
Simvastatin: In Vitro Metabolic Profiling of a Potent Competitive HMG-CoA Reductase Inhibitor


Abstract

1. Introduction
2. Materials and Methods
2.1. General
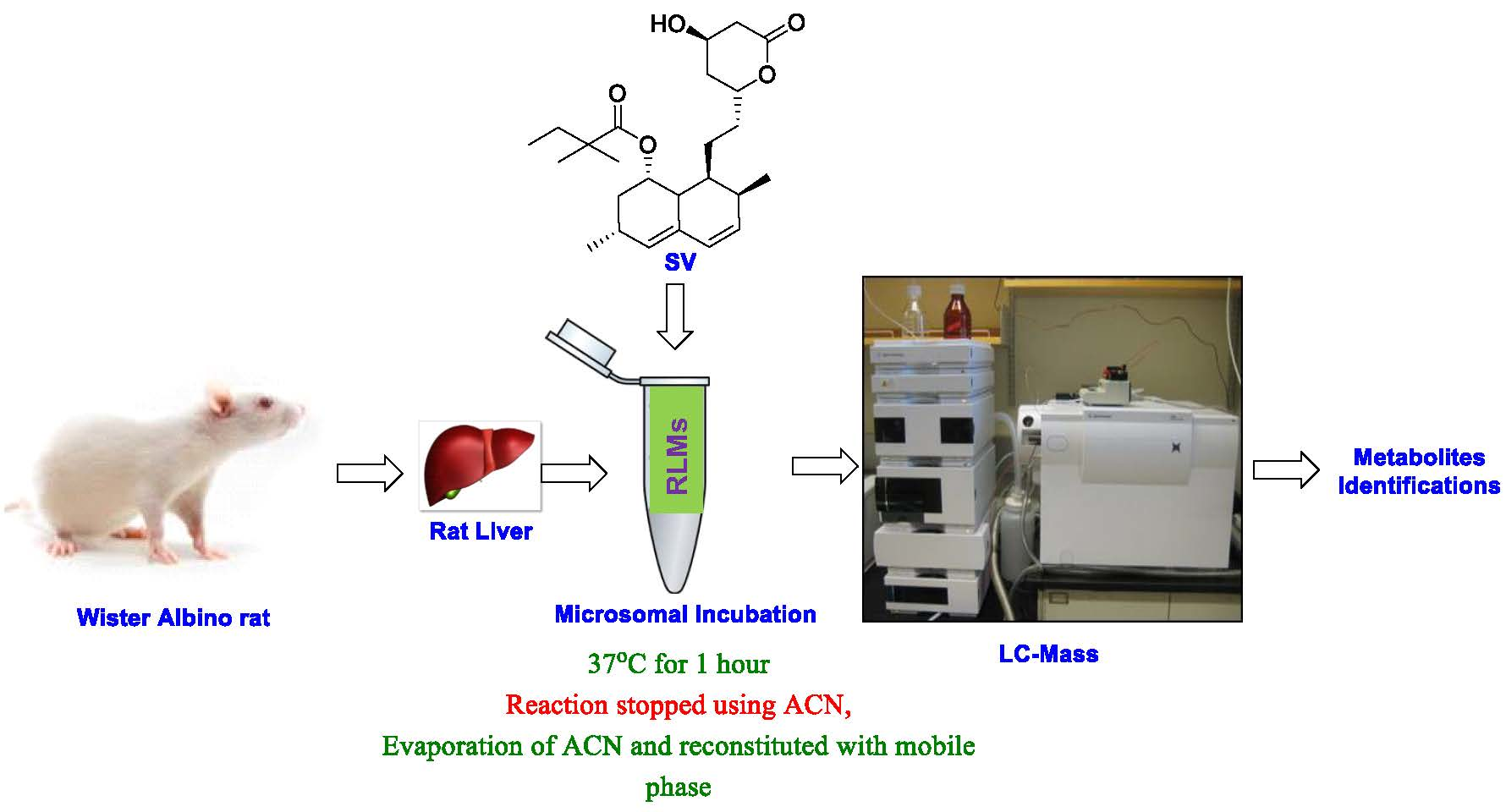
2.2. Verification of Microsomal CYP450 Activity
2.3. Determination of Protein Content of the Prepared RLMs
2.4. Instrumentation
2.5. Preparation of RLMs
2.5.1. Differential Centrifugation Method
2.5.2. Calcium Aggregation Method [41]
2.5.3. Incubation of Simvastatin with Rat Liver Microsomes
2.6. Isolated Perfused Rat Liver Hepatocytes
Perfusion of Rat Liver Hepatocyte Protocol
2.7. Incubation of Simvastatin with Isolated Perfused Rat Liver Hepatocytes
2.8. Mass Spectrometric Conditions
3. Results and Discussion
3.1. LC–MS Data of SV
3.2. Cytochrome P450-Dependent Metabolism
3.3. LC–MS Data of Metabolites
3.3.1. Identification of Phase-I Metabolites
Monohydroxylated Metabolites (M1–M6)
Dihydroxylated Metabolites (M7–M13)
Exomethylene SV (M14 and/or M15)
Carboxyl-SV (M16–M19)
Dihydrodiol Metabolite (M20)
Exomethylene-SVA Metabolites (M21–M23)
Hydroxl-SVA Metabolites (M24–M26)
Dihydrodiol-SVA Metabolites (M27–M28)
Hydrolyzed-SV (SVA) Metabolites (M29)
Summary of Detected Phase-I Metabolites in LC–MS
3.3.2. Identification of Phase-II Metabolites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wen, B.; Zhu, M. Applications of mass spectrometry in drug metabolism: 50 years of progress. Drug Metab. Rev. 2015, 47, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tang, W. Drug metabolism in drug discovery and development. Acta Pharm. Sin. B 2018, 8, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Nelson, S.D. Common Biotransformation Reactions. In Mass Spectrometry in Drug Metabolism and Disposition: Basic Principles and Applications, 1st ed.; Lee, M.S., Zhu, M., Eds.; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2011; pp. 13–41. [Google Scholar]
- Obach, R.S. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 1999, 27, 1350–1359. [Google Scholar] [PubMed]
- Carlile, D.J.; Stevens, A.J.; Ashforth, E.I.; Waghela, D.; Houston, J.B. In vivo clearance of ethoxycoumarin and its prediction from In vitro systems. Use Of drug depletion and metabolite formation methods in hepatic microsomes and isolated hepatocytes. Drug Metab. Dispos. 1998, 26, 216–221. [Google Scholar]
