Abstract
In the countercurrent chromatography and centrifugal partition chromatography, separation method transfer and scale-up is often described as an easy and straightforward procedure. Separation methods are usually developed on lab scale columns and subsequently transferred using linear scale-up factors to semi-preparative or preparative columns of the same column design. However, the separation methods described in the literature have been developed on various columns of different design and size. This is accompanied by differences in the separation behavior of the columns and therefore makes separation method transfer difficult. In the current study, the separation performances of different columns were evaluated and compared. Linear correlations of stationary phase retention and column efficiency as a function of flow rate were found to be applicable for the calculation of separation resolution in the typical operating range of each column. In this context, a two-point short-cut approach for a fast column characterization is recommended. This allows a quick prediction of the separation method transferability between columns, which saves experimental time and effort. In the current study, the transferability between five different columns from lab scale countercurrent chromatography (CCC) (18 mL) to semi-preparative centrifugal partition chromatography (CPCs) (250 mL) with different cell numbers and design is investigated.
1. Introduction
Solid support-free liquid-liquid chromatography (LLC) is a powerful technique with increasing popularity for the separation of active compounds from plant extracts and biotechnological products. In general, two fundamental column designs exist: hydrodynamic columns better known as countercurrent chromatography (CCC), and hydrostatic columns known as centrifugal partition chromatography (CPC). CCC columns consist of a coil that is wound on a bobbin that rotates around its own axis and revolves around a second axis of a centrifuge [1,2]. CPC columns have only one axis of rotation with a constant centrifugal field. Conventional CPC columns consist of several identical annular disks that are connected in series, with polytetrafluoroethylene (PTFE) plates in between acting as seals. Each disk contains a certain number of cells that are circumferentially engraved and interconnected by narrow short channels, referred to as ducts [3,4,5]. Due to continuous developments, many subgroups with various column designs emerged for each of the two categories [3,4,5]. CCC columns, for example, differ in its rotary motion, ratios of the rotation radii (β-values), and different coil or tubing diameters and geometries, while CPC columns often have different cell and duct design as well as size. In literature, a huge number of applications have been established on many different columns. However, the columns often differ significantly in column design as well as size. Consequently, suitable operating parameters (e.g., flow rate, rotational speed) and separation performances (e.g., stationary phase retention, column efficiency) between the columns are also different. Therefore, efficient and systematic strategies are needed to quickly transfer separation methods between different apparatuses.
In conventional liquid chromatography (LC), e.g., High-performance liquid chromatography (HPLC), clear rules exist on how to transfer batch separation methods between columns. Here, columns are of cylindrical shape and packed with a solid stationary phase. The design parameters are column diameter, length and stationary phase particle size. In general, sample load, i.e., the feed sample concentration and relative volumetric column loading, is kept constant and the same or a better number of theoretical stages is aimed for. To achieve these criteria when transferring a separation method, usually the particle size and the column length are kept the same, while the cross sectional area and the mobile phase flow rate are adjusted in order to guarantee the same mobile phase velocity in both columns [6]. In literature, this approach was studied also for applicability in CCC and CPC. The same mobile phase linear velocity was used in both columns when transferring the separation method between columns [7]. However, this is difficult, especially in CPC, since the cross-sectional area of the CPC cells is not constant and changes along the cells of different geometries. It was shown, that an average theoretical cross sectional area calculated as the ratio of cell volume and height does not give satisfactory results when cells clearly differ in design and/or size [7].
So far, LLC applications are mostly developed at lab scale columns with a volume between 10 and 300 mL. As commonly known, a good separation performance is related to a high stationary phase retention [4]. This is probably the reason, why the separation method transfer in CCC and CPC is commonly done by adjusting operating parameters, i.e., rotational speed and flow rate, in order to keep the stationary phase retention in both columns the same. Then, usually linear scale-up factors are used for calculating the injection volume or mass load, e.g., the ratio of column volumes in CPC and CCC, or coil lengths in CCC columns [7,8,9,10,11]. Although this has been often used as a fast and relatively satisfactory strategy, it is not a generally valid separation method transfer approach.
Another approach is the model-based methodology for optimizing a CPC column size for a particular separation introduced by Chollet et al. [12]. Varying hydrodynamics and mass transfer efficiency in different columns are taken into account enabling the calculation of an optimal column length. This approach allows the design of an optimized tailor-made column for a particular application, but requires preliminary tests for the determination of the model parameters as well as experience in modelling. Commercial columns are usually standardized and have a fix size. Therefore, this approach is not directly applicable for the separation method transfer in standardized and non-adjustable columns.
Bouju et al. proposed the free-space between the peaks method for the intra-apparatus method transfer between differently sized CPC columns with same cell geometry [13]. With this strategy, a separation method developed at lab scale can be easily transferred to large scale columns using the relation of the space between the peaks determined by pulse injections with low injection volumes ( on the two columns. Then, the injection volume is maximized on the first column and the maximum injection volume on the second column is directly calculated using the previously determined ratio of the space between the peaks. However, this method assumes in advance that the transfer of the separation method between the two columns is possible and no adjustment or selection of suitable operating parameters is provided. Hence, this approach can only be directly applied, if a higher separation resolution occurs on the column where the method is intended to be transferred.
According to literature, columns of same size but different design or cell geometries behave differently as a function of operating conditions such as flow rate and rotational speed (-field) [14,15,16,17,18]. Differences in stationary phase retention and column efficiency have a clear influence on the separation resolution and process performances, including product purity, recovery, achievable productivity, and solvent consumption [16,19,20,21,22,23,24]. Hence, a reliable and easy strategy for the separation method transfer between columns independent of their size and type is needed that considers the separation performances of different column designs.
In this sense, two general questions arise before the actual transfer of a separation method: 1. is the method transferable at all; 2. which operating conditions have to be selected in order to achieve the highest possible productivity and at the same time a preferably low solvent consumption for pre-set purity and/or recovery requirements. At this point, the current work addresses the prediction and evaluation of separation method transferability in LLC. Hence, the separation method transferability between different columns is discussed. A systematic approach for the column characterization and method transfer is proposed in order to achieve comparable resolution of the separation on different CCC/CPC devices. For the column characterization a mixture of four parabens and the common aqueous-organic solvent based biphasic solvent system Arizona N was used, which is composed of n-heptane/ethyl acetate/methanol/water 1/1/1/1 v/v/v/v. In the study, different LLC columns available in our lab were used, namely a small 18.2 mL lab scale CCC and four different semi-preparative CPC columns with the same nominal total column volume of 250 mL, but different cell design and peripheral setups. It should be noted that the intention of this study was not to compare the separation performance of the different columns, but to establish a fast strategy for the estimation of separation method transferability between two columns.
2. Materials and Methods
2.1. Chemicals
The solvents n-heptane, ethyl acetate and methanol of the biphasic solvent system were all of analytical grade (EMSURE, purity 99%) from Merck KGaA (Darmstadt, Germany). Milli-Q water was obtained from a Milli-Q Direct Water Purification System from Merck Millipore (Darmstadt, Germany). The solutes d/l-tryptophan (racemic mixture), methyl paraben (MP), ethyl paraben (EP), propyl paraben (PP), and butyl paraben (BP) with a purity of ≥99% were purchased from Alfa Aesar GmbH & Co KG (Karlsruhe, Germany).
2.2. Equipment
The experiments were carried out with different liquid–liquid chromatographic columns listed in Table 1. The hydrodynamic CCC-Mini column is a hydrodynamic J-type centrifuge with a 0.8 mm ID PTFE tube, a beta value range between 0.5 and 0.78, and a total column volume of 18.2 mL. The CCC-Mini was connected to a cooling unit (FL 300, Julabo, Seelbach, Germany) and an isocratic HPLC pump (Gilson 306, Gilson, Middleton, WI, USA), which could deliver flow rates of up to 50 mL min-1. The effluent was monitored with a UV-detector (Gilson 171 UV-DAD-detector, Gilson, Middleton, WI, USA) at a wavelength of 255 nm. The sample was introduced through a manual 6-port valve and an injection loop of 72 μL (<1% ).

Table 1.
List of investigated columns, including name, acronym, column type, cell type, total column volume, and total number of cells.
The SCPC-250 column consists of two single columns connected in series with an experimentally determined total volume of 182 mL. Each of the single columns has 10 disks with 90 engraved twin cells, resulting in total 1800 twin cells. The maximum achievable rotational speed is 3000 rpm (715 ) and the whole column can be operated at a maximum pressure drop of 100 bar. The CPC unit was connected to two isocratic preparative HPLC pumps with maximum flow rates of 50 mL min−1, one for filling the column with the stationary phase and the other one for pumping the mobile phase during the separation. The effluent was monitored with a UV detector (ECOM DAD600 2WL 200–600 nm, Prague, Czech Republic) at 255 nm. The feed sample was introduced through a six-port manual injection valve, and an injection loop of 1 mL (<1% ) was used.
The CPC 250 column has an experimentally determined total column volume of 242 mL and consists of eight disks, each disk contains 108 engraved twin cells, in total 864 cells. The disks are made of stainless steel and sealed by PTFE plates. The maximum achievable rotational speed is 3000 rpm (729 ) and the maximum pressure drop is 100 bar. The CPC unit was connected to a PLC 2250 purification system including a preparative HPLC pump with maximum flow rate of 250 mL min−1. The effluent was monitored with an integrated Gilson UV-DAD detector at 255 nm. The feed sample was introduced through an automated injection valve, and an injection loop of 1 mL (<1% ) was used.
