Modern Instrumental Limits of Identification of Ignitable Liquids in Forensic Fire Debris Analysis



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Sample Dilution Scheme
2.3. General Instrumental Analysis
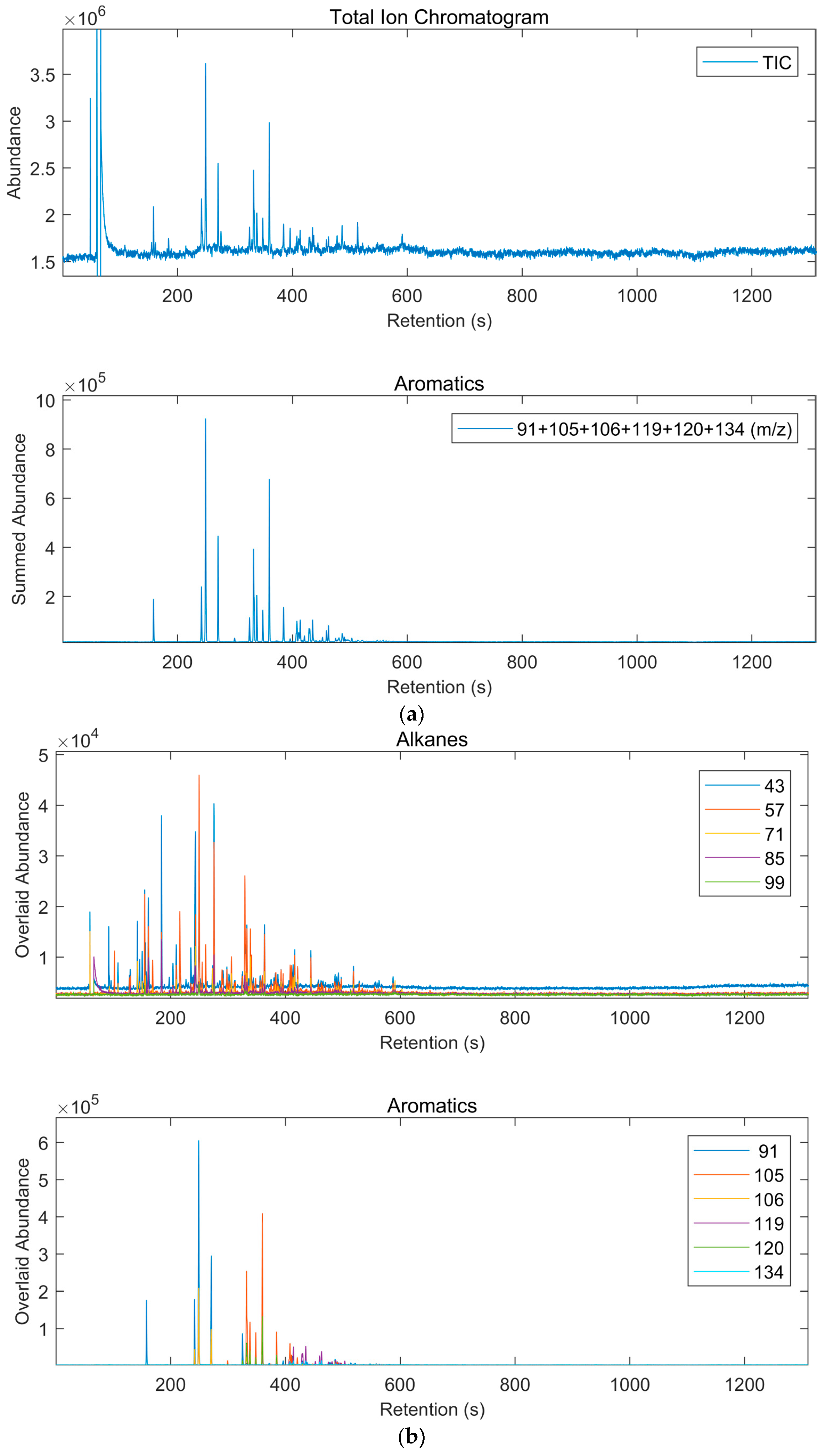
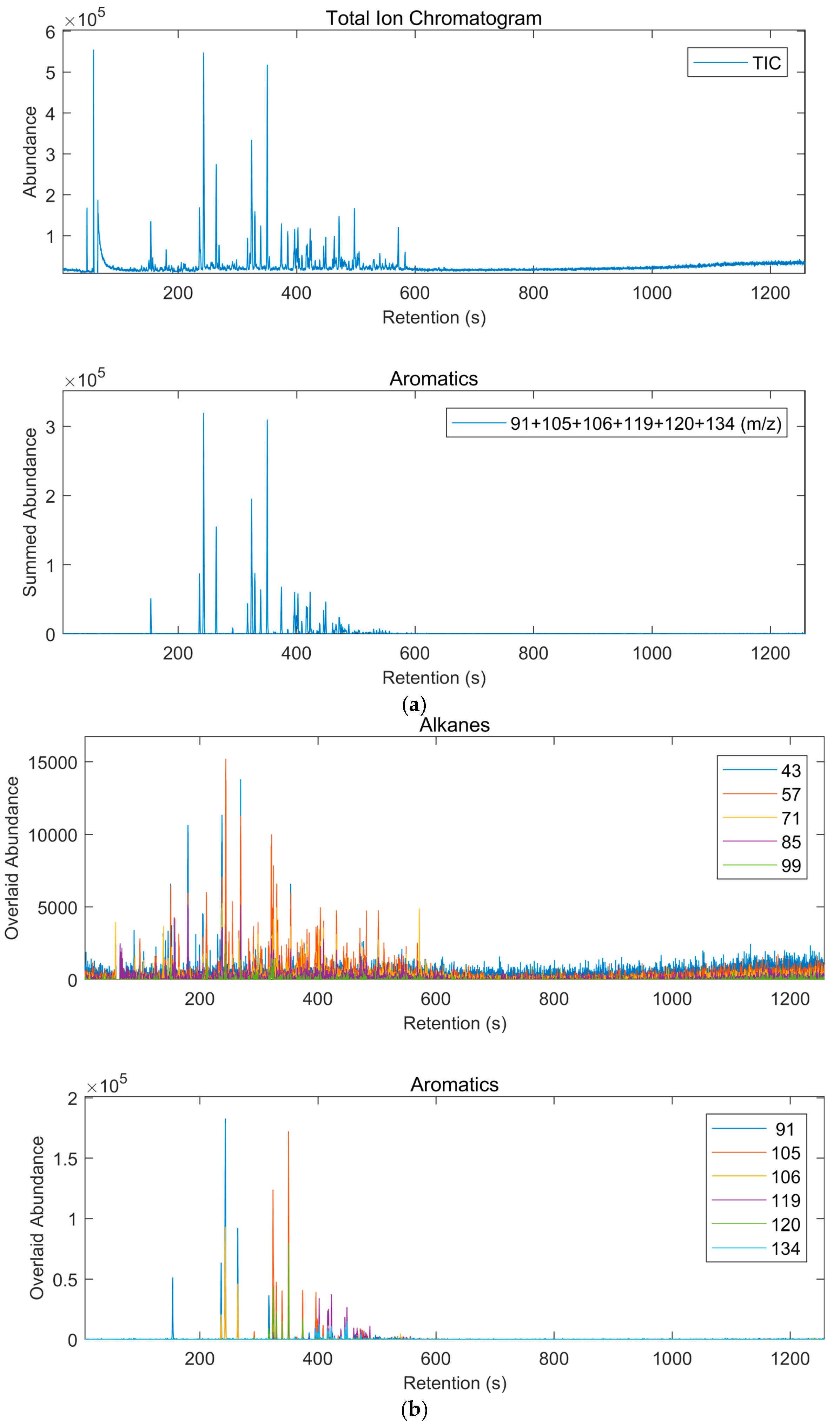
2.4. GC-MSD Analysis
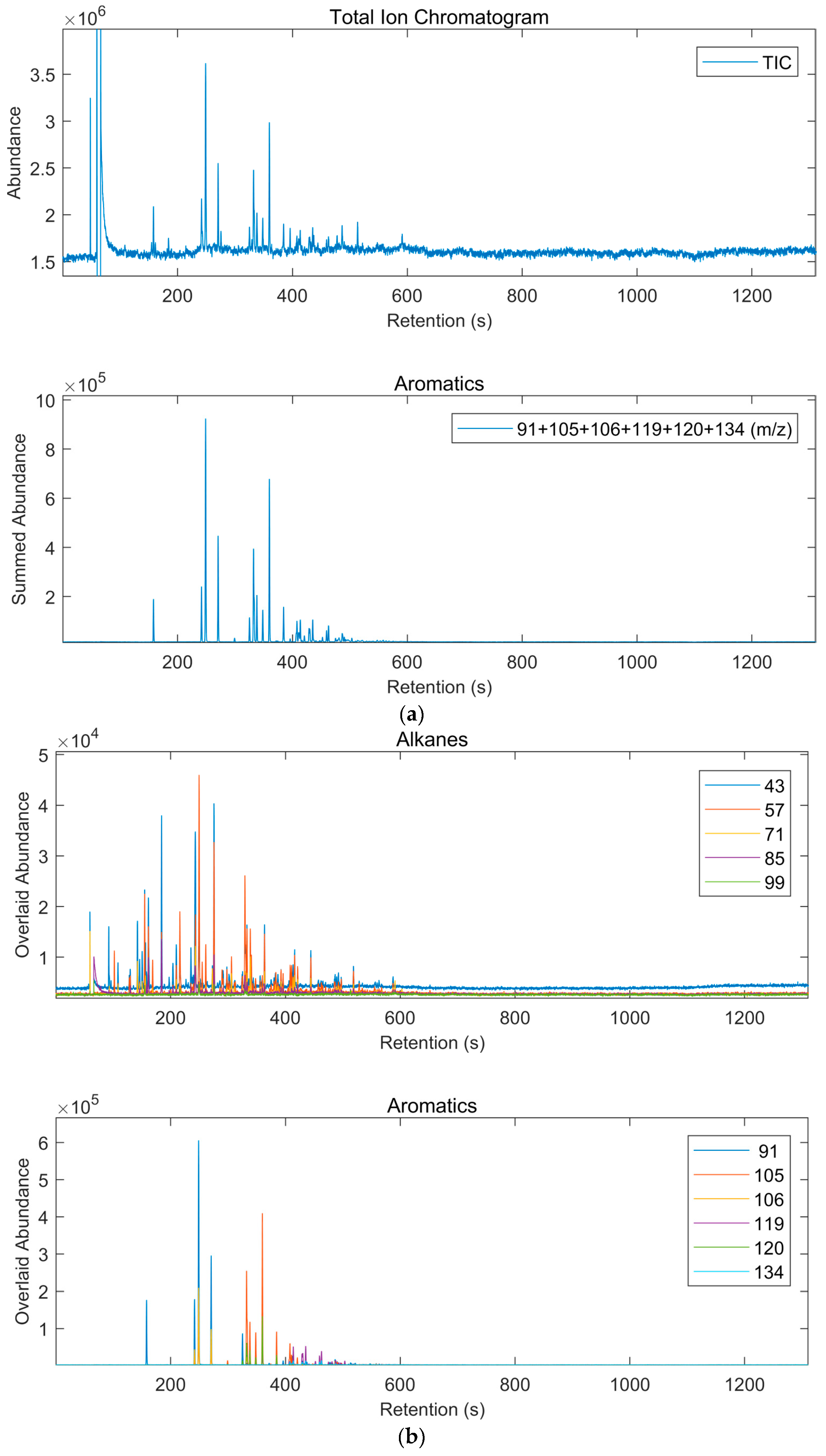
2.5. GC-TOF Analysis
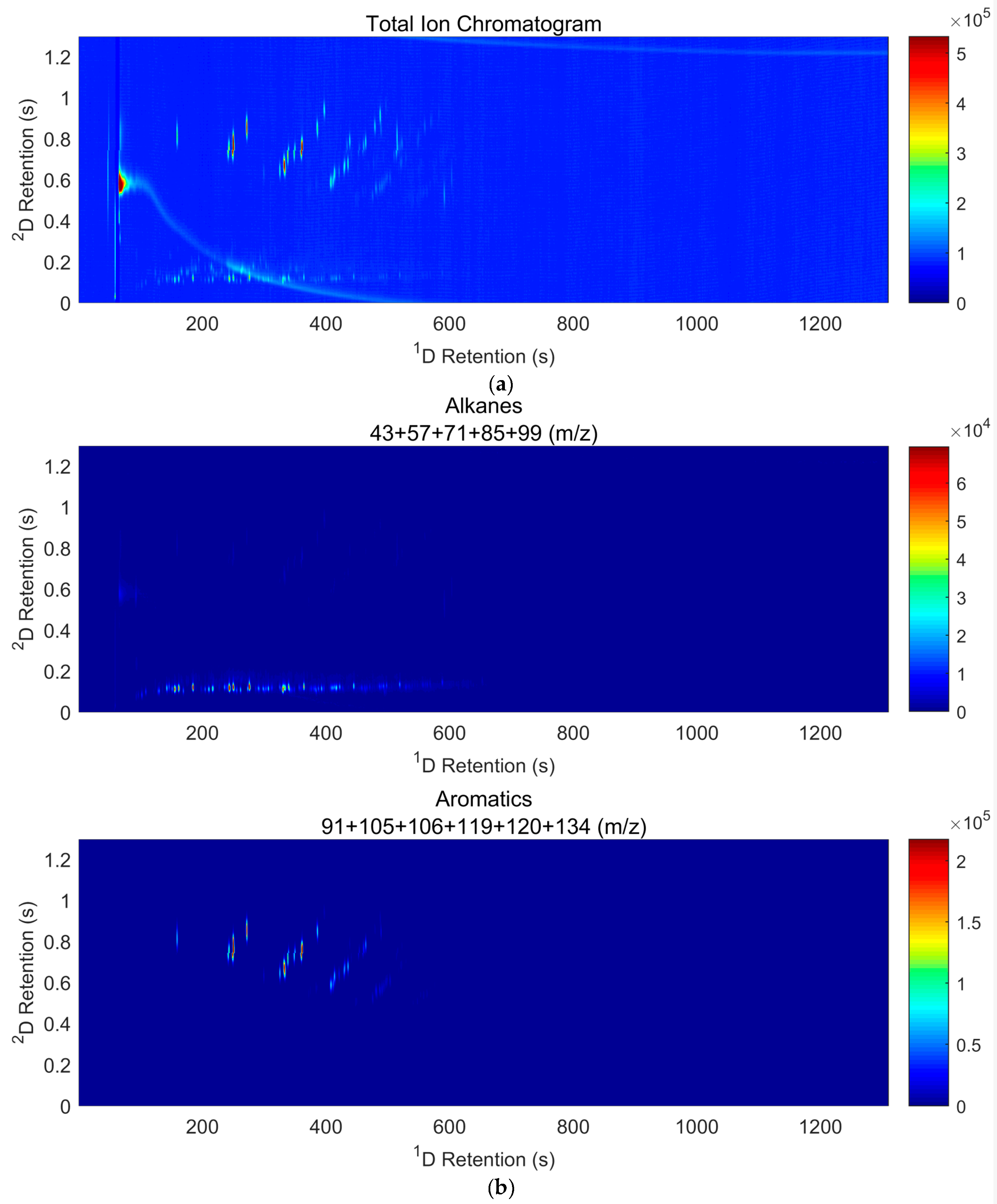
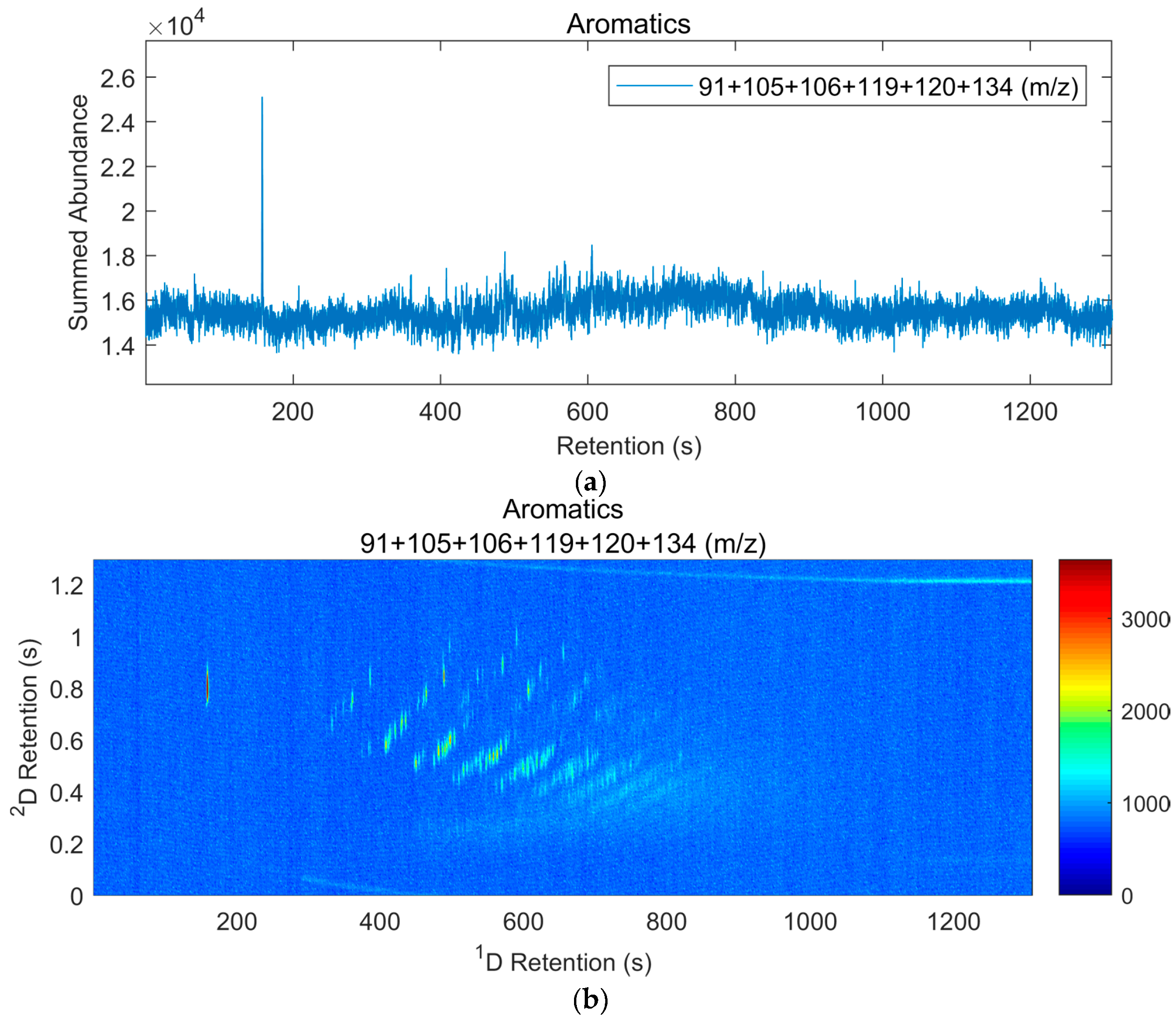
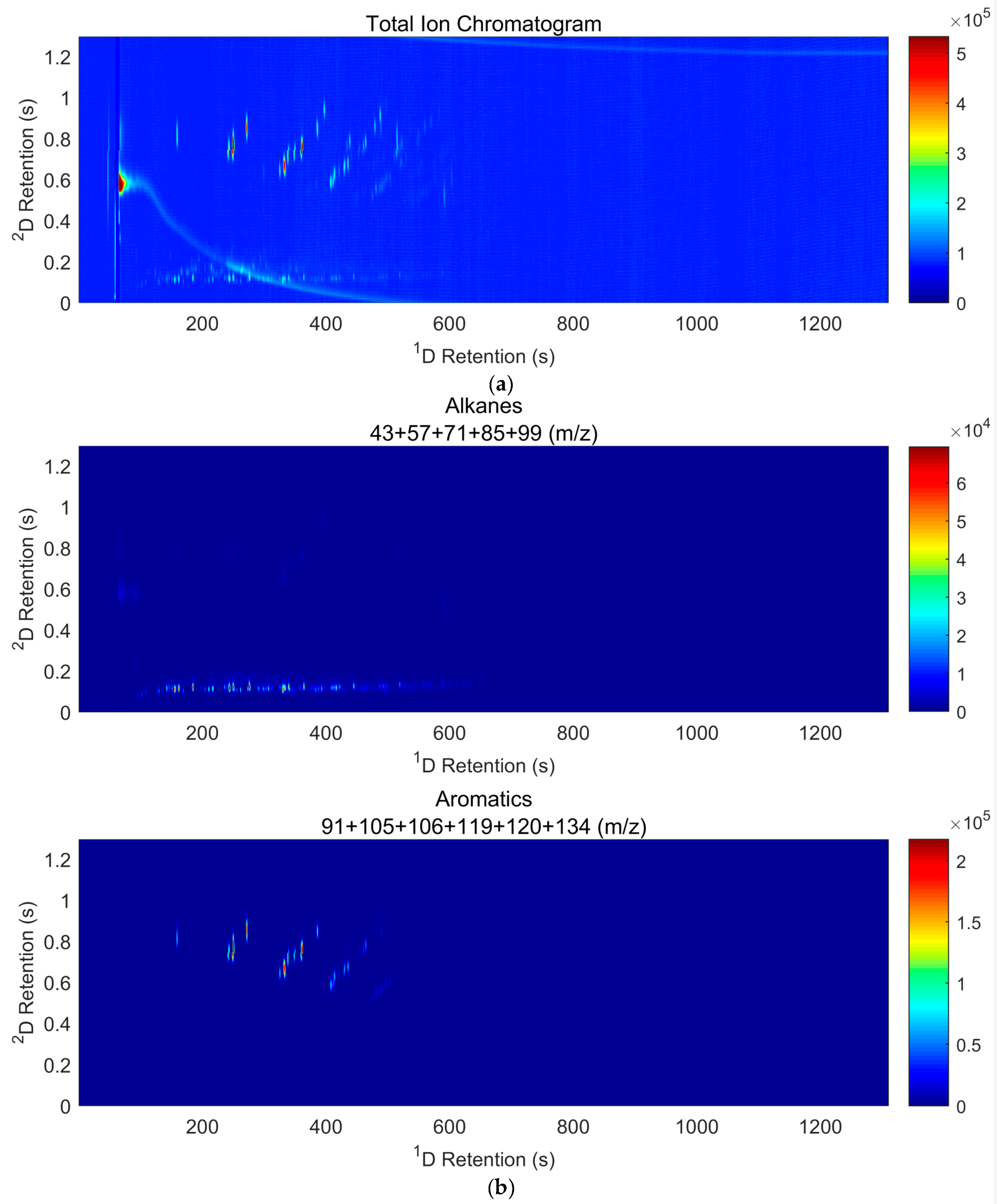
2.6. GC×GC-TOF Analysis
2.7. Interpretation of Results
3. Results
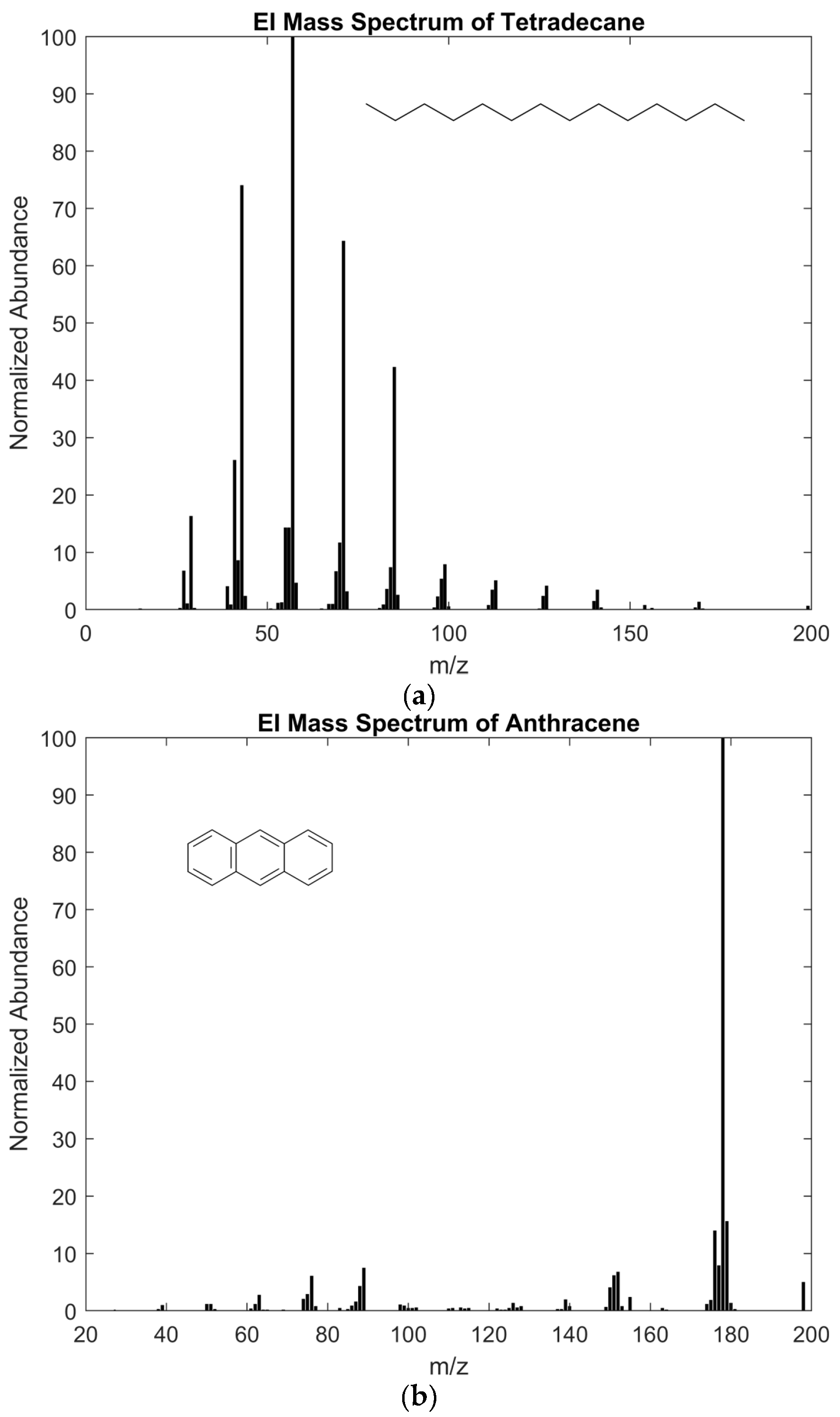
3.1. General Criteria for Identification of Ignitable Liquids
3.2. Results of Interpretation of GC-MSD Analyses
3.2.1. GC-MSD Analysis of Gasoline (Sections SI.3 Pages 27–70)
3.2.2. GC-MSD Analysis of Gasoline with Pyrolysate (Sections SI.3 Pages 94–137)
3.2.3. GC-MSD Analysis of Diesel (Sections SI.3 Pages 7–26)
3.2.4. GC-MSD Analysis of Diesel with Pyrolysate (Sections SI.3 Pages 74–93)
3.3. Results of Interpretation of GC-TOF Analyses
3.3.1. GC-TOF Analysis of Gasoline (Sections SI.3 Pages 165–220)
3.3.2. GC-TOF Analysis of Gasoline with Pyrolysate (Sections SI.3 Pages 254–313)
3.3.3. GC-TOF Analysis of Diesel (Sections SI.3 Pages 139–164)
3.3.4. GC-TOF Analysis of Diesel with Pyrolysate (Sections SI.3 Pages 228–253)
3.4. Results of Interpretation of GC×GC-TOF Analyses
3.4.1. GC×GC-TOF Analysis of Gasoline (Sections SI.3 Pages 331–360)
3.4.2. GC×GC-TOF Analysis of Gasoline with Pyrolysate (Sections SI.3 Pages 382–413)
3.4.3. GC×GC-TOF Analysis of Diesel (Sections SI.3 Pages 315–330)
