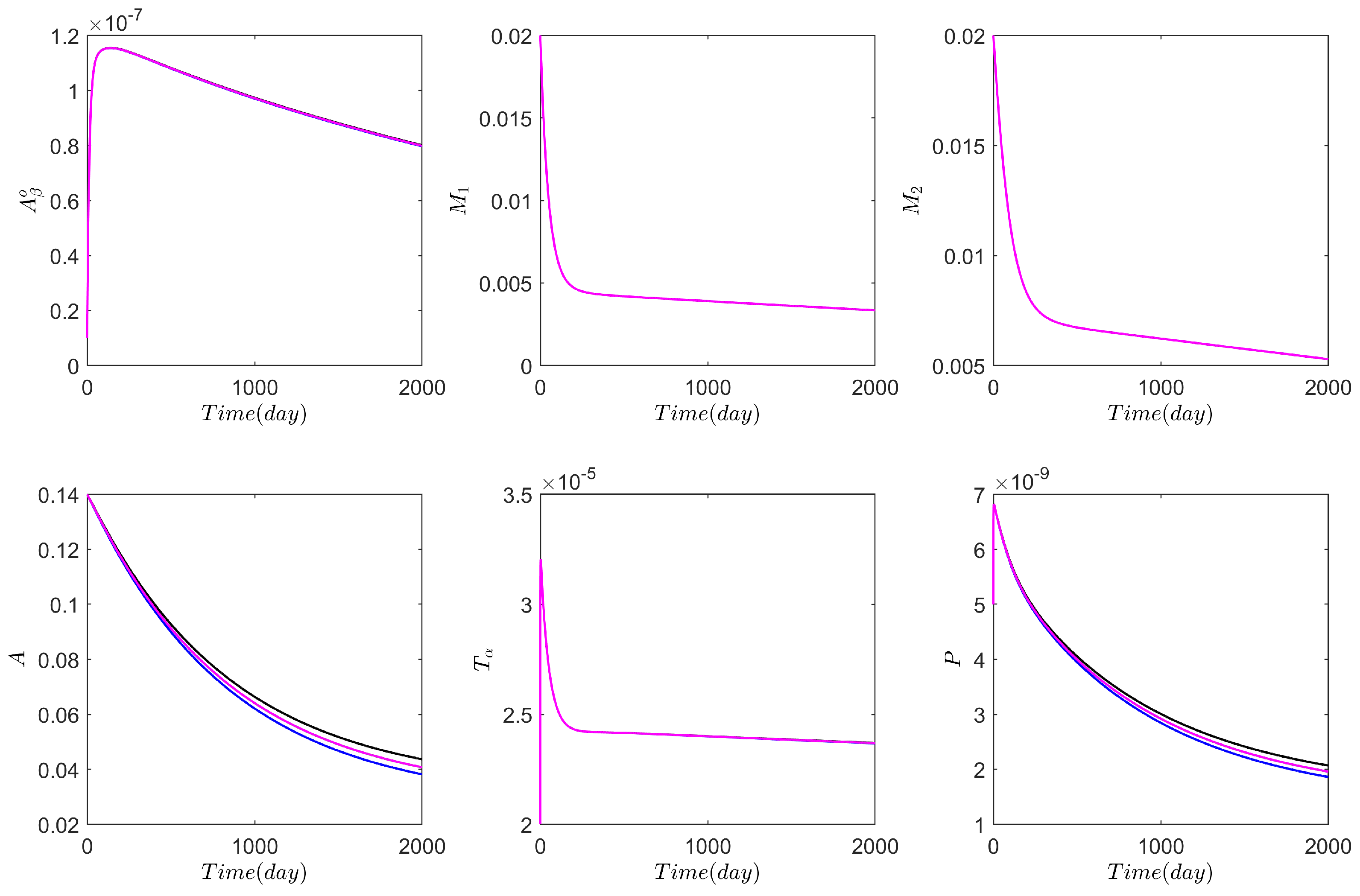
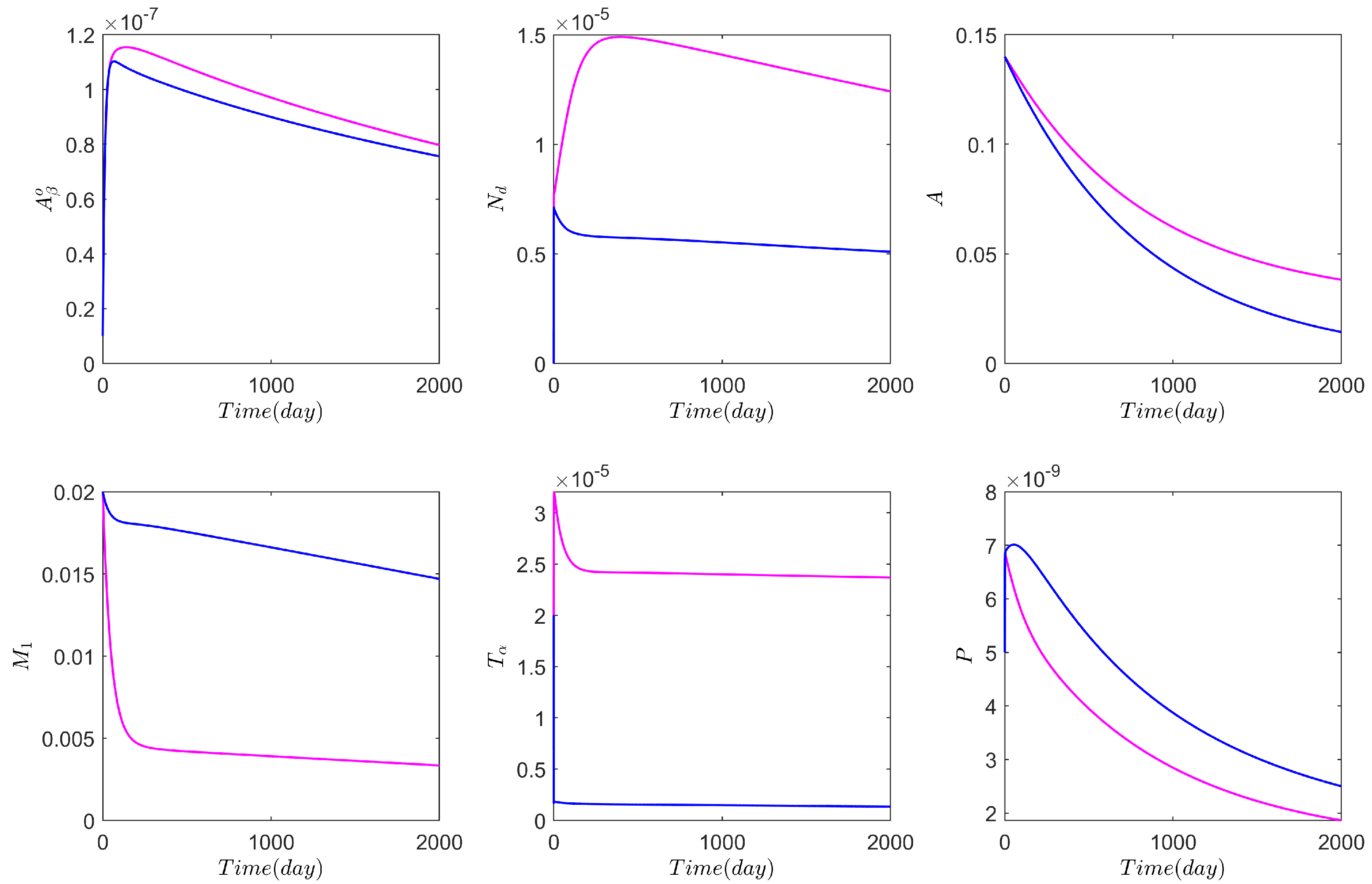
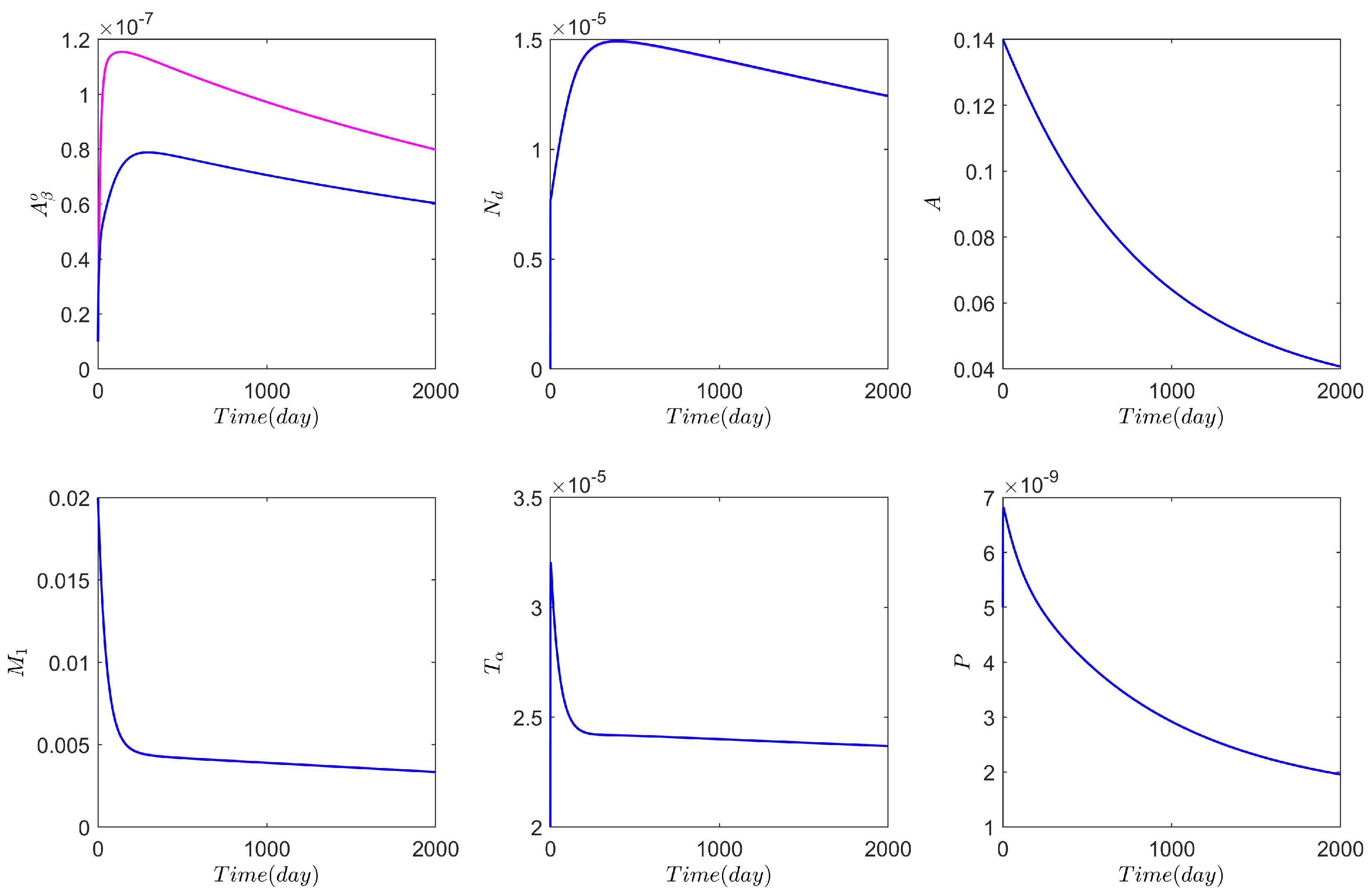
3.1. Computational Experiments on the Role of Cytokines and Astrocytes in AD
3.2. Computational Experiments on AD Drugs
4. Discussion and Future Directions
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nichols, E.; Steinmetz, J.D.; Vollset, S.E.; Fukutaki, K.; Chalek, J.; Abd-Allah, F.; Abdoli, A.; Abualhasan, A.; Abu-Gharbieh, E.; Akram, T.T.; et al. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- Torres-Acosta, N.; O’Keefe, J.H.; O’Keefe, E.L.; Isaacson, R.; Small, G. Therapeutic potential of TNF-α inhibition for Alzheimer’s disease prevention. J. Alzheimer’s Dis. 2020, 78, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A. The Role of Protein Misfolding and Tau Oligomers (TauOs) in Alzheimer’s Disease (AD). Int. J. Mol. Sci. 2019, 20, 4661. [Google Scholar] [CrossRef] [Green Version]
- Frost, G.R.; Li, Y.M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017, 7, 170228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaheen, H.; Singh, S.; Melnik, R. A neuron-glial model of exosomal release in the onset and progression of Alzheimer’s disease. Front. Comput. Neurosci. 2021, 15, 653097. [Google Scholar] [CrossRef]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smit, T.; Deshayes, N.A.; Borchelt, D.R.; Kamphuis, W.; Middeldorp, J.; Hol, E.M. Reactive astrocytes as treatment targets in Alzheimer’s disease—Systematic review of studies using the APPswePS1dE9 mouse model. Glia 2021, 69, 1852–1881. [Google Scholar] [CrossRef]
- Preman, P.; Alfonso-Triguero, M.; Alberdi, E.; Verkhratsky, A.; Arranz, A.M. Astrocytes in Alzheimer’s disease: Pathological significance and molecular pathways. Cells 2021, 10, 540. [Google Scholar] [CrossRef]