The column CPC 250 PRO SPECIAL BIO VERSION with an experimentally determined total volume of 244 mL has 12 disks, where each disk contains 20 engraved twin-cells; in total 240 cells. The disks are made of stainless steel and are additionally coated with PTFE for applications with biomolecules. The disks are sealed by PTFE plates. The column can be operated at a pressure drop of up to 100 bar and a maximal rotational speed of 3000 rpm (715 ). The column was connected to an isocratic HPLC pump (AZURA P2.1L, KNAUER, Berlin, Germany), which could deliver flow rates of up to 220 mL min−1. The effluent was monitored with a UV-detector (AZURA MWD 2.1L, KNAUER, Berlin, Germany) at a wavelength of 255 nm. The sample was introduced through a manual 6-port valve and an injection loop of 1 mL (<1% ).
The CPC 250 PRO SC prototype with an experimentally determined total volume of 240 mL has a monobloc design made of stainless steel and contains 196 spherical cells in total. The cells are connected to each other with small ducts with a total volume of only 5 mL. Due to the different column design, no sealing PTFE plates between the column disks are necessary, allowing an operation of the column with a maximal pressure drop of 200 bar and a rotational speed of 4000 rpm (2450 ). A detailed description of the design and production process can be found in the corresponding patent [25]. The column was connected to an isocratic preparative HPLC pump (PrepStar 218 Solvent Delivery Module, Varian, Palo Alto, CA, USA), which could deliver flow rates of up to 200 mL min−1. The effluent was monitored with a UV-Vis detector (ProStar 325 UV-Vis Detector, Varian, Palo Alto, CA, USA) and a SUPER PREP 4 × 0.15 mm detector cell at a wavelength of 255 nm. The sample was introduced through a six-port manual injection valve. An injection loop of 1 mL (<1% ) was used.
For the sake of clarity, in the following text the columns are referred to by their acronyms introduced in Table 1.
2.3. Methods
2.3.1. Preparation of the Biphasic Liquid Systems and Feed Samples
The biphasic liquid system n-heptane/ethyl acetate/methanol/water 1/1/1/1 (v/v/v/v), Arizona N, was prepared by mixing the corresponding volume portions of the solvents at room temperature. The mixture was vigorously shaken and equilibrated at room temperature for at least two hours. For the experiments the phases were split and placed in two separate containers at room temperature. Feed solutions were prepared by adding appropriate amounts of the solutes to the mobile phase used for the pulse injections, lower phase in descending mode. The pulse injections for the characterization of all columns were performed in descending mode with d/l-tryptophan, methyl paraben, ethyl paraben, propyl paraben and butyl paraben. A sample concentration of 2 mg mL−1 was used for all five solutes, respectively.
2.3.2. Pulse Injection Experiments
Pulse injections of the mixtures in each column were performed in descending mode. For all measurements, the columns were first filled with the intended stationary phase, i.e., the upper phase. After that, the rotational speed was set to the desired value and mobile phase was pumped through the column with the respective flow rates until no more stationary phase was eluting from the column, but at least one column volume. A feed mixture of d/l-tryptophan and four parabens was injected via a sample loop and the effluent was monitored with a UV-Vis detector at the specific wavelength (Note: In the CPE spherical cells experiments, no butyl paraben was injected). The stationary phase retention was calculated from the retention volume of the tracer d/l-tryptophan (partition coefficient equal to 0 in the Arizona N system).
3. Results
3.1. Approach of Column Characterization-Based Separation Method Transferability
In this work, the evaluation of separation method transferability between different columns is studied. The columns differ in type, size and design, but also in the typical operating range in terms of relative centrifugal acceleration (-field) and mobile phase flow rate. Hence, the column performance is different in every single column as a result of differences in flow pattern, mass transfer area and solute residence time distribution [16,19,20,21,22,26,27]. The different column characteristics can be described by a different stationary phase retention, column efficiency and peak resolution.
The objective is to evaluate whether a transfer of the separation from one unit to another is possible by keeping the resolution between the target compounds constant. First, the different columns are characterized with pulse injections of small injection volume (). This is a similar procedure to LC, where an insignificant injection volume and low feed concentrations are used for the characterization of the column so that column load influences on the elution profile can be neglected [6,28,29]. Subsequently, the stationary phase retention, column efficiency and resolution between the peaks is determined in dependence of flow rate at a set rotational speed. Based on the determined resolution, separation method transferability between the columns can be easily foreseen for the selected type of solvent systems. In this manner, three general possibilities can occur: a resolution between the target compounds higher, equal, or lower compared to the original separation method.
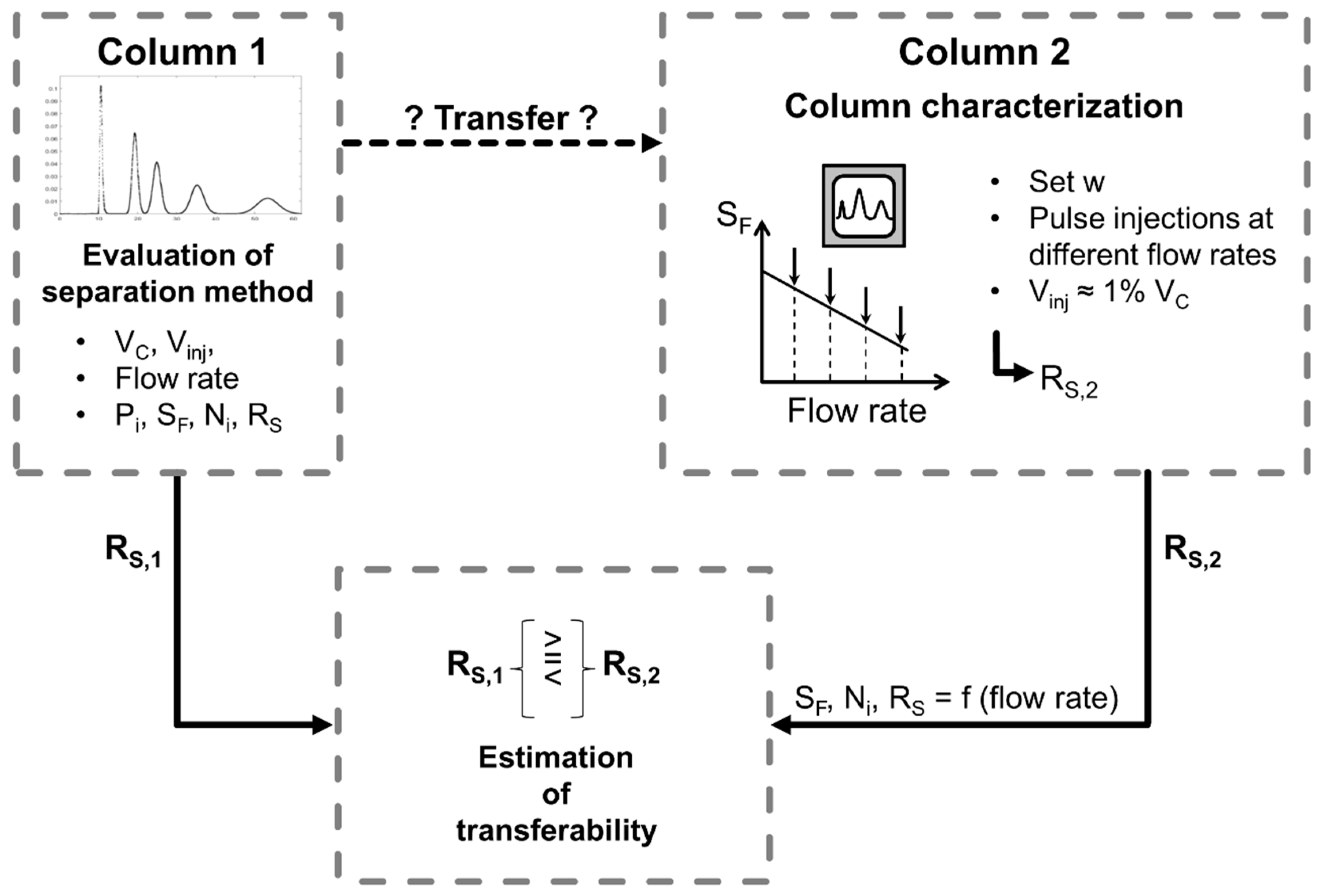
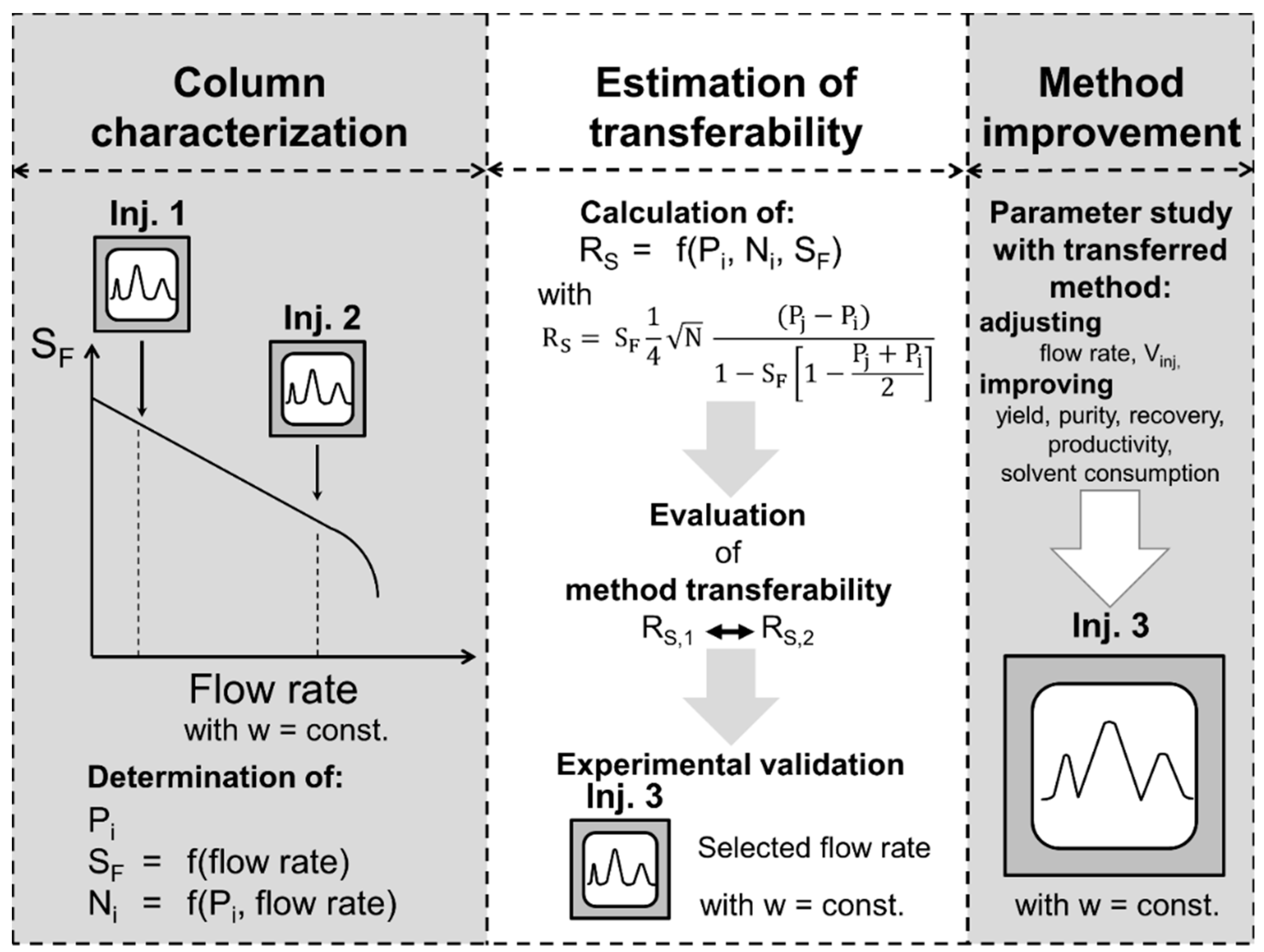
However, the previously described situation, where both columns are extensively characterized with the same solvent system and solute, is usually not given. Often, an application found in literature is intended to be established on the column in the author’s own lab. Still, the separation method transferability could be estimated purposefully as illustrated schematically in Figure 1 without extensive experimental trial and error attempts. First, the separation found in literature is evaluated in terms of separation performance, i.e., resolution. Then, the column where the separation method should be transferred to is characterized: a few pulse injections are performed, in the typical operating range of flow rate and a suitable but constant -field of the column, the resolution is determined, and transferability evaluated. If the obtained resolution is higher than in the original column, further optimization in terms of injection volume, productivity, and solvent consumption could be directly followed. For this purpose, the information about experimentally determined stationary phase retention and column efficiency from the pulse injection experiments and the cell model can be used for simulations [30,31,32]. This approach saves experimental effort and time, and enables a targeted separation method transfer, which is further discussed in Section 3.3.