3.4.4. GC×GC-TOF Analysis of Diesel with Pyrolysate (Sections SI.3 Pages 365–381)
4. Discussion
4.1. Notes on Design and Interpretation
4.2. Observations on GC-MSD Results
4.3. Observations on GC-TOF Results
4.4. Observations on GC×GC-TOF Results
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jackowski, J.P. The incidence of ignitable liquid residues in fire debris as determined by a sensitive and comprehensive analytical scheme. J. Forensic Sci. 1997, 42, 828–832. [Google Scholar] [CrossRef]
- Sandercock, P.M.L. Passive headspace extraction of ignitable liquids using activated carbon cloth. Can. Soc. Forensic Sci. J. 2016, 49, 176–188. [Google Scholar] [CrossRef]
- Lentini, J.J.; Dolan, J.A.; Cherry, C. The petroleum-laced background. J. Forensic Sci. 2000, 45, 968–989. [Google Scholar] [CrossRef] [PubMed]
- DeHaan, J.D. Fire investigations and the forensic lab: What the lab should be doing, or, it’s not about the GC. CAC News 2002, 4, 14–16. [Google Scholar]
- DeHaan, J.D. Our changing world, Part 3: Is more sensitive necessarily more better? and Part 4: A matter of time. Fire Arson Investig. 2002, 52, 20–23. [Google Scholar]
- Almirall, J.R.; Furton, K.G. Analysis and Interpretation of Fire Scene Evidence; CRC Press: Boca Raton, FL, USA, 2004; p. 262. [Google Scholar]
- Hetzel, S.S.; Moss, R.D. How long after waterproofing a deck can you still isolate an ignitable liquid? J. Forensic Sci. 2005, 50, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Lang, T.; Dixon, B.M. The possible contamination of fire scenes by the use of positive pressure ventilation fans. Can. Soc. Forensic Sci. J. 2000, 33, 55–60. [Google Scholar] [CrossRef]