- Raj, A.; Tora, V.; Gao, X.; Cho, H.; Choi, J.Y.; Ryu, Y.H.; Lyoo, C.H.; Franchi, B. Combined Model of Aggregation and Network Diffusion Recapitulates Alzheimer’s Regional Tau-Positron Emission Tomography. Brain Connect. 2021, 11, 624–638. [Google Scholar] [CrossRef]
- Bertsch, M.; Franchi, B.; Raj, A.; Tesi, M.C. Macroscopic modelling of Alzheimer’s disease: Difficulties and challenges. Brain Multiphysics 2021, 2, 100040. [Google Scholar] [CrossRef]
- Planas-Fontanez, T.M.; Sainato, D.M.; Sharma, I.; Dreyfus, C.F. Roles of astrocytes in response to aging, Alzheimer’s disease and multiple sclerosis. Brain Res. 2021, 1764, 147464. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Olabarria, M.; Noristani, H.N.; Yeh, C.Y.; Rodriguez, J.J. Astrocytes in Alzheimer’s disease. Neurotherapeutics 2010, 7, 399–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Birch, A.M. The contribution of astrocytes to Alzheimer’s disease. Biochem. Soc. Trans. 2014, 42, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Habib, N.; Centini, G.; Lazzeri, L.; Amoruso, N.; El Khoury, L.; Zupi, E.; Afors, K. Bowel Endometriosis: Current Perspectives on Diagnosis and Treatment. Int. J. Women’s Health 2020, 12, 35–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Hao, W.; Friedman, A. Mathematical model on Alzheimer’s disease. BMC Syst. Biol. 2016, 10, 108. [Google Scholar] [CrossRef] [Green Version]
- Thompson, T.B.; Chaggar, P.; Kuhl, E.; Goriely, A. Protein-protein interactions in neurodegenerative diseases: A conspiracy theory. PLoS Comput. Biol. 2020, 16, e1008267. [Google Scholar] [CrossRef]
- Pal, S.; Melnik, R. Pathology dynamics in healthy-toxic protein interaction and the multiscale analysis of neurodegenerative diseases. In Computational Science—ICCS 2021; Paszynski, M., Kranzlmüller, D., Krzhizhanovskaya, V.V., Dongarra, J.J., Sloot, P.M., Eds.; Lecture Notes in Computer Science; Springer: Cham, Switzerland, 2021; Volume 12746, pp. 528–540. [Google Scholar]