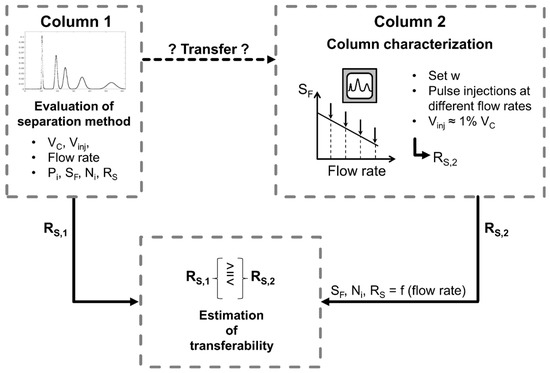
Figure 1.
Schematic procedure of the evaluation of the separation method transferability between two different columns.
In the current study, the solvent system n-heptane/ethyl acetate/methanol/water 1/1/1/1 v/v/v/v, Arizona N, is used and methyl paraben, ethyl paraben, propyl paraben, and butyl paraben are selected as model compounds. In addition, d/l-tryptophan is used as a tracer compound in descending mode, i.e., lower phase as mobile phase.
3.2. Column Characterization
Five columns of different size and design (see Table 1) were characterized in terms of stationary phase retention, column efficiency and resolution as a function of mobile phase flow rate. The rotational speed () for each column was kept constant and individually selected. The selected rotational speeds are in a range where a further increase of the rotational speed has no significant positive influence on the stationary phase retention and column efficiency, and complies with the pressure drop limitations of each column. This is usually known from previous studies with similar solvent systems [17,33] or suggested by the producer of the column. The rotational speed was set as following: CCC (1900 rpm, 216 ), CPC 1800 (1700 rpm, 230 ), CPC 864 (1737 rpm, 230 ), CPE 240 (1700 rpm, 230 ), CPE 196 spherical cells (2000 rpm, 613 ). The experimental data for the characterization of the CPE 240 are taken from Morley et al. [34].
3.2.1. Stationary Phase Retention
The stationary phase retention () is the fraction of the column volume occupied by the stationary phase:
where is the volume of the stationary phase and is the volume of the column.
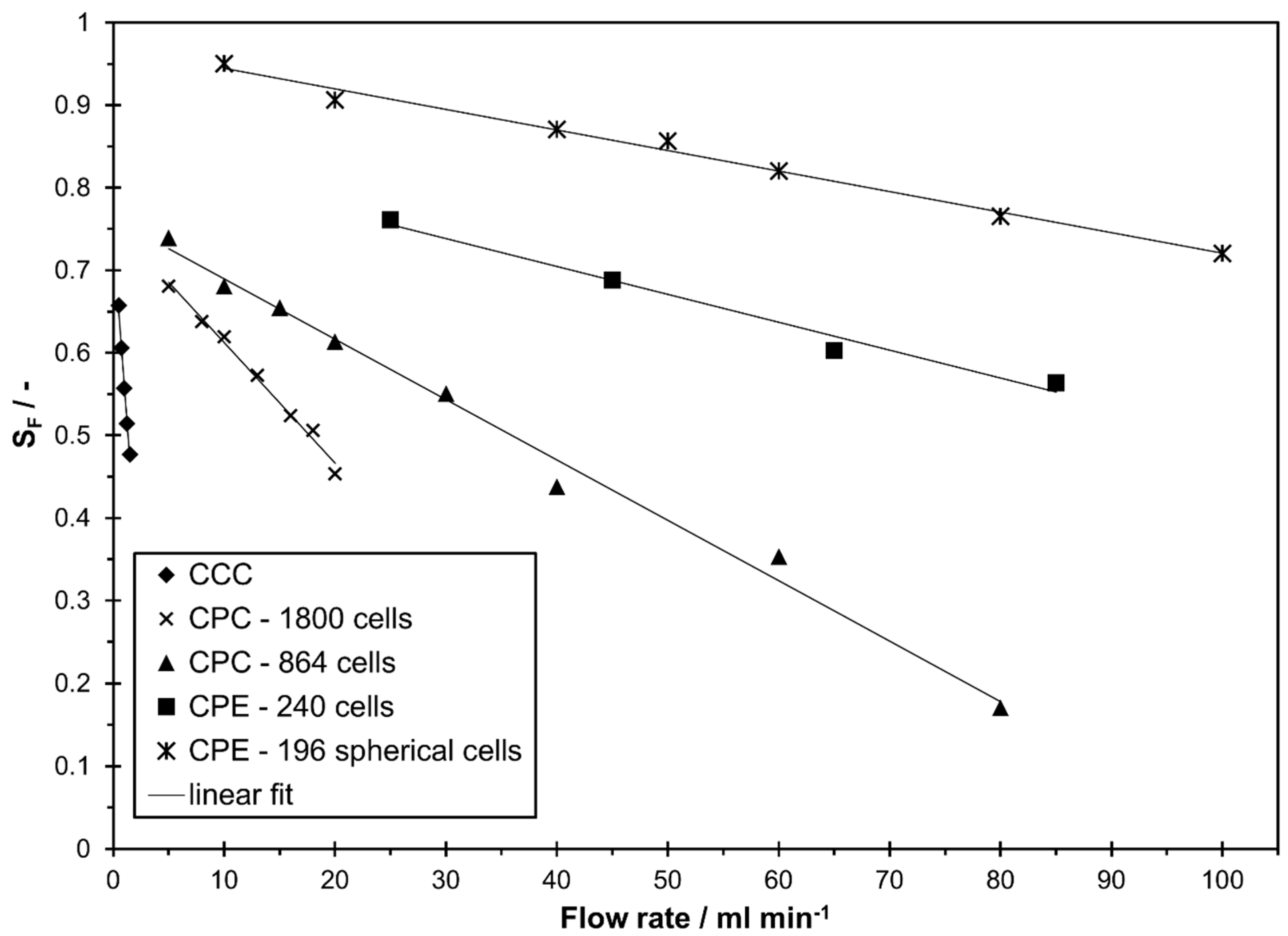
In Figure 2 the stationary phase retention as a function of the mobile phase flow rate is shown for all studied columns in descending mode (lower phase as mobile phase). The maximum studied flow rate was selected according to the maximum flow rate recommended by the manufacturer (CCC), the maximum allowed pressure drop of the column (CPC 1800, CPE 240), the maximum applicable flow rate for the applied detector (CPE 196 spherical cells), or simply due to a low stationary phase that is suboptimal for a sufficient separation in LLC (CPC 864). For all columns, a linear decrease with an increase in flow rate was observed within the studied range. This is in line with similar studies from literature for both, CCC [5,18,33,35,36,37,38,39,40] and CPC [14,16,20,22,24,38,41] of different column sizes and geometries within a certain flow rate range. However, it should be noted that this only applies up to a certain flow rate. Above that, especially in CPC columns, an abrupt drop in stationary phase retention can occur and a linear correlation between and flow rate is no longer possible [14,16,38,41]. In the studied columns, no abrupt decrease in stationary phase retention was observed at a certain flow rate. With the studied solvent system, a good stationary phase retention above 0.6 is achievable in all columns. For the studied operating parameters, the CPE 196 spherical cells column had the highest stationary phase retention while the biggest range of was observed with the CPC 864.