- Endo, A.; Kuroda, M.; Tanzawa, K. Competitive inhibition of 3-hydroxy-3-methylglutaryl coenzyme A reductase by ML-236A and ML-236B fungal metabolites, having hypocholesterolemic activity. FEBS Lett. 1976, 72, 323–326. [Google Scholar] [CrossRef]
- Nováková, L.; Vlčková, H.; Šatínský, D.; Sadílek, P.; Solichová, D.; Bláha, M.; Bláha, V.; Solich, P. Ultra-high-performance liquid chromatography tandem mass spectrometric detection in clinical analysis of simvastatin and atorvastatin. J. Chromatogr. B 2009, 877, 2093–2103. [Google Scholar] [CrossRef]
- Shitara, Y.; Sugiyama, Y. Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: Drug–drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol. Ther. 2006, 112, 71–105. [Google Scholar] [CrossRef]
- Sandhu, S.; Wiebe, N.; Fried, L.F.; Tonelli, M. Statins for improving renal outcomes: A meta-analysis. J. Am. Soc. Nephrol. 2006, 17, 2006–2016. [Google Scholar] [CrossRef]
- Sacks, F.M. The relative role of low-density lipoprotein cholesterol and high-density lipoprotein cholesterol in coronary artery disease: Evidence from large-scale statin and fibrate trials. Am. J. Cardiol. 2001, 88, 14–18. [Google Scholar] [CrossRef]
- Bertolini, S.; Bittolo Bon, G.; Campbell, L.M.; Farnier, M.; Langan, J.; Mahla, G.; Pauciullo, P.; Sirtori, C.; Egros, F.; Fayyad, R.; et al. Efficacy and safety of atorvastatin compared to pravastatin in patients with hypercholesterolemia. Atherosclerosis 1997, 130, 191–197. [Google Scholar] [CrossRef]
- Yang, D.J.; Hwang, L.S. Study on the conversion of three natural statins from lactone forms to their corresponding hydroxy acid forms and their determination in Pu-Erh tea. J. Chromatogr. A 2006, 30, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Jemal, M.; Xia, Y.Q. Bioanalytical method validation design for the simultaneous quantitation of analytes that may undergo interconversion during analysis. J. Pharm. Biomed. Anal. 2000, 22, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Corsini, A.; Maggi, F.M.; Catapano, A.L. Pharmacology of competitive inhibitors of HMG-CoA reductase. Pharmacol. Res. 1995, 31, 9–27. [Google Scholar] [CrossRef]
- Pawan Kumar, B.; Deepti, J. Simvastatin: Review of Updates on Recent Trends in Pharmacokinetics, Pharmacodynamics, Drug–drug Interaction, Impurities and Analytical Methods. Curr. Pharm. Anal. 2012, 8, 135–156. [Google Scholar] [CrossRef]
- Simvastatin. Available online: http://www.drugs.com/monograph/simvastatin.html (accessed on 20 June 2022).