- Koussaifes, P.M. Evaluation of fire scene contamination by using positive-pressure ventilation fans. Forensic Sci. Commun. 2002, 4, 4. [Google Scholar]
- Armstrong, A.; Babrauskas, V.; Holmes, D.L.; Martin, C.; Powell, R.; Riggs, S.; Young, L.D. The evaluation of the extent of transporting or “tracking” an identifiable ignitable liquid (gasoline) throughout fire scenes during the investigative process. J. Forensic Sci. 2004, 49, 741–748. [Google Scholar] [CrossRef]
- Belchior, F.; Andrews, S.P. Evaluation of cross-contamination of nylon bags with heavy-loaded gasoline fire debris and with automotive paint thinner. J. Forensic Sci. 2016, 61, 1622–1631. [Google Scholar] [CrossRef]
- DeHaan, J.D. Canine accelerant detection teams: Validation and certification. CAC News 1994, 2, 17–21. [Google Scholar]
- Kurz, M.E.; Billard, M.; Rettig, M.; Augustiniak, J.; Lange, J.; Larsen, M.; Warrick, R.; Mohns, T.; Bora, R.; Broadus, K.; et al. Evaluation of canines for accelerant detection at fire scenes. J. Forensic Sci. 1994, 39, 1528–1536. [Google Scholar] [CrossRef] [PubMed]
- Tindall, R.; Lothridge, K. An evaluation of 42 accelerant detection canine teams. J. Forensic Sci. 1995, 40, 561–564. [Google Scholar] [CrossRef]
- Katz, S.R.; Midkiff, C.R. Unconfirmed canine accelerant detection: A reliability issue in court. J. Forensic Sci. 1998, 43, 329–333. [Google Scholar] [CrossRef]