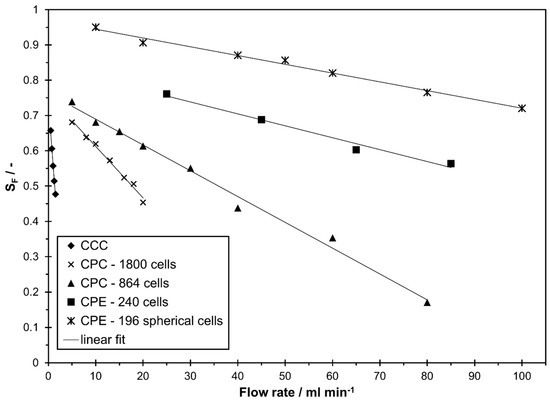
Figure 2.
Stationary phase retention in descending mode in different countercurrent chromatography (CCC) and centrifugal partition chromatography (CPC) columns as a function of mobile phase flow rates for the solvent system n-heptane/ethyl acetate/methanol/water 1/1/1/1 v/v/v/v.
3.2.2. Column Efficiency and Resolution of the Columns
In addition to a high stationary phase retention, a high column efficiency is beneficial for a good separation i.e., high resolution. In all studied columns, the efficiency is determined with pulse injection experiments of the model compounds methyl paraben, ethyl paraben, propyl paraben and butyl paraben at different mobile phase flow rates in descending mode. The column efficiency, also called number of theoretical plates (), was calculated from the recorded UV-Vis signal with Equation (2).
where is the retention time of component i and the corresponding peak variance.
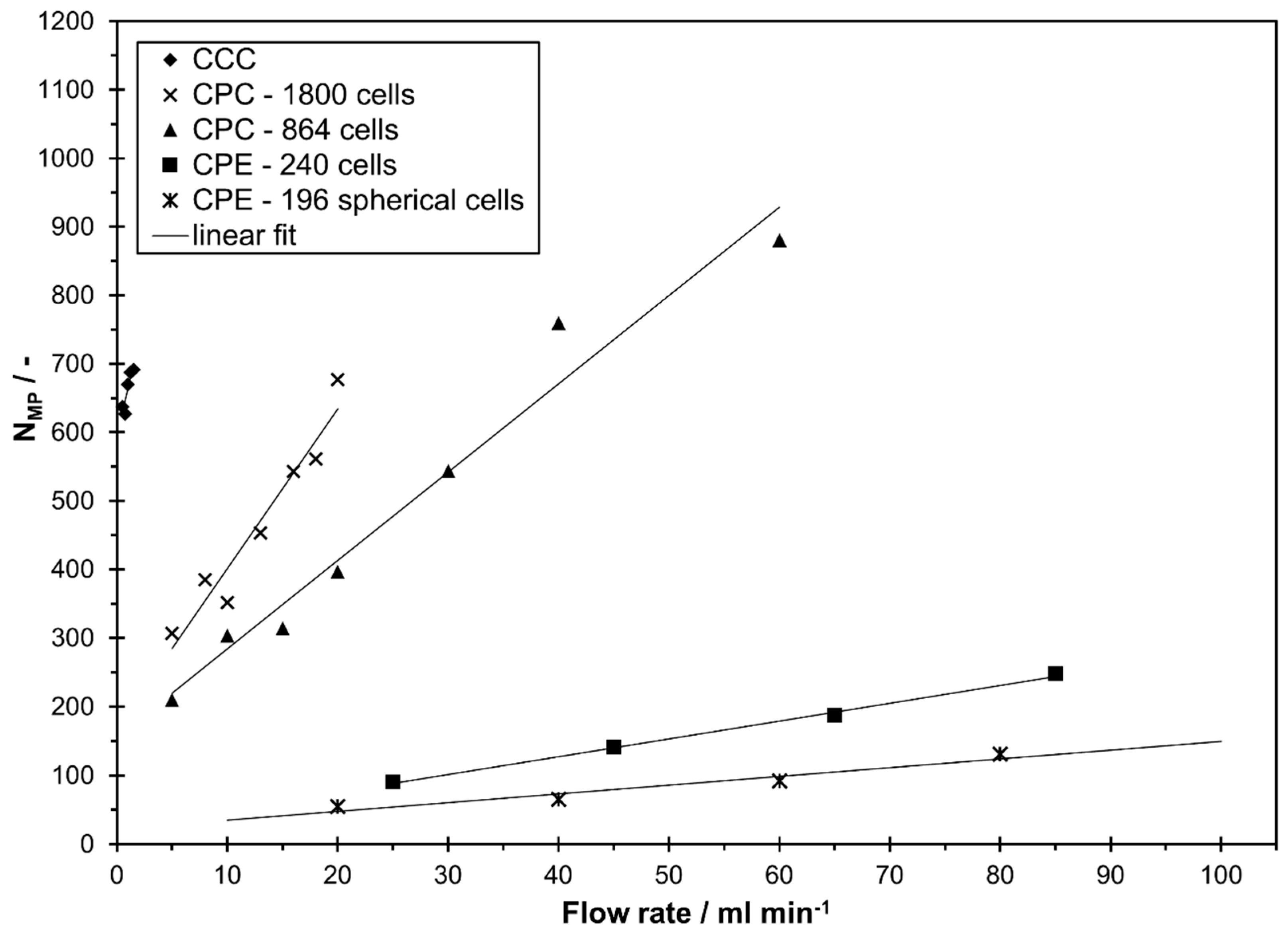
In Figure 3, the efficiency of methyl paraben is shown as a function of flow rate in descending mode. A linear increase with an increase in flow rate was observed for all columns in the studied range. However, all studied columns with different designs clearly differ in column efficiency. As expected, a higher number of theoretical plates is determined in CPC columns with a higher number of physical cells. It is worth mentioning that the CPE 240 and CPE 196 spherical cells have a much higher single cell efficiency compared to the other two CPCs. Still, the total number of theoretical plates of CPC 1800 and CPC 864 is clearly higher than in the two CPEs which is due to the higher number of physical cells. Similarly, the lab scale CCC outperforms the two CPEs in terms of column efficiency and a similar is achievable compared to the two CPC columns. In all columns and at all flow rates, the number of theoretical plates is highest for the first eluting compound methyl paraben and lowest for the last eluting compound butyl paraben.
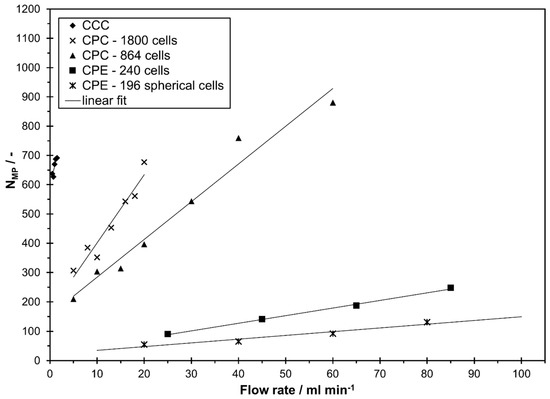
Figure 3.
Number of theoretical plates (efficiency) of methyl paraben in descending mode of different CCC and CPC columns as a function of mobile phase flow rates for the solvent system n-heptane/ethyl acetate/methanol/water 1/1/1/1 v/v/v/v.
The resolution between two peaks is defined according to Equation (3). The resolution in this study was calculated according to Equation (4), assuming a Gaussian peak shape ().
where and are the retention times of the first and second eluting compound; and are the corresponding peak widths at base line; and are the peak standard deviations.
The resolution can also be described by the partition coefficient , column efficiency , and stationary phase retention (Equation (5)):
where is the mean value of the number of theoretical plates of both components [42].
The partition coefficient of a solute is solvent system dependent and described by the thermodynamic equilibrium. Hence, for the same solvent system, and are the same for all columns. The partition coefficient of a compound can be calculated from the chromatogram of a pulse injection at a certain flow rate () according to Equation (6):
As described by Berthod et al. [43] for the homologous series of alkyl benzenes, a linear correlation of the partition coefficients can be applied to the parabens used in this study. Based on the chromatogram of a CCC pulse injection at 1 mL min−1 (see chromatogram in Figure A1 in Appendix A), with representing the number of methylene groups ( 4.22). In contrast, and are solvent system, column and operating parameter dependent, and determine the difference in resolution between different columns. Once and are known, Equation (5) can be used to calculate the resolution.
Using the linear correlations of and , the peak resolution can be calculated according to Equation (5) in the whole flow rate range. As apparent from Figure 4, showing experimental values of as well as calculated values with Equation (5) and the linear correlations of and from Figure 2 and Figure 3, the calculated and experimental values are in good agreement with minor deviations of up to 0.15. Only in terms of the CPE 240 a deviation of up to 0.2 was observed at a flow rate of 85 mL min−1. In this sense, the accuracy of the calculated values is good enough for a first approximation of the resolution when estimating the transferability of a certain separation method.
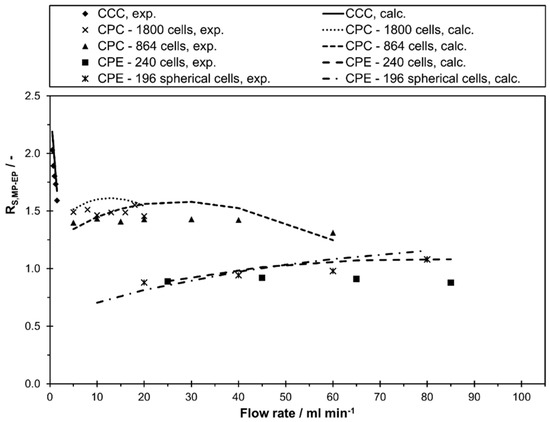
Figure 4.