- Alberts, A.W. Lovastatin and simvastatin—Inhibitors of HMG CoA reductase and cholesterol biosynthesis. Cardiology 1990, 77 (Suppl. 4), 14–21. [Google Scholar] [CrossRef]
- Bybee, K.A.; Lee, J.H.; O’Keefe, J.H. Cumulative clinical trial data on atorvastatin for reducing cardiovascular events: The clinical impact of atorvastatin. Curr. Med. Res. Opin. 2008, 24, 1217–1229. [Google Scholar] [CrossRef] [PubMed]
- Roche, V.F. Antihyperlipidemic Statins: A Self-Contained, Clinically Relevant Medicinal Chemistry Lesson. Am. J. Pharm. Educ. 2005, 69, 77. [Google Scholar] [CrossRef]
- Lennernäs, H.; Fager, G. Pharmacodynamics and pharmacokinetics of the HMG-CoA reductase inhibitors. Similarities and differences. Clin. Pharmacokinet. 1997, 32, 403–425. [Google Scholar] [CrossRef]
- Naci, H.; Brugts, J.; Ades, T. Comparative tolerability and harms of individual statins: A study-level network meta-analysis of 246,955 participants from 135 randomized, controlled trials. Circ. Cardiovasc. Qual. Outcomes 2013, 6, 390–399. [Google Scholar] [CrossRef]
- Di Stasi, S.L.; MacLeod, T.D.; Winters, J.D.; Binder-Macleod, S.A. Effects of statins on skeletal muscle: A perspective for physical therapists. Phys. Ther. 2010, 90, 1530–1542. [Google Scholar] [CrossRef]
- Alkhatatbeh, M.J.; Abdul-Razzak, K.K.; Khasawneh, L.Q.; Saadeh, N.A. Prevalence of musculoskeletal pain in association with serum 25-hydroxyvitamin D concentrations in patients with type 2 diabetes mellitus. Biomed. Rep. 2018, 8, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Weise, W.J.; Possidente, C.J. Fatal rhabdomyolysis associated with simvastatin in a renal transplant patient. Am. J. Med. 2000, 108, 351–352. [Google Scholar] [CrossRef] [PubMed]
- Summaries for Patients. Muscle abnormalities in four patients taking statins to treat unfavorable cholesterol levels. Ann. Intern. Med. 2002, 137, I45. [Google Scholar] [CrossRef] [PubMed]
- Meier, C.R.; Schlienger, R.G.; Kraenzlin, M.E.; Schlegel, B.; Jick, H. HMG-CoA reductase inhibitors and the risk of fractures. JAMA 2000, 283, 3205–3210. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. DNA topoisomerase I inhibitors: Chemistry, biology, and interfacial inhibition. Chem. Rev. 2009, 109, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Prescribing Medicines in Pregnancy Database. Available online: https://www.huidziekten.nl/formularium/documenten/medicines-pregnancy.pdf (accessed on 20 June 2022).