- Ottley, B.L. Beyond the crime laboratory: The admissibility of unconfirmed forensic evidence in arson cases. N. Engl. J. Crim. Civ. Confin. 2010, 36, 263. [Google Scholar]
- Twibell, J.D.; Home, J.M.; Smalldon, K.W. A comparison of the relative sensitivities of the adsorption wire and other methods for the detection of accelerant residues in fire debris. J. Forensic Sci. Soc. 1980, 22, 155–159. [Google Scholar] [CrossRef]
- Loscalzo, P.; DeForest, P.R.; Chao, J. A study to determine the limit of detectability of gasoline vapor from simulated arson residues. J. Forensic Sci. 1980, 25, 162–167. [Google Scholar] [CrossRef]
- Thatcher, P.J. The scientific investigation of fire causes. Forensic Sci. Prog. 1986, 1, 117–152. [Google Scholar]
- Choodum, A.; Nic Daéid, N. Development and validation of an analytical method for hydrocarbon residues using gas chromatography-mass spectrometry. Anal. Methods 2011, 3, 1136–1142. [Google Scholar] [CrossRef]
- Frysinger, G.S.; Gaines, R.B. Forensic analysis of ignitable liquids in fire debris by comprehensive two-dimensional gas chromatography. J. Forensic Sci. 2002, 47, 471–482. [Google Scholar]
- Dolan, J. Recent advances in the applications of forensic science to fire debris analysis. Anal. Bioanal. Chem. 2003, 376, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, E.; Lentini, J.J. ASTM standards for fire debris analysis: A review. Forensic Sci. Int. 2003, 132, 63–67. [Google Scholar] [CrossRef]
- Taylor, C.M.; Rosenhan, A.K.; Raines, J.M.; Rodriguez, J.M. An arson investigation by using comprehensive two-dimensional gas chromatography-quadrupole mass spectrometry. J. Forensic Res. 2012, 3, 169. [Google Scholar]