Separation resolution, experimental and calculated values using Equation (5) and the linear correlations of and , between methyl paraben and ethyl paraben in descending mode of different CCC and CPC columns as a function of mobile phase flow rates for the solvent system n-heptane/ethyl acetate/methanol/water 1/1/1/1 v/v/v/v.
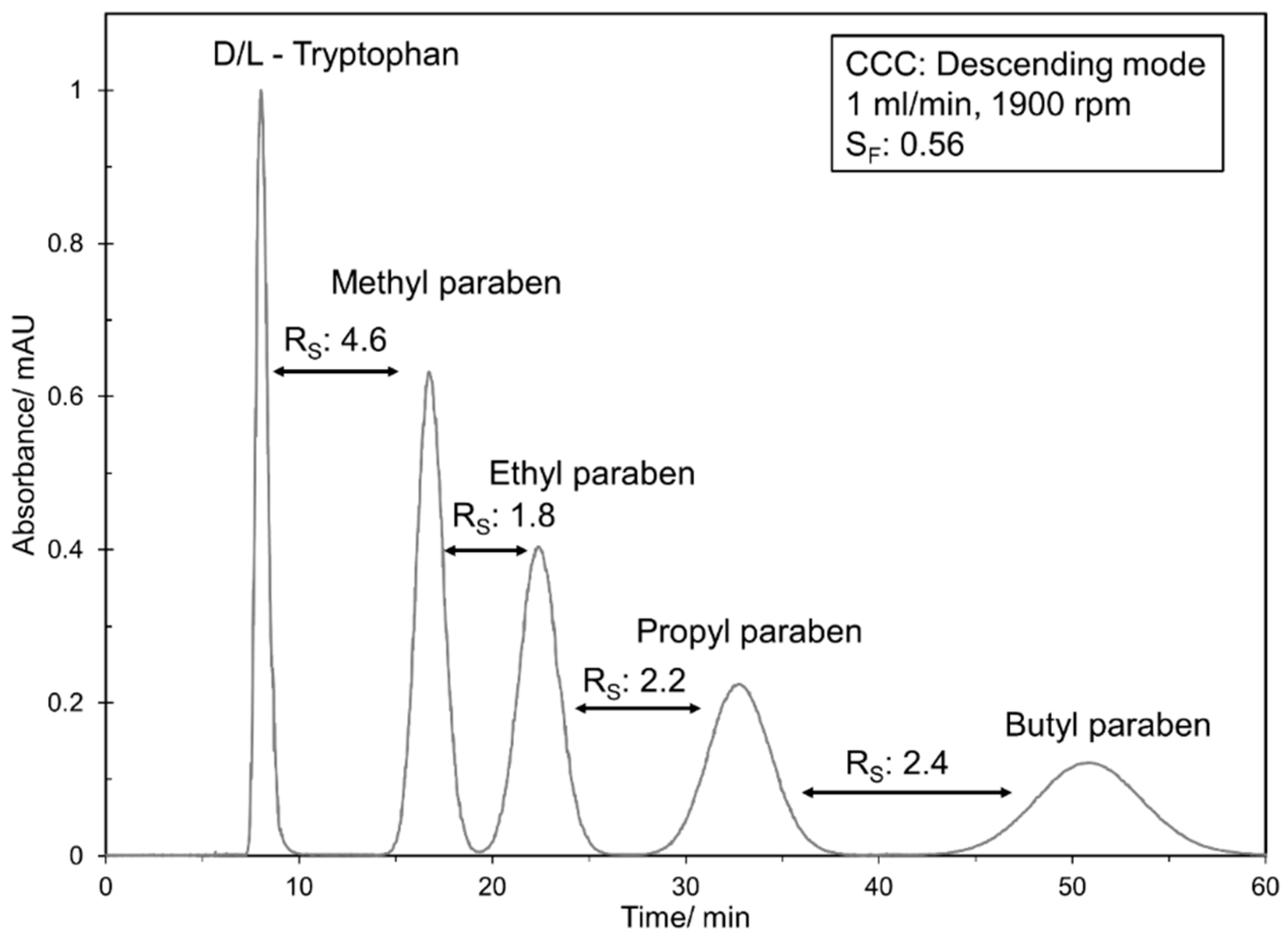
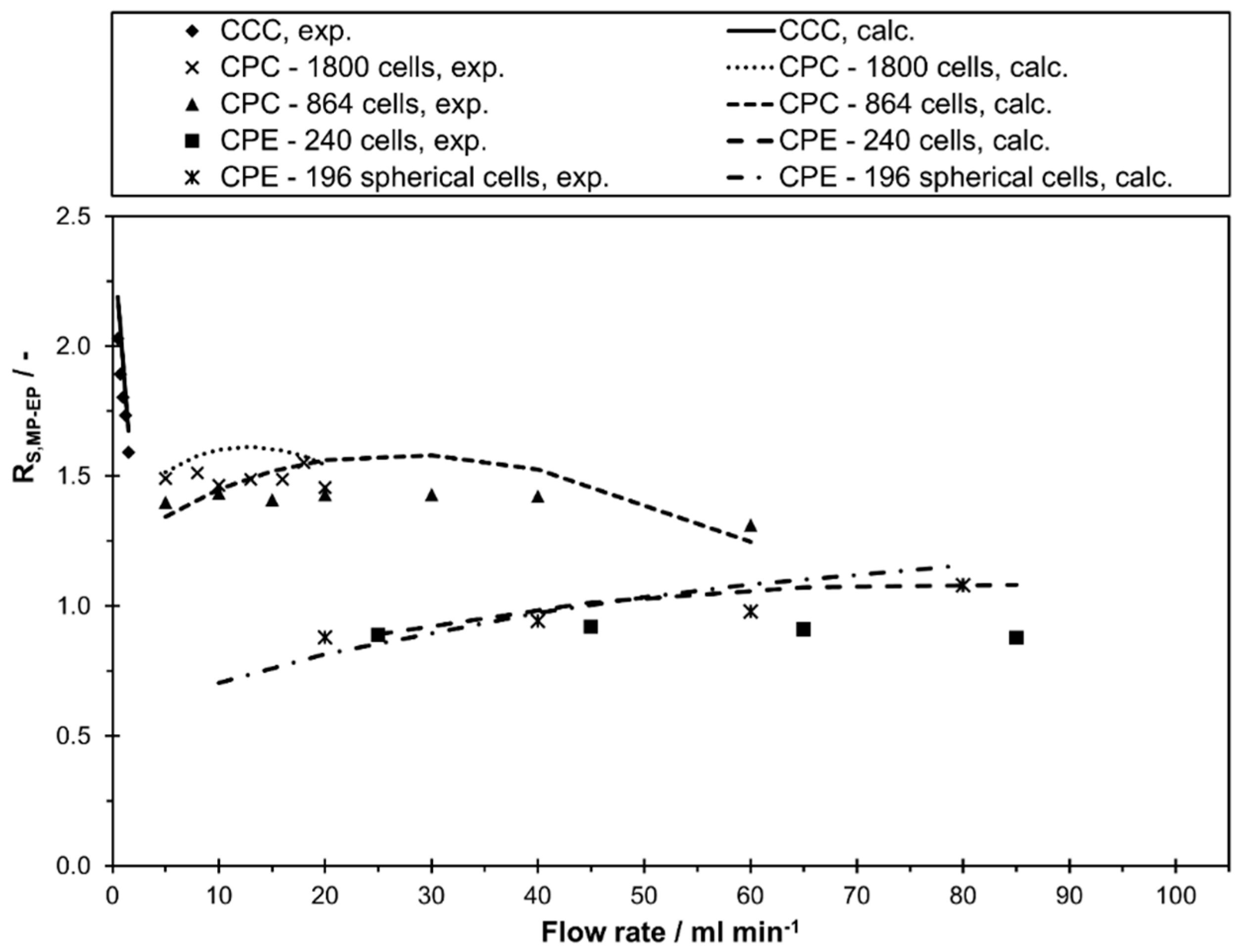
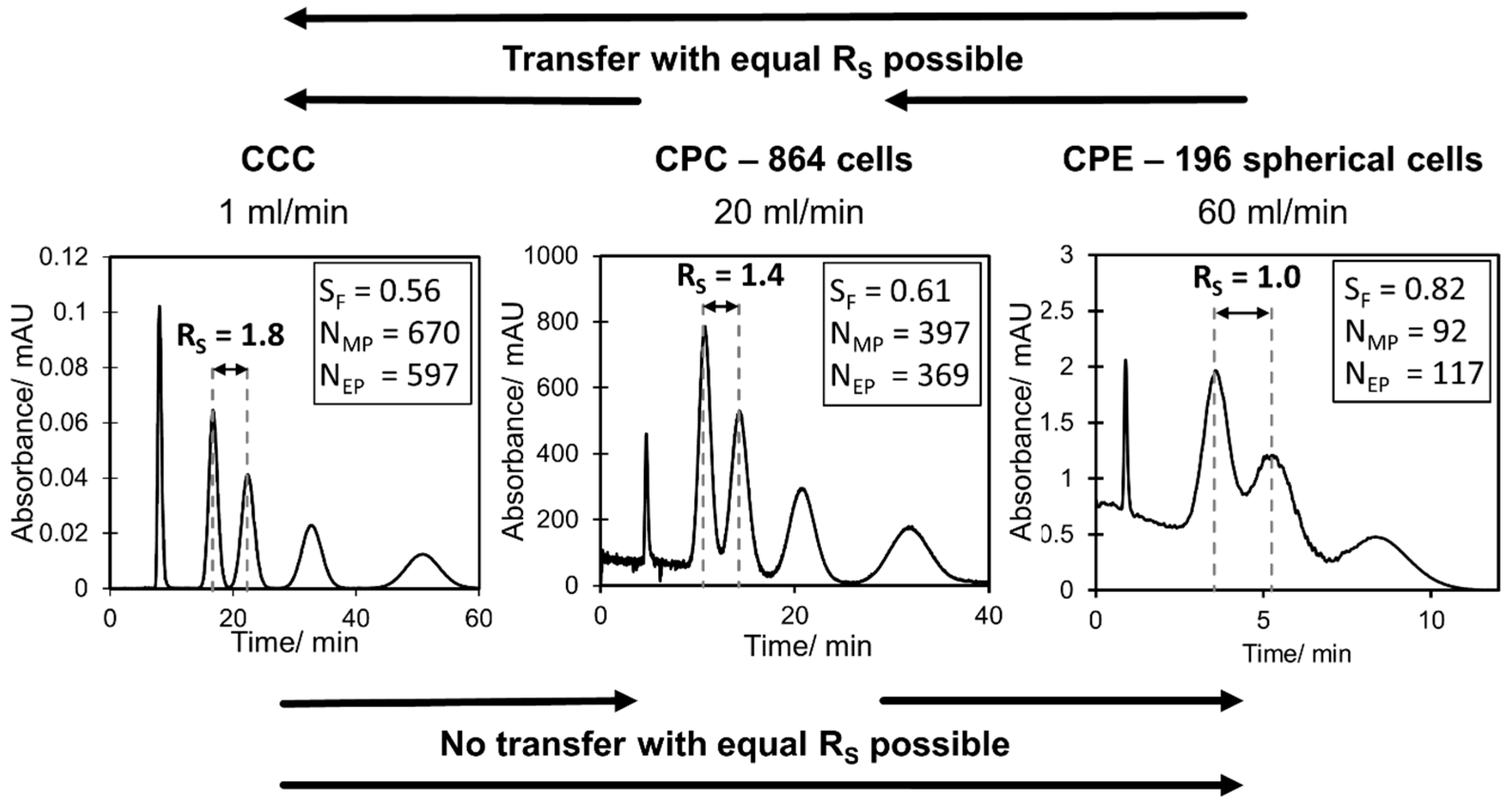
In the current study, the highest resolution could be determined in the lab scale CCC. However, a clear decrease in resolution between methyl paraben and ethyl paraben was determined with an increase in flow rate from 0.5 mL min−1 ( to 1.5 mL min−1 (. Consequently, these two compounds could be separated completely with the lab scale CCC in the whole flow rate range. In contrast, the resolution at CPC 1800 and CPC 864 stayed almost constant with an increase in flow rate. While a complete base line separation ( between methyl paraben and ethyl paraben was still possible in CPC 1800, a small peak overlapping occurred in CPC 864. The CPE 240 and CPE 196 spherical cells show a similar behavior within the studied flow rate range, but with a clearly lower resolution compared to CPC and CCC. Interestingly, the CPE 196 spherical cell even slightly outperforms the CPE 240 at higher flow rates. Additionally, it can be directly anticipated from Figure 4 whether a higher, similar or lower peak resolution can be expected for a certain pulse injection in the investigated columns. In this work, a separation method transfer is possible in the following order of the resolution: CCC > CPC 1800 > CPC 864 > CPE 196 spherical cells > CPE 240. This trend becomes even clearer in Figure 5, where corresponding chromatograms with pulse injections are shown. In terms of CCC, a median flow rate of 1 mL min−1 is shown. For CPC 864 and CPE 196 spherical cells, representative flow rates were arbitrarily selected since does not change (CPC 864) or just slightly increases (CPE 196 spherical cells) within the studied flow rate range (see Figure 4).