- Uchiyama, N.; Kagami, Y.; Saito, Y.; Abe, S.; Ohtawa, M.; Hata, S. Metabolic fate of 2,2-dimethylbutyryl moiety of simvastatin in rats: Identification of metabolites by gas chromatography/ mass spectrometry. Eur. J. Drug Metab. Pharmacokinet. 1991, 16, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Ohtawa, M.; Uchiyama, N. Sex difference in metabolism of simvastatin by rat hepatic microsomes. Eur. J. Drug Metab. Pharmacokinet. 1992, 17, 175–181. [Google Scholar] [CrossRef]
- Uchiyama, N.; Kagami, Y.; Saitoh, Y.; Ohtawa, M. Male-specific metabolism of simvastatin by rat liver microsomes. Chem. Pharm. Bull. 1991, 39, 236–238. [Google Scholar] [CrossRef]
- Prueksaritanont, T.; Gorham, L.M.; Ma, B.; Liu, L.; Yu, X.; Zhao, J.J.; Slaughter, D.E.; Arison, B.H.; Vyas, K.P. In vitro metabolism of simvastatin in humans [SBT] identification of metabolizing enzymes and effect of the drug on hepatic P450s. Drug Metab. Dispos. 1997, 25, 1191–1199. [Google Scholar]
- Fenne, O.S. In Vitro Studies Suggest Involvement of CYP2D6 in the Metabolism of Simvastatin and Active Metabolites. Master’s Thesis, University of Oslo, Oslo, Norway, 2003. [Google Scholar]
- Yin, W.; Al-Wabli, R.I.; Attwa, M.W.; Rahman, A.F.M.M.; Kadi, A.A. Detection and characterization of simvastatin and its metabolites in rat tissues and biological fluids using MALDI high resolution mass spectrometry approach. Sci. Rep. 2022, 12, 4757. [Google Scholar] [CrossRef]
- Kadi, A.A.; Yin, W.; Rahman, A.F.M.M. In-Vitro metabolic profiling study of potential topoisomerase inhibitors ‘pyrazolines’ in RLMs by mass spectrometry. J. Chromatogr. B 2019, 1114, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.F.M.M.; Al-Shakliah, N.S.; Yin, W.; Kadi, A.A. In vitro Investigation of Metabolic Profiling of a Potent Topoisomerase Inhibitors Fluorescein Hydrazones (FLHs) in RLMs by LC-MS/MS. J. Chromatogr. B 2017, 1054, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Kadi, A.A.; Al-Shakliah, N.S.; Yin, W.; Rahman, A.F.M.M. In vitro investigation of metabolic profiling of newly developed topoisomerase inhibitors (ethyl fluorescein hydrazones, EtFLHs) in RLMs by LC–MS/MS. J. Chromatogr. B 2017, 1054, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Billings, R.E. Sex differences in rats in the metabolism of phenytoin to 5-(3,4-dihydroxyphenyl)-5-phenylhydantoin. J. Pharmacol. Exp. Ther. 1983, 225, 630–636. [Google Scholar] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Mountney, A.; Boutte, A.M.; Gilsdorf, J.; Lu, X.C.; Tortella, F.C.; Shear, D.A. Intravenous Administration of Simvastatin Improves Cognitive Outcome following Severe Traumatic Brain Injury in Rats. J. Neurotrauma 2016, 33, 1492–1500. [Google Scholar] [CrossRef]
- Iyer, K.; Walawalkar, P.; Serai, P. Isolation and catalytic competence of different animal liver microsomal fractions prepared by calcium-aggregation method. Indian J. Pharm. Sci. 2006, 2, 262. [Google Scholar] [CrossRef]
- Schenkman, J.B.; Cinti, D.L. Preparation of microsomes with calcium. Methods Enzymol. 1978, 52, 83–89. [Google Scholar]
- Berry, M.N.; Friend, D.S. High-yield preparation of isolated rat liver parenchymal cells: A biochemical and fine structural study. J. Cell Biol. 1969, 43, 506–520. [Google Scholar] [CrossRef]
- Germershausen, J.I.; Hunt, V.M.; Bostedor, R.G.; Bailey, P.J.; Karkas, J.D.; Alberts, A.W. Tissue selectivity of the cholesterol-lowering agents lovastatin, simvastatin and pravastatin in rats in vivo. Biochem. Biophys. Res. Commun. 1989, 158, 667–675. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, J.; Zhang, Y.; Zuo, Z. Role of esterase mediated hydrolysis of simvastatin in human and rat blood and its impact on pharmacokinetic profiles of simvastatin and its active metabolite in rat. J. Pharm. Biomed. Anal. 2019, 168, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Vickers, S.; Duncan, C.A.; Vyas, K.P.; Kari, P.H.; Arison, B.; Prakash, S.R.; Ramjit, H.G.; Pitzenberger, S.M.; Stokker, G.; Duggan, D.E. In vitro and in vivo biotransformation of simvastatin, an inhibitor of HMG CoA reductase. Drug Metab. Dispos. 1990, 18, 476–483. [Google Scholar] [PubMed]