- Roskoski, R. Michaelis-Menten Kinetics. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Seeman, P.; Seeman, N. Alzheimer’s disease: Beta-amyloid plaque formation in human brain. Synapse 2011, 65, 1289–1297. [Google Scholar] [CrossRef]
- Zhao, J.; O’Connor, T.; Vassar, R. The contribution of activated astrocytes to Aβ production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2011, 8, 150. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, P.; Wu, J.; Wang, Y.; Li, P.; Hou, X.; Zhang, Q.; Wei, N.; Zhao, Z.; Liang, H.; et al. The Cascade of Oxidative Stress and Tau Protein Autophagic Dysfunction in Alzheimer’s Disease. Alzheimer’s Dis. Challenges Future 2015, 2, 48347. [Google Scholar]
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondragon-Rodriguez, S.; Perry, G.; Zhu, X.; Boehm, J. Amyloid Beta and tau proteins as therapeutic targets for Alzheimer’s disease treatment: Rethinking the current strategy. Int. J. Alzheimers Dis. 2012, 2012, 630182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wray, S.; Noble, W. Linking amyloid and tau pathology in Alzheimer’s disease: The role of membrane cholesterol in Abeta-mediated tau toxicity. J. Neurosci. 2009, 29, 9665–9667. [Google Scholar] [CrossRef]
- Garwood, C.J.; Pooler, A.M.; Atherton, J.; Hanger, D.P.; Noble, W. Astrocytes are important mediators of Abeta-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011, 2, 167. [Google Scholar] [CrossRef] [Green Version]
- Kevrekidis, P.G.; Thompson, T.B.; Goriely, A. Anisotropic diffusion and traveling waves of toxic proteins in neurodegenerative diseases. Phys. Lett. A 2020, 384, 12935. [Google Scholar] [CrossRef]