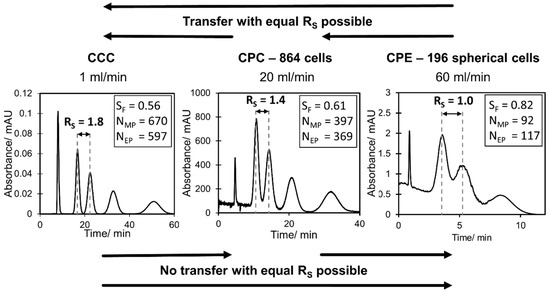
Figure 5.
Representative chromatograms of paraben pulse injections in CCC, CPC 864 and CPE 196 spherical cells at different flow rates.
In summary, the partition coefficient of compound is the same in all columns and independent of operating conditions, since is a thermodynamic parameter. In contrast, linear correlations for and as a function of flow rate can be obtained. decreases and increases with an increase in flow rate in all studied columns. In terms of resolution the columns behaved differently. The shown trends are consistent with previous CCC and CPC studies [14,15,17,18,44]. Although the resolution appears to be mostly linear dependent on the flow rate in Figure 4, this is not necessarily the case for all possible flow rates, since, according to Equation (5), is not a linear function of and . The big difference in the achievable resolution between CPC and CPE can be easily explained by the much higher number of physical cells in the CPC columns.
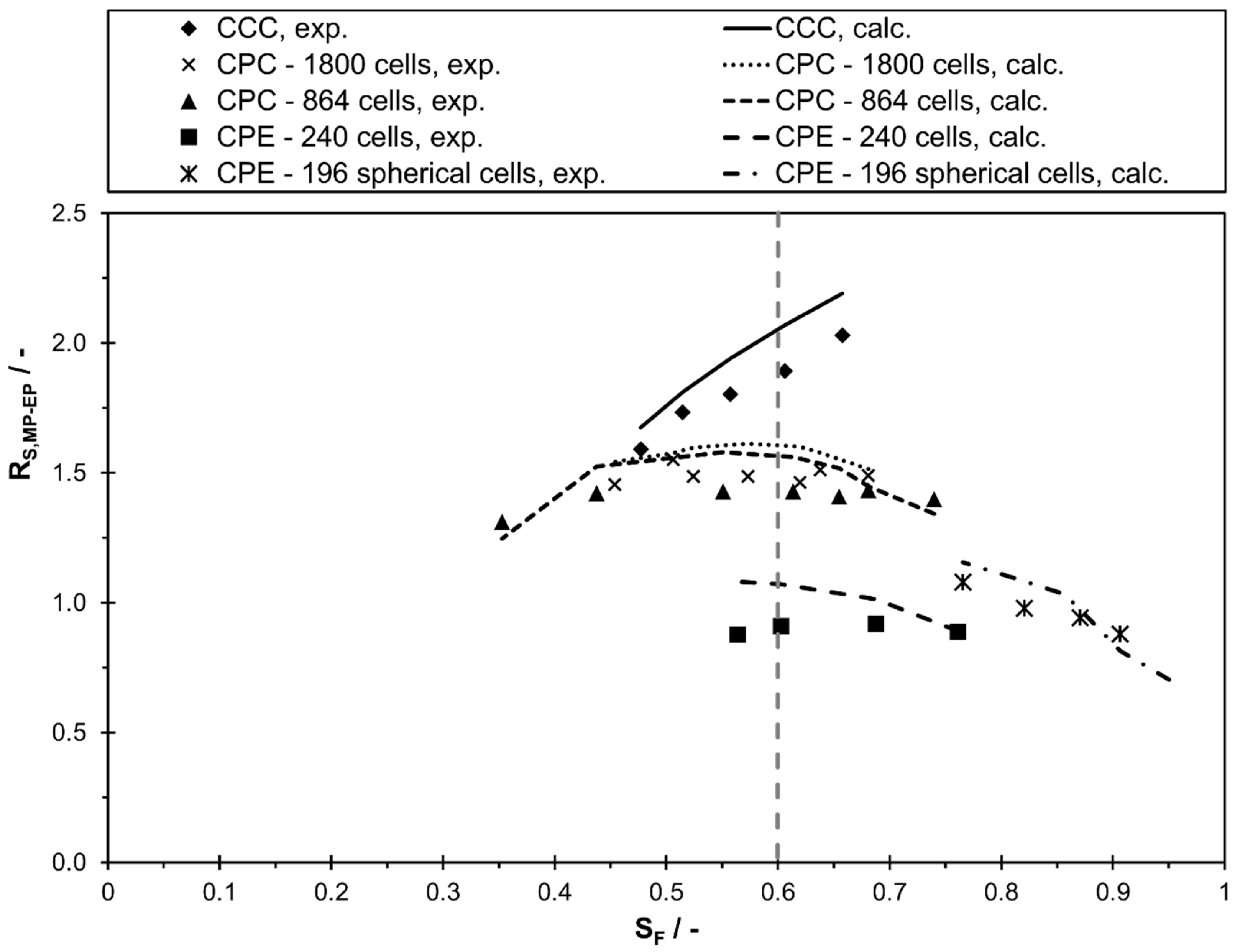
In order to evaluate the applicability of the commonly used separation method transfer between two columns based on the same stationary phase retention, the resolution is presented as a function of stationary phase retention in Figure 6. As is apparent from the figure, between two peaks can clearly differ in the various columns for a certain value of . For example, at a stationary phase retention of 0.6 the resolution varies between 0.9 (CPE 240) and 1.9 (CCC). Consequently, the frequently used method transfer strategy with same and linear scale-up factors between the columns is not generally applicable.
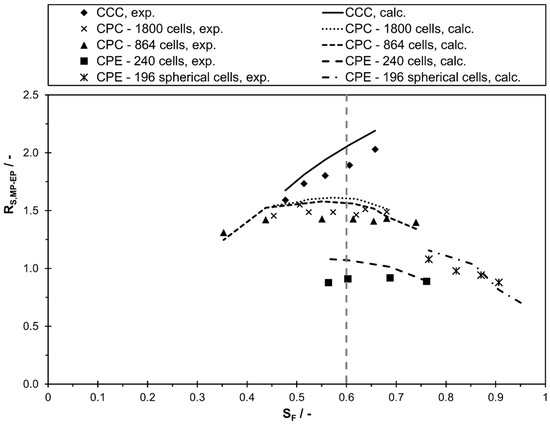
Figure 6.