- Klee, M.S.; Blumberg, L.M. Theoretical and practical aspects of fast gas chromatography and method translation. J. Chromatogr. Sci. 2002, 40, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, L.M.; Klee, M.S. Optimal heating rate in gas chromatography. J. Microcolumn Sep. 2000, 12, 508–514. [Google Scholar] [CrossRef]
- American Society for Testing and Materials. ASTM E1618-14 Standard Test Method for Ignitable Liquid Residues in Extracts from Fire Debris Samples by Gas Chromatography-Mass Spectrometry; ASTM International: West Conshohocken, PA, USA, 2014. [Google Scholar]
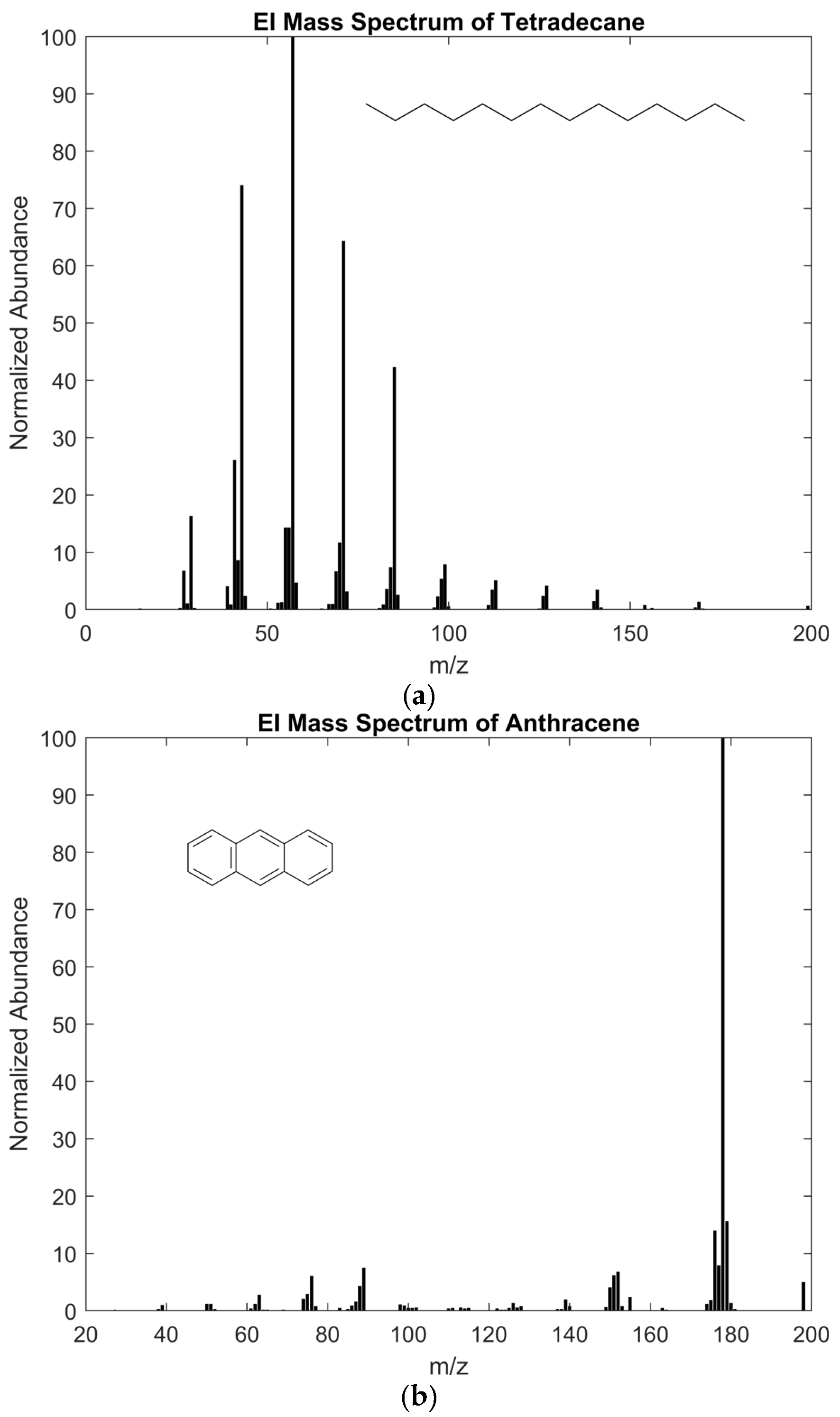
- McLafferty, F.W.; Tureček, F. Interpretation of Mass Spectra, 4th ed.; University Science Books: Mill Valley, CA, USA, 1993; p. 371. [Google Scholar]
- Stauffer, E.; Dolan, J.A.; Newman, R. Fire Debris Analysis, 1st ed.; Elsevier/Academic Press: London, UK, 2008. [Google Scholar]
- Sampat, A.; van Daelen, B.; Lopatka, M.; Mol, H.; van der Weg, G.; Vivó-Truyols, G.; Sjerps, M.; Schoenmakers, P.; van Asten, A. Detection and characterization of ignitable liquid residues in forensic fire debris samples by comprehensive two-dimensional gas chromatography. Separations 2018, 5, 43. [Google Scholar] [CrossRef]
- Doong, R.; Chang, S.; Sun, Y. Solid-phase microextraction for determining the distribution of sixteen US Environmental Protection Agency polycyclic aromatic hydrocarbons in water samples. J. Chromatogr. A 2000, 879, 177–188. [Google Scholar] [CrossRef]