- Bertsch, M.; Franchi, B.; Meschini, V.; Tesi, M.C.; Tosin, A. A sensitivity analysis of a mathematical model for the synergistic interplay of amyloid beta and tau on the dynamics of Alzheimer’s disease. Brain Multiphys. 2021, 2, 1–13. [Google Scholar] [CrossRef]
- Waters, J. The concentration of soluble extracellular amyloid beta protein in acute brain slices from CRND8 mice. PLoS ONE 2010, 5, e15709. [Google Scholar] [CrossRef]
- Gao, H.M.; Zhou, H.; Zhang, F.; Wilson, B.C.; Kam, W.; Hong, J.S. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J. Neurosci. 2011, 31, 1081–1092. [Google Scholar] [CrossRef]
- Zou, J.Y.; Crews, F.T. Release of neuronal HMGB1 by ethanol through decreased HDAC activity activates brain neuroimmune signalling. PLoS ONE 2014, 9, e87915. [Google Scholar]
- Savchenko, V.L.; McKanna, J.A.; Nikonenko, I.R.; Skibo, G.G. Microglia and astrocytes in the adult rat brain: Comparative immunocytochemical analysis demonstrates the efficacy of lipocortin 1 immunoreactivity. Neuroscience 2000, 96, 195–203. [Google Scholar] [CrossRef]
- Hao, W.; Crouser, E.D.; Friedman, A. Mathematical model of sarcoidosis. Proc. Natl. Acad. Sci. USA 2014, 111, 16065–16070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohsfield, L.A.; Humpel, C. Migration of blood cells to beta-amyloid plaques in Alzheimer’s disease. Exp. Gerontol. 2015, 65, 8–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theriault, P.; ElAli, A.; Rivest, S. The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimers Res. Ther. 2015, 7, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalkai, B.; Kerepesi, C.; Varga, B.; Grolmusz, V. High-Resolution Directed Human Connectomes and the Consensus Connectome Dynamics. PLoS ONE 2019, 14, e0215473. [Google Scholar] [CrossRef]