Separation resolution, experimental and calculated values using Equation (5) and the linear correlations of and , between methyl paraben and ethyl paraben in descending mode of different CCC and CPC columns as a function of stationary phase retention for the solvent system n-heptane/ethyl acetate/methanol/water 1/1/1/1 v/v/v/v.
3.3. Short-Cut Approach for Evaluation of Separation Method Transferability
In this section, a two-point short-cut approach for a preliminary evaluation of separation method transferability is proposed (Figure 7). According to the results of the column characterization study (Section 3.2) shown in Figure 2, Figure 3 and Figure 4, linearly correlates with the flow rate at a constant rotational speed. In addition, can be assumed as linearly dependent on the flow rate in the studied range. Therefore, two pulse injections are sufficient to determine the mentioned linear correlations with a satisfactory accuracy. In this context, a typical operating range of the column in terms of flow rate is selected based on previous experience or manufacturer’s information. The suitable rotational speed is chosen to achieve a good compromise between good stationary phase retention and pressure drop. Initially, a pulse injection of the separation method to be transferred with a preferably small injection volume ( is performed at a relatively low flow rate within the common operating range. The flow rate is then increased stepwise and the stationary phase retention is determined at each stage by measuring the eluted stationary phase at hydrodynamic equilibrium. The flow rate is increased in a way that decreases linearly until a stationary phase retention between 0.4 and 0.5 is achieved, which represents a reasonable minimum value of for a good separation resolution in liquid–liquid chromatography [4]. If an abrupt decrease in occurs before or a maximum pressure drop is reached, the column needs to be refilled with stationary phase and equilibrated at a flow rate lower than the one where the abrupt decrease of occurred or below the maximum pressure drop. Subsequently, a second pulse injection is performed at the highest flow rate suitable according to the previous criteria. Finally, the chromatograms of both pulse injections, one at low and the other at a higher flow rate, are analyzed in terms of , , and . In all columns the same values of are obtained, since is a thermodynamic parameter. Then, linear correlations of and as a function of flow rate can be assumed. The resolution can then be calculated according to Equation (5) in the whole flow rate range and one could quickly assess whether a separation method transfer with aimed resolution (e.g., ) is possible between two columns.
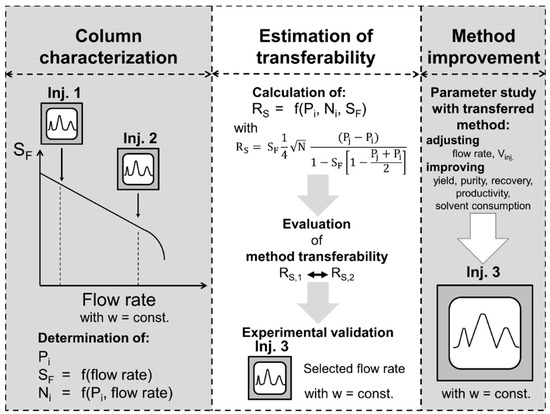
Figure 7.
Scheme of the short-cut method for the evaluation of separation method transferability.
After the transferability of the separation method has been determined, an experimental validation at the particular flow rate at which the previous criteria are fulfilled can be performed and by this time, the separation method is transferred. A further improvement of the transferred method could follow by applying the space between the peaks method [13] or additional simulations as part of theoretical parameter studies. This can be done by a parameter study, where different flow rates and injection volumes can be evaluated by applying the linear correlations of and from the short-cut method as input, e.g., as part of a simulation study based on the equilibrium cell model [30,45]. In this context, individual requirements such as purity, yield, productivity, and solvent consumption can be assessed. For a more comprehensive and reliable model-based optimization of the separation performance, a more extensive characterization of the column like in Section 3.2 with additional pulse injections could be recommended especially for the optimization and fine tuning of operating conditions at preparative or industrial scale [46,47]. Finally, it could be shown that transferability between columns can be determined by performing a two-point short-cut characterization. We believe that the proposed approach saves experimental effort and time.
4. Discussion and Conclusions
The objective of this work was to establish a systematic approach for the estimation of separation method transferability between different columns in countercurrent chromatography and centrifugal partition chromatography. In this sense, the separation method transferability was studied with the solvent system n-heptane/ethyl acetate/methanol/water 1/1/1/1 v/v/v/v and a mixture of four parabens.
The separation performance of five columns was compared in terms of stationary phase retention, column efficiency and resolution in dependence of flow rate. It was shown, that all studied columns behave differently as a function of flow rate. Nevertheless, a linear correlation of stationary phase retention and column efficiency as a function of flow rate was obtained within a certain flow rate range. This can be used to estimate whether a separation method is transferable and in which flow rate range a similar resolution is achievable. Moreover, the results of this study show that the commonly used separation method transfer strategy between two columns, where a flow rate is selected in order to keep the stationary phase retention in both columns the same and linear scale-up factors are chosen, can reach its limits. Evidently, similar stationary phase retention does not guarantee similar separation resolution.
A comprehensive evaluation of the columns in terms of stationary phase retention and column efficiency allows solute and solvent system specific parameters to be obtained and used for the (model-based) optimization of the operating parameters (e.g., injection volume and flow rate). A fast characterization of the columns can be done by a two-point short-cut method based on two pulse injection experiments at different flow rates. This saves not only experimental effort, but also material and time, which is costly, especially on a preparative scale. This approach can be applied during scale-up between two columns available on site as well as for the estimation of transferability of a separation method from literature. Furthermore, it would be useful for the CCC/CPC community to characterize existing commercially available and most frequently used CCC/CPC columns with the most commonly used solvent systems and model components (for example those from GUESSmix). This information would be a helpful contribution for a preliminary evaluation of separation method transferability between columns, i.e., labs, and established methods from literature.
Author Contributions
Both authors contributed substantially to the work reported: Conceptualization, S.R.; Data curation, S.R.; Formal analysis, S.R.; Investigation, S.R.; Methodology, S.R.; Resources, M.M.; Supervision, M.M.; Validation, S.R.; Visualization, S.R.; Writing—original draft, S.R.; Writing—review and editing, S.R. and M.M.
Funding
This work was supported by the German Research Foundation (DFG) and the Technical University of Munich (TUM) in the framework of the Open Access Publishing Program.
Acknowledgments
The authors thank Pfeiffer from AlphaCrom AG (Switzerland) for the loan of the pump and detector for the CPC spherical cells experiments. The authors thank Gilson International B.V. (Germany) for the loan of the CPC-250 and the PLC 2250 purification system setup. We thank Raena Morley for carrying out part of the CPE experiments.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
The following are additional data to this article:
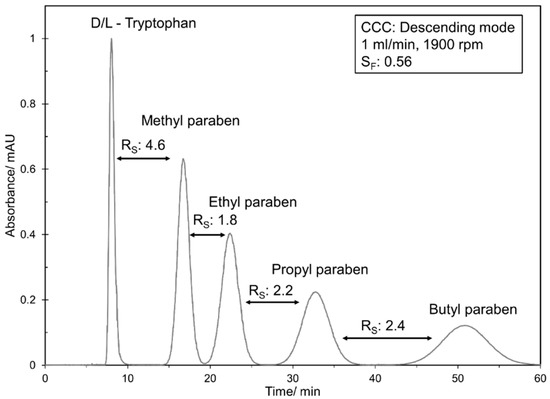
Figure A1.
CCC pulse injection of d/l-tryptophan, methyl paraben, ethyl paraben, propyl paraben and butyl paraben with the solvent system n-heptane/ethyl acetate/methanol/water 1/1/1/1 v/v/v/v in descending mode (1 mL min−1, 1900 rpm, , detector wavelength 255 nm).
Figure A1.
CCC pulse injection of d/l-tryptophan, methyl paraben, ethyl paraben, propyl paraben and butyl paraben with the solvent system n-heptane/ethyl acetate/methanol/water 1/1/1/1 v/v/v/v in descending mode (1 mL min−1, 1900 rpm, , detector wavelength 255 nm).