- Speight, J.G. Handbook of Petroleum Analysis; John Wiley & Sons: New York, NY, USA, 2001; p. 473. [Google Scholar]
- Sandercock, P.M.L. A survey of Canadian gasoline (2004). Can. Soc. Forensic Sci. J. 2007, 40, 105–130. [Google Scholar] [CrossRef]
- Sandercock, P.M.L. Survey of Canadian gasoline (Winter 2010). Can. Soc. Forensic Sci. J. 2012, 45, 64–78. [Google Scholar] [CrossRef]
- Bertsch, W.; Zhang, Q.W.; Holzer, G. Using the tools of chromatography, mass spectrometry, and automated data processing in the detection of arson. J. High Resolut. Chromatogr. 1990, 13, 597–605. [Google Scholar] [CrossRef]
- Lentini, J.J. An improved method of obtaining ion profiles from ignitable liquid residue samples. CAC News 1995, 4, 18. [Google Scholar]
- Kurz, M.E.; Schultz, S.; Griffith, J.; Broadus, K.; Sparks, J.; Dabdoub, G.; Brock, J. Effect of background interference on accelerant detection by canines. J. Forensic Sci. 1996, 41, 868–873. [Google Scholar] [CrossRef] [PubMed]
- American Society for Testing and Materials. ASTM E1412-16 Standard Practice for Separation of Ignitable Liquid Residues from Fire Debris Samples by Passive Headspace Concentration with Activated Charcoal; ASTM International: West Conshohocken, PA, USA, 2016. [Google Scholar]
- Dehaan, J.D.; Bonarius, K. Pyrolysis products of structure fires. J. Forensic Sci. Soc. 1988, 28, 299–309. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source Vial Conc. (μL/mL) | Aliquot from Source Vial (μL) | Volume of Standard in Aliquot (μL) | Diluent (μL) | Resulting Conc. (μL/mL) |
|---|---|---|---|---|
| Neat liquid | 50 | 50 | 950 | 50 |
| 50 | 50 | 2.5 | 450 | 5 |