- Hao, W.; Friedman, A. The LDL-HDL profile determines the risk of atherosclerosis: A mathematical model. PLoS ONE 2014, 9, e90497. [Google Scholar] [CrossRef] [Green Version]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting tumour necrosis factor alpha for Alzheimer’s disease. Curr. Alzheimer. Res. 2017, 14, 412–425. [Google Scholar] [CrossRef] [Green Version]
- Kern, D.M.; Lovestone, S.; Cepeda, M.S. Treatment with TNF-α inhibitors versus methotrexate and the association with dementia and Alzheimer’s disease. Alzheimer’s Dement. 2021, 7, e12163. [Google Scholar] [CrossRef]
- Jayaraman, A.; Htike, T.T.; James, R.; Picon, C.; Reynolds, R. TNF-mediated neuroinflammation is linked to neuronal necroptosis in Alzheimer’s disease hippocampus. Acta Neuropathol. Commun. 2021, 9, 159. [Google Scholar] [CrossRef]
- Pillai, J.A.; Bebek, G.; Khrestian, M.; Bena, J.; Bergmann, C.C.; Bush, W.S.; Leverenz, J.B.; Bekris, L.M. TNFRSF1B gene variants and related soluble TNFR2 levels impact resilience in Alzheimer’s disease. Front. Aging Neurosci. 2021, 13, 638922. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s disease drug development pipeline: 2021. Alzheimer’s Dement. 2021, 7, e12179. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Melnik, R. Nonlocal models in the analysis of brain neurodegenerative protein dynamics with application to Alzheimer’s disease. arXiv 2021, arXiv:2112.11681. [Google Scholar]
- Chang, R.; Yee, K.L.; Sumbria, R.K. Tumour necrosis factor α inhibition for Alzheimer’s disease. J. Cent. Nerv. Syst. Dis. 2017, 9, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Knox, J.; Chang, J.; Derbedrossian, A.; Vasilevko, V.; Cribbs, D.; Boado, R.J.; Pardridge, W.M.; Sumbria, R.K. Blood–Brain Barrier Penetrating Biologic TNF-α Inhibitor for Alzheimer’s Disease. Mol. Pharm. 2017, 14, 2340–2349. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Parameter | Value | Parameter | Value |