References
- Ito, Y. High-speed countercurrent chromatography. Nature 1987, 326, 419–420. [Google Scholar] [CrossRef]
- Ito, Y. Origin and evolution of the coil planet centrifuge: A personal reflection of my 40 years of ccc research and development. Sep. Purif. Rev. 2005, 34, 131–154. [Google Scholar] [CrossRef]
- Berthod, A.; Faure, K. Separations with a Liquid Stationary Phase: Countercurrent Chromatography or Centrifugal Partition Chromatography. In Analytical Separation Science; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015. [Google Scholar]
- Berthod, A. Countercurrent Chromatography; Elsevier: Amsterdam, The Netherlands, 2002; Volume 38. [Google Scholar]
- Foucault, A.P. Centrifugal Partition Chromatography; Chromatographic Science Series; M: Dekker: New York, NY, USA, 1995; Volume 68. [Google Scholar]
- Schmidt-Traub, H.; Schulte, M.; Seidel-Morgenstern, A. Preparative Chromatography, 2nd ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012. [Google Scholar]
- Das Neves Costa, F.; Hubert, J.; Borie, N.; Kotland, A.; Hewitson, P.; Ignatova, S.; Renault, J.H. Schinus terebinthifolius countercurrent chromatography (part iii): Method transfer from small countercurrent chromatography column to preparative centrifugal partition chromatography ones as a part of method development. J. Chromatogr. A 2017, 1487, 77–82. [Google Scholar] [CrossRef]
- Sutherland, I.A.; Audo, G.; Bourton, E.; Couillard, F.; Fisher, D.; Garrard, I.; Hewitson, P.; Intes, O. Rapid linear scale-up of a protein separation by centrifugal partition chromatography. J. Chromatogr. A 2008, 1190, 57–62. [Google Scholar] [CrossRef]
- Wood, P.; Ignatova, S.; Janaway, L.; Keay, D.; Hawes, D.; Garrard, I.; Sutherland, I.A. Counter-current chromatography separation scaled up from an analytical column to a production column. J. Chromatogr. A 2007, 1151, 25–30. [Google Scholar] [CrossRef]
- Das Neves Costa, F.; Vieira, M.N.; Garrard, I.; Hewitson, P.; Jerz, G.; Leitão, G.G.; Ignatova, S. Schinus terebinthifolius countercurrent chromatography (part ii): Intra-apparatus scale-up and inter-apparatus method transfer. J. Chromatogr. A 2016, 1466, 76–83. [Google Scholar] [CrossRef]
- Sutherland, I.; Hewitson, P.; Ignatova, S. Scale-up of counter-current chromatography: Demonstration of predictable isocratic and quasi-continuous operating modes from the test tube to pilot/process scale. J. Chromatogr. A 2009, 1216, 8787–8792. [Google Scholar] [CrossRef]
- Chollet, S.; Marchal, L.; Jeremy, M.; Renault, J.H.; Legrand, J.; Foucault, A. Methodology for optimally sized centrifugal partition chromatography columns. J. Chromatogr. A 2015, 1388, 174–183. [Google Scholar] [CrossRef]
- Bouju, E.; Berthod, A.; Faure, K. Scale-up in centrifugal partition chromatography: The “free-space between peaks” method. J. Chromatogr. A 2015, 1409, 70–78. [Google Scholar] [CrossRef]
- Fumat, N.; Berthod, A.; Faure, K. Effect of operating parameters on a centrifugal partition chromatography separation. J. Chromatogr. A 2016, 1474, 47–58. [Google Scholar] [CrossRef]
- Goll, J.; Audo, G.; Minceva, M. Comparison of twin-cell centrifugal partition chromatographic columns with different cell volume. J. Chromatogr. A 2015, 1406, 129–135. [Google Scholar] [CrossRef]
- Schwienheer, C.; Merz, J.; Schembecker, G. Investigation, comparison and design of chambers used in centrifugal partition chromatography on the basis of flow pattern and separation experiments. J. Chromatogr. A 2015, 1390, 39–49. [Google Scholar] [CrossRef]
- Roehrer, S.; Minceva, M. Characterization of a centrifugal partition chromatographic column with spherical cell design. Chem. Eng. Res. Des. 2019, 143, 180–189. [Google Scholar] [CrossRef]
- Peng, A.; Hewitson, P.; Sutherland, I.; Chen, L.; Ignatova, S. The effect of increasing centrifugal acceleration/force and flow rate for varying column aspect ratios on separation efficiency in counter-current chromatography. J. Chromatogr. A 2018, 1581–1582, 80–90. [Google Scholar] [CrossRef]
- Van Buel, M.; Van Halsema, F.; Van der Wielen, L.; Luyben, K. Flow regimes in centrifugal partition chromatography. AIChE J. 1998, 44, 1356–1362. [Google Scholar] [CrossRef]
- Marchal, L.; Foucaul, A.; Patissier, G.; Rosant, J.M.; Legrand, J. Influence of flow patterns on chromatographic efficiency in centrifugal partition chromatography. J. Chromatogr. A 2000, 869, 339–352. [Google Scholar] [CrossRef]
- Marchal, L.; Legrand, J.; Foucault, A. Mass transport and flow regimes in centrifugal partition chromatography. AIChE J. 2002, 48, 1692–1704. [Google Scholar] [CrossRef]
- Adelmann, S.; Baldhoff, T.; Koepcke, B.; Schembecker, G. Selection of operating parameters on the basis of hydrodynamics in centrifugal partition chromatography for the purification of nybomycin derivatives. J. Chromatogr. A 2013, 1274, 54–64. [Google Scholar] [CrossRef]
- Foucault, A.P.; Frias, E.C.; Bordier, C.G.; Goffic, F.L. Centrifugal partition chromatography: Stability of various biphasic systems and pertinence of the “stoke’s model” to describe the influence of the centrifugal field upon the efficiency. J. Liq. Chromatogr. 1994, 17, 1–17. [Google Scholar] [CrossRef]
- Adelmann, S.; Schembecker, G. Influence of physical properties and operating parameters on hydrodynamics in centrifugal partition chromatography. J. Chromatogr. A 2011, 1218, 5401–5413. [Google Scholar] [CrossRef]
- Couillard, F. Connecting channels and cells for centrifugal partition chromatographs. Wo 2009/066014 a1, 28 May 2009. [Google Scholar]
- Marchal, L.; Intes, O.; Foucault, A.; Legrand, J.; Nuzillard, J.-M.; Renault, J.-H. Rational improvement of centrifugal partition chromatographic settings for the production of 5-n-alkylresorcinols from wheat bran lipid extract. J. Chromatogr. A 2003, 1005, 51–62. [Google Scholar] [CrossRef]
- Peng, A.; Hewitson, P.; Sutherland, I.; Chen, L.; Ignatova, S. How changes in column geometry and packing ratio can increase sample load and throughput by a factor of fifty in counter-current chromatography. J. Chromatogr. A 2018, 1580, 120–125. [Google Scholar] [CrossRef]
- Guiochon, G.; Felinger, A.; Shirazi, D.G.; Katti, A.M. Fundamentals of Preparative and Nonlinear Chromatography; Elsevier: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Nicoud, R.-M. Chromatographic Processes; Cambridge University Press: Cambridge, UK, 2015. [Google Scholar]
- Martin, A.J.P.; Synge, R.L.M. A new form of chromatogram employing two liquid phases. A theory of chromatography. 2. Application to the micro-determination of the higher monoamino-acids in proteins. Biochem. J. 1941, 35, 1358–1368. [Google Scholar] [CrossRef]
- Kostanian, A.E. Modelling counter-current chromatography: A chemical engineering perspective. J. Chromatogr. A 2002, 973, 39–46. [Google Scholar] [CrossRef]
- Kostanyan, A.E. General regularities of liquid chromatography and countercurrent extraction. Found. Chem. Eng. 2006, 40, 587–593. [Google Scholar] [CrossRef]
- Ignatova, S.; Sutherland, I. A fast, effective method of characterizing new phase systems in ccc. J. Liq. Chromatogr. Relat. Technol. 2003, 26, 1551–1564. [Google Scholar] [CrossRef]
- Morley, R.; Minceva, M. Unpublished work. 2018. [Google Scholar]
- He, C.H.; Zhao, C.X. Retention of the stationary phase for high-speed countercurrent chromatography. AIChE J. 2007, 53, 1460–1471. [Google Scholar] [CrossRef]
- Du, Q.Z.; Wu, C.J.; Qian, G.J.; Wu, P.D.; Ito, Y. Relationship between the flow-rate of the mobile phase and retention of the stationary phase in counter-current chromatography. J. Chromatogr. A 1999, 835, 231–235. [Google Scholar] [CrossRef]
- Sutherland, I.; Du, Q.; Wood, P. The relationship between retention, linear flow, and density difference in countercurrent chromatography. J. Liq. Chromatogr. Relat. Technol. 2001, 24, 1669–1683. [Google Scholar] [CrossRef]
- Foucault, A.; Bousquet, O.; Le Goffic, F. Importance of the parameter vm/vc in countercurrent chromatography: Tentative comparison between instrument designs. J. Liq. Chromatogr. 1992, 15, 2691–2706. [Google Scholar] [CrossRef]
- Sutherland, I.A.; Booth, A.J.; Brown, L.; Kemp, B.; Kidwell, H.; Games, D.; Graham, A.S.; Guillon, G.G.; Hawes, D.; Hayes, M.; et al. Industrial scale-up of countercurrent chromatography. J. Liq. Chromatogr. Relat. Technol. 2001, 24, 1533–1553. [Google Scholar] [CrossRef]
- Wood, P.L.; Hawes, D.; Janaway, L.; Sutherland, I.A. Stationary phase retention in ccc: Modelling the j-type centrifuge as a constant pressure drop pump. J. Liq. Chromatogr. Relat. Technol. 2003, 26, 1373–1396. [Google Scholar] [CrossRef]
- Kotland, A.; Chollet, S.; Autret, J.M.; Diard, C.; Marchal, L.; Renault, J.H. Modeling ph-zone refining countercurrent chromatography: A dynamic approach. J. Chromatogr. A 2015, 1391, 80–87. [Google Scholar] [CrossRef]
- Berthod, A.; Faure, K. Revisiting resolution in hydrodynamic countercurrent chromatography: Tubing bore effect. J. Chromatogr. A 2015, 1390, 71–77. [Google Scholar] [CrossRef]
- Berthod, A.; Bully, M. High-speed countercurrent chromatography used for alkylbenzene liquid-liquid partition coefficient determination. Anal. Chem. 1991, 63, 2508–2512. [Google Scholar] [CrossRef]
- Bousquet, O.; Foucault, A.P.; Goffic, F.L. Efficiency and resolution in countercurrrent chromatography. J. Liq. Chromatogr. 1991, 14, 3343–3363. [Google Scholar] [CrossRef]
- Völkl, J.; Arlt, W.; Minceva, M. Theoretical study of sequential centrifugal partition chromatography. AIChE J. 2013, 59, 241–249. [Google Scholar] [CrossRef]
- Goll, J.; Minceva, M. Continuous fractionation of multicomponent mixtures with sequential centrifugal partition chromatography. AIChE J. 2017, 63, 1659–1673. [Google Scholar] [CrossRef]
- Morley, R.; Minceva, M. Operating mode selection for the separation of intermediately-eluting components with countercurrent and centrifugal partition chromatography. J. Chromatogr. A 2019. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).