- Vickers, S.; Duncan, C.A.; Chen, I.W.; Rosegay, A.; Duggan, D.E. Metabolic disposition studies on simvastatin, a cholesterol-lowering prodrug. Drug Metab. Dispos. 1990, 18, 138–145. [Google Scholar]
- Prueksaritanont, T.; Ma, B.; Yu, N. The human hepatic metabolism of simvastatin hydroxy acid is mediated primarily by CYP3A, and not CYP2D6. Br. J. Clin. Pharmacol. 2003, 56, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Luo, G.; Ding, X.; Lu, C. Preclinical experimental models of drug metabolism and disposition in drug discovery and development. Acta Pharm. Sin. B 2012, 2, 549–561. [Google Scholar] [CrossRef]
- Prueksaritanont, T.; Subramanian, R.; Fang, X.; Ma, B.; Qiu, Y.; Lin, J.H.; Pearson, P.G.; Baillie, T.A. Glucuronidation of Statins in Animals and Humans: A Novel Mechanism of Statin Lactonization. Drug Metab. Dispos. 2002, 30, 505–512. [Google Scholar] [CrossRef]
- Fish, R.; Danneman, P.; Brown, M.; Karas, A. Anesthesia and Analgesia in Laboratory Animals; Academic Press: London, UK, 1997. [Google Scholar]
- Strober, W. Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. 2001, 3, A3B1–A3B2. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Detected Metabolites | No. | Ion Trap | QqQ | ESI (+) (m/z) | ||
|---|---|---|---|---|---|---|
| Rt (min) | Detected | Rt (min) | Detected | |||
| Monohydroxy-SV | M1 | 13.4 | √ | 13.5 | √ | 435 |
| M2 | 16.6 | √ | 16.4 | √ | ||
| M3 | 21.3 | √ | 21.2 | √ | ||
| M4 | - | - | 12 | √ | ||
| M5 | - | - | 14 | √ | ||
| M6 | - | - | 17.2 | √ | ||
| Dihydroxy-SV | M7 | 8.7. | √ | 9.9 | √ | 451 |
| M8 | 9.7 | √ | 10.2 | √ | ||
| M9 | 10.7 | √ | 11.2 | √ | ||
| M10 | 12.6 | √ | - | - | ||
| M11 | 13.1 | √ | - | - | ||
| M12 | 15.4 | √ | - | - | ||
| M13 | 16.9 | √ | - | - | ||
| Exomethylene-SV | M14 | 16.2 | √ | 15.3 | √ | 417 |
| M15 | - | - | 21.3 | √ | ||
| Carboxyl-SV | M16 | 14.1 | √ | 13 | √ | 449 |
| M17 | 14.8 | √ | 14 | √ | ||
| M18 | 15.5 | √ | - | - | ||
| M19 | 16.9 | √ | 16.7 | √ | ||
| Dihydrodiol-SV | M20 | - | - | 12 | √ | 453 |
| Detected Metabolites | No. | Ion Trap | QqQ | ESI (−) (m/z) | ||
|---|---|---|---|---|---|---|
| Rt (min) | Detected | Rt (min) | Detected | |||
| Exomethylene-SVA | M21 | 21.8 | √ | - | - | 433 |
| M22 | - | - | 11.1 | √ | ||
| M23 | - | - | 13.3 | √ | ||
| Monohydroxy-SVA | M24 | 13.3 | √ | - | - | 451 |
| M25 | - | - | 6.2 | √ | ||
| M26 | - | - | 6.9 | √ | ||
| Dihydrodiol-SVA | M27 | 8.1 | √ | - | - | 469 |
| M28 | - | - | 4.9 | √ | ||
| SVA | M29 | 22.9 | √ | - | - | 435 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, W.; Alwabli, R.I.; Attwa, M.W.; Rahman, A.F.M.M.; Kadi, A.A. Simvastatin: In Vitro Metabolic Profiling of a Potent Competitive HMG-CoA Reductase Inhibitor. Separations 2022, 9, 400. https://doi.org/10.3390/separations9120400
Yin W, Alwabli RI, Attwa MW, Rahman AFMM, Kadi AA. Simvastatin: In Vitro Metabolic Profiling of a Potent Competitive HMG-CoA Reductase Inhibitor. Separations. 2022; 9(12):400. https://doi.org/10.3390/separations9120400
Chicago/Turabian StyleYin, Wencui, Reem I. Alwabli, Mohamed W. Attwa, A. F. M. Motiur Rahman, and Adnan A. Kadi. 2022. "Simvastatin: In Vitro Metabolic Profiling of a Potent Competitive HMG-CoA Reductase Inhibitor" Separations 9, no. 12: 400. https://doi.org/10.3390/separations9120400
APA StyleYin, W., Alwabli, R. I., Attwa, M. W., Rahman, A. F. M. M., & Kadi, A. A. (2022). Simvastatin: In Vitro Metabolic Profiling of a Potent Competitive HMG-CoA Reductase Inhibitor. Separations, 9(12), 400. https://doi.org/10.3390/separations9120400