|---|---|---|---|---|---|
| 9.51 | 9.51 | 0.03 | |||
| 0.017 | |||||
| 0.04 | |||||
| 0.007 | |||||
| 0.47 | |||||
| 333 | |||||
| 0.14 | 0.47 | ||||
| 6 | 0.05 | ||||
| 0.47 | 0.14 | ||||
| Parameter | Value | Parameter | Value | Parameter | Value |
|---|---|---|---|---|---|
| 5 | 10 | 1 |
| Variable | Function | Variable | Function |
|---|---|---|---|
| Amyloid beta inside neurons | Amyloid beta outside neurons | ||
| hyperphosphorylated tau protein | Neurofibrillary tangle inside neurons | ||
| Neurofibrillary tangle outside neurons | N | Live neurons | |
| A | Astrocytes | Dead neurons | |
| Amyloid beta oligomer | H | High mobility group box 1 | |
| Proinflammatory microglias | Anti-inflammatory microglias | ||
| Peripheral proinflammatory macrophages | Peripheral anti-inflammatory macrophages | ||
| Tumour necrosis factor alpha | Transforming growth factor beta | ||
| Interleukin 10 | P | Monocyte chemoattractant protein-1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pal, S.; Melnik, R. Coupled Neural–Glial Dynamics and the Role of Astrocytes in Alzheimer’s Disease. Math. Comput. Appl. 2022, 27, 33. https://doi.org/10.3390/mca27030033
Pal S, Melnik R. Coupled Neural–Glial Dynamics and the Role of Astrocytes in Alzheimer’s Disease. Mathematical and Computational Applications. 2022; 27(3):33. https://doi.org/10.3390/mca27030033
Chicago/Turabian StylePal, Swadesh, and Roderick Melnik. 2022. "Coupled Neural–Glial Dynamics and the Role of Astrocytes in Alzheimer’s Disease" Mathematical and Computational Applications 27, no. 3: 33. https://doi.org/10.3390/mca27030033
APA StylePal, S., & Melnik, R. (2022). Coupled Neural–Glial Dynamics and the Role of Astrocytes in Alzheimer’s Disease. Mathematical and Computational Applications, 27(3), 33. https://doi.org/10.3390/mca27030033