| 50 | 10 | 0.5 | 490 | 1 |
| 5 | 50 | 0.25 | 450 | 0.5 |
| 1 | 50 | 0.05 | 450 | 0.1 |
| 0.5 | 50 | 0.025 | 450 | 0.05 |
| 1 | 10 | 0.01 | 390 | 0.025 |
| 0.1 | 50 | 0.005 | 450 | 0.01 |
| 0.05 | 50 | 0.0025 | 450 | 0.005 |
| 0.01 | 50 | 0.0005 | 450 | 0.001 |
| 0.005 | 50 | 0.00025 | 450 | 0.0005 |
| Ignitable Liquid | Solution Conc. (μL/mL) | Volume on-Column (pL) | Number of Injections by Technique | ||
|---|---|---|---|---|---|
| GC-MSD | GC-TOF | GC×GC-TOF | |||
| Gasoline 1 | 0.001 | 0.0125 | N/A | N/A | 3 |
| (Esso) | 0.005 | 0.0625 | N/A | 3 | 3 |
| 75% Evaporated | 0.01 | 0.125 | 3 | 3 | 3 |
| 0.05 | 0.625 | 3 | 3 | 3 | |
| 0.1 | 1.25 | 3 | 3 | 1 | |
| 0.5 | 6.25 | 1 | 1 | 1 | |
| 1 | 12.5 | 1 | 1 | 1 | |
| Gasoline 2 | 0.001 | 0.0125 | N/A | N/A | 3 |
| (7-Eleven) | 0.005 | 0.0625 | N/A | 3 | 3 |
| 75% Evaporated | 0.01 | 0.125 | 3 | 3 | 3 |
| 0.05 | 0.625 | 3 | 3 | 3 | |
| 0.1 | 1.25 | 3 | 3 | 1 | |
| 0.5 | 6.25 | 1 | 1 | 1 | |
| 1 | 12.5 | 1 | 1 | 1 | |
| Diesel | 0.005 | 0.0625 | N/A | N/A | 3 |
| (7-Eleven) | 0.01 | 0.125 | N/A | 3 | 3 |
| 25% Evaporated | 0.05 | 0.625 | 3 | 3 | 3 |
| 0.1 | 1.25 | 3 | 3 | 3 | |
| 0.5 | 6.25 | 3 | 3 | 3 | |
| 1 | 12.5 | 1 | 1 | 1 | |
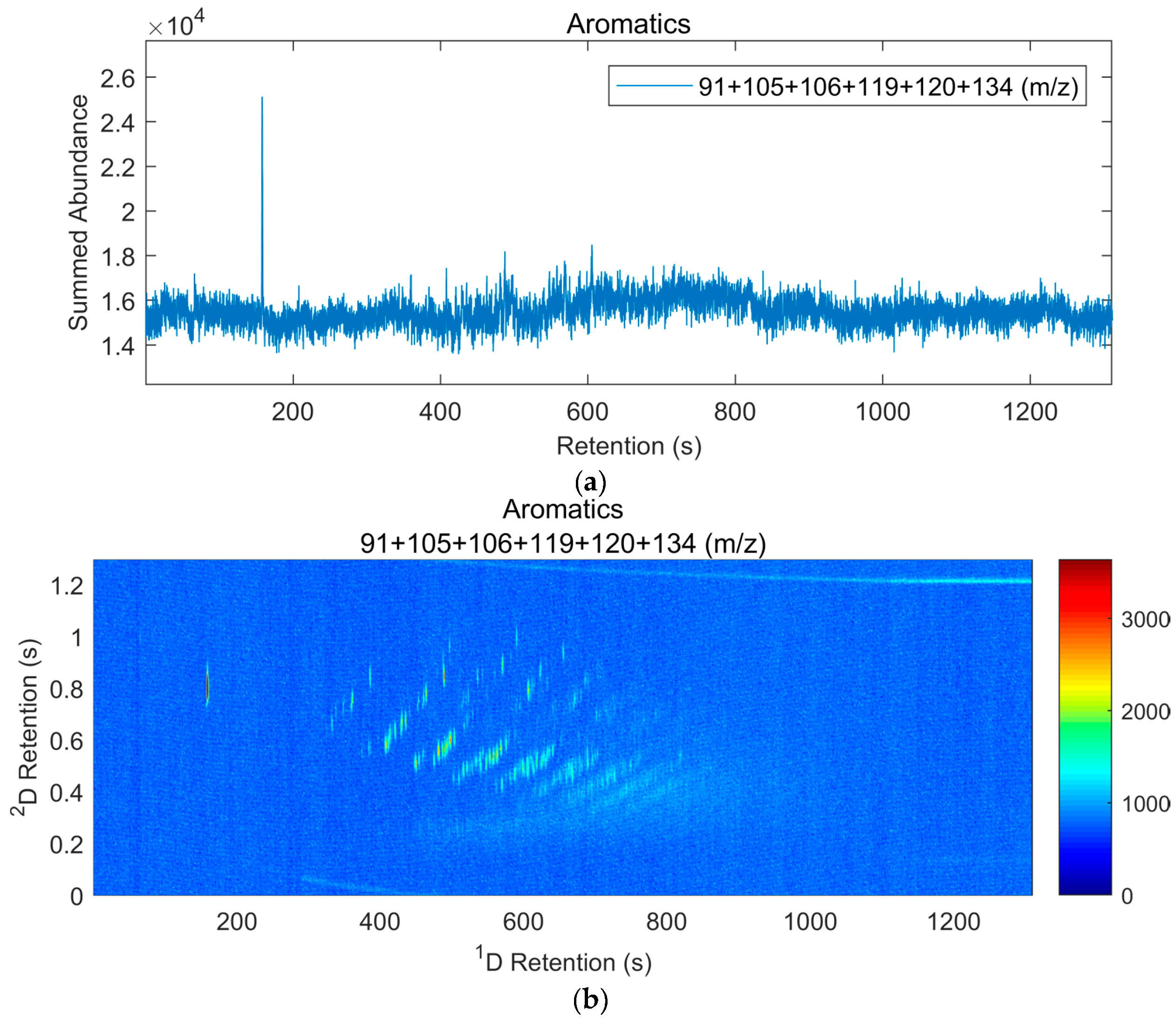
| Class of Compounds | Overlapping Classes 1 | Ions (m/z) |
|---|---|---|
| Alkanes | Most Organics | 43, 57, 71, 85, 99 |
| Cycloalkanes | Alkenes | 41, 55, 69, 83, 97 |
| Aromatics | Terpenes | 91, 105, 106, 119, 120, 134 |
| Indanes | Tetralins, Styrenes, Aromatics | 117, 118, 131, 132, 145, 146 |
| Naphthalenes | Decalins | 128, 141, 142, 155, 156, 170 |
| pL on Column | Neat Dilutions | Pyrolysate-Doped | |||||
|---|---|---|---|---|---|---|---|
| GC-MSD | GC-TOF | GC×GC | GC-MSD | GC-TOF | GC×GC | ||
| Gasoline 1 | 0.0125 | ||||||
| 0.0625 | |||||||
| 0.125 | |||||||
| 0.625 | |||||||
| 1.25 | |||||||
| 6.25 | |||||||
| Gasoline 2 | 0.0125 | ||||||
| 0.0625 | |||||||
| 0.125 | |||||||
| 0.625 | |||||||
| 1.25 | Contested | ||||||
| 6.25 | |||||||
| Diesel | 0.0625 | ||||||
| 0.125 | |||||||
| 0.625 | |||||||
| 1.25 | |||||||
| 6.25 | Misclass. | ||||||
| 12.5 | Misclass. | ||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abel, R.J.; Zadora, G.; Sandercock, P.M.L.; Harynuk, J.J. Modern Instrumental Limits of Identification of Ignitable Liquids in Forensic Fire Debris Analysis. Separations 2018, 5, 58. https://doi.org/10.3390/separations5040058
Abel RJ, Zadora G, Sandercock PML, Harynuk JJ. Modern Instrumental Limits of Identification of Ignitable Liquids in Forensic Fire Debris Analysis. Separations. 2018; 5(4):58. https://doi.org/10.3390/separations5040058
Chicago/Turabian StyleAbel, Robin J., Grzegorz Zadora, P. Mark L. Sandercock, and James J. Harynuk. 2018. "Modern Instrumental Limits of Identification of Ignitable Liquids in Forensic Fire Debris Analysis" Separations 5, no. 4: 58. https://doi.org/10.3390/separations5040058
APA StyleAbel, R. J., Zadora, G., Sandercock, P. M. L., & Harynuk, J. J. (2018). Modern Instrumental Limits of Identification of Ignitable Liquids in Forensic Fire Debris Analysis. Separations, 5(4), 58. https://doi.org/10.3390/separations5040058